

Abstract
Brain metastases are the most common fatal complication of systemic cancer, especially of lung (40–50%) and breast (20–30%) cancers. In this era of personalized therapy, there is a critical need to uncover the signaling architecture of brain metastases; however, little is known about what signaling pathways are activated in the context of the brain microenvironment. Using a unique study set of 42 brain metastases from patients with breast or nonsmall cell lung cancer (NSCLC), the phosphorylation/activation states of 128 key signaling proteins involved in cancer signaling were measured in laser capture microdissected tumor epithelium using reverse phase protein microarray (RPMA) technology. Distinct pathway activation subgroups from both breast and lung metastases were underpinned by, among others, ERBB2, AKT, mTOR, EGFR, SMAD, and ERKp38 signaling. Breast cancer metastases showed significantly (p < 0.05) higher activation of the c-ERBB2/IGFR-AKT pathway network compared to NSCLC metastases, whereas NSCLC metastases to the brain exhibited higher relative levels of many members of the EGFR-ERK signaling network. Protein pathway activation mapping using RPMA revealed both the heterogeneity of signaling networks in brain metastases that would require a prior stratification to targeted therapies as well as the requirement of direct analysis of the metastatic lesion.
Keywords: brain metastasis, breast cancer, lung cancer, laser-capture microdissection, protein microarray, proteomics, targeted therapeutics
Graphical abstract
INTRODUCTION
Brain metastases are the most serious and fatal complication of systemic cancer, affecting approximately 170 000 patients/year in the United States, an incidence 10 times higher than primary brain tumors.1,2 The majority of brain metastases arise from lung (40–50%) and breast (20–30%) primary tumors,3,4 and their incidence is rising, mostly due to the use of therapeutic strategies that improve control of systemic extracranial disease, resulting in increased patient survival and thus allowing time for lesions in this sanctuary site to emerge. Many current chemotherapeutic agents are not able to effectively penetrate the blood–brain barrier (BBB), which remains a formidable obstacle for most therapeutic agents in current use. Despite current strategies for treating brain metastases: surgery in the case of operable isolated lesions and whole brain radiation therapy (WBRT) for patients who present with multiple brain metastases, survival prognosis is poor (<1 year).5,6
Metastatic spread of cancer cells is a multistep phenomenon involving molecular and genetic changes described as the “metastatic cascade”.7 According to the “Seed and Soil” theory of Paget,8 there is an organ selectivity based on a successful interaction between the tumor cells (the “seed”) and the microenvironment of the secondary location (the “soil”) which supports the extravasation, survival, and growth or resistance to therapy of the metastatic colony.9,10 A number of recent studies have identified primary tumor expression signatures, either gene or protein-based, that are associated with metastatic potential, in some cases to specific organ sites such as lung, liver, bone, and brain.11–17 These results indicate that certain aggressive tumors may have early “hard-wired” molecular changes that predestine the tumor cells for successful distant metastasis.
Most current strategies involving molecularly targeted therapies used for treating advanced metastatic disease are often based on the assumption that the primary tumor and metastatic lesions are identical at the molecular level and will respond similarly to therapy. However, specific molecular changes have been identified between a variety of primary and metastatic tumors that call this assumption into question.18–24 Consequently, because brain metastasis is often the terminal fatal event for many cancers such as lung and breast cancer, and if the metastatic lesion is driven by different molecular events from the primary tumor, then it is critical that these molecular changes are fully characterized. While recent efforts have focused on molecular analysis of metastatic lesions at the genetic level,18 few studies have focused on a proteomic-based approach to characterize brain metastasis. Moreover, since past analyses of gene and protein expression concordance have revealed little correlation between the two information archives, a direct analysis of protein expression is required.25–27 Because most of the newer classes of molecularly targeted inhibitors act on deranged and hyperactivated protein signaling events, it is necessary to characterize and map the activated signaling architecture of brain metastasis. These signaling events are driven by and through post-translational modifications, principally phosphorylation, which control kinase regulated biochemical events through specific protein–protein interactions.28
Clues about important signaling events in metastatic progression are beginning to emerge, however. Activation of the epidermal growth factor receptor (EGFR) is known to be a significant factor in the development, growth, and metastasis of many types of cancer, including lung and breast cancer. The EGFR signal transduction network plays an important role in multiple tumorigenic processes, contributing to cancer cell proliferation, angiogenesis, and protection from apoptosis, as well as to metastasis.29 There have been reports of higher incidence of brain metastasis for patients receiving trastuzumab therapy targeting the overexpression of HER-2 in breast cancer, probably due to an increase in survival and systemic control of disease allowing for the development and growth of brain metastases where they may be less sensitive to the trastuzumab administered, or that HER-2 positive breast cancers have a greater affinity to metastasize to the brain.30 Other factors, such as high EGFR expression, low CK5/6/19 expression, p53-positivity and low BCL-2 expression, although less widely studied, are also reported to be associated with brain metastases from breast cancer.31,32 Another small molecule, able to cross the BBB and used for the treatment of breast cancer brain metastases is lapatinib, which inhibits the phosphorylation of EGFR, HER2, and downstream signaling proteins.33
While these efforts have begun to characterize protein signaling activation in the context of brain metastasis, a broad-scale mapping of the activated signaling events of brain metastasis has not been realized. In the present study, we analyzed the activated protein signaling architecture in laser capture microdissected (LCM) NSCLC and breast epithelial cells from individual biopsy specimens using reverse phase protein microarray (RPMA) technology15,34,35 to interrogate the activation state of over 100 key signaling proteins known to be involved in tumorigenesis and metastatic progression. RPMAs have emerged as a powerful proteomics tool to quantitatively measure hundreds of proteins and phosphoproteins from tiny input samples, and are an especially useful technology platform for the analysis of clinical specimens. The goals of this study were to use a proteomics based approach to develop a pilot brain metastasis signaling activity knowledge base in the context of two different “seeds” (breast and lung cancer cells) with the same “soil” (brain) and attempt to understand which protein signaling networks are activated in order to rationally identify therapeutic targets and companion diagnostic opportunities for clinical testing.
