

Abstract
The Rho GTPase family member RhoE regulates actin filaments partly by binding to and inhibiting ROCK I, a serine/threonine kinase that induces actomyosin contractility. Here, we show that ROCK I can phosphorylate multiple residues on RhoE in vitro. In cells, ROCK I-phosphorylated RhoE localizes in the cytosol, whereas unphosphorylated RhoE is primarily associated with membranes. Phosphorylation has no effect on RhoE binding to ROCK I, but instead increases RhoE protein stability. Using phospho-specific antibodies, we show that ROCK phosphorylates endogenous RhoE at serine 11 upon cell stimulation with platelet-derived growth factor, and that this phosphorylation requires an active protein kinase C signalling pathway. In addition, we demonstrate that phosphorylation of RhoE correlates with its activity in inducing stress fibre disruption and inhibiting Ras-induced transformation. This is the first demonstration of an endogenous Rho family member being phosphorylated in vivo and indicates that phosphorylation is an important mechanism to control the stability and function of this GTPase-deficient Rho protein.
Keywords: PDGF, phosphorylation, RhoE, Rnd3, ROCK I
Introduction
Members of the Ras superfamily of GTP-binding proteins regulate multiple aspects of cell behaviour. They cycle between an active, GTP-bound conformation, and, after GTP hydrolysis, an inactive, GDP-bound conformation. The Rho subfamily GTPases are involved in regulating cytoskeleton dynamics, thereby controlling cytoskeletal organization and cell motility responses. The Rnd proteins, Rnd1, Rnd2, and RhoE/Rnd3, form a distinct branch of Rho proteins. In addition to a Rho-like core, these proteins contain extended carboxyl-termini, and Rnd1 and RhoE also have short extensions at their amino-termini. In contrast to RhoA, which increases actin filament formation, transient expression of Rnd1 or RhoE induces loss of actin stress fibres, rounding of the cell body, and production of extensively branching processes (Guasch et al, 1998; Nobes et al, 1998). Expression of RhoE also increases cell migration speed (Guasch et al, 1998) and blocks cell cycle progression (Villalonga et al, 2004).
Rnd proteins bind only weakly to GDP and have very low if any intrinsic GTPase activity (Foster et al, 1996; Guasch et al, 1998; Nobes et al, 1998). Consequently, they exist predominantly in a GTP-bound state. Rnd protein activity is presumably regulated differently from GTP hydrolysis, for example, by altering protein expression levels. Indeed, RhoE expression has been shown to be upregulated upon platelet-derived growth factor (PDGF) (Riento et al, 2003) or hepatocyte growth factor (Tanimura et al, 2002) stimulation of Swiss 3T3 or MDCK cells, respectively, or upon Raf activation in MDCK cells (Hansen et al, 2000). In each case, increased RhoE levels coincide with cell morphological changes, such as loss of actin stress fibres. RhoE functions by binding to and inhibiting the RhoA effector ROCK I (Riento et al, 2003), a serine/threonine kinase that regulates the contractility of actin stress fibres (Riento and Ridley, 2003). In addition, RhoE binds to p190 RhoGAP and increases the GAP activity of p190 towards RhoA, thus promoting the formation of inactive GDP-bound RhoA (Wennerberg et al, 2003).
Another way of regulating protein function is by post-translational modifications. In particular, phosphorylation has been suggested to control the localization and function of Ras, Rab, Rad/Gem/Kir (RGK), and Rho family members (see, for example, Ballester et al, 1987; Kawata et al, 1989; Lang et al, 1996; Moyers et al, 1997; Ribeiro-Neto et al, 2002; Ding et al, 2003; Ellerbroek et al, 2003; Ward et al, 2004). The best evidence so far for phosphorylation of an endogenous small GTPase comes from studies on Gem, which is phosphorylated via a Cdc42/protein kinase Cζ-dependent pathway (Ward et al, 2004). This phosphorylation is required for Gem-mediated cytoskeletal reorganization and it also increases Gem protein half-life (Ward et al, 2004).
Here, we have studied how RhoE function is regulated. We demonstrate that serine 11 of RhoE is phosphorylated in vivo by ROCK I upon physiological stimulation. Our data indicate that phosphorylation is an important mechanism to control the function of this GTPase-deficient Rho protein.
Results
RhoE is phosphorylated by ROCK I in vitro and in cells
We have previously shown that RhoE binds to the serine/threonine kinase ROCK I (Riento et al, 2003). In order to investigate if RhoE is phosphorylated by the two ROCK isoforms, ROCK I and ROCK II, myc-tagged ROCKs expressed in COS7 cells and immunoprecipitated with anti-myc antibody were incubated with purified, recombinant RhoE in an in vitro kinase assay. Using 32P-labelled ATP, RhoE was shown to be phosphorylated by the full-length and constitutivelyactive (Δ1) forms of ROCK I (Figure 1A). Neither the kinase-dead form of ROCK I nor the active form of closely related citron kinase was able to phosphorylate RhoE. Surprisingly, ROCK II, which has 65% overall amino-acid identity to ROCK I, phosphorylated RhoE only weakly. Therefore, we tested if ROCK II interacted with RhoE in a GST pull-down assay. myc-ROCK II expressed in COS7 cells did not bind detectably to GST-RhoE (Figure 1B). As a positive control, the active form of RhoA, GST-V14RhoA, interacted with myc-ROCK II.
Figure 1.
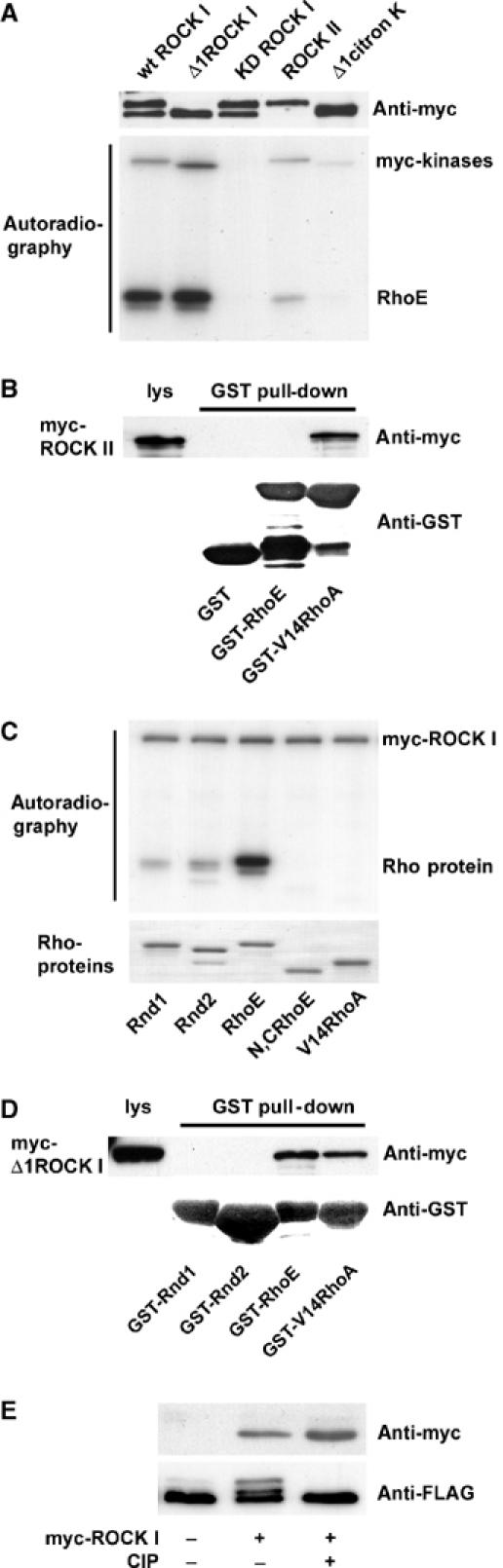
RhoE is phosphorylated by ROCK I. (A) Purified recombinant RhoE protein (2 μg) was incubated with the indicated myc-tagged kinases on beads in an in vitro kinase assay in the presence of [γ-32P]ATP. Proteins were resolved by SDS–PAGE and protein phosphorylation was detected by autoradiography. The presence of myc-tagged kinases was verified by immunoblotting. A fraction of wild-type (wt) ROCK I and kinase-dead (KD) ROCK I is C-terminally cleaved resulting in a smaller protein species (Coleman et al, 2001). (B) Lysates of COS7 cells expressing myc-ROCK II were incubated with the indicated GST-tagged proteins on beads, and the complexes were analysed by immunoblotting. (C) The constitutively active form of ROCK I, myc-Δ1ROCK I, on beads was incubated with 2 μg of each of the purified, recombinant Rho proteins in an in vitro kinase assay. Proteins were visualized by Coomassie staining, and protein phosphorylation was detected by autoradiography. N,CRhoE: His-ΔN,CRhoE (residues 16–200). (D) Lysates of COS7 cells expressing myc-Δ1ROCK I were incubated with GST-tagged proteins on beads and analysed by immunoblotting. (E) FLAG-RhoE was expressed in COS7 cells either alone or with myc-Δ1ROCK I, and immunoprecipitated with anti-FLAG antibody. For phosphatase treatment, the immunoprecipitates were incubated with calf intestinal phosphatase. FLAG-RhoE in the immunoprecipitates and the expression levels of myc-Δ1ROCK I in cell lysates were analysed by SDS–PAGE and immunoblotting.
