

Abstract
IMPORTANCE
Hereditary cancer syndromes infer high cancer risks and require intensive cancer surveillance, yet the prevalence and spectrum of these conditions among unselected patients with early-onset colorectal cancer (CRC) is largely undetermined.
OBJECTIVE
To determine the frequency and spectrum of cancer susceptibility gene mutations among patients with early-onset CRC.
DESIGN, SETTING, AND PARTICIPANTS
Overall, 450 patients diagnosed with colorectal cancer younger than 50 years were prospectively accrued from 51 hospitals into the Ohio Colorectal Cancer Prevention Initiative from January 1, 2013, to June 20, 2016. Mismatch repair (MMR) deficiency was determined by microsatellite instability and/or immunohistochemistry. Germline DNA was tested for mutations in 25 cancer susceptibility genes using next-generation sequencing.
MAIN OUTCOMES AND MEASURES
Mutation prevalence and spectrum in patients with early-onset CRC was determined. Clinical characteristics were assessed by mutation status.
RESULTS
In total 450 patients younger than 50 years were included in the study, and 75 gene mutations were found in 72 patients (16%). Forty-eight patients (10.7%) had MMR-deficient tumors, and 40 patients (83.3%) had at least 1 gene mutation: 37 had Lynch syndrome (13, MLH1 [including one with constitutional MLH1 methylation]; 16, MSH2; 1, MSH2/monoallelic MUTYH; 2, MSH6; 5, PMS2); 1 patient had the APC c.3920T>A, p.I1307K mutation and a PMS2 variant; 9 patients (18.8%) had double somatic MMR mutations (including 2 with germline biallelic MUTYH mutations); and 1 patient had somatic MLH1 methylation. Four hundred two patients (89.3%) had MMR-proficient tumors, and 32 patients (8%) had at least 1 gene mutation: 9 had mutations in high-penetrance CRC genes (5, APC; 1, APC/PMS2; 2, biallelic MUTYH; 1, SMAD4); 13 patients had mutations in high- or moderate-penetrance genes not traditionally associated with CRC (3, ATM; 1, ATM/CHEK2; 2, BRCA1; 4, BRCA2; 1, CDKN2A; 2, PALB2); 10 patients had mutations in low-penetrance CRC genes (3, APC c.3920T>A, p.I1307K; 7, monoallelic MUTYH). Importantly, 24 of 72 patients (33.3%) who were mutation positive did not meet established genetic testing criteria for the gene(s) in which they had a mutation.
CONCLUSIONS AND RELEVANCE
Of 450 patients with early-onset CRC, 72 (16%) had gene mutations. Given the high frequency and wide spectrum of mutations, genetic counseling and testing with a multigene panel could be considered for all patients with early-onset CRC.
Colorectal cancer (CRC) is the third most common cancer diagnosed in the United States, excluding nonmelanoma skin cancers.1 The median age of CRC diagnosis is 69 years in males and 73 years in females; 10% of patients with CRC are diagnosed when they are younger than 50 years.1 Early-onset cancer is a hallmark of inherited cancer predisposition. Identification of hereditary cancer syndromes has significant implications for patients and families, as it facilitates risk assessment, directs clinical management, and can guide treatment options.
Lynch syndrome, caused by germline mutations in the mismatch repair (MMR) genes MLH1, MSH2, MSH6 and PMS2, or EPCAM, is the most common known cause of hereditary CRC and accounts for 4% to 13.5% of patients with early-onset CRC.2–6 Patients with tumors exhibiting characteristics of MMR deficiency are more likely to have Lynch syndrome; therefore, professional guidelines recommend all patients with CRC receive tumor screening for Lynch syndrome, with referral to genetic counseling for those with MMR deficiency.7,8 The National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology for Genetic/Familial High-Risk Assessment: Colorectal (NCCN Guidelines) suggests that all patients with CRC diagnosed younger than 50 years consider genetic testing for Lynch syndrome.9
The prevalence of other hereditary cancer syndromes among patients with early-onset CRC is largely unknown because previous studies are limited and have been confined to selected (high-risk) patient populations.5,6 With the advent of next-generation sequencing (NGS), genetic testing for hereditary CRC has shifted from phenotype-specific single gene assessment to broad panels providing simultaneous assessment of multiple genes implicated in various hereditary cancer syndromes. Previous studies have shown that multigene panel testing for hereditary CRC is feasible, timely, and more cost-effective than single gene testing.10 However, the clinical utility of multigene panel testing among patients with early-onset CRC is not known.
Using multigene panel testing, we determined the prevalence and spectrum of germline mutations in 25 genes associated with various hereditary cancer syndromes in 450 patients with CRC diagnosed at younger than 50 years, unselected for family history or MMR status of the tumor.
Methods
Patients
As of June 20, 2016, 2785 patients who had surgical resection in Ohio for newly diagnosed invasive colorectal adenocarcinoma between the ages of 17 and 96 years, on or after January 1, 2013, were prospectively enrolled into the ongoing Ohio Colorectal Cancer Prevention Initiative (OCCPI). The OCCPI was created to decrease CRC incidence in Ohio by identifying patients with hereditary predisposition (statewide universal tumor screening for newly diagnosed patients with CRC), increasing colonoscopy compliance for first-degree relatives of patients with CRC and encouraging future research through the creation of a biorepository. The 51 Ohio hospitals participating in the OCCPI (eTable 1 in the Supplement) were selected to represent a cross-section of clinical centers in the state based on high reported volume of patients with CRC, affiliation with a high volume hospital, or interest in participation. Institutional review board approval was obtained by the individual hospitals, Community Oncology Programs, or by ceding review to the Ohio State University (OSU) institutional review board. Written informed consent was obtained.
Of the total patients enrolled, 594 were diagnosed at younger than 50 years, and 450 of those ostensibly unrelated patients had all of their testing completed in time for inclusion in this analysis (75.8% of the total patients enrolled younger than 50 years). Using tumor registry numbers from 2013, there were an estimated 1207 patients diagnosed with CRC at younger than 50 years in the state of Ohio between January 2013 and June 2016.11 Therefore, this analysis includes 37.3% (450 of 1207) of eligible patients.