MATERIAL AND METHODS
Samples, Patient Data, and Laser Capture Microdissection (LCM)
A total of 42 samples (10 breast and 32 lung metastases to the brain) and relevant clinical data were obtained from the Medical College of Virginia Hospital, Virginia Commonwealth University in accordance with protocols approved by the institution’s Institutional Review Board. The snap-frozen samples were embedded in Optimal Cutting Temperature (OCT) cryoprotective material (Sakura Finetek, Torrance, CA) and stored at −80 °C. A number of 8 μm cryostat sections were made of each specimen onto uncharged glass slides and stored at −80 °C. A representative section of each case was stained with hematoxylin and eosin (Sigma-Aldrich, St. Louis, MO), and a board-certified pathologist verified the presence and location of tumor cells within each sample. The frozen section slides were fixed briefly in 70% ethanol, rinsed in water, stained with hematoxylin (Sigma-Aldrich), and dehydrated in an ethanol gradient (70, 95, and 100%) with a final rinse in xylene (Sigma-Aldrich). The sections were left to air-dry briefly prior to LCM. Microdissected cells were obtained using a PixCell IIe instrument (Life Technologies, Carlsbad, CA), and the LCM caps stored at −80 °C prior to microarray construction.
Reverse Phase Protein Microarray Construction
Microdissected samples were lysed directly from the LCM caps using extraction buffer (equal volumes of 2× Tris-Glycine SDS Sample Buffer (Invitrogen, Carlsbad, CA), and T-PER Tissue Protein Extraction Reagent (Pierce/Thermo-Fisher, Rockford, IL) plus 2.5% 2-β-mercaptoethanol (Sigma-Aldrich) at an approximate ratio of 750 cells/μL extraction buffer. The resulting lysates were printed on glass-backed nitrocellulose array slides (Nexterion Slides, Schott, Elmsford, NY) using an Aushon 2470 arrayer equipped with 185-μm solid pins (Aushon Biosystems, Billerica, MA). Each lysate was printed in triplicate on the array. Additionally, we have developed a series of reference lysate standard curves for the arrays that maintain a fixed total protein concentration and vary in analyte concentration by fixed percentages. These reference lysate curves were also printed in triplicate, and served as positive and negative controls for analyte detection as well as to establish the linear dynamic range for analyte detection. Slides were stored desiccated at −20 °C before immunostaining.
Immunostaining
Printed slides were prepared for staining by treating with 1× ReBlot (Millipore, Billerica, MA) for 15 min, followed by 2 × 5 min washes with PBS. Slides were treated for 1 h with blocking solution (1 g of I-block (Applied Biosystems, Bedford, MA), 0.5% Tween-20 in 500 mL of PBS) with constant rocking at room temperature. Immunostaining was completed on an automated slide stainer using a catalyzed signal amplification kit (CSA kit, Dako, Carpinteria, CA). Slides were stained with a library of 128 antibodies against phosphorylated and total proteins involved in cell proliferation, survival, motility, and apoptosis signaling. Each antibody was subjected to validation by immunoblotting prior to use on the reverse phase arrays. Antibody validation criteria included detection of a single band at the appropriate molecular weight in positive control lysates and the absence or significantly reduced presence of a band in negative control lysates by immunoblotting. A complete list of antibodies, sources, and dilutions used for these experiments is available in Supplementary Table 1. Primary antibody was incubated on each array for 30 min at room temperature, and binding was detected using a biotinylated goat anti-rabbit IgG H+L antibody (1:5000) (Vector Laboratories, Burlingame, CA) or a rabbit anti-mouse IgG antibody (1:10) (Dako), followed by streptavidin-conjugated IRDye680 (LI-COR Biosciences, Lincoln, NE). Negative control slides, incubated with biotinylated secondary antibody only, were included in each staining run.
Total protein for each sample was determined by probing arrays selected at regular intervals throughout the print run with SYPRO Ruby blot stain (Invitrogen) according to the manufacturer’s directions. Digital images for each stained slide (antibody and total protein) were collected using a Vidar Revolution 4550 Scanner (Vidar Systems Corporation, Herndon, VA). Scanned images were evaluated for both staining and printing artifacts which interfered with data analysis and affected analytes were repeated as necessary to obtain reliable data. Images were analyzed using MicroVigene v2.9.9.9 (Vigenetech, Carlisle, MA) which performed spot finding followed by local background subtraction from raw spot intensities, negative control intensity subtraction from each spot, replicate averaging, and last, total protein normalization, generating a single data value for each sample. Total protein values used for normalization were obtained in Microvigene as described above except for the elimination of the negative control intensity subtraction and normalization steps. For this study, a threshold staining level for analysis was set at 2 SDs above local background. Additional QA/QC measures for antibody staining included evaluation of reference lysate staining by visual inspection of scanned images and examination of data analysis results for the positive and negative reference lysates. Final data results for the 42 samples analyzed are provided in Supplementary Table 2.
Statistical Methods
The Ward method for two-way unsupervised hierarchical clustering was performed using JMP v5.1 (SAS Institute, Cary, NC). GraphPad Prism v5.02 (GraphPad Software, Inc., La Jolla, CA) was used to generate scatter plots. t testing or Wilcoxon two sample rank sum test using R v2.9.2 (R Foundation for Statistical Computing, Vienna, Austria) was employed to compare values between two groups, depending on normalcy distribution values. P-values < 0.05 were considered statistically significant.
