

Abstract
Objectives
The aims of this study were to develop a population pharmacokinetic model for intravenous paracetamol in preterm and term neonates and to assess the generalizability of the model by testing its predictive performance in an external dataset.
Methods
Nonlinear mixed-effects models were constructed from paracetamol concentration–time data in NONMEM 7.2. Potential covariates included body weight, gestational age, postnatal age, postmenstrual age, sex, race, total bilirubin, and estimated glomerular filtration rate. An external dataset was used to test the predictive performance of the model through calculation of bias, precision, and normalized prediction distribution errors.
Results
The model-building dataset included 260 observations from 35 neonates with a mean gestational age of 33.6 weeks [standard deviation (SD) 6.6]. Data were well-described by a one-compartment model with first-order elimination. Weight predicted paracetamol clearance and volume of distribution, which were estimated as 0.348 L/h (5.5 % relative standard error; 30.8 % coefficient of variation) and 2.46 L (3.5 % relative standard error; 14.3 % coefficient of variation), respectively, at the mean subject weight of 2.30 kg. An external evaluation was performed on an independent dataset that included 436 observations from 60 neonates with a mean gestational age of 35.6 weeks (SD 4.3). The median prediction error was 10.1 % [95 % confidence interval (CI) 6.1–14.3] and the median absolute prediction error was 25.3 % (95 % CI 23.1–28.1).
Conclusions
Weight predicted intravenous paracetamol pharmacokinetics in neonates ranging from extreme preterm to full-term gestational status. External evaluation suggested that these findings should be generalizable to other similar patient populations.
1 Introduction
Inadequate management of neonatal pain is associated with poor short- and long-term neurodevelopmental outcomes [1]. In neonates and infants, mild to moderate pain is commonly treated with paracetamol (N-acetyl-p-aminophenol, acetaminophen) [2, 3]. Although paracetamol has been widely used for nearly a century, intravenous formulations only became available recently [4], with regulatory approval for use in patients aged 2 years or older occurring as late as 2010 in the case of the US [5]. Intravenous paracetamol has rapidly gained favor for applications in which enteral delivery is not possible, such as postoperative pain relief [4], and several recent studies have reported on the pharmacokinetics of intravenous paracetamol in neonates [6–8]. However, there is still a lack of consensus regarding optimal dosing guidelines [3, 6, 9–11] and, in many cases, administration of intravenous paracetamol to neonates is limited to off-label use [3]. Furthermore, pharmacokinetic data from extremely preterm neonates (<28 weeks’ gestation) remain scarce [3, 6, 8].
Appropriate dose selection for neonates is complicated by developmental changes that occur during early life [12]. Paracetamol is primarily eliminated by hepatic metabolism; therefore, measures of hepatic maturation and function are critical for explanation of between-subject variability (BSV) in paracetamol pharmacokinetics [6–8, 13, 14]. Population pharmacokinetic modeling is a powerful tool for analyzing sparse neonatal data because the approach simultaneously utilizes information from all subjects to characterize the study population, BSV, and influential patient characteristics (covariates) [15, 16]. Unfortunately, neonatal population pharmacokinetic studies are frequently limited by small numbers of subjects. Ideally, the external generalizability of population pharmacokinetic models should be evaluated with independent data that were not used in model development; however, due to a paucity of neonatal studies, this is rarely possible [15–18].
The aims of this study were to develop a population pharmacokinetic model for intravenous paracetamol in preterm and term neonates, and to assess generalizability of the model by testing its predictive performance when applied to an external dataset.
2 Methods
2.1 Study 1: Model-Building Dataset
2.1.1 Ethics Approval and Study Registration
This was a prospective, single-center, open-label study of the pharmacokinetics of intravenous paracetamol in neonates. The study was approved by the Institutional Review Board at the Children’s National Health System (Washington, DC, USA) and was carried out in concordance with International Conference on Harmonisation (ICH) Guidelines for Good Clinical Practice [19]. The study was registered at ClinicalTrials.gov (NCT01328808) [20].