To analyse if ROCK I phosphorylated the other Rnd subfamily members, recombinant Rnd1 and Rnd2 were incubated with myc-Δ1ROCK I in the in vitro kinase assay. Compared to RhoE, both Rnd1 and Rnd2 were considerably poorer substrates for ROCK I-mediated phosphorylation (Figure 1C). In addition, Rnd1 and Rnd2 were not able to bind myc-Δ1ROCK I in a GST pull-down assay (Figure 1D). V14RhoA and a RhoE construct that lacks both the N-terminal and the C-terminal extensions were not phosphorylated by ROCK I (Figure 1C). This indicates that ROCK I phosphorylates residues in these extensions, and these are not present in RhoA and have low homology between RhoE and Rnd1 or Rnd2.
Expression of FLAG-RhoE together with myc-Δ1ROCK I in COS7 cells resulted in a RhoE mobility shift on SDS–PAGE. To analyse if this is due to phosphorylation, immunoprecipitated FLAG-RhoE was incubated with calf intestinal phosphatase. This treatment abolished the ROCK I-induced mobility shift on RhoE, demonstrating that ROCK I phosphorylated RhoE in cells (Figure 1E).
In vitro phosphorylation sites of RhoE
To identify the ROCK I-phosphorylated residues, in vitro myc-Δ1ROCK I-phosphorylated recombinant RhoE was subjected to mass spectrometric analysis and Edman degradation. Figure 2A and B shows the mass spectra of 32P-labelled RhoE peptides. The masses corresponded to two singly phosphorylated RhoE peptides: the N-terminal peptide (including residues GSPGIP from GST) (amino acids 2–16) and the C-terminal peptide (amino acids 216–235 or 215–235; trypsin cuts this peptide at multiple sites). Two radiolabelled serines on both the peptides were mapped by Edman degradation (Figure 2C and D). These were S7 and S11 in the N-terminal peptide, and S218 and S222 in the C-terminal peptide. Mutation of these four serines to alanines reduced but did not abolish ROCK I-mediated phosphorylation of FLAG-RhoE in vitro (Figure 2E), indicating that there were further ROCK I phosphorylation sites on RhoE. All the ROCK I-phosphorylated residues are known to be located in the N- and C-terminal extensions of RhoE (Figure 1C), but of the other S/T residues in these extensions, we were not able to analyse sequences covering S210, T214, and S240, as they were located in peptides that could not be detected by mass spectrometry. We therefore used site-directed mutagenesis to mutate these sites. RhoE with alanine mutations on S210, T214, and S240, in addition to the four phosphorylation sites identified by mass spectrometry, was not phosphorylated by ROCK I in vitro (Figure 2E). RhoE containing any single S/T of the seven identified phosphorylation sites with the other six sites mutated was still phosphorylated by ROCK I (Figure 2E and data not shown), suggesting that ROCK I could phosphorylate each of the seven residues on RhoE independently of the other sites.
Figure 2.
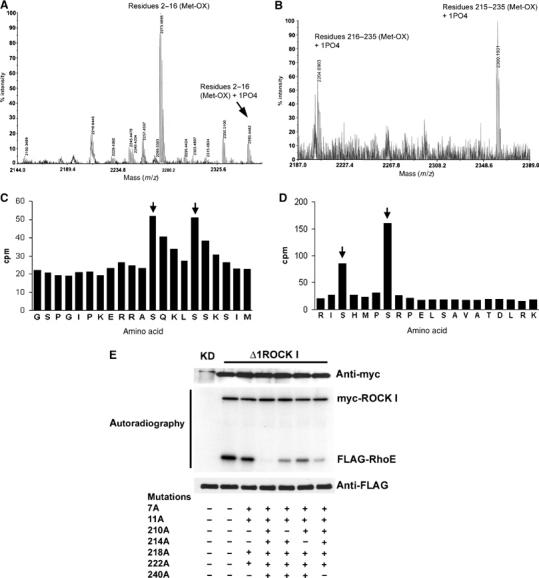
Identification of RhoE phosphorylation sites. (A, B) RhoE phosphorylated in vitro with myc-Δ1ROCK I was digested, the peptides were separated by reverse-phase chromatography, and the radioactive fractions were analysed by mass spectrometry. Masses of N-terminal (residues 2–16 plus six amino acids from GST (A)) and C-terminal (residues 216–235 or 215–235 (B)) singly phosphorylated peptides are shown. In both peptides, the methionine residue is oxidized. (C, D) Edman degradation of the radiolabelled N- and C-terminal RhoE peptides. The radioactive peaks of serines 7 and 11 (C) and serines 218 and 222 (D) are indicated by arrows. (E) Active myc-Δ1ROCK I or kinase-dead (KD) myc-ROCK I was incubated with wild-type or serine-to-alanine-mutated immunoprecipitated FLAG-RhoE in an in vitro kinase assay. Proteins were resolved by SDS–PAGE and protein phosphorylation was detected by autoradiography. The presence of myc-tagged kinases and FLAG-RhoE constructs was verified by immunoblotting.
pS7 and pS11 antibodies specifically recognize phosphorylated RhoE
S7 or S11 mutation to alanine abolished the Δ1ROCK I-induced RhoE mobility shift on SDS–PAGE (Figure 3A), suggesting that phosphorylation of S7 and S11 affects the conformation of RhoE. Mutation of any of the other five ROCK I-phosphorylated residues to alanines had no effect on FLAG-RhoE mobility (data not shown). Rabbit polyclonal antibodies were therefore raised against N-terminal peptides containing the sequences KERRA pS QKL (pS7) and QKL pS SKSI (pS11), corresponding to residues 2–10 and 8–15 of RhoE, respectively. Antibodies recognizing nonphosphorylated peptides were removed and the phospho-specific antibodies were affinity purified using peptide columns. To elucidate the phospho-specificity of these antibodies, FLAG-RhoE was expressed either alone or with myc-Δ1ROCK I in COS7 cells and immunoprecipitated with anti-FLAG antibody. S11 phosphorylation increased massively on ROCK I coexpression (Figure 3B). In contrast, S7 phosphorylation changed less upon ROCK I expression, and inhibition of endogenous ROCK activity with Y-27632 reduced but did not prevent pS7 antibody from detecting RhoE. This implies that S7 of RhoE is phosphorylated by another kinase in addition to ROCK I in cells, or that Y-27632 does not completely inhibit ROCK activity. Neither of the antibodies detected protein phosphatase 1-treated RhoE, or serine 7/11 to alanine-mutated RhoE, demonstrating phospho-specificity and site specificity of the antibodies (Figure 3B).
Figure 3.