Samples
Blood and a paraffin-embedded tumor block or unstained slides were submitted for each patient. Study pathologists confirmed the tumor histology and marked areas with at least 30% tumor and normal adjacent tissue. Blood and tissue (tumor and normal) underwent DNA extraction using standard methods.12
Tumor Screening for Lynch Syndrome
All tumors were screened for MMR deficiency by microsatellite instability (MSI) testing and/or immunohistochemical (IHC) analysis. Tumor screening was performed centrally at OSU, if not already completed in a Clinical Laboratory Improvement Amendments–approved laboratory for clinical care. Microsatellite instability testing was completed using tumor and normal DNA to detect a size change in microsatellites using the Promega MSI Analysis System version 1.2 (Promega Corporation). This included fluorescently labeled primers for coamplification of 7 repeat markers (BAT-25, BAT-26, NR-21, NR-24, MONO-27, Penta C, Penta D). Tumors showing MSI at 0 markers were classified as microsatellite stable (MSS). Tumors showing MSI at 1 marker were classified as microsatellite low. Tumors showing MSI at 2 or more markers were classified as microsatellite high (MSI-H). Immunohistochemistry of the MMR proteins was performed using the 2-stain method as previously described.13 Staining for all 4 MMR proteins was attempted if MSI could not be performed. Antibodies included MLH-1 Clone: Leica ES05 (Mouse: NCL-L-MLH1), MSH-2 Clone: Calbiochem FE11 (Mouse: NA27), MSH-6 Clone: Epitomics EP49 (Rabbit: AC-0047), PMS-2 Clone: BD Pharmingen A16-4 (Mouse: 556415). Proteins with convincing stain in more than 1% of cells were considered present. Methylation of the MLH1 promoter was assessed at 4 CpG sites between −209 and −188 using pyrosequencing14 when tumors were MSI-H and/or absent MLH1 and PMS2 proteins on IHC. The average percent of methylation detected at the 4 CpG sites was used to classify tumors as methylated (≥10% methylation) or not (<10% methylation).
Germline Genetic Testing
The testing strategy is detailed in the Figure. All patients underwent germline testing for 25 cancer susceptibility genes: APC, ATM, BARD1, BMPR1A, BRCA1, BRCA2, BRIP1, CDH1, CDK4, CDKN2A, CHEK2, EPCAM, MLH1, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, PTEN, RAD51C, RAD51D, SMAD4, STK11, TP53. Two clinical laboratories were used for germline testing based on the MMR status of the tumor. Genetic testing for patients with MMR deficient tumors (MSI-H and/or abnormal IHC without MLH1 methylation) was performed at the University of Washington (UW). Genomic regions were captured using biotinylated RNA oliognucleotides (SureSelect; Agilent Technologies) and sequenced on an Illumina HiSeq2000 instrument (Illumina Inc).15 Large rearrangements were detected.16 Genetic testing for patients with MMR-proficient tumors (MSS and/or normal IHC) or MLH1 methylation was performed at Myriad Genetics Inc, and ultra-deep targeted sequencing was performed using the RainDance Thunder-Storm platform (RainDance Technologies) for DNA amplification and Illumina MiSeq and HiSeq2000 instruments.17 Large rearrangements were detected. The Clinical Laboratory Improvement Amendments–approved laboratories adjudicated the pathogenicity of all mutations using criteria established by the American College of Medical Genetics and International Agency for Research on Cancer guidelines.18,19
Figure. Testing Strategy.
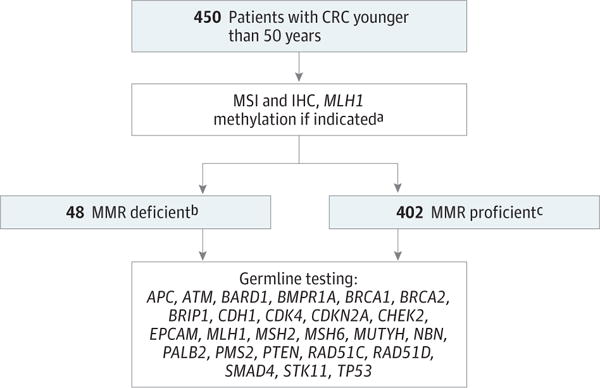
CRC indicates colorectal cancer; IHC, immunohistochemistry; MMR, mismatch repair; MSI, microsatellite instability.
aMLH1 methylation indicated if tumor MSI-high and/or absent MLH1/PMS2 proteins on IHC.
bMMR-deficient tumor indicates MSI-high and/or abnormal IHC.
cMMR-proficient tumor indicates microsatellite stability and/or normal IHC.
Tumor Genetic Testing
Select patients underwent additional testing of the MMR genes in tumor DNA at UW using methods previously described.20 For patients with unexplained MMR deficiency (MMR-deficient tumor, no germline MMR mutation or MLH1 methylation), tumors were assessed for 2 MMR mutations or 1 MMR mutation with loss of heterozygosity of the opposite allele (double somatic MMR mutations), which has been shown to cause sporadic MMR deficiency.20 For patients with MMR-deficient tumors and a germline MMR variant of uncertain significance, tumors were assessed for additional MMR mutations or loss of heterozygosity to attempt to clarify the pathogenicity of the variant. Variants were reclassified as likely pathogenic when tumor screening results supported pathogenicity and 1 additional pathogenic mutation was identified in the tumor using methods previously described.20
Statistical Analysis
Descriptive statistics were provided. Wilson score intervals with continuity correction were used to compute confidence intervals. Pearson χ2 tests with continuity correction and Fisher exact test were used to estimate P values; all tests were 2-sided, and level of significance was set at .05.
Results
Patient characteristics are shown in Table 1. Men accounted for 52.2% (n = 235) of patients; the mean age at CRC diagnosis was 42.5 years. Over 85% of patients self-reported their race as white (n = 385) and 9.1% of patients (n = 41) self-reported their race as black. Among the patients, 8.2% (n = 37) had an additional malignancy. Overall, patients reported a family history of at least 1 first-degree relative with cancer(s) of the colon (n = 86 [19.1%]), endometrium (n = 19 [4.2%]), breast (n = 42 [9.3%]), ovary (n = 11 [2.4%]), and/or pancreas (n = 10 [2.2%]).