2.1.2 Study Population
Patients <28 days postnatal age with an indwelling arterial line and a clinical indication for intravenous analgesia who were admitted to intensive care units at the Children’s National Health System (Washington, DC, USA) were considered for inclusion. Exclusion criteria were severe asphyxia, grade III or IV intraventricular hemorrhage, major congenital malformations, neurological disorders, receipt of neuromuscular blockers, and hepatic or renal failure, including systemic hypoperfusion.
2.1.3 Dosing and Sampling Schedule
Intravenous paracetamol (Ofirmev, 10 mg/mL; Mallinckrodt Pharmaceuticals, Dublin, Ireland) was administered by 30-min infusions at 15 mg/kg/dose. The dosing schedule was based on gestational age. Neonates <28 weeks’ gestation received five doses at 12-h intervals; neonates ≥28 weeks’ gestation received seven doses at 8-h intervals. Blood samples (0.2 mL) were obtained from arterial lines at approximately 0, 1, 2, 4, 6, 8, 12, and 24 h after the first and last paracetamol doses. Patients were randomly assigned to one of two sampling schedules, each consisting of 9–10 collection times. Blood was collected in sodium heparin Vacutainer tubes (BD, Franklin Lakes, NJ, USA) and centrifuged for 10–15 min at 1500×g at 4 °C. Plasma was transferred to cryovials and stored at −70 °F. Study samples were shipped on dry ice to the Center for Human Toxicology at the University of Utah, and stored at −80 °C prior to analysis.
2.1.4 Analytical Method
Paracetamol plasma concentrations were determined using high-performance liquid chromatography–tandem mass spectrometry (HPLC–MS/MS). Plasma samples (10 μL) were fortified with paracetamol-d4 internal standard (Toronto Research Chemicals, Toronto, ON, Canada) and prepared using protein precipitation with acetonitrile (600 μL). Sample supernatants were evaporated to dryness and reconstituted in 0.1 % aqueous formic acid (400 μL). Reconstituted samples were injected (100 μL) onto an Agilent 1260 Infinity HPLC system interfaced with an Agilent 6460 triple-quadrupole mass spectrometer (Agilent Technologies, Santa Clara, CA, USA). The autosampler was maintained at 5 °C, and the autosampler needle was washed with methanol/water (1/1, v/v) between injections. Chromatographic separation was achieved with an Agilent Poroshell 120 EC-C18 column (2.1 × 100 mm, 2.7 μm particle size; Agilent Technologies, Santa Clara, CA, USA) maintained at 40 °C using a gradient mobile phase consisting of 10 mM aqueous ammonium acetate, pH 3.5 (A) and methanol (B) at a flow rate of 0.25 mL/min. Mobile phase was maintained at 3 % B for 6 min, increased linearly to 35 % B over 3 min, maintained at 95 % B for 3 min, decreased linearly to 3 % B over 0.5 min, and re-equilibrated at 3 % B for 7.5 min. The mass spectrometer was operated in positive electrospray ionization + Agilent Jet Stream mode with multiple reaction monitoring (MRM). The following settings were applied: 350 °C gas temperature, 10 L/min gas flow, 30 psi nebulizer pressure, 350 °C sheath gas temperature, 9 L/min sheath gas flow, 3500 V capillary voltage, 500 V nozzle voltage, 250 V delta EMV, 80 V fragmentor voltage, 14 V collision energy, and 100 ms dwell time. MRM transitions for paracetamol and paracetamol-d4 were 152.1 → 110.0 and 156.1 → 114.1 (precursor → product m/z), respectively.
The lower limit of quantification (LLOQ) was 0.05 mg/ L [mean accuracy 92 %, coefficient of variation (CV) 5.3 %, n = 6], and the calibration curve maintained linearity up to 50 mg/L (1/x2 weighting). Triplicate sets of quality control samples at concentrations of 0.15, 0.80, 8.0, and 40 mg/L accompanied each study sample batch. At these concentrations, mean intra-assay (n = 5) and inter-assay (n = 20) accuracy ranged from 98 to 104 %, and intra- and inter-assay imprecision was <10 % CV.
