

Abstract
CDK7 phosphorylates the RNA polymerase II (pol II) CTD and activates the P-TEFb-associated kinase, CDK9, but its regulatory roles remain obscure. Here, using human CDK7 analog-sensitive (CDK7as) cells, we observed reduced capping enzyme recruitment, increased pol II promoter-proximal pausing, and defective termination at gene 3′-ends upon CDK7 inhibition. We also noted CDK7 regulates chromatin modifications downstream of transcription start sites. H3K4me3 spreading was restricted at gene 5′-ends and H3K36me3 was displaced toward gene 3′-ends in CDK7as cells. Mass spectrometry identified factors that bound TFIIH-phosphorylated vs. P-TEFb-phosphorylated CTD (vs. unmodified); capping enzymes and H3K4 methyltransferase complexes, SETD1A/B, selectively bound phosphorylated CTD and the H3K36 methyltransferase SETD2 specifically bound P-TEFb-phosphorylated CTD. Moreover, TFIIH-phosphorylated CTD stimulated SETD1A/B activity toward nucleosomes, revealing a mechanistic basis for CDK7 regulation of H3K4me3 spreading. Collectively, these results implicate a CDK7-dependent “CTD code” that regulates chromatin marks in addition to RNA processing and pol II pausing.
Keywords: RNA-Seq, proteomics, Mediator, THZ1, chromatin, epigenetic, TFIIH, P-TEFb, H3K4me3, H3K36me3
Graphical abstract
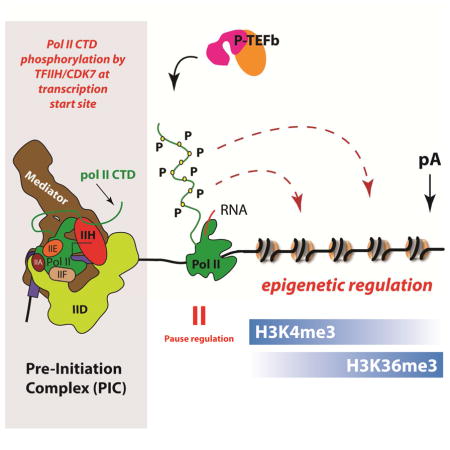
CDK7 is a subunit within the general transcription factor TFIIH. TFIIH is an integral component of the RNA polymerase II (pol II) pre-initiation complex, but the regulatory functions of CDK7 remain poorly defined, especially in metazoans. During transcription initiation, CDK7 phosphorylates the C-terminal domain (CTD) of the pol II subunit RPB1. Whereas TFIIH appears to be required for pol II transcription genome-wide (Chen et al., 2015a), CDK7 kinase activity is not strictly required for transcription in vivo or in vitro (Kanin et al., 2007; Serizawa et al., 1993). The highly conserved mammalian pol II CTD comprises heptad repeats with a general consensus sequence YSPTSPS. The pol II CTD is dynamically phosphorylated in synchrony with the cycle of transcription initiation, elongation, and termination (Buratowski, 2009; Harlen and Churchman, 2017; Heidemann et al., 2013). CDK7 phosphorylates the CTD on Ser5 and Ser7 (Akhtar et al., 2009; Glover-Cutter et al., 2009; Roy et al., 1994), and phosphorylation at these sites is enriched at the 5′-ends of genes (Harlen and Churchman, 2017; Komarnitsky et al., 2000). The importance of elucidating CDK7 function is highlighted by the fact that a CDK7 inhibitor, THZ1, is a promising new therapeutic strategy that specifically inhibits growth of multiple cancer cell types in pre-clinical studies (Kwiatkowski et al., 2014). A caveat with interpreting the cellular and in vivo effects of THZ1 is that, while it is most effective at inhibiting CDK7, it also inhibits other kinases (Kwiatkowski et al., 2014).
In metazoans, CDK7 may indirectly control CTD phosphorylation at Ser2 through activation of CDK9, via T-loop phosphorylation (Larochelle et al., 2012). CDK9 functions within P-TEFb (CDK9, CCNT1) as a Ser2 CTD kinase, but it can also phosphorylate Ser5 and Ser7 in vitro (Czudnochowski et al., 2012). CDK9 is a positive regulator of transcription elongation that antagonizes pol II promoter-proximal pausing (Kwak and Lis, 2013). The ability of CDK7 to regulate CDK9 suggests that CDK7 may help control pol II pausing, but the evidence is limited. Although activation of CDK9 by CDK7 is expected to inhibit pausing (Larochelle et al., 2012), in vitro and ChIP assays at individual genes suggested that CDK7 inhibition by THZ1 or other chemical inhibitors may enhance pausing (Kelso et al., 2014; Nilson et al., 2015). To help resolve this conundrum, we report here a genome-wide analysis of how specific inhibition of CDK7 kinase activity affects pol II promoter-proximal pausing.
The role of human CDK7 in pre-mRNA capping is also unclear. In yeast, CTD Ser5 phosphorylation by the CDK7 homolog Kin28 is required for capping enzyme (CE) recruitment and efficient mRNA capping (Hong et al., 2009; Kanin et al., 2007; Komarnitsky et al., 2000; Schroeder et al., 2000; Schwer and Shuman, 2011; Suh et al., 2016). In mammals, an alternative role has been proposed for CDK7 in which it counters an unknown antagonist of the CE guanylyltransferase (Nilson et al., 2015). The role of CTD phosphorylation in CE recruitment to metazoan genes has not been tested, although in vitro, mammalian CE is allosterically activated upon binding Ser5 phosphorylated CTD (Ho and Shuman, 1999). Furthermore, it has been proposed that pol II promoter-proximal pausing helps ensure that mRNA capping is completed prior to transcript elongation (Mandal et al., 2004; Pei and Shuman, 2002).
Studies in yeast and mammalian cells have provided evidence that phosphorylated forms of the pol II CTD interact with histone H3K4 and H3K36 methyltransferases (Kizer et al., 2005; Lee and Skalnik, 2008; Ng et al., 2003; Yoh et al., 2008). In mammals, co-transcriptional histone H3 methylation on K4 and K36 residues is carried out by the SETD1A/SETD1B complexes and the SETD2 protein, respectively. H3K4me3 is deposited predominantly on the nucleosomes flanking transcriptional start sites (Schneider et al., 2004) but on a subset of genes this modification spreads further downstream. What controls H3K4me3 spreading is poorly understood, but it has been correlated with elevated transcription and high pol II occupancy at transcription start sites (TSS) (Chen et al., 2015c). H3K36me3 is deposited in regions through which pol II has elongated, typically in a broad gradient that increases 5′-3′ across transcription units. In yeast, the Set2 H3K36 methyltransferase interacts directly with Ser2-phosphorylated CTD (Kizer et al., 2005) and the Ser2 kinase, Ctk1, facilitates K36 methylation (Xiao et al., 2003). In metazoans, the relationship between CTD phosphorylation and co-transcriptional deposition of H3K36me3 has not been investigated extensively, although in vitro formation of a pol II complex with SETD2 correlated with CTD Ser2 phosphorylation (Yoh et al., 2008).
A more complete understanding of human pol II CTD function requires an improved description of its interactome, including physiologically relevant phosphorylated isoforms. Previous studies addressed this challenge by pol II CTD IP-MS from yeast extracts (Harlen et al., 2016) or through analysis of short CTD peptides for affinity chromatography (Carty et al., 2000; Carty and Greenleaf, 2002; Morris et al., 1999). Here, we completed comparative MS analyses of human proteins bound to unmodified vs. TFIIH- or P-TEFb-phosphorylated full-length (i.e. 52 heptad repeats) mammalian pol II CTD. We identified factors that specifically bind TFIIH- vs. P-TEFb-phosphorylated CTD, in support of the CTD code hypothesis (Buratowski, 2003). A set of phospho-CTD-specific interactions were pursued further in cells using a well-tested chemical genetics strategy to selectively inactivate human CDK7 (Larochelle et al., 2007). Our results reveal expanded roles for CDK7, many of which appear to be metazoan-specific, including regulation of epigenetic modifications (the term epigenetic here refers to post-translational histone modifications). We identify CDK7 as a key regulator of H3K4me3 spreading in human cells, which has been linked to essential RNA processing (Suzuki et al., 2017) and developmental processes in mammals (Dahl et al., 2016; Liu et al., 2016; Zhang et al., 2016). We further describe how selective inhibition of CDK7 kinase activity broadly affects pol II promoter-proximal pausing, termination, and capping enzyme recruitment. Among other things, these results have important implications for developmental diseases linked to CDK7 activity (Coin et al., 1999) and for emerging anti-cancer strategies that target CDK7 (Kwiatkowski et al., 2014).