Table 1.
Clinicopathologic Characteristics by Mutation Status
| Characteristic | Mutations | Study Population (n = 450) |
||||
|---|---|---|---|---|---|---|
| All (n = 72) | MMR Only (n = 37)a,b |
Other CRC (n = 22)c,d |
Non-CRC (n = 13)e |
No Mutation or VUS (n = 378) |
||
| Average age at CRC diagnosis, y | 41.1 | 41.7 | 38.9 | 43.2 | 42.8 | 42.5 |
| Diagnosed, No. (%) | ||||||
| Younger than 20 y | 0 | 0 | 0 | 0 | 1 (0.3) | 1 (0.2) |
| Between 20–29 y | 4 (5.6) | 2 (5.4) | 2 (9.0) | 0 | 14 (3.7) | 18 (4.0) |
| Between 30–39 y | 21 (29.2) | 9 (24.3) | 10 (45.5) | 2 (15.4) | 77 (20.4) | 98 (21.8) |
| Between 40–49 y | 47 (65.3) | 26 (70.3) | 10 (45.5) | 11 (84.6) | 286 (75.7) | 333 (74.0) |
| CRC site, No. (%) | ||||||
| Right colon | 25 (34.7) | 19 (51.4) | 4 (18.2) | 2 (15.4) | 76 (20.1) | 101 (22.4) |
| Left colon | 22 (35.6) | 9 (24.3) | 9 (40.9) | 4 (30.8) | 178 (47.1) | 200 (44.4) |
| Transverse | 5 (6.9) | 4 (10.8) | 0 | 1 (7.7) | 11 (2.9) | 16 (3.6) |
| Rectum | 20 (27.8) | 5 (13.5) | 9 (40.9) | 6 (46.2) | 105 (27.8) | 125 (27.8) |
| Not specifiedf | 0 | 0 | 0 | 0 | 8 (2.1) | 8 (1.8) |
| Stage, No. (%) | ||||||
| I | 12 (16.7) | 8 (21.6) | 3 (13.6) | 1 (7.7) | 37 (9.8) | 49 (10.9) |
| II | 16 (22.2) | 11 (29.7) | 3 (13.6) | 2 (15.3) | 77 (20.4) | 93 (20.7) |
| III | 29 (40.3) | 15 (40.5) | 9 (40.9) | 5 (38.5) | 168 (44.4) | 197 (43.8) |
| IV | 15 (20.8) | 3 (8.1) | 7 (31.8) | 5 (38.5) | 93 (24.6) | 108 (24.0) |
| Unavailable | 0 | 0 | 0 | 0 | 3 (0.8) | 3 (0.6) |
| Sex, No. (%) | ||||||
| Male | 37 (51.4) | 20 (54.1) | 12 (54.5) | 5 (38.5) | 198 (52.4) | 235 (52.2) |
| Female | 25 (48.6) | 17 (45.9) | 10 (45.5) | 8 (61.5) | 180 (47.6) | 215 (47.8) |
| Self-reported race, No. (%) | ||||||
| White | 62 (86.1) | 31 (83.8) | 19 (86.4) | 12 (92.3) | 323 (85.5) | 385 (85.6) |
| African American/black | 8 (11.1) | 6 (16.2) | 1 (4.5) | 1 (7.7) | 33 (8.7) | 41 (9.1) |
| Asian | 0 | 0 | 0 | 0 | 8 (2.1) | 8 (1.8) |
| Other | 2 (2.8) | 0 | 2 (9.0) | 0 | 8 (2.1) | 10 (2.2) |
| Not reported | 0 | 0 | 0 | 0 | 6 (1.6) | 6 (1.3) |
| Hispanic, No. (%) | ||||||
| Yes | 2 (2.8) | 2 (5.4) | 0 | 0 | 3 (0.8) | 5 (1.1) |
| No | 70 (97.2) | 35 (94.6) | 22 (100) | 13 (100) | 375 (99.2) | 445 (98.9) |
| Other self-reported malignancy, No. (%) | ||||||
| Cancer | ||||||
| Synchronous (colon) | 6 (8.3) | 5 (13.5) | 1 (4.5) | 0 | 7 (1.9) | 13 (2.9) |
| Metachronous (colon) | 2 (2.8) | 2 (5.4) | 0 | 0 | 1 (0.3) | 3 (0.7) |
| Endometrial | 1 (1.4) | 1 (2.7) | 0 | 0 | 1 (0.3) | 2 (0.4) |
| Breast | 1 (1.4) | 0 | 0 | 1 (7.7) | 1 (0.3) | 2 (0.4) |
| Ovarian | 0 | 0 | 0 | 0 | 0 | 0 |
| Pancreatic | 0 | 0 | 0 | 0 | 0 | 0 |
| Other | 4 (5.6) | 2 (5.4) | 2 (9.0) | 0 | 14 (3.7) | 18 (4.0) |
| None | 59 (81.9) | 28 (75.7) | 19 (86.4) | 12 (92.3) | 354 (93.7) | 413 (91.8) |
| Self-reported family cancer history, No. (%)g | ||||||
| Cancer | ||||||
| Colon | 33 (45.8) | 27 (73.0) | 5 (22.7) | 1 (7.7) | 53 (14) | 86 (19.1) |
| Endometrial | 8 (11.1) | 8 (21.6) | 0 | 0 | 11 (2.9) | 19 (4.2) |
| Breast | 8 (11.1) | 3 (8.1) | 2 (9.1) | 3 (23.1) | 34 (9.0) | 42 (9.3) |
| Ovarian | 2 (2.8) | 1 (2.7) | 0 | 1 (7.7) | 9 (2.4) | 11 (2.4) |
| Pancreatic | 2 (2.8) | 1 (2.7) | 1 (4.5) | 0 | 8 (2.1) | 10 (2.2) |
Abbreviations: CRC, colorectal cancer; MMR, mismatch repair; VUS, variant of uncertain significance.
MMR genes include MLH1, MSH2, MSH6, PMS2.