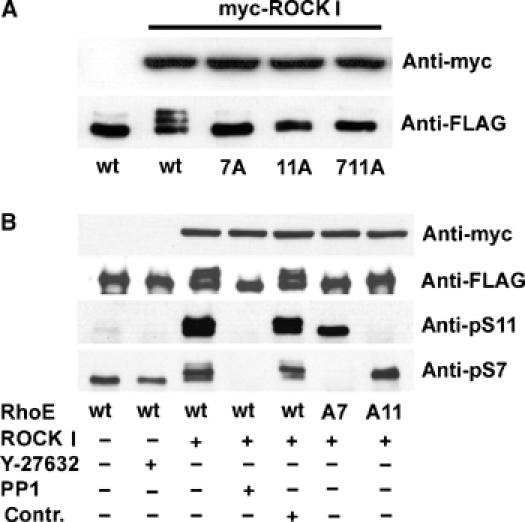
Specificity of anti-pS7 and anti-pS11 antibodies. Wild-type or serine 7/11 to alanine-mutated FLAG-RhoE was expressed in COS7 cells either with or without myc-Δ1ROCK I. (A) The lysates were resolved by SDS–PAGE and immunoblotted with anti-FLAG and anti-myc antibodies. (B) FLAG-RhoE was immunoprecipitated with anti-FLAG antibody and detected by anti-pS7 or anti-pS11 antibodies. The protein expression was verified using anti-myc or anti-FLAG antibodies (cell lysates for ROCK and immunoprecipitates for RhoE). For ROCK inhibition, the cells were incubated with 10 μM Y-27632 for 12 h. For phosphatase treatment, the immunoprecipitates were incubated with protein phosphatase 1 (PP1) for 60 min, or under the same conditions without PP1 (Contr.).
RhoE phosphorylation has no effect on ROCK I binding but alters RhoE localization
Next, we studied the effect of RhoE phosphorylation on ROCK I binding. GST-RhoE binds to both the active and kinase-dead forms of myc-ROCK I (amino acids 1–727) (Figure 4A). Incubation of RhoE/ROCK I complexes under conditions in which ROCK I phosphorylates at least 60% of RhoE (data not shown) did not dissociate the complex (Figure 4A). Moreover, a RhoE construct in which all the seven ROCK I phosphorylation sites were mutated to alanines (allA-RhoE) was still able to bind ROCK I. Taken together, these data suggest that RhoE/ROCK I complex association is not regulated by protein phosphorylation. Similarly, ROCK I-mediated phosphorylation of RhoE did not alter its binding to p190-B RhoGAP (data not shown).
Figure 4.
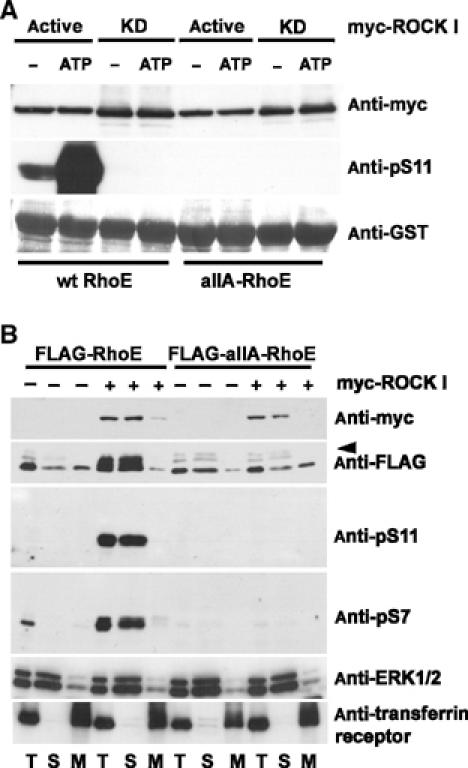
Phosphorylated RhoE localizes in the cytosol but phosphorylation is not required for the RhoE/ROCK I complex formation. (A) Wild-type (wt) GST-RhoE or a GST-RhoE construct in which all the seven ROCK I-phosphorylated residues were mutated to alanines (allA) were used for a pull-down assay of lysates of COS7 cells expressing active or kinase-dead (KD) myc-ROCK I (residues 1–727). After washes, the complexes were analysed directly (−) or after incubation under kinase assay conditions (ATP). The levels of GST-RhoE proteins and RhoE phosphorylation were elucidated by immunoblotting with anti-GST and anti-pS11 antibodies, respectively. The ROCK proteins associated with GST-RhoE constructs were detected by anti-myc antibody. (B) FLAG-tagged wild-type (wt) RhoE or allA-RhoE was expressed in COS7 cells together with myc-Δ1ROCK I. The cell membranes (M) were separated by membrane flotation, and analysed by immunoblotting with the phospho-specific RhoE antibodies. ERK1/2 and transferrin receptor are indicators of soluble and membrane fractions, respectively. Two nonspecific bands were often detected with anti-FLAG antibody (arrowhead). S: soluble fraction; T: postnuclear fraction.
RhoE has been shown to localize both in the cytosol and on intracellular membranes, such as the trans-Golgi network (Riento et al, 2003). To determine if phosphorylation by ROCK I alters RhoE localization, wild-type and allA FLAG-RhoE were expressed in COS7 cells together with myc-Δ1ROCK I. Cellular membranes were isolated by membrane flotation, and the cytosolic and membrane fractions were analysed by immunoblotting. Anti-pS7 and anti-pS11 antibodies detected the phosphorylated RhoE only in the cytosolic fraction, whereas the nonphosphorylated allA-RhoE was present both in the membrane and cytosolic fractions (Figure 4B). Subcellular fractionation was validated by detecting ERK1/2 and transferrin receptor in the cytosolic and membrane fractions, respectively.
ROCK I-mediated phosphorylation increases RhoE protein stability
We frequently observed that expression of ROCK I increased the amount of wild-type RhoE protein, but not of allA-RhoE, in cells (Figure 4B). We therefore investigated if phosphorylation of RhoE by ROCK I has an effect on RhoE protein stability. FLAG-RhoE was expressed in COS7 cells either alone or with myc-Δ1ROCK I, and cells were metabolically pulse-labelled with 35S-Met, Cys and then chased for 12 h (Figure 5A and B) or 24 h (Figure 5B) before immunoprecipitation with anti-FLAG antibody. Quantitation of RhoE protein levels revealed that expression of ROCK I increased the half-life of RhoE compared to RhoE expressed alone. The degree of allA-RhoE degradation was not affected by ROCK I expression, and it was similar to that of wild-type RhoE without ROCK I (Figure 5A). Incubation of FLAG-RhoE-expressing cells with a proteosome inhibitor MG132 prevented RhoE degradation, suggesting that an active proteosome machinery is required for RhoE degradation (Figure 5A). Similarly, proteosome inhibitors (MG132, aLLnL) increased the level of endogenous RhoE protein (data not shown).
Figure 5.
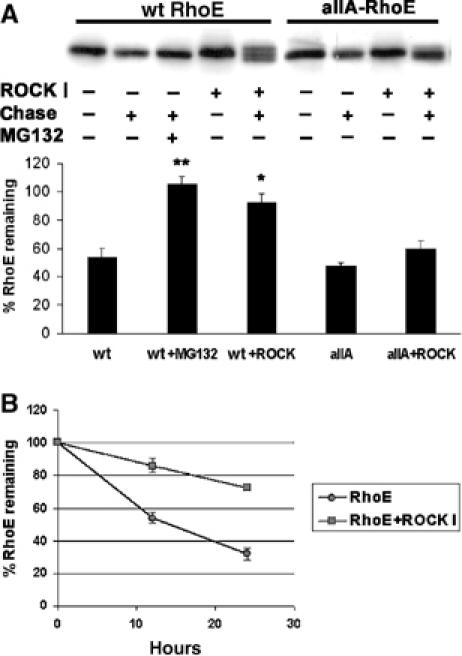
Phosphorylation increases RhoE protein stability. FLAG-tagged wild-type (wt) RhoE or allA-RhoE was expressed in COS7 cells with or without myc-Δ1ROCK I. Cellular proteins were metabolically labelled with 35S-Met, Cys, chased for 12 h (A, B) or 24 h (B), and FLAG-RhoE proteins were immunoprecipitated before and after the chase with anti-FLAG antibody. The immunoprecipitated proteins were resolved by SDS–PAGE and analysed by autoradiography. Bars represent percentage (±s.e.m., n=3) of FLAG-RhoE radioactivity remaining after the chase. The difference of wild-type (wt) RhoE expressed with ROCK I to that of RhoE expressed alone and the difference of RhoE in MG132-treated cells to that of nontreated cells are statistically significant (Student's t-test; *P<0.05, **P<0.01).