2.2 Study 2: External Evaluation Dataset
2.2.1 Ethics Approval and Study Registration
Data for external model evaluation were obtained from a previously published, prospective, single-center, open-label study on the pharmacokinetics of intravenous paracetamol in neonates (PARANEO) [6]. The study was approved by the Ethics Committee at University Hospitals Leuven (Leuven, Belgium) and was carried out in concordance with ICH Guidelines for Good Clinical Practice [19]. The study was registered at ClinicalTrials.gov (NCT00969176) [21] and with the European Clinical Trials Database (EUdraCT) (2009-011243-39).
2.2.2 Study Population
Patients ≤28 days postnatal age with a clinical indication for intravenous paracetamol who were admitted to the Neonatal Intensive Care Unit at University Hospitals Leuven (Leuven, Belgium) were considered for inclusion. Exclusion criteria were recent receipt of paracetamol (<48 h) or clinical contraindication for paracetamol administration (i.e. hepatic failure).
2.2.3 Dosing and Sampling Schedule
Intravenous paracetamol (10 mg/mL; Sintetica SA, Mendrisio, Switzerland) was administered by 15-min infusions with a loading dose of 20 mg/kg, followed by up to seven maintenance doses at 6-h intervals. Each maintenance dose consisted of 5.0, 7.5, or 10.0 mg/kg, respectively, for neonates with postmenstrual ages <32, 32–36, or >36 weeks. Samples were obtained from an arterial line over the 48 h following paracetamol loading doses. The sampling schedule focused on the periods shortly after each loading dose (<2 h; peak concentrations) and just before maintenance doses (5–6 h after the previous dose; trough concentrations). Blood was centrifuged, and plasma samples were stored at −20 °C until analysis.
2.2.4 Analytical Method
Paracetamol plasma concentrations were determined using HPLC coupled with ultraviolet detection. Details of the analytical method have been previously reported [22]. The LLOQ was 0.08 mg/L (<20 % CV), and intra- and interday imprecision was <15 % CV.
2.3 Pharmacometric Model Development
The paracetamol pharmacokinetic model was developed in NONMEM (nonlinear mixed-effects modeling) 7.2 interfaced with PDx-Pop 5.0 (ICON Development Solutions, Ellicott City, MD, USA) using the first-order conditional estimation with interaction method. Processing and visualization of NONMEM output were performed in R 3.0.1 (CRAN.R-project.org). Throughout model development, standard diagnostic plots were generated to evaluate model fit, including observed concentrations versus population-and individual-predicted concentrations, conditional weighted residuals versus time and population-predicted concentrations, and histograms of individual random effects. Models were also compared based on the precision of parameter estimates (parametric standard errors) and condition number [23]. Hierarchical models were compared using the objective function value (OFV). Non-hierarchical models were compared using the Bayesian Information Criterion (BIC) [24].
Based on visual data inspection and a review of the literature, one- and two-compartment structural models were considered. Models were parameterized with clearance and volume terms. Structural models also incorporated the rate and duration of intravenous paracetamol infusion for each dose. Random effects were classified as either between-subject variability (BSV) or residual unexplained variability (RUV). Individual pharmacokinetic parameters were assumed to be log-normally distributed, and BSV was modeled with an exponential function (Eq. 1):
| (1) |
where θi is the individual model-predicted pharmacokinetic parameter (e.g. clearance), θpop is the population mean for the pharmacokinetic parameter θ, and ηi is the between-subject random effect on θ for the ith individual; ηi is normally distributed with a mean of zero and a variance of ω2. Additive, proportional, and combined additive and proportional functions were tested for incorporation of RUV [25].