RESULTS
To understand signaling activation networks in the context of two different “seeds” (breast and lung) within the same “soil” (brain), LCM was used to collect pure populations of tumor epithelial cells from the brain metastasis specimens. This provided for accurate cell signaling analysis of metastatic cancer cells within the context of the brain microenvironment. The requirement for LCM to obtain accurate protein pathway information has been recently demonstrated using both colorectal and breast cancer tissue clinical study sets.35,36
The signaling architecture of LCM-procured metastatic breast cancer cells to the brain revealed somewhat heterogeneous signaling activation patterns among the samples in unsupervised hierarchical clustering (Figure 1). Some tumors demonstrated generally higher relative levels of signaling activity across many end points (Figure 1). A subset of tumors had higher relative levels and activation of the ERBB receptor family, particularly phospho- and total ERBB2 as well as downstream end points, suggestive of full pathway activation. However, high relative AKT and ERK activation was not strictly correlated with high relative activity of ERBB signaling (correlation coefficients <0.30), indicating that these downstream proteins were activated either by other upstream events or through direct activation by conformational changes caused by mutations.
Figure 1.

Signaling pathway activation mapping of breast cancer metastases to the brain by unsupervised hierarchical clustering. Clustering analysis of 128 signaling proteins (horizontal axis) whose activation/expression levels determined by RPMA is shown for 10 patient tumor samples obtained by LCM (vertical axis). In the map, red color represents higher relative levels of activity/expression, black represents intermediate levels, and green represents lower relative levels of activity/expression.
Unsupervised hierarchical clustering of metastatic lung cancer cells to the brain (Figure 2A) revealed, much like the breast cancer metastases, heterogeneous patterns of signaling activation among the individual tumors. Five unique subgroups of tumors could be identified based on their similar signaling profiles (Figure 2A, B, Table 1). Clustering analysis of the within group averages for each protein showed that tumors within Group A had high relative levels of phosphorylation of several EGFR sites, such as EGFR Y1068, Y1045, and Y992 as well as total ERBB4/HER4 and downstream substrates such as ACK1 Y857/858 (Figure 2B, Table 1). In the same subgroup, we also found higher relative levels of several proteins involved with DNA damage response pathways and apoptosis, such as p53, CHK2 S33/35, BCL-2 S70, and T56, phosphorylation of the IGF receptor, and higher relative levels of proteins belonging to ERK, AKT and mTOR, signaling pathways. Tumors within Subgroup B had lower relative levels for most of the signaling proteins examined except for EGFR S1046/1047 and VEGFR2 Y951 along with total EGFR, and proteins involved with apoptosis and DNA repair such as cleaved Caspase 3 D175, cleaved Caspase 9 D315, and cleaved PARP D214. Subgroup C tumors were marked by high relative levels of many downstream proteins in major signaling pathways, such as phospho-VEGFR, ERK, mTOR, AKT, and MAPK. Furthermore, high levels of many apoptotic factors were found in this group. The tumors in subgroup D demonstrated low relative levels of activation for most end points but had high relative activation of ErbB family receptors with their downstream substrates, including AKT and ERK. Subgroup E revealed high relative levels of activation for most proteins examined, in particular many tyrosine kinase membrane receptors and their downstream substrates and many proteins related to AKT, ERK, mTOR, PTEN, and integrin signaling pathways.
Figure 2.

Signaling pathway activation mapping of brain metastases from lung cancer by unsupervised hierarchical clustering. Clustering analysis of 128 signaling proteins (horizontal axis) whose activation/expression levels determined by RPMA is shown for 32 patient tumor samples obtained by LCM (vertical axis). In the map, red color represents higher relative levels of activity/expression, black represents intermediate levels, and green represents lower relative levels of activity/expression. (A) Cluster map of all 32 cases with 5 main signaling subgroups indicated (A–E) on the right. (B) Clustering analysis as shown in panel A using the average value for each signaling end point within each subgroup A–E.
Table 1.
Phosphoprotein Activation Patterns in Tumor Signaling Subgroups of Brain Metastases from Lung Cancera
| ERBB signaling | apoptosis/DNA damage response | AKT-mTOR signaling | AMPK signaling | MAPK signaling | IGFR signaling | VEGFR signaling | |
|---|---|---|---|---|---|---|---|
| Group A | EGFR Y1068↑ | p53↑ | mTOR S2481↔ | IGF1R-IR | |||
| EGFR Y1045↑ | BCL-2 T56↑ | p70S6K S371↑ | Y1131–Y1146↑ | ||||
| EGFR Y992↑ | BCL-2 S70↑ | IGF1R-IR | |||||
| Total ERBB4↑ | Y1135/6-Y1150/1↑ | ||||||
| ACK1 Y857/858↑ | IRS1 S612↑ | ||||||
| Group B | Total EGFR↑ | Cleaved Caspase 3 D175↑ | mTOR S2481↔ | ELK1 S383↑ | VEGFR2 Y951↑ | ||
| EGFR | Cleaved Caspase 9 D315↑ | mTOR S2448↑ | |||||
| S1046/1047↑ | Cleaved PARP D214↑ p53↑ | ||||||
| Group C | Total ERBB2↑ | Cleaved Caspase 3 D175↑ | mTOR S2481↔ | AMPKα S485↑ | MEK1/2 S217/221↑ | VEGFR2 Y951↑ | |
| EGFR Y1148↔ | Cleaved Caspase 9 D330↑ | mTOR S2448↑ | LKB1 S334↑ | ERK1/2 T202/Y204↑ | VEGFR2 Y996↑ | ||
| EGFR Y1173↔ | Cleaved PARP D214↑ | p38 MAPK T180/Y182↑ | VEGFR2 | ||||
| BAD S136↑ | CREB S133↑ | Y1175↑ | |||||
| BAD S155↑ | |||||||
| Group D | ERBB2 Y1248↑ | Cleaved Caspase 9 D315↑ | mTOR S2481↔ | AMPKα S485↑ | A-RAF S299↑ | VEGFR2 Y996↑ | |
| Total ERBB2↑ | mTOR S2448↔ | AMPKβ S108↑ | MEK1/2 S217/221↑ | ||||
| EGFR Y1148↑ | LKB1 S334↑ | ERK1/2 T202/Y204↑ | |||||
| EGFR Y1173↑ | p38 MAPK T180/Y182↑ | ||||||
| ELK1 S383↑ | |||||||
| CREB S133↔ | |||||||
| Group E | EGFR Y1068↑ | Cleaved Caspase 3 D175↑ | AKT T308↑ | AMPKα S485↑ | A-RAF S299↑ | IGF1R-IR | |
| EGFR Y1045↑ | Cleaved Caspase 9 D315↑ | AKT S473↑ | AMPKβ S108↑ | B-RAF S445↑ | Y1131–Y1146↑ | ||
| EGFR Y992↑ | Cleaved Caspase 9 D330↑ | mTOR S2481↑ | LKB1 S334↑ | MEK1/2 S217/221↑ | IGF1R-IR | ||
| EGFR Y1148↑ | p53↑ | mTOR S2448↑ | Y1135/6-Y1150/1↑ | ||||
| EGFR Y1173↑ | CHK2 S33/35↑ | PTEN S380↑ | IRS1 S612↑ | ||||
| ERBB3 Y1197↑ | BCL-2 T56↑ | p70S6K S371↑ | |||||
| ERBB3 Y1289↑ | BCL-2 S70↑ | p70S6K T389↑ | |||||
| Total ERBB4↑ | BAD S112↑ | ||||||
| Total ERBB3↑ | BAD S136↑ | ||||||
| Total ERBB2↑ | BAD S155↑ |
↑ = high relative expression in a subgroup; ↔ = intermediate relative expression in a subgroup.