Endogenous RhoE is phosphorylated on S11 upon PDGF stimulation
To determine if endogenous RhoE is phosphorylated in cells, anti-pS7 and anti-pS11 antibodies were used for immunoblotting of Swiss 3T3 cell lysates. Neither of the phospho-specific antibodies detected RhoE in growing or starved cells. However, stimulation of starved Swiss 3T3 cells with PDGF induced RhoE phosphorylation on serine 11, reaching maximal levels at 30 min (Figure 6A). Approximately 20% of endogenous RhoE was phosphorylated upon PDGF stimulation of Swiss 3T3 cells. Similar results were obtained for NIH 3T3 cells (data not shown). Of the RhoE doublet detected by the mouse anti-RhoE antibody, the anti-pS11 antibody recognized only the higher molecular weight species, possibly because phosphorylation alters RhoE mobility on SDS–PAGE (Figure 3A). Phosphorylation of serine 11 of RhoE was reduced by adding either of two ROCK inhibitors, Y-27632 or H-1152 (Ikenoya et al, 2002), to Swiss 3T3 cells before stimulating with PDGF (Figure 6A and B). Inhibition of RhoE phosphorylation in three independent experiments was approximately 40% with Y-27632 and 65% with H-1152 (data from one representative experiment are shown in Figure 6A and B). Interestingly, S11 RhoE was not detectably phosphorylated in Swiss 3T3 cells stimulated with lysophosphatidic acid (LPA) (Figure 6C), although LPA stimulated stress fibre formation (data not shown). This suggests that an active RhoA pathway is not sufficient for ROCK-mediated RhoE phosphorylation. Neither PDGF nor LPA induced detectable levels of S7 RhoE phosphorylation (data not shown).
Figure 6.
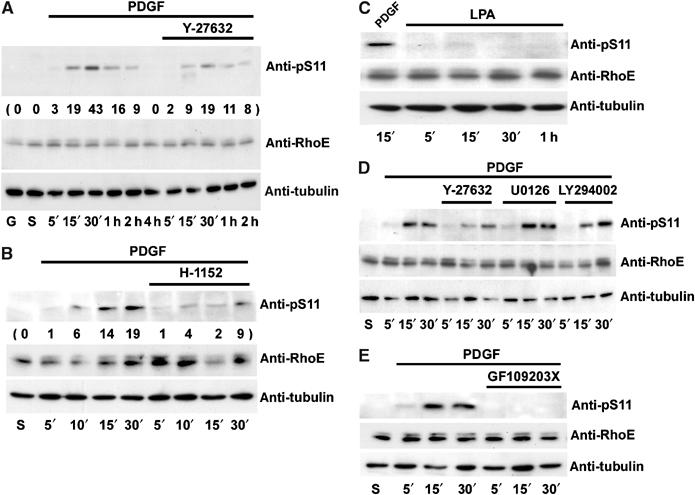
PDGF induces S11 phosphorylation on endogenous RhoE. Starved Swiss 3T3 cells were treated with PDGF (A–E) or LPA (C) for the indicated times, and after cell lysis, equal amounts of RhoE in cell lysates were analysed with anti-pS11 antibody. (A, D) Y-27632 or (B) H-1152 was added to the cells before PDGF stimulation to inhibit ROCK activity. Quantitation of S11 phosphorylation is shown in parentheses. (D) For MAPK inhibition, U0126, for PI3-kinase inhibition, LY294002, and (E) for PKC inhibition, GX109203X were added to the cells before cell stimulation. G: growing cells; S: starved cells.
To analyse which of the PDGF-stimulated signalling pathways were required for ROCK I activation and RhoE phosphorylation, Swiss 3T3 cells were incubated with various kinase inhibitors before PDGF treatment. The protein kinase C (PKC) inhibitor GF109203X (Figure 6E), but not the mitogen-activated protein (MAP) kinase inhibitor U0126 or the phosphatidylinositol 3- (PI3-) kinase inhibitor LY294002 (Figure 6D), abrogated PDGF-induced S11 RhoE phosphorylation.
PMA induces S11 RhoE phosphorylation that coincides with increased RhoE protein levels
To verify that PKC activation results in RhoE phosphorylation, starved Swiss 3T3 cells were stimulated with phorbol myristate acetate (PMA). PMA-induced PKC activation led to a substantial increase in S11 RhoE phosphorylation (Figure 7A), which peaked at around 30 min and was abrogated by preincubation of the cells with the PKC inhibitor GF109203X (Figure 7B), and reduced by the ROCK inhibitor Y-27632 or H-1152 (Figure 7C). In addition, PMA treatment resulted in increased RhoE protein levels (Figure 7A), leading to approximately 55% more RhoE protein after 2 h of cell stimulation with PMA (three independent experiments, data not shown). Stimulation of Swiss 3T3 cells with bombesin, which has been shown to activate PKC (Rey et al, 2001), also resulted in S11 RhoE phosphorylation (data not shown). As PKC targets are phosphorylated within minutes of cell stimulation with PDGF or PMA (see, for example, Rosen et al, 1990; Gilligan et al, 2002), the relatively slow increase in RhoE phosphorylation induced by PMA and PDGF implies that in vivo RhoE is not a direct target for PKC. Indeed, the PKC target MARCKS (Rosen et al, 1990) was maximally phosphorylated by 5 min of cell stimulation with PDGF or PMA, at a time point where RhoE was not yet significantly phosphorylated (Figure 7A). In addition, the ROCK inhibitors had no effect on PDGF- or PMA-induced MARCKS phosphorylation (data not shown), further demonstrating that the effect of these inhibitors on S11 RhoE phosphorylation was not due to PKC but ROCK inhibition.
Figure 7.
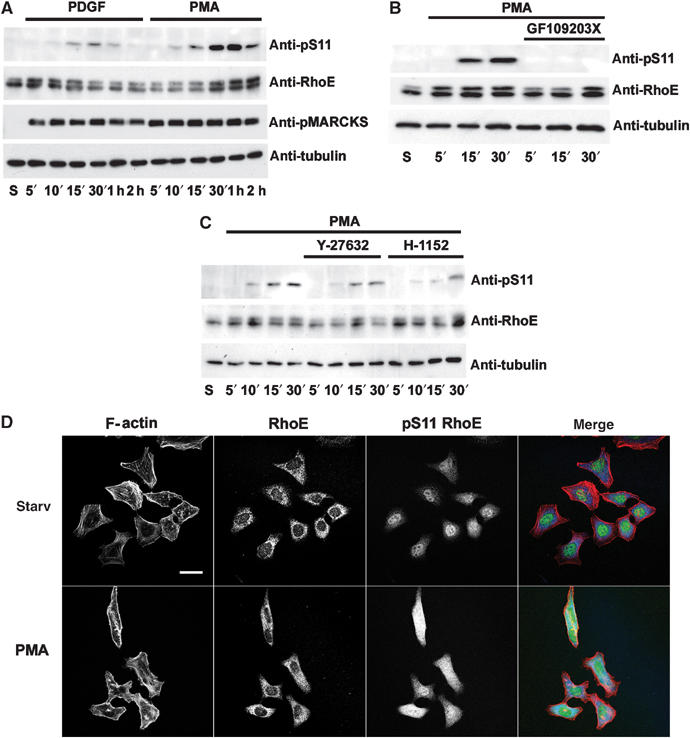
PMA stimulation induces RhoE phosphorylation. (A) Starved Swiss 3T3 cells were treated with PDGF or PMA (20 ng/ml) for the indicated times. Equal amounts of total cell proteins were analysed by anti-pS11 and anti-RhoE antibodies. Activation of PKC was determined by detecting phosphorylation of the PKC target MARCKS. (B) GX109203X, a PKC inhibitor, or (C) Y-27632 or H-1152, ROCK inhibitors, were added to the cells before cell stimulation with 5 ng/ml PMA. (D) Starved HeLa cells were treated with 20 ng/ml PMA for 30 min and stained with phalloidin (red) to visualize filamentous (F-) actin, and with mouse anti-RhoE (blue) and rabbit anti-pS11 (green) antibodies. The bar represents 30 μm.