Includes 1 patient with both MSH2/MUTYH mutations.
Other CRC genes include APC, APC p.I1307K, biallelic MUTYH, monoallelic MUTYH, and SMAD4.
Includes 1 patient with both APC/PMS2 mutations (tumor was microsatellite stable) and a separate patient with both APC p.I1307K/PMS2 VUS (tumor was microsatellite unstable).
Non-CRC genes include ATM, BRCA1, BRCA2, CDKN2A, CHEK2, PALB2.
Stage IV or metastatic biopsy.
First-degree relatives.
Tumor Screening for Lynch syndrome
Among the patients, 10.7% (n = 48) had MMR-deficient tumors (n = 8 [16.7%], stage I; n = 13 [27.1%], stage II; n = 23 [47.9%], stage III; n = 4 [8.3%], stage IV). Three patients with MMR-deficient tumors had MLH1 methylation. All tumor screening results (MSI and IHC) were concordant for each patient, except for 1 (MSI-H, normal IHC). In total, 402 patients (89.3%) had MMR-proficient tumors (n = 41 [10.2%], stage I; n = 80 [19.9%] stage II; n = 174 [43.3%], stage III; n = 104 [25.9%], stage IV; n = 3 [0.7%], stage unavailable).
Overall Germline Genetic Testing
Among 450 patients with early-onset CRC, 75 pathogenic or likely pathogenic cancer susceptibility gene mutations were found in 72 patients (16%; 95% CI, 12.8%–19.8%). The spectrum of mutations is shown in Table 2. Thirty-six patients (8%) had Lynch syndrome only; 2 patients (0.4%) had Lynch syndrome and another hereditary cancer syndrome; 34 patients (7.6%) had a different hereditary cancer syndrome (including a third patient with 2 syndromes). Sixty-one patients (13.6%) had mutations in high- or moderate-penetrance genes, and 11 patients (2.4%) had mutations in low-penetrance genes.
Table 2.
Germline Mutations Identified and Associated Syndromes
| Gene | Associated Syndrome or Cancer(s) | Overall Penetrance | Patients With Mutation, No. (%) | (95% CI) |
|---|---|---|---|---|
| Any pathogenic or likely pathogenic mutation | 72 (16) | (12.8–19.8) | ||
| Genes associated with colon cancer | 59 (13.1) | (10.2–16.7) | ||
| MLH1 | Lynch syndrome | High | 13 (2.9) | (1.6–5.0) |
| MSH2 | Lynch syndrome | High | 16 (3.6) | (2.1–5.8) |
| MSH2/monoallelic MUTYH | Lynch syndrome/colon cancer | High/low | 1 (0.2) | (0.01–1.4) |
| MSH6 | Lynch syndrome | Moderate | 2 (0.4) | (0.08–1.8) |
| PMS2 | Lynch syndrome | Moderate | 5 (1.1) | (0.4–2.7) |
| APC | Familial adenomatous polyposis (FAP) | High | 5 (1.1) | (0.4–2.7) |
| APC p.I1307K | Colon cancer | Low | 4 (0.9) | (0.3–2.4) |
| MUTYH | ||||
| Biallelic | MUTYH-associated polyposis (MAP) | High | 4 (0.9) | (0.3–2.4) |
| Monoallelic | Colon cancer | Low | 7 (1.6) | (0.7–3.3) |
| SMAD4 | Juvenile polyposis syndrome | High | 1 (0.2) | (0.01–1.4) |
| APC/PMS2 | FAP/Lynch syndrome | High/moderate | 1 (0.2) | (0.01–1.4) |
| Genes not traditionally associated with colon cancer | 13 (2.9) | (1.6–5.0) | ||
| BRCA1 | Hereditary breast-ovarian cancer syndrome | High | 2 (0.4) | (0.08–1.8) |
| BRCA2 | Hereditary breast-ovarian cancer syndrome | High | 4 (0.9) | (0.3–2.4) |
| ATM | Breast cancer, pancreatic cancer | Moderate | 3 (0.7) | (0.2–2.1) |
| ATM/CHEK2 | Breast cancer, pancreatic cancer | Moderate | 1 (0.7) | (0.01–1.4) |
| PALB2 | Breast cancer, pancreatic cancer | Moderate | 2 (0.4) | (0.08–1.8) |
| CDKN2A | Melanoma, pancreatic cancer | High | 1 (0.2) | (0.01–1.4) |
Genotype and phenotype data (including family history) for patients with pathogenic mutations are provided in eTable 3 in the Supplement. Patients with pathogenic mutations were more likely to report a family history of colon cancer (45.8% vs 14%; P < .001) and endometrial cancer (11.1% vs 2.9%; P = .005)compared with patients without mutations. Nine patients came from unique families with a known mutation in a cancer susceptibility gene but had not undergone predictive testing for the familial mutation prior to their CRC diagnosis. Patients with Lynch syndrome (19 of 37) were diagnosed at earlier stages (I, II) compared with patients with another hereditary cancer syndrome (9 of 35) (51.4% vs 25.7%; P = .047).
Germline Results for Patients With MMR-Deficient Tumors
Forty (83.3%) of the 48 patients with MMR-deficient tumors had at least 1 mutation in a cancer susceptibility gene. Thirty-seven patients had Lynch syndrome (13, MLH1; 16, MSH2; 1, MSH2/monoallelic MUTYH; 2, MSH6; 5, PMS2), 2 had biallelic MUTYH mutations, and 1 had the common low-penetrance APC c.3920T>A, p.I1307K mutation21–23 and a variant of uncertain significance in PMS2 (c.322G>T, p.G108W). Nine patients’ MMR-deficient tumors were explained by double somatic MMR mutations (including 2 patients with germline biallelic MUTYH mutations) (eTable 4 in the Supplement). Of the 3 patients with MLH1 methylation, 1 was found to have a MSH2 mutation (the tumor showed absence of all 4 MMR proteins on IHC), 1 was found to have constitutional MLH1 methylation by testing blood, and the third patient did not have any germline mutations and constitutional methylation was ruled out.