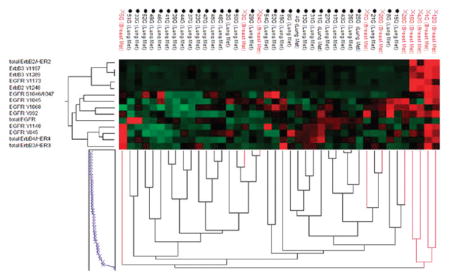
It is believed that many primary lung cancers tend to be driven predominantly by EGFR signaling,37–39 whereas breast cancers are more commonly driven by signaling through the HER2 receptor pathways.40,41 We were curious to determine if brain metastatic lesions from these different primary origins shared the same characteristics, thus maintaining certain “seed” characteristic hallmarks. Unsupervised hierarchical clustering of our study set using the various total and phosphorylated ERBB family receptor end points included in this study revealed a subgroup composed of 4 metastases from breast that had dramatically elevated relative levels of total and phospho-ERBB2 (Figure 3, top red cases). This subgroup also had high relative levels of total and phospho-ERBB3 as well as high total ERBB4 and was also marked by low relative levels of EGFR and heterogeneous levels of various phospho-EGFR end points. The remaining cases, including all of the metastases from lung, had markedly lower relative levels of total and phospho-ERBB2 and organized into subgroups based on more heterogeneous patterns of receptor activation. One subgroup comprising metastases from both breast and lung had high relative levels of EGFR Y992, Y1045, and Y1068 phosphorylation, while another group of metastases from lung cancers had very high relative levels of total EGFR. A third small grouping of cases had high relative levels of total ERBB3 and/or ERBB4 accompanied by higher relative activation of combinations of EGFR phosphorylation sites.
Figure 3.

ERBB receptor family expression and activation mapping in brain metastases by unsupervised hierarchical clustering. Clustering analysis of 42 brain metastases from breast and lung cancers (vertical axis) by the total and phospho-ERBB receptor family end points included in this study (horizontal axis). End points from left to right are: total ERBB2, ERBB3 Y1197, ERBB3 Y1289, EGFR Y1173, ERBB2 Y1248, EGFR S1046/1047, EGFR Y1045, EGFR Y1068, EGFR Y992, total EGFR, EGFR Y1148, EGFR Y845, total ERBB4, and total ERBB3. Brain metastases from breast are indicated in red and from lung are in black.
We were interested in determining if our analyses provided evidence for downstream pathway activation in brain metastases that could lead to new therapeutic strategies or targets that could be used in combination with currently approved agents. Statistical analysis of our data comparing brain metastases from breast with those from lung cancer resulted in 30 statistically different signaling proteins with p < 0.05 (Table 2). Metastases from breast cancer had higher average intensity values for 21 of the 30 different signaling proteins, including total and phospho-ERBB2, phospho-EGFR Y992, Y1045, and Y1068 and various downstream proteins. In particular, as shown in Figure 4A, breast cancer metastases to the brain showed high levels of four signaling proteins, which provide evidence for pathway activation downstream of receptor tyrosine kinases such as ERBB2 and the insulin/IGF1R receptor. In fact, statistically significant high levels of ERBB2 Y1248 (p = 0.01), AKT T308 (p = 0.001), PRAS40 T246 (p = 0.014), and IGF-1R Y1135/1136/IR Y1150/1151 (p = 0.006) were found, all attributable to the same signaling pathway or to common networks. In the brain metastases from lung cancer, we found that certain downstream proteins related to EGFR signaling were statistically higher than in the metastases from breast (Figure 4B). Higher levels of activation of p90RSK S380 (p = 0.001) and RSK3 T356/360 (p = 0.004), followed by their downstream substrate, CREB S133 (p = 0.033), an important nuclear transcription factor, all of which are downstream of EGFR, suggest that this pathway of demonstrated relevance in primary NSCLC is also active in brain metastases from lung cancer. Interestingly, levels of total EGFR were higher in metastases from lung versus breast, but several phospho-EGFR end points were higher in breast (Table 2, Figure 4B). We also observed that different phospho-AKT sites were statistically higher in each group: AKT S473 was higher in metastases from lung and AKT T308 was higher in the breast metastases (Table 2, Figure 4A).
Table 2.