To analyse the localization of phosphorylated RhoE in PMA-treated cells, we used HeLa cells, which express higher levels of RhoE than Swiss 3T3 fibroblasts (data not shown). PMA stimulation of HeLa cells resulted in increased phosphorylation of S11 of RhoE, as detected with anti-pS11 antibody by immunoblotting (data not shown) and immunofluorescence (Figure 7D). RhoE had a predominantly perinuclear localization prior to stimulation, as previously described (Riento et al, 2003). In PMA-treated cells, the anti-pS11 antibody showed a more diffuse cytoplasmic staining, consistent with phosphorylated RhoE localizing primarily in the cytosol (Figure 4B). The nuclear staining with the anti-pS11 antibody is nonspecific and was observed with the preimmune serum.
Phosphorylation of RhoE correlates with its activity in stress fibre disruption and inhibition of Ras-induced transformation
To test the effect of phosphorylation on RhoE function, we used an NIH 3T3 cell line that expresses HA-RhoE in an inducible manner (Villalonga et al, 2004). Removal of tetracycline induced RhoE protein expression within 3 h (Figure 8A), which resulted in a loss of stress fibres (Figure 8B). However, by 18 h of RhoE expression, the cells reverted to a normal morphology and reassembled stress fibres (Figure 8B; Villalonga et al, 2004). Interestingly, S11 of RhoE was phosphorylated only at time points where stress fibres were disassembled, and dephosphorylation of RhoE coincided with the reappearance of stress fibres (Figure 8A and B). This strongly suggests that nonphosphorylated RhoE is not able to disrupt actin filaments, and that phosphorylation regulates RhoE activity.
Figure 8.
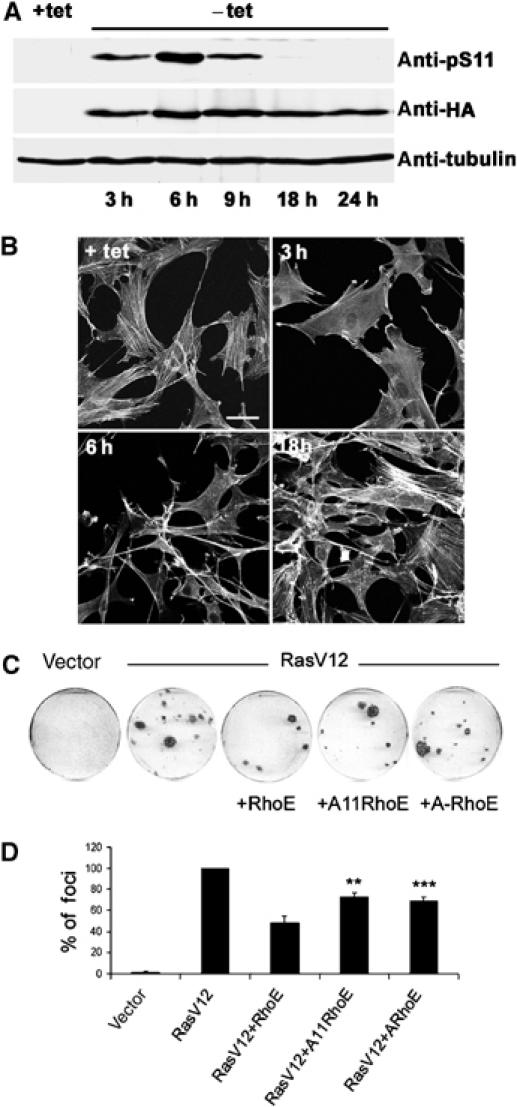
Phosphorylation of RhoE correlates with its activity. (A, B) RhoE-3T3 cells were grown in the presence (+tet) or absence (−tet) of tetracycline. HA-RhoE expression and S11 RhoE phosphorylation were analysed by immunoblotting using anti-HA and anti-pS11 antibodies, respectively (A). F-actin was detected by phalloidin staining (B). The bar represents 10 μm. (C, D) NIH 3T3 cells were transfected with the indicated expression plasmids and maintained in 5% serum for 12–15 days, replacing the medium every 2 days. After 12–15 days, cells were stained with crystal violet and the number of foci counted. (C) Plates from a single representative experiment, conducted in duplicate. (D) The mean values from three independent experiments as in (C), each conducted in duplicate, are shown in the graph, representing the percentage (±s.e.m.) of transformed foci relative to RasV12 transfectants. The differences in inhibition of Ras-induced transformation by the alanine-mutated RhoE forms (ARhoE: allA-RhoE) to that of wild-type RhoE are statistically significant (Student's t-test; **P<0.01, ***P<0.001).
RhoE has also been shown to block Ras-induced cell transformation (Villalonga et al, 2004). To determine if phosphorylation regulates RhoE function in inhibition of transformation, we transfected NIH 3T3 cells with H-RasV12 together with Ala11 RhoE or allA-RhoE. Focus formation assays revealed that nonphosphorylated RhoE forms were not able to inhibit Ras-induced transformation as well as wild-type RhoE (Figure 8C and D), indicating that phosphorylation of RhoE is required for its optimal activity in this assay.
Discussion
Unlike other Rho family members, RhoE does not act as a classic GTPase switch, as it does not detectably hydrolyse GTP. Here, we have investigated how RhoE function is regulated. We show that the serine/threonine kinase ROCK I can phosphorylate RhoE in vitro and in cells, and that this phosphorylation increases RhoE stability and correlates with its activity in inducing stress fibre disruption and inhibiting Ras-induced cell transformation.
Both ROCK I and ROCK II bind to RhoA (Leung et al, 1995; Ishizaki et al, 1996; Matsui et al, 1996). Our results show that only ROCK I binds to RhoE, and that RhoE is a substrate for ROCK I and not ROCK II, providing the first evidence that these two highly homologous kinases have different targets. As ROCKs phosphorylate several proteins in vitro, it is possible that the kinase–substrate interaction determines their substrate specificity in vivo. ROCK I and ROCK II kinase domains are 92% identical, but the N-terminal regions upstream of the kinase domains show little homology. Since this region in ROCK I is important for RhoE binding (Riento et al, 2003), it is likely that the lack of interaction with ROCK II reflects these sequence differences at the N-terminus. In addition, Rnd1 and Rnd2 do not bind to ROCK I, and thus it will be interesting to determine which regions of RhoE confer specificity of binding.
Other Ras superfamily proteins have been reported to be phosphorylated, but most studies have been with overexpressed GTPases, and the results therefore remain controversial. For example, using phospho-specific antibodies, Ellerbroek et al showed that overexpressed RhoA was phosphorylated in cells on S188 (which corresponds to S240 on RhoE), but they, and others, were not able to detect this phosphorylation on the endogenous protein (Essler et al, 1999; Sauzeau et al, 2000; Sawada et al, 2001; Ellerbroek et al, 2003). Similarly, S7 of overexpressed RhoE is phosphorylated, but so far we have not detected its phosphorylation on endogenous protein. Using phospho-specific antibodies, endogenous RGK family member Gem, however, has been shown to be phosphorylated in cells. Gem phosphorylation altered its localization and half-life but, surprisingly, occurred only after 2 h of cell stimulation with PMA (Ward et al, 2004). The signalling pathways leading to Gem phosphorylation are therefore not clear.
PDGF is a major mitogen for fibroblasts and certain other cell types. In vivo, it has important roles during the embryonic development and wound healing (reviewed in Hoch and Soriano, 2003). PDGF binding to its receptor leads to stimulation of cell growth, but also to changes in cell shape and motility by inducing reorganization of the actin cytoskeleton (Ferns et al, 1990). Activation of PDGF receptor stimulates several signalling pathways (reviewed in Heldin and Westermark, 1999), of which PKC, but not PI3-kinase or the Ras pathway, is involved in S11 RhoE phosphorylation. PDGF-induced phosphorylation on serine 11 of RhoE is mediated at least partly by ROCK I, as the ROCK inhibitor Y-27632 or H-1152 reduces the PDGF-induced S11 RhoE phosphorylation. Inhibition of PKC abrogates RhoE phosphorylation, whereas inhibition of ROCKs only reduces RhoE phosphorylation. These data suggest that PKC is upstream of ROCK I, and that, in addition to ROCK I, there may be another kinase that can phosphorylate RhoE. RhoE phosphorylation upon PDGF or PMA treatment of Swiss 3T3 cells reaches its maximum at 30 min, indicating that the kinases involved are not direct targets of PDGF receptor or PKC.