Results
Targeted proteomics identifies factors bound to unmodified vs. TFIIH-phosphorylated pol II CTD
We purified GST-tagged, full-length murine pol II CTD (Figure 1A) and incubated it with ATP and purified human TFIIH (Figure 1B) to make the hyper-phosphorylated form (Figure 1C). The murine pol II CTD varies from the human CTD at just a single residue within the 52 heptad repeats (T4A in repeat 38). Using antibodies against Ser2- or Ser5-phosphorylated CTD, the TFIIH-phosphorylated CTD showed evidence for Ser5 but not Ser2 CTD phosphorylation (Figure 1D).
Figure 1.

Pol II CTD phosphorylation by human TFIIH. (A) Coomassie-stained gel of purified, full-length murine GST-Pol II CTD (GST-CTD). (B) Silver-stained gel of purified human TFIIH. (C) Time course showing TFIIH-dependent phosphorylation of full-length GST-CTD; the single band after 60 minutes suggests a homogeneous phosphorylated state. (D) Western blot corresponding to TFIIH-dependent CTD phosphorylation time course shown in panel C. TFIIH also phosphorylates Ser7, as shown in Figure S3B. See also Figure S1.
Each immobilized affinity resin (TFIIH-phosphorylated CTD or unmodified CTD) was incubated with HeLa nuclear extract (NE), washed, and eluted as outlined in Figure 2A. In each experiment, the NE was supplemented with phosphatase inhibitors to prevent loss of CTD phosphorylation during the incubation (Figure S1). Samples were also treated with Benzonase to ensure that no pol II CTD-associated factors were bound via any nucleic acid tether. Samples eluted from each binding experiment (unmodified CTD: n = 7 biological replicates or TFIIH-phosphorylated CTD: n = 5 biological replicates) were digested with trypsin and worked up for subsequent LC-MS/MS analysis (see Methods). As controls, we completed parallel experiments using GST only as well as experiments with the proximal CTD (heptad repeats 1–24, unmodified or TFIIH-phosphorylated, n = 2 biological replicates each) or distal CTD (heptad repeats 25–52, unmodified or TFIIH-phosphorylated, n = 2 biological replicates each); we also completed experiments in which the unmodified CTD or TFIIH-phosphorylated CTD (proximal, distal, or full-length) was incubated with a HeLa nuclear pellet fraction, which is presumed to represent chromatin-associated material (Wuarin and Schibler, 1994).
Figure 2.

Pol II CTD interactome data. (A) Experimental workflow. (B) Network view generated from MS data and the STRING database (Szklarczyk et al., 2015) for proteins bound to the unmodified pol II CTD. (C) STRING network view of proteins bound to the TFIIH-phosphorylated CTD. All data shown meet a false discovery rate (FDR) q ≤ 0.01 threshold. See also Figure S1, S2.
MS analyses identified dozens of proteins that bound to the full-length unmodified CTD or the TFIIH-phosphorylated CTD (Table S1; Figure 2B, C). Consistent with previous studies, the Mediator complex bound the unmodified CTD but not the TFIIH-phosphorylated CTD (Max et al., 2007; Naar et al., 2002), and subunits that comprise the CDK8/CDK19 module (e.g. CDK8, CCNC, MED12, MED13) were not detected in any of the CTD-bound samples, including the unmodified pol II CTD (Figure 2B, C, Table S2). Factors such as PIN1 and mRNA capping enzymes (CE) were observed only in TFIIH-phosphorylated CTD samples, in agreement with previous results (Ho et al., 1998; Morris et al., 1999; Pillutla et al., 1998). We also observed that the SETD1A/B histone H3K4 methyltransferase complex specifically bound the TFIIH-phosphorylated CTD. Whereas most proteins/protein complexes bound either the unmodified CTD or the phosphorylated CTD (Figure 2B, C; Table S1), an exception was the multi-subunit Integrator complex, which appeared to bind the unmodified and TFIIH-phosphorylated CTD equally well. Comparison of the interactomes of full-length CTD with proximal (heptad repeats 1–24) or distal (repeats 25–52) CTD, showed few differences (Table S1, Figure S2); similarly, few differences were noted from the nuclear pellet vs. nuclear extract results (Table S1, Figure S2).
CDK7 inhibition reduces CTD Ser5 phosphorylation and capping enzyme (CE) recruitment
To follow up on the MS data, we took advantage of a well-characterized human cell line with homozygous mutations in the CDK7 ATP binding site (Larochelle et al., 2007). The CDK7 “analog sensitive” (hereafter called CDK7as) mutant retains CDK7 kinase activity in cells and in vitro; kinase activity is only inhibited upon addition of an ATP analog competitive inhibitor, such as NM-PP1 (Larochelle et al., 2006; Larochelle et al., 2007). Moreover, because NM-PP1 is too bulky to bind wild-type CDK7, it has negligible effects on CDK7 activity in WT cells (Larochelle et al., 2007). Western blotting of extracts from WT and CDK7as HCT116 cells, each treated with the inhibitor NM-PP1, confirmed that whereas total pol II remained unchanged (Figure S3), global levels of Ser-5 phosphorylated CTD decreased about 50% in CDK7as cells (Figure 3A), in agreement with previous results (Larochelle et al., 2007). To monitor genome-wide Ser5 phosphorylation of pol II, we performed ChIP-Seq in WT and CDK7as cells, each treated with NM-PP1. These experiments showed that CDK7 inhibition diminished CTD-Ser5-P near the TSS at thousands of genes relative to the WT control (Figure 3B). This reduction corresponded to reduced CTD phosphorylation after normalizing to total pol II ChIP signal (Figure 3C).
Figure 3.

Inhibition of CDK7 reduces CTD Ser5 phosphorylation and CE (RNGTT) recruitment. (A) Quantitative western data showing global phospho-Ser5 CTD levels in wild-type (WT) and CDK7as cells. Top: representative data with increasing total protein (3, 6, 12 micrograms); bottom: quantitation (± s.e.m) from n=3 technical replicates taken from nuclear extracts prepared from NM-PP1-treated WT or CDK7as cells, normalized to TBP. (B) Metaplots of mean anti-Pol II CTD Ser5-Phospho ChIP-Seq signals (normalized to yeast spike-in) in WT and CDK7as cells, each treated with NM-PP1 inhibitor (10 μM, 24h). Results are plotted for 100-base bins −2000 to +2000 bp from the TSS of highly expressed genes separated by over 2kb. (C) Metaplots of mean pol II CTD Ser5-Phospho ChIP signals normalized to total pol II in WT and CDK7as cells, each treated with NM-PP1, as in B. (D) UCSC genome browser screenshot of anti-capping enzyme (CE) ChIP-Seq signals at EEF1A1 gene in WT and CDK7as cells, each treated with NM-PP1, as in B. Blue vertical line corresponds to the poly(A) site. (E) Metaplots, as in B, for anti-CE ChIP signals in WT and CDK7as cells. 100-base bins are shown in flanking regions −1.5kb to +0.5kb relative to the TSS and −0.5kb to +3.5kb relative to poly(A) site. Gene body regions between +500 relative to the TSS and −500 relative to the poly(A) site were divided into 20 variable-length bins. (F) Metaplots of mean CE ChIP signals normalized to total pol II in WT and CDK7as cells, each treated with NM-PP1, as in B. See also Figure S3.
Because the MS experiments identified CE subunits (RNGTT, CMTR1) specifically in the TFIIH-phosphorylated samples (Figure 2B; Table S2B), we next assessed whether CDK7 kinase activity affected CE recruitment. We performed CE ChIP-Seq (anti-RNGTT) in WT and CDK7as cells, each treated with NM-PP1. In agreement with our previous study of selected genes (Glover-Cutter et al., 2008), ChIP-Seq revealed that CE was recruited to gene 5′ ends and associated throughout the gene body and downstream of poly(A) sites (Figure 3D). CDK7 inhibition reduced CE occupancy at the 5′ ends of thousands of transcribed genes (Figure 3E). To determine whether this reduction was an indirect effect of diminished pol II levels, we normalized CE ChIP signals to total pol II. The results confirmed the specific reduction of CE recruitment when CDK7 was inhibited (Figure 3F). Together, the data shown in Figure 3 indicate that the human CDK7 kinase functions to enhance CE recruitment to gene 5′ ends and that this function is likely mediated by Ser5 CTD phosphorylation as suggested by in vitro binding studies (Ho and Shuman, 1999).