Statistically Different End Points between Brain Metastases from Breast and Lung Primary Tumorsa
| Protein | p-value | relative differences |
|---|---|---|
| 4E-BP1 S65 | 0.048 | ↑B |
| AKT T308 | 0.001 | ↑B |
| A-Raf S299 | 0.001 | ↑B |
| Chk2 S33/35 | 0.019 | ↑B |
| Cyclin E | 0.036 | ↑B |
| EGFR Y1045 | 0.022 | ↑B |
| EGFR Y1148 | 0.027 | ↑B |
| EGFR Y992 | 0.002 | ↑B |
| ErbB2 Y1248 | 0.010 | ↑B |
| ErbB2/HER2 | 0.001 | ↑B |
| Estrogen Rec Alpha | 0.029 | ↑B |
| Estrogen Rec Alpha S118 | 0.040 | ↑B |
| GSK3 Alpha-Beta S21/9 | 0.035 | ↑B |
| IGF-1R (Y1135/36)/IR (Y1150/51) | 0.006 | ↑B |
| p70 S6 Kinase S371 | 0.037 | ↑B |
| p70 S6 Kinase T389 | 0.037 | ↑B |
| PDGF Rec Beta Y751 | 0.038 | ↑B |
| PDK1 S241 | 0.003 | ↑B |
| PRAS40 T246 | 0.014 | ↑B |
| Ret Y905 | 0.000 | ↑B |
| Stat5 Y694 | 0.040 | ↑B |
| Adducin S662* | 0.042 | ↓B |
| AKT S473 | 0.001 | ↓B |
| CC9 D315 | 0.006 | ↓B |
| CREB S133* | 0.033 | ↓B |
| EGFR | 0.006 | ↓B |
| LKB1 S334* | 0.023 | ↓B |
| MARCKS S152/156 | 0.014 | ↓B |
| p90 RSK S380* | 0.001 | ↓B |
| RSK3 (T356/S360) | 0.004 | ↓B |
Asterisk (*) = Wilcoxon rank sum test; p-value = probability value; B = breast met to the brain.
Figure 4.

One-way scatterplots of selected statistically different signaling end points between brain metastases from breast and lung cancers. (A) Selected signaling end points from Table 2 with higher levels of expression in brain metastases from breast cancer illustrating activation of proteins downstream of receptor tyrosine kinase signaling. (B) Selected signaling end points from Table 2 with higher levels of expression in brain metastases from lung cancers suggesting phosphorylation of downstream proteins in the EGFR signaling pathway.
DISCUSSION
Metastatic brain tumors have tremendous morbidity and mortality associated with their occurrence, due largely to the fact that many current agents cannot cross the BBB, and thus, any metastatic cells that successfully breach this barrier can more easily survive and colonize in this sanctuary site. It is imperative that we identify both new molecular targets and design appropriate therapeutic agents that can attack this lethal complication of cancer. Because most FDA-approved molecularly targeted therapeutic agents for cancer target protein enzymatic activity, proteomic analysis of the kinase-driven signal transduction activation patterns in these brain metastatic lesions could likely reveal new potential therapeutic targets or suggest novel combinations of existing therapeutics for use in the adjuvant setting. Also, these analyses may help to identify targets that could be useful in the chemoprevention setting, as many breast cancer and NSCLC patients remain at high risk for developing brain metastasis, even after achieving complete pathologic response from adjuvant therapy.42–44
The data presented in this report represent the first broad scale, protein-based signaling pathway activation mapping analyses performed on brain metastases from breast and lung cancer, which represent two of the most common tumors that metastasize to the brain. The study employed the RPMA platform, which has been extensively used by us in other analyses of human malignancies.15,34,35 The study set examined was a collection of frozen specimens that were carefully chosen based on the control of preanalytical variables such as sample collection, handling, storage, and time-to-freezing after surgical removal. Further, based on past evidence that upfront cellular enrichment from cellular tissue compartments is required for accurate protein measurement and activation status determination,35,36 we utilized LCM to greatly enrich for tumor epithelium (>95% purity based on pre- and post-LCM microscopic visualization) as the cellular input for all analyses.
Our broad-scale mapping results of the ongoing signaling architecture reveal heterogeneity in the patterns of signaling pathway protein activation among individual breast and lung tumors, allowing the lung tumors to be divided into five subgroups based on signaling activation patterns. The subgroups were underpinned by signaling activation of a number of important drug targets such as EGFR, ERBB4/HER4, ACK1, p53, CHK2, BCL-2, VEGFR2, AKT, ERK, mTOR, PTEN, and integrin signaling, which indicated the possibility for stratification of NSCLC patients with brain metastasis for therapies that target these pathway activations. Similar signaling heterogeneity was also observed in several of our laboratory’s previous studies of primary human cancers.15,20–22,35,45 We did identify a subset of metastases from breast tumors in our study set (4 of 10) that exhibited high levels of ERBB2/HER2 expression and activation. HER2-overexpressing and triple-negative breast tumors have been shown to have higher frequencies of brain metastasis than other breast cancers, with occurrences as high as 30–35%, compared to 10–16% among patients with advanced breast cancers.9,44,46 Our data confirm these observations, even though the size of our study set was small.
Signaling activation of the ERBB family of receptor tyrosine kinases has unquestioned relevance for tumor progression and metastasis in both breast and lung cancer.37–41 In an experimental breast cancer brain metastasis model system, it has been shown that HER2 overexpression increases the outgrowth of metastatic breast tumor cells in the brain and suggests that signaling through this receptor pathway is active and results in cell proliferation in the brain microenvironment.47 Interestingly, the highest relative levels of ERK and AKT phosphorylation did not appear to strongly correlate with ERBB2 activation in our study set but more so with phospho-EGFR Y1045, Y1068, and Y1173 and suggests the potential for active signaling through the EGFR receptor in these tumors, which is often overexpressed in triple negative and basal type breast cancers, both of which have a poor prognosis. In contrast, the brain metastases from lung cancer were more heterogeneous in their patterns of signaling activation, which may indicate the ways in which invasion, extravasation, and metastatic adaption to the new microenvironment for NSCLC are more varied than for breast cancer, and thus, the molecular derangements that underpin this process are more heterogeneously selected.