Germline Results for Patients With MMR-Proficient Tumors
Thirty-two (8%) of 402 patients with MMR-proficient tumors had at least 1 mutation in a cancer susceptibility gene. Nine patients had mutations in high-penetrance genes with established CRC risk (5, APC; 1, APC/PMS2; 2, biallelic MUTYH; 1, SMAD4), 13 had mutations in high- or moderate-penetrance genes not traditionally associated with CRC risk (3, ATM; 1, ATM/CHEK2; 2, BRCA1; 4, BRCA2; 1, CDKN2A; 2, PALB2), and 10 had mutations in low-penetrance CRC genes (3, APC c.3920T>A, p.I1307K; 7, monoallelic MUTYH).
Variants of Uncertain Significance
One hundred seventy-eight variants of uncertain significance were found in 145 patients (32.2%) (eTable 2 in the Supplement). The genes most likely to have a variant discovered included ATM (n = 30 [16.9%]), APC (n = 19 [10.7%]), and CHEK2 (n = 18 [10.1%]). Five variants were upgraded to pathogenic (or likely pathogenic) after additional testing. One patient with double somatic MMR mutations had a germline pathogenic MUTYH mutation and MUTYH variant c.698G>A, p.G233D, that was reclassified to likely pathogenic based on segregation analysis and clinical history. Tumor sequencing clarified that PMS2 c.215G>A, p.G72E and MSH2 c.1832T>A, p.V611E were likely pathogenic owing to the presence of the germline variant plus 1 additional pathogenic somatic mutation, in addition to supportive tumor screening results. RNA studies showing altered MLH1 splicing (exon 2 skipping) in a majority of transcripts and cosegregation with disease proved that MLH1 c.207 + 5G>C was pathogenic. Cosegregation with disease proved that MSH6 c.1109T>C, p.L370S was pathogenic.
Discussion
This prospective, statewide study indicates that 1 of every 6 patients with CRC diagnosed younger than 50 years has at least 1 pathogenic cancer susceptibility gene mutation (16%). While the prevalence of Lynch syndrome reported herein (8.4%) is consistent with previous publications,2,3 this is the first study to our knowledge to determine the prevalence and spectrum of other hereditary cancer syndromes (8%) found in an unselected series of patients with early-onset CRC. All patients found to have pathogenic mutations received genetic counseling and current evidence-based guidelines for intensive cancer surveillance based on their mutation status.9 For some patients, the identification of MMR tumor status and/or gene mutation(s) provided actionable therapeutic targets for their current CRC (eg, PARP [poly adenosine diphosphate–ribose polymerase] inhibitors and anti-PD1 [programmed cell death protein 1] immunotherapy).24,25 At-risk family members benefited from genetic counseling and cascade testing to determine the necessity of potentially life-saving cancer surveillance and prevention options.
Multigene panel testing facilitated identification of hereditary cancer syndromes in patients who may have otherwise been missed. Importantly, 24 of 72 patients (33.3%) with pathogenic mutations did not meet NCCN Guidelines for at least 1 of the gene(s) in which they were found to have a mutation.9 Forty-four of the 72 patients (61.1%) with pathogenic mutations did meet NCCN Guidelines owing to having MMR-deficient tumors and/or the presence of more than 10 adenomatous polyps; however, they likely would have only received phenotype-specific genetic testing for Lynch syndrome or polyposis had testing been done outside of this study.9 In that scenario, 4 patients (5.6%) would have had at least 1 of their mutations missed without the use of a broad multigene panel. Three patients with MMR-deficient tumors were found to have additional mutations in genes that would not have been assessed: 1 had biallelic MUTYH mutations (without polyposis), 1 had a monoallelic MUTYH mutation, and 1 had the APC c.3920T>A, p.I1307K mutation. One patient with polyposis (due to a known APC mutation) and an MMR-proficient tumor was unexpectedly found to also have Lynch syndrome caused by a pathogenic PMS2 mutation.
While many of the detected mutations were in genes with established CRC risk, 13 of 72 patients (18.1%) had mutations in genes not traditionally associated with CRC: 3, ATM;1, ATM/CHEK2; 2, BRCA1; 4, BRCA2; 1, CDKN2A; and 2, PALB2. Notably, 6 patients had mutations in BRCA1/2 (known to cause hereditary breast-ovarian cancer syndrome [HBOC]). Four patients with BRCA1/2 mutations met NCCN genetic testing criteria for HBOC, and 2 patients with BRCA1/2 mutations did not have personal or family history of breast or ovarian cancer and did not meet NCCN genetic testing criteria for HBOC. Previous studies have reported early-onset CRC in women with BRCA1 mutations26 and BRCA2 mutations in families with familial colorectal cancer type X.27 It is possible that these 13 mutations are incidental findings; however, the cancer spectrum and penetrance for many well-established and newly discovered genes will likely be redefined now that multigene panel testing is becoming more routine.
Another novel aspect of this study was the use of tumor sequencing to elucidate the etiology of patients with early-onset CRC with unexplained MMR deficiency and clarify the pathogenicity of MMR variants of uncertain significance. It was previously reported that 68% of patients with CRC and endometrial cancer with unexplained MMR deficiency do not have Lynch syndrome, and their MMR-deficient tumors were the result of double somatic MMR mutations.20 In our study, 100% (9/9) of patients with early-onset CRC and unexplained MMR deficiency were found to have double somatic MMR mutations (including 2 patients with germline biallelic MUTYH mutations). This finding has significant clinical implications and underscores the importance of somatic testing for patients with early-onset CRC and unexplained MMR deficiency. Traditionally, patients with CRC (particularly those diagnosed at a young age) with unexplained MMR deficiency would be medically managed as if they had Lynch syndrome with an unidentifiable germline MMR mutation. Proving that their MMR deficiency was the result of double somatic mutations likely means that they do not have Lynch syndrome and may not need to follow intensive Lynch syndrome surveillance guidelines. It is possible that these patients have something undiscovered that predisposes them to the development of somatic MMR mutations, but most likely, their CRC was sporadic and it would be appropriate for their family members to follow screening guidelines based on the family history of early-onset CRC. In addition, this is the first time that we know of that somatic testing was used specifically to aid in MMR variant reclassification, assisting in the reclassification of 2 variants among patients with MMR-deficient tumors.