CDK7 kinase activity regulates H3K4 trimethylation at gene 5′-ends
The proteomics data showed that all subunits of the H3K4 methyltransferases SETD1A/B bound the TFIIH-phosphorylated CTD but not the unmodified CTD (Figure 2B, C; Table S2). To investigate the functional significance of SETD1A/B interaction with TFIIH-phosphorylated CTD, we examined how CDK7 inhibition affected H3K4 trimethylation, genome-wide, using ChIP-Seq in WT and CDK7as cells, each treated with NM-PP1. These experiments revealed that CDK7 inhibition reduced the level of H3K4me3 at over 1200 genes (Table S3) and reduced spreading of H3K4me3 into gene bodies (Figure 4A–D; replicates in Figure S4B–E) while global levels of H3K4me3 and SETD1A largely remained constant (Figure S3). Reduced H3K4me3 at these genes was not due to indirect effects from reduced total histone H3 occupancy (Figure 4E) or reduced pol II occupancy, as shown by anti-pol II ChIP-Seq (Figure S6E). Interestingly, CDK7 inactivation appeared to reduce H3K4-trimethylation at nucleosomes downstream of the TSS far more than upstream (Figure 4A). These experiments show that human CDK7 functions to modulate H3K4-trimethylation at gene 5′-ends, and that CDK7 activity regulates H3K4me3 spreading into gene bodies.
Figure 4.

CDK7 inhibition reduces H3K4me3 spreading; TFIIH-phosphorylated CTD stimulates SETD1A/B methyltransferase activity on nucleosomal templates. (A) Metaplots of mean anti-H3K4me3 ChIP-Seq signals in WT and CDK7as cells, each treated with NM-PP1 inhibitor (10 μM, 24h). Shown are data for a subset of genes with strongly reduced H3K4me3 signals upon CDK7 inhibition. (B–D) UCSC genome browser screen shots of anti-H3K4me3 ChIP-Seq signals in WT and CDK7as cells, each treated with NM-PP1 inhibitor, as in A. CDK7 inhibition limits H3K4me3 spreading into gene bodies (red arrows). (E) Metaplots of mean anti-H3 ChIP-Seq signals in WT and CDK7as cells for the subset of genes with strongly reduced H3K4me3 signals shown in panel A. (F) Quantitative western blots show elevated H3K4me3 levels when SETD1A or SETD1B enzyme reactions with nucleosomal substrates were supplemented with TFIIH-phosphorylated CTD vs. equivalent amounts of unmodified pol II CTD (none = no pol II CTD added). Data are from 3 biological replicates (± s.e.m.). See also Figure S3, S4, S5.
TFIIH-phosphorylated CTD activates SETD1A/B methyltransferase activity in vitro
Previous reports have shown that the phosphorylated pol II CTD can stimulate CE activity (Ho and Shuman, 1999), thereby ensuring that CE is most active when associated with pol II. Similarly, we wondered whether TFIIH-phosphorylated pol II CTD might stimulate SETD1A/B histone methyltransferase activity. To test this idea, we completed enzymatic assays with purified nucleosomal templates, the SETD1A or SETD1B complex, ± pol II CTD in its unmodified or TFIIH-phosphorylated forms. As shown in Figure 4F (see also Figure S5), the activity of both the SETD1A and SETD1B complex was stimulated by the TFIIH-phosphorylated CTD (but not unmodified CTD), suggesting that, upon binding the phosphorylated pol II CTD, the SETD1A/B H3K4 methyltransferases are allosterically activated. Notably, the activation affected only H3K4me3, suggesting the TFIIH-phosphorylated CTD specifically activates H3K4 trimethylation from mono- or di-methylated intermediates (Figure S5).
CDK7 inhibition increases pol II pausing index and delays termination
Human CDK7 activity has been implicated in both positive and negative control of early pol II elongation on several genes in cells and in vitro (Glover-Cutter et al., 2009; Kelso et al., 2014; Larochelle et al., 2012; Nilson et al., 2015), but its effects have yet to be examined genome-wide. We carried out ChIP-Seq experiments to determine how pol II occupancy was affected by CDK7 inhibition. Biological replicate experiments show that CDK7 inhibition caused a widespread increase in pol II occupancy at the TSS relative to gene bodies. This is evident from inspection of individual genes (Figure 5A–C, replicates in Figure S6A–C) and in meta-plots of thousands of genes (Figure 5D). These results demonstrated that a major genome-wide effect of CDK7 inhibition is increased pol II pausing index (the ratio of pol II density at the promoter/gene body), as shown in Figure 5E (replicate in Figure S6D). Consistent with enhanced pol II pausing upon CDK7 inhibition, anti-histone H3 ChIP-seq demonstrated greater displacement of nucleosomes from the TSS (Figure 4E and Figure S4A) (Gilchrist et al., 2008). Although CDK7 inhibition did not reduce pol II occupancy at most promoters, we found that it markedly diminished recruitment to super-enhancers in CDK7as cells (Figure S6F). This supports the concept that expression of genes driven by super-enhancers is highly dependent on CDK7 (Kwiatkowski et al., 2014).
Figure 5.

CDK7 inhibition increases pol II pause index and alters pol II occupancy at gene 3′-ends. (A–C) UCSC genome browser screenshots of anti-pol II ChIP-Seq signals (normalized to yeast spike-in) in WT and CDK7as cells, each treated with NM-PP1. (D) Metaplots of mean anti-pol II ChIP-Seq signals (normalized to total number of mapped reads) in WT and CDK7as cells (as in A–C), each treated with NM-PP1 inhibitor for well-expressed genes. (E) CDK7 inhibition increases pol II pausing index. Cumulative index plots of pausing index (log2 pol II promoter density/pol II gene body density; promoter defined as −30 to +300 bases relative to TSS and gene body as +301 to poly(A) site), calculated from anti-pol II ChIP signals in WT and CDK7as cells, each treated with NM-PP1. Rightward shift curve in CDK7as cells noted by red arrow. P-value calculated by two-sided Kolmogorov-Smirnov test. (F) THZ1 increases pol II pausing index. Cumulative index plots (as in E) for anti-pol II ChIP-Seq of HCT116 cells treated with THZ1 (1 μM) or DMSO for 1 hr. Slight rightward shift in THZ1-treated cells noted by red arrow. (G) UCSC genome browser screenshot of anti-pol II ChIP-Seq signals in cells treated with THZ1 as in F, or DMSO control. (H) CDK7 inhibition appears to delay transcription termination. Metaplots of mean anti-pol II ChIP-Seq signals (normalized to poly(A) site) in WT and CDK7as cells, each treated with NM-PP1, for well-expressed genes in the region −500 to the poly(A) site (blue vertical line). See also Figure S3, S4, S6, S7.
As an additional control, we investigated whether THZ1, a covalent kinase inhibitor with specificity for CDK7 (Kwiatkowski et al., 2014), similarly affected pol II pausing. HCT116 cells were treated with THZ1 (1 μM) or DMSO for 1 hour and subjected to anti-pol II ChIP-Seq. This showed that the pol II pausing index increased even with this short THZ1 treatment, although the effect was more modest than in CDK7as cells (Figure 5F). Moreover, a redistribution of pol II toward 5′ ends was detected on individual genes upon THZ1 treatment (Figure 5G, Figure S6G–I). The response to THZ1 therefore independently supported the conclusion that CDK7 inhibition generally increases pol II pausing.
The pol II ChIP-Seq data in NM-PP1-treated WT vs. CDK7as cells also revealed that pol II occupancy extended further downstream of the 3′ poly(A) site in CDK7as cells, consistent with delayed transcription termination. This was evident on individual genes (red arrows, Figure 5A–C) and in metagene plots representing thousands of genes (Figure 5H). In sum, these data reveal that human CDK7 kinase activity regulates promoter-proximal pol II pausing and impacts late stages of pol II transcription by delaying termination at gene 3′-ends.
Effects of CDK7 inhibition on mRNA levels
We conducted RNA-Seq analyses of WT and CDK7as cells, each treated with NM-PP1, to determine how global mRNA levels were affected by CDK7 inhibition (Table S4). After 24h NM-PP1 treatment, we observed 1430 up-regulated and 3755 down-regulated genes in CDK7as cells (p < 0.001; Figure S7A). In agreement with impaired termination downstream of genes (Figure 5H), slightly more reads mapped outside the coding region in CDK7as cells, as shown in Figure S7B. As expected, many, but not all, genes with lower (1.5x) Pol II gene body ChIP-Seq signal in CDK7as cells (Table S5) showed reduced mRNA levels (Figure S7C; p < 2.22E−16). We also observed a significant (p < 2.22E−16) overlap between down-regulated genes and those with reduced H3K4 trimethylation at the promoter (Figure S7D). A correlation between mRNA down-regulation and reduced CE recruitment was not observed, which may reflect compensatory mechanisms involving mRNA stability, as reported in yeast upon Kin28 inactivation (Rodriguez-Molina et al., 2016).