Comparison of ERBB receptor signaling activity in brain metastases from breast and lung cancers suggests the possibility of differential clinical response to inhibitors targeting these receptors. On the basis of our results, it would appear that brain metastases from lung cancer would be more likely to respond to treatment with EGFR inhibitors as opposed to inhibitors of ERBB2 activity which would be a preferred target in metastases from breast cancer. Though several phospho-EGFR end points were more highly activated in metastases from breast cancer in this study set, levels of total EGFR averaged higher in the metastases from lung. Mean comparison results also provided evidence for differential activation of various signaling proteins downstream of the ERBB family receptors in metastases from breast and lung primary tumors. Indeed, the small molecule dual EGFR/ERBB2 kinase inhibitor, lapatinib, has been shown to have antitumor activity in preclinical models of brain metastasis from breast cancer,48 and has demonstrated modest clinical benefit alone and in combination with capecitabine in patients with brain metastases from HER2-positive breast cancer.49 Moreover, we found that activation of IGFR and downstream signaling was higher in the brain metastasis from breast cancer patients compared to NSCLC patients. If validated in subsequent study sets, this result could point to a rationale for clinical testing of the many new anti-IGFR kinase inhibitor drugs in breast cancer patients with brain metastasis. Small molecule inhibitors of EGFR, such as erlotinib and gefitinib, have been tested in NSCLC patients with CNS metastases and clinical responses have been observed, particularly in tumors carrying EGFR mutations that predict sensitivity to the drug,50 but significant toxicities have been observed in some studies combining erlotinib treatment with radiation therapy.51 Our studies also revealed that breast and lung metastases to the brain each had differentially elevated levels of unique phospho-AKT sites, T308 and S473, respectively. Phosphorylation of AKT in breast cancer is a poor prognostic marker for response to hormone therapy and in HER2-overexpressing tumors. In lung cancer, phosphorylation of AKT on S473 has been associated with improved response rate to gefitinib.52 It could be possible that combination therapeutic strategies merging small molecule RTK inhibitors with inhibitors of downstream effectors, such as AKT, could improve clinical efficacy of these therapies against brain metastases.
These analyses have produced what we believe is an important preliminary data set of protein signaling activation maps of brain metastasis that will require further efforts to verify the signaling subtypes identified. Despite the powerful methodology utilized, one caveat of this study is that a small set of tumors (n = 42) was used to generate final conclusive data about which pathway activation phenotypes underpin brain metastasis from breast and lung cancer and thus, aggressive protein pathway activation signatures. However, based on the methodological workflow used, we expect that this pilot data can be confidently used as a launch-point for further hypothesis-driven analysis and verification of the results herein. Also, while the signaling analysis was extensive, the full repertoire of the human kinome was certainly not surveyed, and many specific signaling proteins were not specifically queried. We fully expect that even larger coverage of the signal transduction machinery will yield new discoveries. However, our analysis rationale for this study was to carefully select key signaling proteins known to be involved in tumorigenesis and metastasis, regulating growth and energy metabolism, survival, apoptosis, differentiation, motility, and inflammation and which are key surrogates and direct targets for the many kinase inhibitors that largely populate the current phase I–III pipeline. Because many of the proteins we measured are located at key “hubs” within the signal transduction circuitry, we predict that most perturbations in the signaling architecture can be detected despite the inability to measure the full signaling repertoire of the cell. Furthermore, while signaling can be regulated by a number of post-translational modification driven events (e.g., glycosylation, acetylation, etc.), we chose to study these proteins at the functional level by measuring protein phosphorylation, which is the principal regulator of signal transduction and the main analyte end point for the recording of ongoing cellular kinase activity, so that we could generate an accurate snapshot of the ongoing signaling cascades within the tumor cells.
CONCLUSION
Our results, while preliminary, point to organ-type specific signaling architecture maintained within the brain microenvironment. These results could indicate the presence of the aggressive “seed” signatures of protein signaling activation that occur early in the primary tumor and are maintained in the metastatic lesion itself. Recent analysis of colorectal cancer signaling networks within the context of metastatic progression has indicated that this occurs.15 While the causal significance of the pathway activation modules identified in this initial survey of the pathway architecture of brain metastasis remains unknown, we hope that this study of protein pathway activation architecture will serve as a starting point for mapping signaling events in clinical specimens which could be used as rationale for making targeted therapy choices and even prioritize combinations of therapeutics. Our ongoing validation work in larger independent study sets will provide the basis for selection and ranking those signaling networks that will be chosen for further analysis in both animal models for functional workup as well as in prospective clinical assay development for personalized medicine-based opportunities.
Given the poor accrual of brain metastases from clinical studies due to patient morbidity issues, we hope that this study will provide further rationale for prospective collection of brain metastatic tissues so that in-depth molecular analysis of cancer within this unique microenvironment can continue. Even more rare is the collection of patient-matched primary tumors and brain metastases. These types of tissue study sets will be critical to detail the molecular changes that take place during metastatic progression. We also do not know how the signaling architecture of brain metastasis from breast and lung compare to brain metastasis from other solid tumors such as colon, prostate, and melanoma. On the basis of the “seed” specific signaling differences we observed herein between patients with lung and breast cancers, we postulate that new and different signaling activation maps will emerge from new analyses for other cancer cohorts.
Supplementary Material
Acknowledgments
The authors appreciate the generous support of Dr. Vikas Chandhoke and the College of Life Sciences at George Mason University. This work was partly supported by the Italian Istituto Superiore di Sanita within the framework of the Italy/USA cooperation agreement between the U.S. Department of Health and Human Services, George Mason University, and the Italian Ministry of Public Health.
Footnotes
Supporting Information available: Supplementary Tables 1 and 2. This material is available free of charge via the Internet at: http://pubs.acs.org.