Limitations
While the recruiting personnel were strongly discouraged from preferentially enrolling patients with a strong family history of cancer or young age of diagnosis, it is possible that this occurred. Patient’s self-reported family history is a limitation; medical record review would have ensured accuracy but was not feasible. We had 3-generation pedigrees for patients who tested positive and received genetic counseling. However, we only had first-degree relative cancer history for patients who tested negative, so we were unable to provide complete risk assessment or determine who met NCCN genetic testing criteria among patients who did not have a mutation.
The 16% prevalence of hereditary cancer syndromes among patients with early-onset CRC reported herein is likely an underestimate for several reasons. There are likely other CRC susceptibility genes, some of which have not yet been discovered, for which our patients may not have been tested. Additionally, some of the variants of uncertain significance may eventually be found to be pathogenic. For example, at the time of publication there were 2 mutations (CHEK2 c.470T>C, p.I157T and MLH1 c.1897-2A>G) with discrepant pathogenicity classifications from various clinical laboratories (ranging from variant of uncertain significance to pathogenic). For this study, they were considered variants of uncertain significance.
Conclusions
Overall, 75 pathogenic cancer susceptibility gene mutations were found in 72 patients of 450 diagnosed with CRC younger than 50 years (16%). While it is important to continue MMR tumor screening for all patients with CRC for treatment purposes (ie, checkpoint inhibitors, if initial findings are validated in subsequent trials), genetic counseling and testing with a broad multigene panel should be considered for all patients with early-onset CRC due to their high prevalence of hereditary cancer syndromes.
Supplementary Material
Key Points.
Question
What is the frequency and spectrum of cancer susceptibility gene mutations among patients with colorectal cancer diagnosed at younger than 50 years?
Findings
In this cohort study of 450 patients with early-onset colorectal cancer, 72 (16%) had a pathogenic mutation. Panel testing identified mutations in patients that may have otherwise been missed; specifically, 24 of 72 patients (33.3%) who were mutation positive did not meet testing criteria for the gene(s) in which they had a mutation.
Meaning
Multigene panel testing should be considered for all patients with early-onset colorectal cancer.
Acknowledgments
Funding/Support: The Ohio Colorectal Cancer Prevention Initiative (OCCPI) is supported by a grant from Pelotonia, an annual cycling event in Columbus, Ohio, that supports cancer research at The Ohio State University Comprehensive Cancer Center–James Cancer Hospital and Solove Research Institute (Hampel, Paskett, Shields). This study was also supported in part by the National Cancer Institute (grant P30CA16058).
Role of the Funder/Sponsor: The funders/sponsors had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Author Contributions: Mss Pearlman and Hampel had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Concept and design: Hampel, Pearlman, Goldberg, Paskett, Shields, Freudenheim, de la Chapelle.
Acquisition, analysis, or interpretation of data: All authors.
Drafting of the manuscript: Pearlman, Hampel.
Critical revision of the manuscript for important intellectual content: All authors.
Statistical analysis: Liyanarachchi, Pearlman.
Obtained funding: Hampel, Goldberg, Paskett, Shields, Freudenheim, de la Chapelle.
Administrative, technical, or material support: Frankel, Swanson, Zhao, Yilmaz, Miller, Bacher, Bigley, Nelsen, Allen, Pritchard, Shirts, Jacobson.
Study supervision: Hampel, Pearlman, Goldberg, Paskett, Shields, Freudenheim, de la Chapelle.
Conflict of Interest Disclosures: Myriad Genetics Inc donated the next-generation sequencing testing for mismatch repair–proficient patients with CRC diagnosed younger than 50 years for this study. Dr Kalady discloses honorarium from Helomics and Transenterix and is a member of the speakers’ bureau for Helomics. Ms Heald discloses a consulting or advising role with Invitae and Myriad Genetics Inc and is a member of the speakers’ bureau for Myriad Genetics Inc. Dr Paquette has received honoraria from Medtronic and has served on the speakers’ bureau. Dr Kuebler discloses employment with Columbus Oncology Associates, LLC. Dr Mahesh discloses stock in Merck, Pfizer, Kite pharmaceuticals, Oncothyreon, and Exelixis. Ms Buie discloses research funding from Merck, Galera, Amgen, and Celgene. Dr Haut discloses research funding from Amgen, Celgene, Incyte, Conversant Bio, Evidera, EMD Serono, Vector Oncology, Bristol Myers, GlaxoSmithKline, Eli Lilly, Luitpold pharmaceuticals, and Sanofi pharmaceuticals. Mr Allen discloses employment and stock with Myriad Genetics Inc. Dr de la Chapelle discloses a patent or intellectual property interest with Genzyme and Ipsogen. Ms Hampel discloses a consulting or advising role with Invitae, research funding from Myriad Genetics Inc, and honoraria from Beacon LBS. No other conflicts are reported.
Group Information: A full list of the OCCPI study group members can be found at https://cancer.osu.edu/research-and-education/pelotonia-funded-research/statewide-colon-cancer-initiative/occpi-work-group.
Additional Contributions: The authors are grateful to the patients, families, and network of 51 hospitals across Ohio (eTable 1 in the Supplement) who participated in the OCCPI study. In particular, the authors would like to thank the following hospitals for enrolling at least 1 patient included in this study: Adena Regional Medical Center, Akron General Medical Center, Atrium Medical Center, Aultman Hospital, Bethesda North Hospital-TriHealth, Blanchard Valley Health System, Cleveland Clinic Foundation, Doctors Hospital, Fairfield Medical Center, Fairview Hospital, Genesis HealthCare System, Good Samaritan Hospital (Dayton), Good Samaritan Hospital-TriHealth, Grady Memorial Hospital, Grant Medical Center, Hillcrest Hospital, Kettering Medical Center, Licking Memorial Hospital, Marietta Memorial Hospital, Mercy Fairfield Hospital, Mercy Medical Center, MetroHealth Medical Center, Miami Valley Hospital, Mount Carmel East Hospital, Mount Carmel St Ann’s, Mount Carmel West Hospital, ProMedica Flower Hospital, Riverside Methodist Hospital, Springfield Regional Medical Center, St Luke’s Hospital, St Rita’s Medical Center, Summa Akron City Hospital, The Christ Hospital, The Ohio State University, Upper Valley Medical Center, Wayne HealthCare, and Wright-Patterson Medical Center. The authors also thank Ohio State University undergraduate student interns Angela Onorato, James Miner, Jessica Purnell, Emily McDowell, Hannah Datz, Chloe Kent and Michael Bigley, who assisted with database entry and sample processing. They received no compensation beyond their hourly payment as part-time Ohio State University Comprehensive Cancer Center employees. The authors would also like to thank the National Cancer Institute Community Oncology Research Program and the Dayton Clinical Oncology Program for their collaboration on this research study. They received reimbursement for their expenses performing oversite for the study. Written permission was obtained for all persons named in the Additional Contribution section.