The H3K36 methyltransferase SETD2 specifically binds P-TEFb-phosphorylated pol II CTD
CDK9, as part of P-TEFb, phosphorylates the pol II CTD with a specificity distinct from CDK7. To compare/contrast CDK7 vs. CDK9 in the recruitment of factors to the phosphorylated pol II CTD, we completed a series of biochemical and proteomics experiments with the P-TEFb phosphorylated pol II CTD. As with TFIIH, P-TEFb (Figure 6A) efficiently phosphorylated the full-length pol II CTD in vitro to generate a single super-shifted, hyper-phosphorylated CTD (Figure 6B). Unlike the TFIIH-modified CTD, western blot experiments showed both Ser2- and Ser5-phosphorylation with P-TEFb, consistent with previous observations (Figure 6C) (Czudnochowski et al., 2012).
Figure 6.

The P-TEFb phosphorylated CTD interactome. (A) Coomassie-stained gel of P-TEFb. (B) Time course showing P-TEFb-dependent phosphorylation of full-length GST-CTD. This phosphorylated product was used for binding assays and MS analyses. (C) Western blot data corresponding to P-TEFb-dependent CTD phosphorylation time course shown in panel B. P-TEFb also phosphorylates Ser7, as shown in Figure S3B. (D) MS experimental workflow. (E) STRING plot summary of MS data from proteins bound to the P-TEFb-phosphorylated CTD. All data shown meet a false discovery rate (FDR) q ≤ 0.01 threshold. See also Figure S1, S2.
Using the same protocol described for the TFIIH-phosphorylated pol II CTD (Figure 6D), we isolated factors bound to the P-TEFb-modified pol II CTD (Figure S1E, F) and identified them with LC-MS/MS. The data are summarized in Table S1 (n = 3 biological replicates), which include experiments with the proximal (repeats 1–24) and distal (repeats 25–52) portions of the pol II CTD (n = 2 biological replicates each; Table S1, Figure S2) and the nuclear pellet material (Table S1, Figure S2). Although distinct proteins bound P-TEFb vs. TFIIH-phosphorylated CTD (Table S6), many factors were shared. For example, PCIF, PIN1, and subunits of the SETD1A/B, PAF1, Integrator, and CE each appeared to bind the TFIIH-modified or P-TEFb-modified CTD equally well (Figure 6E). One clear exception was the SETD2 protein, which was not detected in any TFIIH-phosphorylated CTD binding experiment, but was abundant in the P-TEFb-phosphorylated CTD sample (Figure 6E and Table S6).
CDK7 inhibition reduces CTD Ser2 phosphorylation and displaces H3K36me3 toward gene 3′-ends
CDK7 activates the P-TEFb kinase (CDK9) via T-loop phosphorylation (Larochelle et al., 2012); therefore, SETD2 binding to the CTD in a P-TEFb-dependent manner suggested that H3K36 methylation could potentially be modulated by CDK7 kinase activity. We probed pol II CTD Ser2 phosphorylation and found that global levels of Ser2 CTD phosphorylation decreased about 30–40% in CDK7as cells vs. WT controls (Figure 7A) consistent with previous results (Larochelle et al., 2012). Anti-Phospho-Ser2 CTD ChIP-Seq in WT and CDK7as cells, each treated with the inhibitor NM-PP1, revealed a widespread reduction in the Ser2-P CTD ChIP signal at the 3′ end of genes relative to the 5′ end (Figure 7B) in CDK7as cells. These data also revealed an extension of the Ser2-P signal into 3′ flanking regions (Figure 7C) in CDK7as cells, consistent with delayed termination as shown in Figure 5A–C, H.
Figure 7.

CDK7 inhibition reduces pol II CTD Ser2 phosphorylation and distributes H3K36me3 toward gene 3′-ends. (A) Quantitative western data showing global phospho-Ser2 CTD levels in wild-type (WT) and CDK7as cells, each treated with NM-PP1. Top: representative data with increasing total protein (3, 6, 12 micrograms); bottom: quantitation (± s.e.m) from n=3 technical replicates taken from nuclear extracts prepared from NM-PP1 treated WT or CDK7as cells, normalized to TBP. (B) Metaplots of mean anti-Pol II CTD Ser2-Phospho ChIP-Seq signals (normalized to yeast spike-in) in WT and CDK7as cells, each treated with NM-PP1 inhibitor. (C) UCSC genome browser screen shot of anti-pol II CTD Ser2-Phospho ChIP-Seq signals on the ACTG1 gene in WT and CDK7as cells (as in B), each treated with NM-PP1 inhibitor. (D) Metaplots of mean anti-H3K36me3 ChIP-Seq signals in WT and CDK7as HCT116 cells, each treated with NM-PP1 inhibitor. (E, F) UCSC genome browser screen shots of anti-H3K36me3 ChIP-Seq signals in WT and CDK7as cells, each treated with NM-PP1 inhibitor. A 3′ shifting of H3K36me3 beyond the poly(A) site is noted by red arrows. See also Figure S3, S4.
The data in Figure 7A–C further support the findings of Fisher et al. that human CDK7 activates CDK9 (Larochelle et al., 2012). Given that SETD2 associated specifically with the P-TEFb-phosphorylated pol II CTD (Figure 6E), we next asked whether CDK7 affected H3K36 trimethylation in cells. Quantitative western blots showed no global change in H3K36me3 levels when CDK7 was inhibited (Figure S3); however, anti-H3K36me3 ChIP-Seq revealed a general re-positioning toward 3′ ends of transcribed genes (Figure 7D–F). Furthermore, we observed that the H3K36me3 mark extended further into 3′ flanking regions, as shown in the metaplots (Figure 7D) and from inspection of individual genes (Figure 7E, F; replicates in Figure S4F, G). This widespread H3K36me3 relocalization was not due to changes in histone H3 occupancy, as shown by anti-H3 ChIP-Seq experiments (Figure S4A), suggesting that human CDK7 activity regulates the deposition of this transcription-coupled chromatin mark.
Discussion
The “CTD code” hypothesis suggests that distinct patterns of post-translational modifications direct the binding of specific factors during different stages of pol II transcription (Buratowski, 2003). By combining cell-based assays (e.g. ChIP-Seq) with biochemical and MS experiments, we were able to link CDK7 kinase-dependent changes in cells to biochemically validated interactions with the pol II CTD. This study advances the field by probing the mammalian pol II CTD interactome using native, full-length 52-heptad repeat phosphorylated CTD substrates and distinguishing TFIIH- vs. P-TEFb-specific effects. The data provide evidence for a CTD code while indicating that it is not entirely kinase-specific. These results are in general accord with recent findings that suggest the phospho-specific CTD code is of restricted complexity (Harlen et al., 2016; Schuller et al., 2016; Suh et al., 2016).
CDK7 is linked to developmental disorders and is a promising target for anti-cancer therapeutics; thus, it is important to understand how CDK7 kinase activity regulates human gene expression. Experiments with the yeast CDK7 homologue, Kin28, have contributed greatly to our understanding of human CDK7 (Hong et al., 2009; Kanin et al., 2007; Komarnitsky et al., 2000; Schroeder et al., 2000; Schwer and Shuman, 2011; Suh et al., 2016); however, transcription regulatory mechanisms are distinct between yeast and humans (Levine et al., 2014). For example, studies with Kin28 cannot address promoter-proximal pausing or CDK7-dependent activation of CDK9, which appear to be metazoan-specific (Larochelle et al., 2012; Mayer et al., 2010). Whereas some of our findings showed conserved functions for human CDK7 (e.g. TFIIH-phosphorylated CTD recruits CE), others were completely the opposite of what could be predicted based upon yeast data (e.g. CDK7-dependent regulation of H3K4me3 spreading) or go beyond what has been done in yeast or other model organisms (e.g. delineation of TFIIH- vs. P-TEFb-phosphorylated CTD interactome or allosteric activation of SETD1A/B by TFIIH-phosphorylated CTD). Some of the key findings and implications of this study are described further below.
CDK7, promoter-proximal pol II pausing, and termination at gene 3′-ends
Our results have revealed functions for human CDK7 in pol II transcription that are mediated, at least in part, by differential factor association with the phosphorylated pol II CTD. We observed a genome-wide increase in the pol II pausing index when CDK7 was inhibited, either by NM-PP1 in CDK7as cells or THZ1 in WT cells (Figure 5). These results are consistent with CDK7-mediated activation of the pause release factor, P-TEFb, by CDK9 T-loop phosphorylation (Larochelle et al., 2012). Interestingly, P-TEFb was observed to interact with the CTD in both its TFIIH-phosphorylated and unphosphorylated forms (Figure 2B, C). Whereas this would not impact the CTD phosphorylation state in our MS or biochemical experiments (e.g. ATP is absent in nuclear extracts), it suggests P-TEFb could associate with pol II at early stages of transcription, including pre-initiation complex assembly. This is consistent with results that suggest promoter-associated P-TEFb remains in a latent state that requires subsequent activation (Larochelle et al., 2012; McNamara et al., 2016). Reduced PAF1 complex association with the pol II CTD in CDK7-inhibited cells might also contribute to the increased pol II pause index. The PAF1 complex regulates pol II promoter-proximal pausing (Chen et al., 2015b; Yu et al., 2015) and subunits of this complex (CDC73, CTR9, PAF1) were enriched in the TFIIH-phosphorylated CTD samples (Figure 2C).