References
- 1.Landis SH, Murray T, Bolden S, Wingo PA. Cancer statistics, 1999. CA Cancer J Clin. 1999;49(1):8–31. doi: 10.3322/canjclin.49.1.8. [DOI] [PubMed] [Google Scholar]
- 2.Nathoo N, Chahlavi A, Barnett GH, Toms SA. Pathobiology of brain metastases. J Clin Pathol. 2005;58(3):237–242. doi: 10.1136/jcp.2003.013623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patel RR, Mehta MP. Targeted therapy for brain metastases: improving the therapeutic ratio. Clin Cancer Res. 2007;13(6):1675–1683. doi: 10.1158/1078-0432.CCR-06-2489. [DOI] [PubMed] [Google Scholar]
- 4.Gril B, Evans L, Palmieri D, Steeg PS. Translational research in brain metastasis is identifying molecular pathways that may lead to the development of new therapeutic strategies. Eur J Cancer. 2010;46(7):1204–1210. doi: 10.1016/j.ejca.2010.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gondi V, Mehta MP. Novel insights into the management of brain metastases. Curr Opin Neurol. 2010;23(6):556–562. doi: 10.1097/WCO.0b013e32833f8cb5. [DOI] [PubMed] [Google Scholar]
- 6.Sun MH, et al. HER family receptor abnormalities in lung cancer brain metastases and corresponding primary tumors. Clin Cancer Res. 2009;15(15):4829–4837. doi: 10.1158/1078-0432.CCR-08-2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yokota J. Tumor progression and metastasis. Carcinogenesis. 2000;21(3):497–503. doi: 10.1093/carcin/21.3.497. [DOI] [PubMed] [Google Scholar]
- 8.Paget S. The distribution of secondary growths in cancer of the breast. Lancet. 1889;1:571–573. [PubMed] [Google Scholar]
- 9.Palmieri D, et al. The biology of metastasis to a sanctuary site. Clin Cancer Res. 2007;13(6):1656–1662. doi: 10.1158/1078-0432.CCR-06-2659. [DOI] [PubMed] [Google Scholar]
- 10.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9(4):239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grinburg-Rashi H, et al. The expression of three genes in primary non-small cell lung cancer is associated with metastatic spread to the brain. Clin Cancer Res. 2009;15(5):1755–1761. doi: 10.1158/1078-0432.CCR-08-2124. [DOI] [PubMed] [Google Scholar]
- 12.Kang Y, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3(6):537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 13.Minn AJ, et al. Genes that mediate breast cancer metastasis to the lung. Nature. 2005;436(7050):518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bos PD, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459(7249):1005–1009. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pierobon MP, et al. Multiplexed cell signaling analysis of metastatic and nonmetastatic colorectal cancer reveals COX2-EGFR signaling activation as a potential prognostic pathway biomarker. Clin Colorectal Cancer. 2009;8(2):110–117. doi: 10.3816/CCC.2009.n.018. [DOI] [PubMed] [Google Scholar]
- 16.Klein A, et al. Identification of brain- and bone-specific breast cancer metastasis genes. Cancer Lett. 2009;276(2):212–220. doi: 10.1016/j.canlet.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 17.Weigelt B, et al. Molecular portraits and 70-gene prognosis signature are preserved throughout the metastatic process of breast cancer. Cancer Res. 2005;65(20):9155–9158. doi: 10.1158/0008-5472.CAN-05-2553. [DOI] [PubMed] [Google Scholar]
- 18.Steeg PS, Theodorescu D. Metastasis: a therapeutic target for cancer. Nat Rev Clin Oncol. 2008;5(4):206–219. doi: 10.1038/ncponc1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kikuchi T, et al. Expression profiles of metastatic brain tumor from lung adenocarcinomas on cDNA microarray. Int J Oncol. 2006;28(4):799–805. [PubMed] [Google Scholar]
- 20.Grubb RL, et al. Pathway biomarker profiling of localized and metastatic human prostate cancer reveal metastatic and prognostic signatures. J Proteome Res. 2009;8(6):3044–3054. doi: 10.1021/pr8009337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petricoin EF, III, et al. Mapping molecular networks using proteomics: a vision for patient-tailored combination therapy. J Clin Oncol. 2005;23(15):3614–3621. doi: 10.1200/JCO.2005.02.509. [DOI] [PubMed] [Google Scholar]
- 22.Sheehan KM, et al. Use of reverse phase protein microarrays and reference standard development for molecular network analysis of metastatic ovarian carcinoma. Mol Cell Proteomics. 2005;4(4):346–355. doi: 10.1074/mcp.T500003-MCP200. [DOI] [PubMed] [Google Scholar]
- 23.Gomez-Roca C, et al. Differential expression of biomarkers in primary non-small cell lung cancer and metastatic sites. J Thorac Oncol. 2009;4(10):1212–1220. doi: 10.1097/JTO.0b013e3181b44321. [DOI] [PubMed] [Google Scholar]
- 24.Italiano A, et al. Comparison of the epidermal growth factor receptor gene and protein in primary non-small-cell-lung cancer and metastatic sites: implications for treatment with EGFR-inhibitors. Ann Oncol. 2006;17(6):981–985. doi: 10.1093/annonc/mdl038. [DOI] [PubMed] [Google Scholar]
- 25.Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19(3):1720–1730. doi: 10.1128/mcb.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson L, Seilhamer J. A comparison of selected mRNA and protein abundances in human liver. Electrophoresis. 1997;18(3–4):533–537. doi: 10.1002/elps.1150180333. [DOI] [PubMed] [Google Scholar]
- 27.Shankavaram UT, et al. Transcript and protein expression profiles of the NCI-60 cancer cell panel: an integromic microarray study. Mol Cancer Ther. 2007;6(3):820–832. doi: 10.1158/1535-7163.MCT-06-0650. [DOI] [PubMed] [Google Scholar]
- 28.Scott JD, Pawson T. Cell signaling in space and time: where proteins come together and when they’re apart. Science. 2009;326(5957):1220–1224. doi: 10.1126/science.1175668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang SM, Harari PM. Epidermal growth factor receptor inhibition in cancer therapy: biology, rationale and preliminary clinical results. Invest New Drugs. 1999;17(3):259–269. doi: 10.1023/a:1006384521198. [DOI] [PubMed] [Google Scholar]