Interestingly, LPA, a known activator of RhoA (Ridley and Hall, 1992), which in turn is thought to stimulate ROCK activity (Ishizaki et al, 1996; Matsui et al, 1996), has no effect on S11 RhoE phosphorylation. This implies that RhoA activation is not sufficient for stimulation of ROCK I kinase activity towards RhoE. This is supported by a report showing that, in addition to interaction with an active RhoA, PKC is required for full activation of ROCKs (Barandier et al, 2003). We have observed that inhibition of PKC has no effect on LPA-induced stress fibre formation (data not shown), suggesting that PKC is not involved in RhoA-mediated, LPA-induced actin filament formation. It is also possible that in LPA-stimulated cells, RhoA-activated proteins other than ROCK I mediate induction of stress fibres and that ROCK I is not sufficiently activated to induce RhoE phosphorylation, as LPA treatment induces formation of actin filaments and focal contacts in the presence of ROCK inhibitors (Riveline et al, 2001; Kole et al, 2004).
Although RhoE localizes both to membrane and cytosolic fractions (Riento et al, 2003), ROCK I-phosphorylated RhoE localizes only in the cytosol. In addition, a higher proportion of RhoE (but not allA-RhoE) was cytosolic upon ROCK I coexpression. This suggests that the membrane association of RhoE is regulated by phosphorylation of its N- and/or C-terminal extensions, in addition to prenylation (Foster et al, 1996). Other small GTP-binding proteins have also been reported to be dissociated from membranes upon phosphorylation (Lang et al, 1996; Ding et al, 2003; Ward et al, 2004). Overexpressed RhoA, for example, can be phosphorylated on a C-terminal serine (S188) adjacent to its prenylation site (Lang et al, 1996). Interestingly, one of the RhoE phosphorylation sites is in a similar position (S240), and future research will be aimed at determining how the different phosphorylation sites on RhoE affect its localization.
We have previously shown that PDGF treatment of starved Swiss 3T3 cells alters RhoE protein levels (Riento et al, 2003). During the first hour of PDGF stimulation, RhoE levels are increased and this coincides with RhoE-like cell morphological changes such as cell rounding and loss of stress fibres. The effect of PDGF on RhoE levels is only moderate, and it is more clearly seen upon PMA stimulation, probably because PMA results in higher levels of RhoE phosphorylation. RhoE phosphorylation upon cell stimulation with PDGF or PMA provides an explanation for the increased RhoE protein levels, as ROCK I-mediated phosphorylation increases RhoE protein half-life. It is likely that the increased stability also results in increased RhoE activity on actin filament disruption. We cannot, however, rule out that other proteins in addition to RhoE are required for PDGF- and PMA-induced effects on actin filaments. LPA does not induce phosphorylation or increased levels of RhoE, which may be important to allow the cells to form stress fibres. The increased RhoE protein stability is not due to binding to ROCK I, as allA-RhoE, which cannot be phosphorylated by but still binds to ROCK I, is degraded at a similar rate in the absence or presence of ROCK I. This insensitivity of allA-RhoE to ROCK I-induced stabilization may account for the reduced ability of allA-RhoE to inhibit Ras-induced transformation.
Here, we provide the first demonstration of growth factor-induced phosphorylation of a Rho family member. ROCK I-mediated phosphorylation and subsequent stabilization of RhoE may provide a negative feedback loop to limit ROCK I activity, since RhoE binds to ROCK I and inhibits its phosphorylation of other targets (Riento et al, 2003). We propose that phosphorylation is an important mechanism to control the function of RhoE on actin filament disruption.
Materials and methods
Antibodies
Rabbit polyclonal antisera were raised against phosphorylated S7 peptide (KERRA pS QKL) and S11 peptide (QKL pS SKSI) by Eurogentec, Seraing, Belgium. Nonspecific antibodies were removed using a non-phospho-peptide (KERRASQKLSSKSI) column, and the phospho-specific antibodies were affinity purified with the corresponding phospho-peptides. Polyclonal and monoclonal RhoE antibodies have been previously described (Riento et al, 2003). Rabbit polyclonal anti-myc (A-14) and ERK1/2 (K-23) antibodies, and mouse monoclonal anti-GST (B-14) antibody were from Santa Cruz. Mouse monoclonal anti-FLAG (M2) and anti-β-tubulin antibodies were purchased from Sigma, anti-human transferrin receptor antibody was from Zymed, anti-HA antibody was from Covance, and rabbit anti-phospho-MARCKS antibody (S152/156) was from Cell Signaling Technology.
Expression vectors and site-directed mutagenesis
Full-length myc-ROCK I, myc-ROCK II, and myc-ROCK KD-IA, and C-terminally truncated constructs myc-Δ1ROCK I, myc-Δ3ROCK I (amino acids 1–727), and myc-Δ1citron kinase were kindly provided by Shuh Narumiya (Kyoto University, Kyoto, Japan). The coding region for residues 1–727 of ROCK I KD (kinase dead) was cloned into KpnI and XhoI sites of pCAG expression vector with N-terminal myc tag. pcDNA3-H-RasV12 was a gift of Julian Downward. The expression vector encoding FLAG-RhoE (described in Riento et al, 2003) was mutagenized using the QuickChange system (Stratagene). The nucleotide changes were verified by DNA sequencing (MWG-Biotech, Milton Keynes, UK).
COS7 cell electroporation
COS7 cells grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% bovine fetal calf serum and penicillin/streptomycin (Invitrogen) were transfected by electroporation in 250 μl of cold electroporation buffer (120 mM KCl, 10 mM K2PO4/KH2PO4, pH 7.6, 25 mM Hepes, pH 7.6, 2 mM MgCl2, and 0.5% Ficoll). A 5 μg portion of DNA was electroporated into cells at 250 V and 960 μF using 0.4 cm Gene Pulser Cuvettes (Bio-Rad). The cells were then grown in normal medium for 24 h.
GST pull-down
GST-Rnd1 and GST-Rnd2 were generated by cloning human Rnd1 and Rnd2 (kindly provided by Catherine Nobes; Nobes et al, 1998) coding regions into BamHI and EcoRI sites of pGEX-4T (Amersham Biosciences). Recombinant Rnd1, Rnd2, RhoE, and V14RhoA were expressed as GST fusion proteins and purified as previously described (Guasch et al, 1998). Transfected COS7 cells were lysed in LB buffer (1% Triton X-100, 20 mM Tris–HCl, pH 8, 130 mM NaCl, 10 mM NaF, 1% aprotinin, 10 μg/ml leupeptin, 1 mM dithiothreitol (DTT), 0.1 mM Na3VO4, and 1 mM PMSF). After removal of insoluble material, the cell lysates were incubated at 4°C for 2 h with the recombinant GST-tagged proteins on glutathione beads (Amersham Biosciences). Beads were washed with LB buffer before eluting the proteins in Laemmli sample buffer. The proteins were resolved by SDS–PAGE and analysed by immunoblotting using Immobilon-P (Millipore) transfer membranes.
Protein purification and in vitro kinase assay
The small GTPases were digested from GST with thrombin according to the manufacturer's protocol (Amersham Biosciences). Purification of His-ΔN,CRhoE (residues 16–200) has been previously described (Garavini et al, 2002). myc-tagged kinases were expressed in COS7 cells, the cells lysed in LB buffer, and the proteins were immunoprecipitated with anti-myc antibody beads (Santa Cruz). The beads were washed with high salt (0.5 M NaCl) to remove co-precipitated proteins. Purified, recombinant Rho proteins were incubated with myc-tagged kinases in kinase buffer (50 mM Tris–HCl, pH 7.5, 10 mM MgCl2, 1 mM DTT, 30 μM adenosine 5′-triphosphate (ATP), and 0.1 μCi/μl [γ-32P]ATP (Amersham Biosciences)) at 30°C for 30 min. The proteins were eluted with Laemmli sample buffer, resolved by SDS–PAGE, and protein phosphorylation was analysed by autoradiography of dried gels.