In addition to altered pol II pausing, we observed elevated pol II occupancy beyond poly(A) sites in gene 3′ flanking regions, consistent with delayed termination (Figure 5H). How human CDK7 activity might promote termination is unclear, but it could potentially stimulate cleavage/polyadenylation or slow pol II elongation to facilitate termination by the torpedo mechanism (Fong et al., 2015). CDK7 effects on termination may also be mediated via recruitment of SETD1A/B or PAF1 complexes (Figure 2C) that have both been linked to regulation of pol II termination (Austenaa et al., 2015; Yang et al., 2016). In contrast to CDK7 inhibition in human cells, Kin28 inhibition in yeast showed depleted pol II occupancy at gene 3′-ends, likely due to premature termination (Kim et al., 2010; Rodriguez-Molina et al., 2016). These results suggest distinct regulatory roles for human CDK7 vs. yeast Kin28 at gene 3′-ends, although the precise molecular mechanisms remain to be determined.
Human CDK7 governs CE recruitment
Our targeted proteomics data revealed that the guanylyltransferase/phosphatase (RNGTT) and the 2′-hydroxyl methyltransferase (CMTR1) capping enzymes bound the phosphorylated pol II CTD, suggesting that synthesis of cap0 and cap1 structures could be coupled through the human pol II CTD. ChIP-Seq experiments (anti-RNGTT) revealed a decrease in CE and pol II CTD Ser5 phosphorylation at gene 5′-ends upon CDK7 inhibition (Figure 3), in agreement with results in yeast (Ho and Shuman, 1999; Komarnitsky et al., 2000; Schroeder et al., 2000; Schwer and Shuman, 2011; Viladevall et al., 2009). Our findings with human CDK7 suggest a conserved mechanism whereby pol II CTD Ser5 phosphorylation promotes CE recruitment but leaves open the possibility that CDK7 may also antagonize a CE-inhibitory factor (Nilson et al., 2015). Combined with ChIP-Seq data that link CDK7 kinase activity to pol II pausing (Figure 3), it appears that human CDK7 may control a proposed checkpoint that coordinates co-transcriptional capping with the pol II transition to productive elongation (Mandal et al., 2004; Pei and Shuman, 2002).
CDK7 regulates H3K4me3 spreading at gene 5′-ends
CDK7 inhibition markedly reduced spreading of H3K4me3 into gene bodies (Figure 4). We also observed that the CDK7 effect on H3K4me3 spreading was asymmetric; that is, CDK7 inhibition reduced H3K4me3 spreading downstream of the TSS more than upstream (Figure 4). Others have noted similar H3K4me3 asymmetry at bidirectional promoters, with increased H3K4me3 (vs. H3K4me2) in the downstream, or sense, direction (Duttke et al., 2015). Interestingly, we observed that the TFIIH-phosphorylated CTD activated SETD1A/B and specifically elevated H3K4me3 levels (vs. H3K4me2) on nucleosomal templates in vitro (Figure S5). Taken together, these findings suggest a mechanism whereby CDK7 may help preferentially establish H3K4me3 marks downstream of bidirectional promoters.
Reduced H3K4me3 spreading upon CDK7 inhibition may involve a combination of CDK7-dependent functions. Because a comprehensive understanding of CDK7 substrates is lacking, we cannot exclude other potential kinase targets. However, our proteomics data and in vitro enzymatic assays provide a mechanistic basis for how CDK7 kinase activity may regulate H3K4me3 spreading that involves both the recruitment and subsequent activation of human SETD1A/B complexes. Results in yeast have linked Set1 binding to Ser5-phosphorylated CTD and Kin28 activity (Ng et al., 2003) and thus support our conclusion that human CDK7 regulates H3K4me3. However, H3K4me3 spreading appears to be regulated by completely different mechanisms in yeast. H3K4 methylation is regulated by the yeast pol II CTD Ser2 kinase Ctk1 (Wood et al., 2007; Xiao et al., 2003), whose human ortholog is CDK12/13. Upon deletion of yeast Ctk1, H3K4me3 spreading was shown to increase (Xiao et al., 2007). Thus, a different kinase, CDK7, regulates H3K4me3 spreading in human cells and kinase inhibition has opposing effects compared with yeast (i.e. deletion of Ctk1 increased spreading in yeast whereas inhibition of CDK7 decreased H3K4me3 spreading in human cells).
Previous studies have shown that H3K4me3 spreading correlates with elevated transcription and high pol II occupancy at the TSS (Chen et al., 2015c). Moreover, H3K4me3 spreading is dynamic and correlates with transcriptional activation of key developmental genes in mouse embryos (Dahl et al., 2016; Liu et al., 2016; Zhang et al., 2016) and with sites of active microRNA processing in human cells (Suzuki et al., 2017). Our results now link CDK7 kinase activity to these biological processes, although further study is needed to precisely define potential CDK7-dependent regulatory roles.
CDK7, P-TEFb, and H3K36me3
The proteomics data revealed that SETD2, which is responsible for global deposition of H3K36me3 (Edmunds et al., 2008), associated with the pol II CTD phosphorylated by P-TEFb, but not TFIIH. SETD2 therefore appears to recognize a P-TEFb-specific CTD phosphorylation pattern. Although SETD2 occupancy cannot be tested directly because reliable ChIP antibodies remain unavailable, we observed a global shift in H3K36me3 distribution toward 3′-ends of transcribed genes upon CDK7 inhibition (Figure 6D–F). Because CDK7 is known to activate P-TEFb via CDK9 T-loop phosphorylation (a mechanism that appears to be metazoan-specific) (Larochelle et al., 2012), we hypothesize that CDK7-dependent changes in H3K36me3 result from reduced CDK9 activity. Consistent with this hypothesis, we observed reduced levels of phospho-Ser2 CTD in CDK7as cells vs. controls using quantitative western blotting and ChIP-Seq (Figure 6A–C). In yeast, H3K36me3 is dependent upon a different pol II CTD kinase, Ctk1 (Xiao et al., 2003), whose human ortholog is CDK12/13. CDK7-dependent regulation of H3K36me3 therefore appears to be specific to metazoans, although downstream chromatin modifications have yet to be investigated in Kin28-inhibited yeast cells.
Concluding remarks
By linking human CDK7 activity to changes in H3K4me3 and H3K36me3 throughout the genome, our results suggest an expanded role for this kinase as an upstream regulator of transcription-associated epigenetic modifications. These findings point to additional mechanistic links with co-transcriptional mRNA processing and DNA methylation, which is a heritable epigenetic mark. Both H3K4me3 and H3K36me3 are implicated in splicing (Kolasinska-Zwierz et al., 2009; Simon et al., 2014; Sims et al., 2007; Spies et al., 2009), and it will be interesting in future studies to determine how CDK7 inhibition may affect mRNA processing. H3K4me3 and H3K36me3 levels also correlate with DNA methylation in opposing ways; H3K4me3 is anti-correlated whereas H3K36me3 directly correlates with DNA methylation (Morselli et al., 2015; Simon et al., 2014). In fact, the DNA methyltransferase DNMT3A associates with H3K36me3 and mutations in SETD2 induce regions of DNA hypo-methylation (Cancer Genome Atlas Research, 2013). Our results also have important implications for emerging anti-cancer therapeutic strategies that target CDK7 (Kwiatkowski et al., 2014), as transient or prolonged CDK7 inhibition could result in heritable epigenetic changes. Furthermore, developmental diseases such as xeroderma pigmentosum or trichothiodystrophy display reduced CDK7 kinase activity (Coin et al., 1999), and our results suggest that epigenetic changes may contribute to the pathology of these diseases.
Experimental Procedures
Details of cell lines, protein purifications, antibodies, and biochemical assays are provided in Supplemental Experimental Procedures.
Mass spectrometry and data analyses
Samples were separated on a 0.075 × 250mm 1.7uM 130A C18 nanoAcquity column at 300nl/min with a nanoAcquity UPLC (Waters) using mobile phases 0.1%(v/v) formic acid in LCMS water and 0.1%(v/v) formic acid in acetonitrile. The entire tryptic peptide sample was analyzed with an LTQ-Orbitrap (ThermoScientific) over a two hour gradient. All rawfiles were processed using MaxQuant version 1.5.2.8 (Cox and Mann, 2008) searching at 1% FDR at the peptide and protein levels against the uniprot human database reviewed 20150401. MaxQuant intensities were input into the algorithm Significance Analysis of INTeractome (SAINT) (Choi et al., 2012), and interactors with a SAINT score ≥0.5 was input into the STRING database data visualization package (Szklarczyk et al., 2015) to create the STRING diagrams.