- 30.Ono M, et al. Brain metastases in patients who receive trastuzumab-containing chemotherapy for HER2-overexpressing meta-static breast cancer. Int J Clin Oncol. 2009;14(1):48–52. doi: 10.1007/s10147-008-0797-8. [DOI] [PubMed] [Google Scholar]
- 31.Tham YL, et al. Primary breast cancer phenotypes associated with propensity for central nervous system metastases. Cancer. 2006;107(4):696–704. doi: 10.1002/cncr.22041. [DOI] [PubMed] [Google Scholar]
- 32.Hicks DG, et al. Breast cancers with brain metastases are more likely to be estrogen receptor negative, express the basal cytokeratin CK5/6, and overexpress HER2 or EGFR. Am J Surg Pathol. 2006;30(9):1097–1104. doi: 10.1097/01.pas.0000213306.05811.b9. [DOI] [PubMed] [Google Scholar]
- 33.Rusnak DW, et al. The effects of the novel, reversible epidermal growth factor receptor/Erb-B-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol Cancer Ther. 2001;1(2):85–94. [PubMed] [Google Scholar]
- 34.Paweletz CP, et al. Reverse phase proteomic microarrays which capture disease progression show activation of pro-survival pathways at the cancer invasion front. Oncogene. 2001;20(16):1981–1989. doi: 10.1038/sj.onc.1204265. [DOI] [PubMed] [Google Scholar]
- 35.Wulfkuhle JD, et al. Multiplexed cell signaling analysis of human breast cancer applications for personalized therapy. J Proteome Res. 2008;7(4):1508–1517. doi: 10.1021/pr7008127. [DOI] [PubMed] [Google Scholar]
- 36.Silvestri A, et al. Protein pathway biomarker analysis of human cancer reveals requirement for upfront cellular-enrichment processing. Lab Invest. 2010;90(5):787–796. doi: 10.1038/labinvest.2010.47. [DOI] [PubMed] [Google Scholar]
- 37.Piyathilake CJ, et al. Differential expression of growth factors in squamous cell carcinoma and precancerous lesions of the lung. Clin Cancer Res. 2002;8(3):734–744. [PubMed] [Google Scholar]
- 38.Scagliotti GV, Selvaggi G, Novello S, Hirsch FR. The biology of epidermal growth factor receptor in lung cancer. Clin Cancer Res. 2004;10(12 Pt 2):4227S–4232S. doi: 10.1158/1078-0432.CCR-040007. [DOI] [PubMed] [Google Scholar]
- 39.Ohasak Y, et al. Epidermal growth factor receptor expression correlates with poor prognosis in non-small cell lung cancer patients with p53 overexpression. Oncol Rep. 2000;7(3):603–607. doi: 10.3892/or.7.3.603. [DOI] [PubMed] [Google Scholar]
- 40.Slamon DJ, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244(4905):707–712. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 41.Press MF, et al. Diagnostic evaluation of HER-2 as a molecular target: an assessment of accuracy and reproducibility of laboratory testing in large, prospective, randomized clinical trials. Clin Cancer Res. 2005;11(18):6598–6607. doi: 10.1158/1078-0432.CCR-05-0636. [DOI] [PubMed] [Google Scholar]
- 42.Chen AM, et al. Risk of cerebral metastases and neurological death after pathological complete response to neoadjuvant therapy for locally advanced nonsmall-cell lung cancer: clinical implications for the subsequent management of the brain. Cancer. 2007;109(8):1668–1675. doi: 10.1002/cncr.22565. [DOI] [PubMed] [Google Scholar]
- 43.Omuro AM, et al. High incidence of disease recurrence in the brain and leptomeninges in patients with nonsmall cell lung carcinoma after response to gefitinib. Cancer. 2005;103(11):2344–2348. doi: 10.1002/cncr.21033. [DOI] [PubMed] [Google Scholar]
- 44.Lin NU, Winer EP. Brain metastases: the HER2 paradigm. Clin Cancer Res. 2007;13(6):1648–1655. doi: 10.1158/1078-0432.CCR-06-2478. [DOI] [PubMed] [Google Scholar]
- 45.Petricoin EF, III, et al. Phosphoprotein signal pathway mapping: Akt/mTOR pathway activation association with childhood rhabdomyosarcoma survival. Cancer Res. 2007;67(7):3431–3434. doi: 10.1158/0008-5472.CAN-06-1344. [DOI] [PubMed] [Google Scholar]
- 46.Lin NU, et al. Sites of distant recurrence and clinical outcomes in patients with metastatic triple-negative breast cancer: high incidence of central nervous system metastases. Cancer. 2008;113(10):2638–2645. doi: 10.1002/cncr.23930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Palmieri D, et al. Her-2 overexpression increases the metastatic outgrowth of breast cancer cells in the brain. Cancer Res. 2007;67(9):4190–4198. doi: 10.1158/0008-5472.CAN-06-3316. [DOI] [PubMed] [Google Scholar]
- 48.Gril B, et al. Effect of lapatinib on the outgrowth of metastatic breast cancer cells to the brain. J Natl Cancer Inst. 2008;100(15):1092–1103. doi: 10.1093/jnci/djn216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin NU, et al. Multicenter phase II study of lapatinib in patients with brain metastases from HER2-positive breast cancer. Clin Cancer Res. 2009;15(4):1452–1459. doi: 10.1158/1078-0432.CCR-08-1080. [DOI] [PubMed] [Google Scholar]
- 50.Katayama T, et al. Efficacy of erlotinib for brain and leptomeningeal metastases in patients with lung adenocarcinoma who showed initial good response to gefitinib. J Thorac Oncol. 2009;4(11):1415–1419. doi: 10.1097/JTO.0b013e3181b62572. [DOI] [PubMed] [Google Scholar]
- 51.Olmez I, et al. Clinical outcomes in extracranial tumor sites and unusual toxicities with concurrent whole brain radiation (WBRT) and erlotinib treatment in patients with non-small cell lung cancer (NSCLC) with brain metastases. Lung Cancer. 2010;70(2):174–179. doi: 10.1016/j.lungcan.2010.01.018. [DOI] [PubMed] [Google Scholar]
- 52.Cicenas J. The potential role of Akt phosphorylation in human cancers. Int J Biol Markers. 2008;23(1):1–9. doi: 10.1177/172460080802300101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.