Contributor Information
Rachel Pearlman, The Ohio State University Comprehensive Cancer Center, Department of Internal Medicine, The Ohio State University Wexner Medical Center, Columbus.
Wendy L. Frankel, Department of Pathology, The Ohio State University Wexner Medical Center, Columbus.
Benjamin Swanson, Department of Pathology, The Ohio State University Wexner Medical Center, Columbus.
Weiqiang Zhao, Department of Pathology, The Ohio State University Wexner Medical Center, Columbus.
Ahmet Yilmaz, Department of Pathology, The Ohio State University Wexner Medical Center, Columbus.
Kristin Miller, Department of Pathology, The Ohio State University Wexner Medical Center, Columbus.
Jason Bacher, Department of Pathology, The Ohio State University Wexner Medical Center, Columbus.
Christopher Bigley, The Ohio State University Comprehensive Cancer Center, Department of Internal Medicine, The Ohio State University Wexner Medical Center, Columbus.
Lori Nelsen, The Ohio State University Comprehensive Cancer Center, Department of Internal Medicine, The Ohio State University Wexner Medical Center, Columbus.
Paul J. Goodfellow, The Ohio State University Comprehensive Cancer Center, Department of Obstetrics and Gynecology, The Ohio State University Wexner Medical Center, Columbus.
Richard M. Goldberg, The Ohio State University Comprehensive Cancer Center, Department of Internal Medicine, The Ohio State University Wexner Medical Center, Columbus.
Electra Paskett, The Ohio State University Comprehensive Cancer Center, Department of Internal Medicine, The Ohio State University Wexner Medical Center, Columbus.
Peter G. Shields, The Ohio State University Comprehensive Cancer Center, Department of Internal Medicine, The Ohio State University Wexner Medical Center, Columbus.
Jo L. Freudenheim, Department of Epidemiology and Environmental Health, University at Buffalo, Buffalo, New York.
Peter P Stanich, Department of Internal Medicine, The Ohio State University Wexner Medical Center, Columbus.
Ilene Lattimer, The Ohio State University Comprehensive Cancer Center, Department of Internal Medicine, The Ohio State University Wexner Medical Center, Columbus.
Mark Arnold, Department of Surgery, The Ohio State University Wexner Medical Center, Columbus.
Sandya Liyanarachchi, The Ohio State University Comprehensive Cancer Center, Department of Cancer Biology and Genetics, The Ohio State University Wexner Medical Center, Columbus.
Matthew Kalady, Department of Colorectal Surgery, Cleveland Clinic, Cleveland, Ohio.
Brandie Heald, Department of Colorectal Surgery, Cleveland Clinic, Cleveland, Ohio.
Carla Greenwood, Department of Digestive Diseases and Surgery, Cleveland Clinic, Cleveland, Ohio.
Ian Paquette, Department of Surgery, University of Cincinnati College of Medicine, Cincinnati, Ohio.
Marla Prues, Cancer Center Research, The Christ Hospital Health Network, Cincinnati, Ohio.
David J. Draper, TriHealth Cancer Institute, Good Samaritan Hospital, Cincinnati, Ohio.
Carolyn Lindeman, TriHealth Cancer Institute, Good Samaritan Hospital, Cincinnati, Ohio.
J. Philip Kuebler, Columbus Oncology and Hematology Associates, Columbus, Ohio.
Kelly Reynolds, Department of Cancer Services, Riverside Methodist Hospital, Columbus, Ohio.
Joanna M. Brell, Department of Medicine, MetroHealth Medical Center, Cleveland, Ohio.
Amy A. Shaper, Research Institute, MetroHealth Medical Center, Cleveland, Ohio.
Sameer Mahesh, Department of Internal Medicine, Summa Cancer Institute, Summa Akron City Hospital, Akron, Ohio.
Nicole Buie, Summa Center for Clinical Trials, Summa Akron City Hospital, Akron, Ohio.
Kisa Weeman, Department of Hematology/Oncology, Aultman Hospital, Canton, Ohio.
Kristin Shine, Department of Oncology Clinical Trials, Aultman Hospital, Canton, Ohio.
Mitchell Haut, Mercy Medical Center, Canton, Ohio.
Joan Edwards, Mercy Medical Center, Canton, Ohio.
Shyamal Bastola, Department of Oncology and Hematology, Genesis HealthCare System, Zanesville, Ohio.
Karen Wickham, Department of Oncology and Hematology, Genesis HealthCare System, Zanesville, Ohio.
Karamjit S. Khanduja, Division of Colon and Rectal Surgery, Mount Carmel East Hospital, Columbus, Ohio.
Rosemary Zacks, Department of Clinical Trials, Mount Carmel East Hospital, Columbus, Ohio.
Colin C. Pritchard, Department of Laboratory Medicine, University of Washington, Seattle.
Brian H. Shirts, Department of Laboratory Medicine, University of Washington, Seattle.
Angela Jacobson, Department of Laboratory Medicine, University of Washington, Seattle.
Brian Allen, Myriad Genetics Inc, Salt Lake City, Utah.
Albert de la Chapelle, The Ohio State University Comprehensive Cancer Center, Department of Cancer Biology and Genetics, The Ohio State University Wexner Medical Center, Columbus.
Heather Hampel, The Ohio State University Comprehensive Cancer Center, Department of Internal Medicine, The Ohio State University Wexner Medical Center, Columbus.