ChIP-seq
ChIP from human extracts was completed as described (Kim et al., 2011; Schroeder et al., 2000). Enzymatic steps and size fractionation of libraries were done on AMPure XP SPRI beads (Beckman Coulter Genomics) and sequenced on the Illumina Hi-Seq platform. Reads were mapped to the hg19 UCSC human genome (Feb. 2009) with Bowtie version 0.12.5 (Langmead et al., 2009) (Table S7). We generated bed and wig profiles using 50bp bins and 200bp windows assuming a 180bp fragment size shifting effect. Results were viewed with the UCSC genome browser and metaplots were generated using R. For metaplots of gene bodies, the region between +500 relative to the transcription start site (TSS) and −500 relative to the poly (A) site was divided into 20 variable length bins. Metaplots of human genes were from a list of 5507 well-expressed genes separated from their neighbors by >2kb (Brannan et al., 2012). Metaplots include all genes in common between the datasets for which a minimum ChIP signal was obtained. Unless noted otherwise, mean read counts/bin were plotted in the metaplots.
RNA-Seq library preparation
Ribosomal RNA was removed using the NEB ribosome removal kit and libraries were prepared using the NEB ultra directional kit, with RNA fragmentation for 8 min and 15 PCR cycles. Single-end RNA-Seq of 1×151 were sequenced on an Illumina Hi-Seq with an average of 60 million reads per sample.
RNA-Seq
RNA-Seq reads from three biological replicates of WT CDK7 and CDK7as HCT116 cells treated with 3NM-PP1 (10 μM, 24hr) were mapped using HISAT2 (Pertea et al., 2016) to the Ensembl Grch37 human transcriptome (Table S7). Read coverage (number of overlapping reads per bp) was calculated over non-overlapping exons. The exon counts were aggregated for each gene to build a gene counting table. Using edgeR (Robinson et al., 2010), we tested for significant differences in mRNA expression between CDK7 and CDK7as cells. GLM-based tag-wise dispersions were estimated with a default option and used in the likelihood ratio test. The significance threshold, p-value = 0.001 was used. The significance of overlaps between regulated mRNAs and those genes with altered pol II and H3K4me3 density was calculated by hypergeometric test using phyper function in R with an assumption of 40k total genes.
Supplementary Material
Table S1. All MS data. Related to Figure 2 and Figure 6.
Table S2. (A) List of proteins that differentially bind unmodified CTD vs. TFIIH-phosphorylated CTD. (B) List of proteins that differentially bind TFIIH-phosphorylated CTD vs. unmodified CTD. Related to Figure 2.
Table S3. Genes with 1.5X reduction in H3K4me3 ChIP-Seq signal in the region +1 to +2kb relative to the TSS in CDK7as cells relative to WT, each treated with NM-PP1, in replicate experiments. Related to Figure 4.
Table S4. RNA-Seq data. Related to Figure 4, 5, S7.
Table S5. Genes with1.5X reduction or increase in pol II ChIP-Seq signal in the gene body region in CDK7as relative to WT (each treated with NM-PP1) in replicate experiments. Related to Figure 5.
Table S6. List of proteins that differentially bind P-TEFb-phosphorylated CTD vs. TFIIH-phosphorylated CTD. Related to Figure 6.
Table S7. Summary of ChIP-Seq and RNA-Seq experiments. Related to Figure 3, 4, 5, 7, S7.
Acknowledgments
We thank R. Fisher for HCT116 CDK7as cells, R. Tjian for ERCC3 antibodies, and J. Goodrich for critical reading of the manuscript; we thank the UC Cancer Center for STR analysis and K. Diener and B. Gao at the UC-Denver sequencing facility. We thank K. Luger and U. Muthurajan for providing purified nucleosome templates. This work was supported by the NSF (MCB-1244175 to DJT) and NIH grants GM063873 and R35GM118051 (to DLB), GM110064 (to DJT), T32 GM07135 (to CCE), and T32 GM08759 (to BLA).
Footnotes
Accession numbers. RNA-Seq and ChIP-Seq data have been deposited under accession code GSE100040. The accession number for the proteomics data reported in this paper is ProteomeXchange PRIDE: PXD006485.
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and seven tables.
Author Contributions
C.C.E. designed, performed and helped analyze all proteomics-related experiments, and generated key reagents. B.E. performed and helped analyze all ChIP-Seq experiments and generated key reagents. B.L.A. completed enzyme assays and quantitative westerns. M.A.A. completed RNA-Seq experiments. H.K. N.F. and J.R.J. assisted with data analysis. K.L. and A.S. provided key reagents. R.D.D. W.M.O. D.L.B. and D.J.T. designed experiments and analyzed data. D.L.B. and D.J.T. wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akhtar MS, Heidemann M, Tietjen JR, Zhang DW, Chapman RD, Eick D, Ansari AZ. TFIIH kinase places bivalent marks on the carboxy-terminal domain of RNA polymerase II. Mol Cell. 2009;34:387–393. doi: 10.1016/j.molcel.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austenaa LM, Barozzi I, Simonatto M, Masella S, Della Chiara G, Ghisletti S, Curina A, de Wit E, Bouwman BA, de Pretis S, et al. Transcription of Mammalian cis-Regulatory Elements Is Restrained by Actively Enforced Early Termination. Mol Cell. 2015;60:460–474. doi: 10.1016/j.molcel.2015.09.018. [DOI] [PubMed] [Google Scholar]
- Brannan K, Kim H, Erickson B, Glover-Cutter K, Kim S, Fong N, Kiemele L, Hansen K, Davis R, Lykke-Andersen J, et al. mRNA decapping factors and the exonuclease Xrn2 function in widespread premature termination of RNA polymerase II transcription. Mol Cell. 2012;46:311–324. doi: 10.1016/j.molcel.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratowski S. The CTD code. Nat Struct Biol. 2003;10:679–680. doi: 10.1038/nsb0903-679. [DOI] [PubMed] [Google Scholar]
- Buratowski S. Progression through the RNA polymerase II CTD cycle. Mol Cell. 2009;36:541–546. doi: 10.1016/j.molcel.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carty SM, Goldstrohm AC, Sune C, Garcia-Blanco MA, Greenleaf AL. Protein-interaction modules that organize nuclear function: FF domains of CA150 bind the phosphoCTD of RNA polymerase II. Proc Natl Acad Sci U S A. 2000;97:9015–9020. doi: 10.1073/pnas.160266597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carty SM, Greenleaf AL. Hyperphosphorylated C-terminal repeat domain-associating proteins in the nuclear proteome link transcription to DNA/chromatin modification and RNA processing. Mol Cell Proteomics. 2002;1:598–610. doi: 10.1074/mcp.m200029-mcp200. [DOI] [PubMed] [Google Scholar]
- Chen F, Gao X, Shilatifard A. Stably paused genes revealed through inhibition of transcription initiation by the TFIIH inhibitor triptolide. Genes Dev. 2015a;29:39–47. doi: 10.1101/gad.246173.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FX, Woodfin AR, Gardini A, Rickels RA, Marshall SA, Smith ER, Shiekhattar R, Shilatifard A. PAF1, a Molecular Regulator of Promoter-Proximal Pausing by RNA Polymerase II. Cell. 2015b;162:1003–1015. doi: 10.1016/j.cell.2015.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Chen Z, Wu D, Zhang L, Lin X, Su J, Rodriguez B, Xi Y, Xia Z, Chen X, et al. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nature genetics. 2015c;47:1149–1157. doi: 10.1038/ng.3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H, Liu G, Mellacheruvu D, Tyers M, Gingras AC, Nesvizhskii AI. Analyzing protein-protein interactions from affinity purification-mass spectrometry data with SAINT. Current protocols in bioinformatics/editoral board, Andreas D. Baxevanis … [et al.] 2012;Chapter 8(Unit8):15. doi: 10.1002/0471250953.bi0815s39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coin F, Bergmann E, Tremeau-Bravard A, Egly JM. Mutations in XPB and XPD helicases found in xeroderma pigmentosum patients impair the transcription function of TFIIH. EMBO J. 1999;18:1357–1366. doi: 10.1093/emboj/18.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Czudnochowski N, Bosken CA, Geyer M. Serine-7 but not serine-5 phosphorylation primes RNA polymerase II CTD for P-TEFb recognition. Nature communications. 2012;3:842. doi: 10.1038/ncomms1846. [DOI] [PubMed] [Google Scholar]