Mass spectrometry and Edman degradation
In vitro-phosphorylated recombinant RhoE was resolved by SDS–PAGE, Coomassie-stained, and subjected to in-gel proteolysis at 37°C for 4 h with 100 ng of either trypsin (Promega) or endoproteinase Asp-N (Sigma). The peptides were separated by reverse-phase chromatography (Vydac C18 0.8 × 150 mm). Radioactive fractions were analysed by MALDI TOF-TOF (ABI 4700). Peptide masses were matched against an ‘in silico' digest of RhoE (MS Digest; Protein Prospector, UCSF). Solid-phase Edman sequencing (ABI Procise) was performed using sequelon acrylamine membranes.
Immunoprecipitation and phosphatase treatments
COS7 cells expressing FLAG-RhoE were lysed in LB buffer, and after clearing the insoluble material, the lysates were incubated with mouse anti-FLAG antibody covalently linked to agarose (M2, Sigma). The immunoprecipitates were washed with LB buffer containing 0.5 M NaCl, and the proteins were eluted with Laemmli sample buffer. ROCK activity was inhibited in FLAG-RhoE-overexpressing COS7 cells with 10 μM Y-27632 (Merck Biosciences) 12 h prior to immunoprecipitation with anti-FLAG antibody. For phosphatase treatments, FLAG-RhoE bound to beads was washed with phosphatase buffer (100 mM NaCl, Tris–HCl, pH 7.9, 10 mM MgCl2, and 1 mM DTT) and incubated with 20 U calf intestinal phosphatase (New England Biolabs) at 37°C for 60 min. Alternatively, the beads were washed with 50 mM Tris–HCl, pH 7.0, 5 mM DTT, 200 μM MnCl2, 100 μM EDTA, and 200 μg/ml BSA and incubated with 2 U Protein phosphatase 1 α-isoform (Merck Biosciences) at 30°C for 60 min.
Complex binding assay
GST-RhoE and GST-allA-RhoE fusion proteins attached to glutathione-Sepharose were incubated with COS7 cell lysates expressing myc-ROCK I (residues 1–727) or myc-ROCK I KD (kinase dead, residues 1–727) as described above for GST pull-downs. After washes, the protein complexes were incubated in kinase buffer at 30°C for 30 min. The complexes were washed with LB buffer and bound proteins eluted in Laemmli sample buffer.
Membrane flotation
COS7 cells expressing FLAG-RhoE and myc-Δ1ROCK I for 12 h were scraped into 20 mM Tris–HCl, pH 7.5, 10 mM KCl, 250 mM sucrose, 10 mM NaF, 1% aprotinin, 10 μg/ml leupeptin, 1 mM DTT, 0.1 mM Na3VO4, and 1 mM PMSF. The cells were broken by repeated passages through a 21-G needle, and after removal of intact cells and nuclei (500 g, 10 min), sucrose was added to the postnuclear supernatant to 2 M. This was overlaid with 1.7 and 0.8 M sucrose in 20 mM Tris–HCl (pH 7.5) and centrifuged for 20 h at 130 000 g, 10°C. The 1.7/0.8 M interface membrane peak was collected, diluted two-fold with 20 mM Tris–HCl, and the membranes were pelleted at 150 000 g for 30 min. Proteins from the soluble 2 M fraction were, after two-fold dilution with 20 mM Tris–HCl, precipitated with 10% trichloroacetic acid on ice for 60 min.
RhoE stability assay
Electroporated COS7 cells expressing FLAG-RhoE and myc-Δ1ROCK I for 4 h were incubated with medium lacking Met and Cys for 30 min. The proteins were metabolically labelled with 15 μCi/ml 35S-Met, Cys for 2 h. After PBS washes, the cells were either lysed in LB buffer or incubated with complete medium for 12 or 24 h before cell lysis. For proteosome inhibition, 10 μM MG132 was added for the incubation. FLAG-RhoE was immunoprecipitated as described above and analysed by SDS–PAGE, autoradiography, and Bio-Rad Molecular Imager FX quantitation.
Swiss 3T3 and RhoE-3T3 cell treatments
Swiss 3T3 cells were grown in DMEM containing 10% bovine fetal calf serum and penicillin/streptomycin (Invitrogen). Cells starved without serum for 24 h were treated with 25 ng/ml recombinant human PDGF-BB (PeproTech EC, UK), 1 μg/ml LPA (Sigma), or 20 or 5 ng/ml PMA (Sigma). Kinase inhibitors (10 μM Y-27632, 10 μM H-1152, 25 μM U0126, 25 μM LY294002, or 1 μM GF109203X; all from Merck Biosciences) were added 30 min before cell stimulation. Cells were lysed in LB buffer, and the insoluble material was removed by centrifugation before adding Laemmli sample buffer. S2–6 cells, which are NIH 3T3 derivatives containing a tetracycline-inducible tTa transactivator, contained an expression vector encoding HA-RhoE (Villalonga et al, 2004). The RhoE 3T3 cell line was grown, protein expression was induced, and cell lysates were produced as previously described (Villalonga et al, 2004).
Immunofluorescence microscopy
RhoE 3T3 cells on coverslips were incubated without tetracycline for various times. HeLa cells were starved for 24 h without serum and stimulated with 20 ng/ml PMA for 30 min. The cells were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.5% Triton X-100 for 5 min, and incubated with mouse anti-RhoE and rabbit anti-pS11 antibodies. Actin filaments were localized by incubating the cells with 0.05 μg/ml of tetramethyl rhodamine isothiocyanate-conjugated phalloidin (Sigma) for 30 min. For secondary antibodies, fluorescein isothiocyanate-conjugated goat anti-rabbit and indodicarbocyanine-conjugated goat anti-mouse antibodies (Jackson ImmunoResearch) were used for 30 min. The specimens were mounted in Mowiol (Merck Biosciences) containing p-phenylenediamine (Sigma) and images were generated with a Zeiss LSM 510 confocal microscope.
Focus formation assays
NIH 3T3 cells were seeded at a cell density of 2 × 105 cells per well in six-well plates the day before transfection. Cells were transfected with the indicated vectors in six-well plates in duplicate. After 24 h, cells were transferred to 10-cm plates and the medium was substituted with 5% FCS-containing DMEM when cells reached confluency (typically, 2–3 days later). After 12–15 days, cells were stained with 0.5% (w/v) crystal violet in 70% ethanol and the number of foci larger than 1 mm in diameter counted.
Acknowledgments
We are grateful to Dr Catherine Nobes for providing pRK5-Rnd1 and pRK5-myc-Rnd2 constructs, to Dr Julian Downward for the pcDNA3-H-RasV12 construct, and to Dr Shuh Narumiya for ROCK and citron constructs. This work was supported by an Academy of Finland grant (KR), a European Commission Marie Curie fellowship (KR), a EMBO postdoctoral fellowship (PV), a BBSRC project grant (AJR), an AICR project grant (AJR), and the Ludwig Institute for Cancer Research.