References
- 1.American Cancer Society. Colorectal Cancer Facts & Figures. 2016 http://www.cancer.org/acs/groups/content/@research/documents/document/acspc-047079.pdf. Accessed July 15, 2016.
- 2.Barnetson RA, Tenesa A, Farrington SM, et al. Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N Engl J Med. 2006;354(26):2751–2763. doi: 10.1056/NEJMoa053493. [DOI] [PubMed] [Google Scholar]
- 3.Hampel H, Frankel WL, Martin E, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol. 2008;26(35):5783–5788. doi: 10.1200/JCO.2008.17.5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer) N Engl J Med. 2005;352(18):1851–1860. doi: 10.1056/NEJMoa043146. [DOI] [PubMed] [Google Scholar]
- 5.Mork ME, You YN, Ying J, et al. High prevalence of hereditary cancer syndromes in adolescents and young adults with colorectal cancer. J Clin Oncol. 2015;33(31):3544–3549. doi: 10.1200/JCO.2015.61.4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yurgelun MB, Allen B, Kaldate RR, et al. Identification of a variety of mutations in cancer predisposition genes in patients with suspected Lynch syndrome. Gastroenterology. 2015;149(3):604–613.e620. doi: 10.1053/j.gastro.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group. Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med. 2009;11(1):35–41. doi: 10.1097/GIM.0b013e31818fa2ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palomaki GE, McClain MR, Melillo S, Hampel HL, Thibodeau SN. EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genet Med. 2009;11(1):42–65. doi: 10.1097/GIM.0b013e31818fa2db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.National Cancer Care Network. Referenced with permission from the NCCN Guidelines for Genetic/Familial High-RiskAssessment: Colorectal v.1.2016 National Comprehensive Cancer Network IArrAJ. https://www.nccn.org/professionals/physician_gls/f_guidelines.asp#detection. Accessed November 17, 2016.
- 10.Gallego CJ, Shirts BH, Bennette CS, et al. Next-generation sequencing panels for the diagnosis of colorectal cancer and polyposis syndromes: a cost-effectiveness analysis. J Clin Oncol. 2015;33(18):2084–2091. doi: 10.1200/JCO.2014.59.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohio Department of Health. Ohio Cancer Incidence Surveillance System (OCISS) https://www.odh.ohio.gov/healthstats/ocisshs/ci_surv1.aspx. Accessed November 28, 2016.
- 12.Buckingham L, Flaws ML. Molecular Diagnostics Fundamentals, Methods, & Clinical Applications. Philadelphia, PA: F. A. Davis Co; 2007. pp. 65–79. [Google Scholar]
- 13.Shia J, Tang LH, Vakiani E, et al. Immunohistochemistry as first-line screening for detecting colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome: a 2-antibody panel may be as predictive as a 4-antibody panel. Am J Surg Pathol. 2009;33(11):1639–1645. doi: 10.1097/PAS.0b013e3181b15aa2. [DOI] [PubMed] [Google Scholar]
- 14.Newton K, Jorgensen NM, Wallace AJ, et al. Tumour MLH1 promoter region methylation testing is an effective prescreen for Lynch syndrome (HNPCC) J Med Genet. 2014;51(12):789–796. doi: 10.1136/jmedgenet-2014-102552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pritchard CC, Smith C, Salipante SJ, et al. ColoSeq provides comprehensive lynch and polyposis syndrome mutational analysis using massively parallel sequencing. J Mol Diagn. 2012;14(4):357–366. doi: 10.1016/j.jmoldx.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nord AS, Lee M, King MC, Walsh T. Accurate and exact CNV identification from targeted high-throughput sequence data. BMC Genomics. 2011;12:184. doi: 10.1186/1471-2164-12-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Judkins T, Leclair B, Bowles K, et al. Development and analytical validation of a 25-gene next generation sequencing panel that includes the BRCA1 and BRCA2 genes to assess hereditary cancer risk. BMC Cancer. 2015;15:215. doi: 10.1186/s12885-015-1224-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richards S, Aziz N, Bale S, et al. ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shirts BH, Casadei S, Jacobson AL, et al. Improving performance of multigene panels for genomic analysis of cancer predisposition. Genet Med. 2016 Feb;:4. doi: 10.1038/gim.2015.212. [DOI] [PubMed] [Google Scholar]
- 20.Haraldsdottir S, Hampel H, Tomsic J, et al. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology. 2014;147(6):1308–1316.e1. doi: 10.1053/j.gastro.2014.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boursi B, Sella T, Liberman E, et al. The APC p.I1307K polymorphism is a significant risk factor for CRC in average risk Ashkenazi Jews. Eur J Cancer. 2013;49(17):3680–3685. doi: 10.1016/j.ejca.2013.06.040. [DOI] [PubMed] [Google Scholar]
- 22.Laken SJ, Petersen GM, Gruber SB, et al. Familial colorectal cancer in Ashkenazim due to a hypermutable tract in APC. Nat Genet. 1997;17(1):79–83. doi: 10.1038/ng0997-79. [DOI] [PubMed] [Google Scholar]
- 23.Liang J, Lin C, Hu F, et al. APC polymorphisms and the risk of colorectal neoplasia: a HuGE review and meta-analysis. Am J Epidemiol. 2013;177(11):1169–1179. doi: 10.1093/aje/kws382. [DOI] [PubMed] [Google Scholar]
- 24.Goyal G, Fan T, Silberstein PT. Hereditary cancer syndromes: utilizing DNA repair deficiency as therapeutic target. Fam Cancer. 2016;15(3):359–366. doi: 10.1007/s10689-016-9883-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Phelan CM, Iqbal J, Lynch HT, et al. Hereditary Breast Cancer Study Group Incidence of colorectal cancer in BRCA1 and BRCA2 mutation carriers: results from a follow-up study. Br J Cancer. 2014;110(2):530–534. doi: 10.1038/bjc.2013.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garre P, Martín L, Sanz J, et al. BRCA2 gene: a candidate for clinical testing in familial colorectal cancer type X. Clin Genet. 2015;87(6):582–58. doi: 10.1111/cge.12427. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.