- Dahl JA, Jung I, Aanes H, Greggains GD, Manaf A, Lerdrup M, Li G, Kuan S, Li B, Lee AY, et al. Broad histone H3K4me3 domains in mouse oocytes modulate maternal-to-zygotic transition. Nature. 2016;537:548–552. doi: 10.1038/nature19360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duttke SH, Lacadie SA, Ibrahim MM, Glass CK, Corcoran DL, Benner C, Heinz S, Kadonaga JT, Ohler U. Human promoters are intrinsically directional. Mol Cell. 2015;57:674–684. doi: 10.1016/j.molcel.2014.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmunds JW, Mahadevan LC, Clayton AL. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 2008;27:406–420. doi: 10.1038/sj.emboj.7601967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong N, Brannan K, Erickson B, Kim H, Cortazar MA, Sheridan RM, Nguyen T, Karp S, Bentley DL. Effects of Transcription Elongation Rate and Xrn2 Exonuclease Activity on RNA Polymerase II Termination Suggest Widespread Kinetic Competition. Mol Cell. 2015;60:256–267. doi: 10.1016/j.molcel.2015.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist DA, Nechaev S, Lee C, Ghosh SK, Collins JB, Li L, Gilmour DS, Adelman K. NELF-mediated stalling of pol II can enhance gene expression by blocking promoter-proximal nucleosome assembly. Genes & Development. 2008;22:1921–1933. doi: 10.1101/gad.1643208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover-Cutter K, Kim S, Espinosa JM, Bentley DL. RNA polymerase II pauses and associates with pre-mRNA processing factors at both ends of genes. Nat Struct Mol Biol. 2008;15:71–78. doi: 10.1038/nsmb1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover-Cutter K, Larochelle S, Erickson B, Zhang C, Shokat K, Fisher RP, Bentley DL. TFIIH-associated Cdk7 kinase functions in phosphorylation of C-terminal domain Ser7 residues, promoter-proximal pausing, and termination by RNA polymerase II. Mol Cell Biol. 2009;29:5455–5464. doi: 10.1128/MCB.00637-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlen KM, Churchman LS. The code and beyond: transcription regulation by the RNA polymerase II carboxy-terminal domain. Nat Rev Mol Cell Biol. 2017;18:263–273. doi: 10.1038/nrm.2017.10. [DOI] [PubMed] [Google Scholar]
- Harlen KM, Trotta KL, Smith EE, Mosaheb MM, Fuchs SM, Churchman LS. Comprehensive RNA Polymerase II Interactomes Reveal Distinct and Varied Roles for Each Phospho-CTD Residue. Cell reports. 2016;15:2147–2158. doi: 10.1016/j.celrep.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidemann M, Hintermair C, Voss K, Eick D. Dynamic phosphorylation patterns of RNA polymerase II CTD during transcription. Biochim Biophys Acta. 2013;1829:55–62. doi: 10.1016/j.bbagrm.2012.08.013. [DOI] [PubMed] [Google Scholar]
- Ho CK, Shuman S. Distinct roles for CTD Ser-2 and Ser-5 phosphorylation in the recruitment and allosteric activation of mammalian mRNA capping enzyme. Mol Cell. 1999;3:405–411. doi: 10.1016/s1097-2765(00)80468-2. [DOI] [PubMed] [Google Scholar]
- Ho CK, Sriskanda V, McCracken S, Bentley D, Schwer B, Shuman S. The guanylyltransferase domain of mammalian mRNA capping enzyme binds to the phosphorylated carboxyl-terminal domain of RNA polymerase II. J Biol Chem. 1998;273:9577–9585. doi: 10.1074/jbc.273.16.9577. [DOI] [PubMed] [Google Scholar]
- Hong SW, Hong SM, Yoo JW, Lee YC, Kim S, Lis JT, Lee DK. Phosphorylation of the RNA polymerase II C-terminal domain by TFIIH kinase is not essential for transcription of Saccharomyces cerevisiae genome. Proc Natl Acad Sci U S A. 2009;106:14276–14280. doi: 10.1073/pnas.0903642106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanin EI, Kipp RT, Kung C, Slattery M, Viale A, Hahn S, Shokat KM, Ansari AZ. Chemical inhibition of the TFIIH-associated kinase cdk7/kin28 does not impair global mRNA synthesis. Proc Natl Acad Sci U S A. 2007;104:5812–5817. doi: 10.1073/pnas.0611505104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelso TW, Baumgart K, Eickhoff J, Albert T, Antrecht C, Lemcke S, Klebl B, Meisterernst M. Cyclin-dependent kinase 7 controls mRNA synthesis by affecting stability of preinitiation complexes, leading to altered gene expression, cell cycle progression, and survival of tumor cells. Mol Cell Biol. 2014;34:3675–3688. doi: 10.1128/MCB.00595-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Erickson B, Luo W, Seward D, Graber JH, Pollock DD, Megee PC, Bentley DL. Gene-specific RNA polymerase II phosphorylation and the CTD code. Nat Struct Mol Biol. 2010;17:1279–1286. doi: 10.1038/nsmb.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim H, Fong N, Erickson B, Bentley DL. Pre-mRNA splicing is a determinant of histone H3K36 methylation. Proc Natl Acad Sci U S A. 2011;108:13564–13569. doi: 10.1073/pnas.1109475108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizer KO, Phatnani HP, Shibata Y, Hall H, Greenleaf AL, Strahl BD. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol Cell Biol. 2005;25:3305–3316. doi: 10.1128/MCB.25.8.3305-3316.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolasinska-Zwierz P, Down T, Latorre I, Liu T, Liu XS, Ahringer J. Differential chromatin marking of introns and expressed exons by H3K36me3. Nature genetics. 2009;41:376–381. doi: 10.1038/ng.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarnitsky P, Cho EJ, Buratowski S. Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 2000;14:2452–2460. doi: 10.1101/gad.824700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak H, Lis JT. Control of transcriptional elongation. Annual review of genetics. 2013;47:483–508. doi: 10.1146/annurev-genet-110711-155440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511:616–620. doi: 10.1038/nature13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larochelle S, Amat R, Glover-Cutter K, Sanso M, Zhang C, Allen JJ, Shokat KM, Bentley DL, Fisher RP. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat Struct Mol Biol. 2012;19:1108–1115. doi: 10.1038/nsmb.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larochelle S, Batliner J, Gamble MJ, Barboza NM, Kraybill BC, Blethrow JD, Shokat KM, Fisher RP. Dichotomous but stringent substrate selection by the dual-function Cdk7 complex revealed by chemical genetics. Nat Struct Mol Biol. 2006;13:55–62. doi: 10.1038/nsmb1028. [DOI] [PubMed] [Google Scholar]
- Larochelle S, Merrick KA, Terret ME, Wohlbold L, Barboza NM, Zhang C, Shokat KM, Jallepalli PV, Fisher RP. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol Cell. 2007;25:839–850. doi: 10.1016/j.molcel.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Skalnik DG. Wdr82 is a C-terminal domain-binding protein that recruits the Setd1A Histone H3-Lys4 methyltransferase complex to transcription start sites of transcribed human genes. Mol Cell Biol. 2008;28:609–618. doi: 10.1128/MCB.01356-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M, Cattoglio C, Tjian R. Looping back to leap forward: transcription enters a new era. Cell. 2014;157:13–25. doi: 10.1016/j.cell.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wang C, Liu W, Li J, Li C, Kou X, Chen J, Zhao Y, Gao H, Wang H, et al. Distinct features of H3K4me3 and H3K27me3 chromatin domains in pre-implantation embryos. Nature. 2016;537:558–562. doi: 10.1038/nature19362. [DOI] [PubMed] [Google Scholar]
- Mandal SS, Chu C, Wada T, Handa H, Shatkin AJ, Reinberg D. Functional interactions of RNA-capping enzyme with factors that positively and negatively regulate promoter escape by RNA polymerase II. Proc Natl Acad Sci U S A. 2004;101:7572–7577. doi: 10.1073/pnas.0401493101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Max T, Sogaard M, Svejstrup JQ. Hyperphosphorylation of the C-terminal repeat domain of RNA polymerase II facilitates dissociation of its complex with mediator. J Biol Chem. 2007;282:14113–14120. doi: 10.1074/jbc.M701345200. [DOI] [PubMed] [Google Scholar]
- Mayer A, Lidschreiber M, Siebert M, Leike K, Soding J, Cramer P. Uniform transitions of the general RNA polymerase II transcription complex. Nat Struct Mol Biol. 2010;17:1272–1278. doi: 10.1038/nsmb.1903. [DOI] [PubMed] [Google Scholar]
- McNamara RP, Reeder JE, McMillan EA, Bacon CW, McCann JL, D’Orso I. KAP1 Recruitment of the 7SK snRNP Complex to Promoters Enables Transcription Elongation by RNA Polymerase II. Mol Cell. 2016;61:39–53. doi: 10.1016/j.molcel.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris DP, Phatnani HP, Greenleaf AL. Phospho-carboxyl-terminal domain binding and the role of a prolyl isomerase in pre-mRNA 3′-End formation. J Biol Chem. 1999;274:31583–31587. doi: 10.1074/jbc.274.44.31583. [DOI] [PubMed] [Google Scholar]