References
- Ballester R, Furth ME, Rosen OM (1987) Phorbol ester- and protein kinase C-mediated phosphorylation of the cellular Kirsten ras gene product. J Biol Chem 262: 2688–2695 [PubMed] [Google Scholar]
- Barandier C, Ming XF, Rusconi S, Yang Z (2003) PKC is required for activation of ROCK by RhoA in human endothelial cells. Biochem Biophys Res Commun 304: 714–719 [DOI] [PubMed] [Google Scholar]
- Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF (2001) Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol 3: 339–345 [DOI] [PubMed] [Google Scholar]
- Ding J, Soule G, Overmeyer JH, Maltese WA (2003) Tyrosine phosphorylation of the Rab24 GTPase in cultured mammalian cells. Biochem Biophys Res Commun 312: 670–675 [DOI] [PubMed] [Google Scholar]
- Ellerbroek SM, Wennerberg K, Burridge K (2003) Serine phosphorylation negatively regulates RhoA in vivo. J Biol Chem 278: 19023–19031 [DOI] [PubMed] [Google Scholar]
- Essler M, Retzer M, Bauer M, Heemskerk JW, Aepfelbacher M, Siess W (1999) Mildly oxidized low density lipoprotein induces contraction of human endothelial cells through activation of Rho/Rho kinase and inhibition of myosin light chain phosphatase. J Biol Chem 274: 30361–30364 [DOI] [PubMed] [Google Scholar]
- Ferns GA, Sprugel KH, Seifert RA, Bowen-Pope DF, Kelly JD, Murray M, Raines EW, Ross R (1990) Relative platelet-derived growth factor receptor subunit expression determines cell migration to different dimeric forms of PDGF. Growth Factors 3: 315–324 [DOI] [PubMed] [Google Scholar]
- Foster R, Hu KQ, Lu Y, Nolan KM, Thissen J, Settleman J (1996) Identification of a novel human Rho protein with unusual properties: GTPase deficiency and in vivo farnesylation. Mol Cell Biol 16: 2689–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garavini H, Riento K, Phelan JP, McAlister MS, Ridley AJ, Keep NH (2002) Crystal structure of the core domain of RhoE/Rnd3: a constitutively activated small G protein. Biochemistry 41: 6303–6310 [DOI] [PubMed] [Google Scholar]
- Gilligan DM, Sarid R, Weese J (2002) Adducin in platelets: activation-induced phosphorylation by PKC and proteolysis by calpain. Blood 99: 2418–2426 [DOI] [PubMed] [Google Scholar]
- Guasch RM, Scambler P, Jones GE, Ridley AJ (1998) RhoE regulates actin cytoskeleton organization and cell migration. Mol Cell Biol 18: 4761–4771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen SH, Zegers MM, Woodrow M, Rodriguez-Viciana P, Chardin P, Mostov KE, McMahon M (2000) Induced expression of Rnd3 is associated with transformation of polarized epithelial cells by the Raf–MEK–extracellular signal- regulated kinase pathway. Mol Cell Biol 20: 9364–9375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldin CH, Westermark B (1999) Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev 79: 1283–1316 [DOI] [PubMed] [Google Scholar]
- Hoch RV, Soriano P (2003) Roles of PDGF in animal development. Development 130: 4769–4784 [DOI] [PubMed] [Google Scholar]
- Ikenoya M, Hidaka H, Hosoya T, Suzuki M, Yamamoto N, Sasaki Y (2002) Inhibition of rho-kinase-induced myristoylated alanine-rich C kinase substrate (MARCKS) phosphorylation in human neuronal cells by H-1152, a novel and specific Rho-kinase inhibitor. J Neurochem 81: 9–16 [DOI] [PubMed] [Google Scholar]
- Ishizaki T, Maekawa M, Fujisawa K, Okawa K, Iwamatsu A, Fujita A, Watanabe N, Saito Y, Kakizuka A, Morii N, Narumiya S (1996) The small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinase. EMBO J 15: 1885–1893 [PMC free article] [PubMed] [Google Scholar]
- Kawata M, Kikuchi A, Hoshijima M, Yamamoto K, Hashimoto E, Yamamura H, Takai Y (1989) Phosphorylation of smg p21, a ras p21-like GTP-binding protein, by cyclic AMP-dependent protein kinase in a cell-free system and in response to prostaglandin E1 in intact human platelets. J Biol Chem 264: 15688–15695 [PubMed] [Google Scholar]
- Kole TP, Tseng Y, Huang L, Katz JL, Wirtz D (2004) Rho kinase regulates the intracellular micromechanical response of adherent cells to rho activation. Mol Biol Cell 15: 3475–3484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang P, Gesbert F, Delespine-Carmagnat M, Stancou R, Pouchelet M, Bertoglio J (1996) Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J 15: 510–519 [PMC free article] [PubMed] [Google Scholar]
- Leung T, Manser E, Tan L, Lim L (1995) A novel serine/threonine kinase binding the Ras-related RhoA GTPase which translocates the kinase to peripheral membranes. J Biol Chem 270: 29051–29054 [DOI] [PubMed] [Google Scholar]
- Matsui T, Amano M, Yamamoto T, Chihara K, Nakafuku M, Ito M, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K (1996) Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. EMBO J 15: 2208–2216 [PMC free article] [PubMed] [Google Scholar]
- Moyers JS, Bilan PJ, Zhu J, Kahn CR (1997) Rad and Rad-related GTPases interact with calmodulin and calmodulin-dependent protein kinase II. J Biol Chem 272: 11832–11839 [DOI] [PubMed] [Google Scholar]
- Nobes CD, Lauritzen I, Mattei MG, Paris S, Hall A, Chardin P (1998) A new member of the Rho family, Rnd1, promotes disassembly of actin filament structures and loss of cell adhesion. J Cell Biol 141: 187–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey O, Young SH, Cantrell D, Rozengurt E (2001) Rapid protein kinase D translocation in response to G protein-coupled receptor activation. Dependence on protein kinase C. J Biol Chem 276: 32616–32626 [DOI] [PubMed] [Google Scholar]
- Ribeiro-Neto F, Urbani J, Lemee N, Lou L, Altschuler DL (2002) On the mitogenic properties of Rap1b: cAMP-induced G(1)/S entry requires activated and phosphorylated Rap1b. Proc Natl Acad Sci USA 99: 5418–5423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, Hall A (1992) The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70: 389–399 [DOI] [PubMed] [Google Scholar]
- Riento K, Guasch RM, Garg R, Jin B, Ridley AJ (2003) RhoE binds to ROCK I and inhibits downstream signaling. Mol Cell Biol 23: 4219–4229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riento K, Ridley AJ (2003) Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol 4: 446–456 [DOI] [PubMed] [Google Scholar]
- Riveline D, Zamir E, Balaban NQ, Schwarz US, Ishizaki T, Narumiya S, Kam Z, Geiger B, Bershadsky AD (2001) Focal contacts as mechanosensors: externally applied local mechanical force induces growth of focal contacts by an mDia1-dependent and ROCK-independent mechanism. J Cell Biol 153: 1175–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen A, Keenan KF, Thelen M, Nairn AC, Aderem A (1990) Activation of protein kinase C results in the displacement of its myristoylated, alanine-rich substrate from punctate structures in macrophage filopodia. J Exp Med 172: 1211–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauzeau V, Le Jeune H, Cario-Toumaniantz C, Smolenski A, Lohmann SM, Bertoglio J, Chardin P, Pacaud P, Loirand G (2000) Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA-induced Ca2+ sensitization of contraction in vascular smooth muscle. J Biol Chem 275: 21722–21729 [DOI] [PubMed] [Google Scholar]
- Sawada N, Itoh H, Yamashita J, Doi K, Inoue M, Masatsugu K, Fukunaga Y, Sakaguchi S, Sone M, Yamahara K, Yurugi T, Nakao K (2001) cGMP-dependent protein kinase phosphorylates and inactivates RhoA. Biochem Biophys Res Commun 280: 798–805 [DOI] [PubMed] [Google Scholar]
- Tanimura S, Nomura K, Ozaki K, Tsujimoto M, Kondo T, Kohno M (2002) Prolonged nuclear retention of activated extracellular signal-regulated kinase 1/2 is required for hepatocyte growth factor-induced cell motility. J Biol Chem 277: 28256–28264 [DOI] [PubMed] [Google Scholar]
- Villalonga P, Guasch RM, Riento K, Ridley AJ (2004) RhoE inhibits cell cycle progression and Ras-induced transformation. Mol Cell Biol 24: 7829–7840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward Y, Spinelli B, Quon MJ, Chen H, Ikeda SR, Kelly K (2004) Phosphorylation of critical serine residues in Gem separates cytoskeletal reorganization from down-regulation of calcium channel activity. Mol Cell Biol 24: 651–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wennerberg K, Forget MA, Ellerbroek SM, Arthur WT, Burridge K, Settleman J, Der CJ, Hansen SH (2003) Rnd proteins function as RhoA antagonists by activating p190 RhoGAP. Curr Biol 13: 1106–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]