- Morselli M, Pastor WA, Montanini B, Nee K, Ferrari R, Fu K, Bonora G, Rubbi L, Clark AT, Ottonello S, et al. In vivo targeting of de novo DNA methylation by histone modifications in yeast and mouse. eLife. 2015;4:e06205. doi: 10.7554/eLife.06205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naar AM, Taatjes DJ, Zhai W, Nogales E, Tjian R. Human CRSP interacts with RNA polymerase II CTD and adopts a specific CTD-bound conformation. Genes Dev. 2002;16:1339–1344. doi: 10.1101/gad.987602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, Robert F, Young RA, Struhl K. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell. 2003;11:709–719. doi: 10.1016/s1097-2765(03)00092-3. [DOI] [PubMed] [Google Scholar]
- Nilson KA, Guo J, Turek ME, Brogie JE, Delaney E, Luse DS, Price DH. THZ1 Reveals Roles for Cdk7 in Co-transcriptional Capping and Pausing. Mol Cell. 2015;59:576–587. doi: 10.1016/j.molcel.2015.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Y, Shuman S. Interactions between fission yeast mRNA capping enzymes and elongation factor Spt5. J Biol Chem. 2002;277:19639–19648. doi: 10.1074/jbc.M200015200. [DOI] [PubMed] [Google Scholar]
- Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nature protocols. 2016;11:1650–1667. doi: 10.1038/nprot.2016.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillutla RC, Yue Z, Maldonado E, Shatkin AJ. Recombinant human mRNA cap methyltransferase binds capping enzyme/RNA polymerase IIo complexes. J Biol Chem. 1998;273:21443–21446. doi: 10.1074/jbc.273.34.21443. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Molina JB, Tseng SC, Simonett SP, Taunton J, Ansari AZ. Engineered Covalent Inactivation of TFIIH-Kinase Reveals an Elongation Checkpoint and Results in Widespread mRNA Stabilization. Mol Cell. 2016;63:433–444. doi: 10.1016/j.molcel.2016.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R, Adamczewski JP, Seroz T, Vermeulen W, Tassan JP, Schaeffer L, Nigg EA, Hoeijmakers JH, Egly JM. The MO15 cell cycle kinase is associated with the TFIIH transcription-DNA repair factor. Cell. 1994;79:1093–1101. doi: 10.1016/0092-8674(94)90039-6. [DOI] [PubMed] [Google Scholar]
- Schneider R, Bannister AJ, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat Cell Biol. 2004;6:73–77. doi: 10.1038/ncb1076. [DOI] [PubMed] [Google Scholar]
- Schroeder SC, Schwer B, Shuman S, Bentley D. Dynamic association of capping enzymes with transcribing RNA polymerase II. Genes Dev. 2000;14:2435–2440. doi: 10.1101/gad.836300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller R, Forne I, Straub T, Schreieck A, Texier Y, Shah N, Decker TM, Cramer P, Imhof A, Eick D. Heptad-Specific Phosphorylation of RNA Polymerase II CTD. Mol Cell. 2016;61:305–314. doi: 10.1016/j.molcel.2015.12.003. [DOI] [PubMed] [Google Scholar]
- Schwer B, Shuman S. Deciphering the RNA polymerase II CTD code in fission yeast. Mol Cell. 2011;43:311–318. doi: 10.1016/j.molcel.2011.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serizawa H, Conaway JW, Conaway RC. Phosphorylation of C-terminal domain of RNA polymerase II is not required in basal transcription. Nature. 1993;363:371–374. doi: 10.1038/363371a0. [DOI] [PubMed] [Google Scholar]
- Simon JM, Hacker KE, Singh D, Brannon AR, Parker JS, Weiser M, Ho TH, Kuan PF, Jonasch E, Furey TS, et al. Variation in chromatin accessibility in human kidney cancer links H3K36 methyltransferase loss with widespread RNA processing defects. Genome research. 2014;24:241–250. doi: 10.1101/gr.158253.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RJ, 3rd, Millhouse S, Chen CF, Lewis BA, Erdjument-Bromage H, Tempst P, Manley JL, Reinberg D. Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol Cell. 2007;28:665–676. doi: 10.1016/j.molcel.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spies N, Nielsen CB, Padgett RA, Burge CB. Biased chromatin signatures around polyadenylation sites and exons. Mol Cell. 2009;36:245–254. doi: 10.1016/j.molcel.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh H, Ficarro SB, Kang UB, Chun Y, Marto JA, Buratowski S. Direct Analysis of Phosphorylation Sites on the Rpb1 C-Terminal Domain of RNA Polymerase II. Mol Cell. 2016;61:297–304. doi: 10.1016/j.molcel.2015.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki HI, Young RA, Sharp PA. Super-Enhancer-Mediated RNA Processing Revealed by Integrative MicroRNA Network Analysis. Cell. 2017;168:1000–1014. e1015. doi: 10.1016/j.cell.2017.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viladevall L, St Amour CV, Rosebrock A, Schneider S, Zhang C, Allen JJ, Shokat KM, Schwer B, Leatherwood JK, Fisher RP. TFIIH and P-TEFb coordinate transcription with capping enzyme recruitment at specific genes in fission yeast. Mol Cell. 2009;33:738–751. doi: 10.1016/j.molcel.2009.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood A, Shukla A, Schneider J, Lee JS, Stanton JD, Dzuiba T, Swanson SK, Florens L, Wash-burn MP, Wyrick J, et al. Ctk complex-mediated regulation of histone methylation by COMPASS. Mol Cell Biol. 2007;27:709–720. doi: 10.1128/MCB.01627-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuarin J, Schibler U. Physical isolation of nascent RNA chains transcribed by RNA polymerase II: evidence for cotranscriptional splicing. Mol Cell Biol. 1994;14:7219–7225. doi: 10.1128/mcb.14.11.7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao T, Hall H, Kizer KO, Shibata Y, Hall MC, Borchers CH, Strahl BD. Phosphorylation of RNA polymerase II CTD regulates H3 methylation in yeast. Genes Dev. 2003;17:654–663. doi: 10.1101/gad.1055503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao T, Shibata Y, Rao B, Laribee RN, O’Rourke R, Buck MJ, Greenblatt JF, Krogan NJ, Lieb JD, Strahl BD. The RNA polymerase II kinase Ctk1 regulates positioning of a 5′ histone methylation boundary along genes. Mol Cell Biol. 2007;27:721–731. doi: 10.1128/MCB.01628-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Li W, Hoque M, Hou L, Shen S, Tian B, Dynlacht BD. PAF Complex Plays Novel Subunit-Specific Roles in Alternative Cleavage and Polyadenylation. PLoS genetics. 2016;12:e1005794. doi: 10.1371/journal.pgen.1005794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoh SM, Lucas JS, Jones KA. The Iws1:Spt6:CTD complex controls cotranscriptional mRNA biosynthesis and HYPB/Setd2-mediated histone H3K36 methylation. Genes Dev. 2008;22:3422–3434. doi: 10.1101/gad.1720008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Yang W, Ni T, Tang Z, Nakadai T, Zhu J, Roeder RG. RNA polymerase II-associated factor 1 regulates the release and phosphorylation of paused RNA polymerase II. Science. 2015;350:1383–1386. doi: 10.1126/science.aad2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Zheng H, Huang B, Li W, Xiang Y, Peng X, Ming J, Wu X, Zhang Y, Xu Q, et al. Allelic reprogramming of the histone modification H3K4me3 in early mammalian development. Nature. 2016;537:553–557. doi: 10.1038/nature19361. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. All MS data. Related to Figure 2 and Figure 6.
Table S2. (A) List of proteins that differentially bind unmodified CTD vs. TFIIH-phosphorylated CTD. (B) List of proteins that differentially bind TFIIH-phosphorylated CTD vs. unmodified CTD. Related to Figure 2.
Table S3. Genes with 1.5X reduction in H3K4me3 ChIP-Seq signal in the region +1 to +2kb relative to the TSS in CDK7as cells relative to WT, each treated with NM-PP1, in replicate experiments. Related to Figure 4.
Table S4. RNA-Seq data. Related to Figure 4, 5, S7.
Table S5. Genes with1.5X reduction or increase in pol II ChIP-Seq signal in the gene body region in CDK7as relative to WT (each treated with NM-PP1) in replicate experiments. Related to Figure 5.
Table S6. List of proteins that differentially bind P-TEFb-phosphorylated CTD vs. TFIIH-phosphorylated CTD. Related to Figure 6.
Table S7. Summary of ChIP-Seq and RNA-Seq experiments. Related to Figure 3, 4, 5, 7, S7.