Potential covariates included current body weight, current body length, current body mass index (BMI), gestational age, postnatal age, postmenstrual age, total bilirubin, estimated glomerular filtration rate (GFR), sex, race (White/Caucasian or Black/African American), ethnicity, and indication (surgical or procedural). Laboratory samples were obtained within 24 h prior to the first paracetamol dose or during the pharmacokinetic sample collection period. Estimated GFR was calculated using the updated Schwartz formula (Eq. 2) [26]:
| (2) |
where eGFR is estimated GFR (mL/min/1.73 m2), length is body length (cm), and Scr is serum creatinine (mg/dL, modified kinetic Jaffe method). Serum creatinine concentrations obtained at ≤3 days postnatal age were considered to reflect maternal renal function and were excluded from analysis. Continuous covariates were normalized to population mean values and were tested for inclusion in linear, power, and exponential functional forms [27]. Categorical covariates were tested for inclusion using additive shift models. Potential covariates were tested using stepwise forward selection followed by stepwise backward elimination. Changes in OFV were considered significant at p < 0.05 (Chi-square distribution, 1 df, ΔOFV > 3.84) during forward selection, and p < 0.01 (ΔOFV > 6.63) during backward elimination [28].
2.4 Internal Model Evaluation
Stability of the final covariate model was evaluated by nonparametric bootstrap [29]. PDx-Pop was used to generate 1000 bootstrap datasets by random sampling with replacement from the original model-building dataset. Additionally, normalized prediction distribution errors (NPDEs) based on 1000 simulations were calculated in NONMEM, and plots were generated for the NPDE distribution and NPDEs versus time, population-predicted concentrations, and influential covariates [30]. Finally, numerical and visual predictive checks were performed to compare observed paracetamol concentrations with concentrations obtained from model-based simulations of 1000 datasets [31]. Predictive checks were performed using Perl-speaks-NONMEM 4.2.0 (PsN, http://psn.sourceforge.net) interfaced with Pirana 2.9.0 (http://pirana-software.com) [32]. Visual predictive check data were prediction corrected [33] and binned based on observation counts.
2.5 External Model Evaluation
An external dataset (Study 2) was used to assess the generalizability of the final covariate model [6]. Predictive performance was assessed as suggested by Sheiner and Beal [34] to quantify bias and precision. Confidence intervals for central tendency measures of bias and precision were obtained via bootstrap techniques (n = 2000). Additionally, external evaluation procedures based on NPDEs and numerical and visual predictive checks were performed as described in Sect. 2.4.
3 Results
Key information from Studies 1 and 2 is summarized in Table 1. The model-building dataset (Study 1) consisted of 267 observations from 35 patients (median 8; range 3–11 concentrations/patient). Of these 267 paracetamol concentrations, one measurement was less than the assay LLOQ and was excluded from the analysis. Six measurements (2 %) were implausible (e.g. peak concentrations observed at trough collection times) and were also excluded. Thus, 260 paracetamol concentrations were used for the development and internal evaluation of the population pharmacokinetic model. The external dataset (Study 2) consisted of 436 paracetamol concentrations from 60 patients (median 7; range 2–11 concentrations/patient). The proportion of patients who received intravenous paracetamol for postoperative analgesia versus a medical condition was similar in the two studies; however, the types of surgery and specific medical indications were more diverse in Study 2. The two studies also had similar proportions of preterm and full-term neonates but the percentage of extremely preterm neo-nates in Study 1 was more than three times that in Study 2 (29 vs. 8 %).
Table 1.
Study information for the model-building and external evaluation datasets
| Study 1, model-building dataset | Study 2, external dataset (PARANEO) [6] | |
|---|---|---|
| NCT identifier | 01328808 | 00969176 |
| Study description | Phase II/III, multiple-dose study of intravenous paracetamol | Phase II/III, multiple-dose study of intravenous paracetamol |
| Study drug | Ofirmev (10 mg/mL) | Paracetamol Sintetica (10 mg/mL) |
| Sampling route | Arterial | Arterial |
| Analytical method | HPLC–MS/MS | HPLC–UV |
| Subjects | 35 neonates | 60 neonates |
| Samples (n) | 260 | 436 |
| N per subject [median (range)] | 8 (3–11) | 7 (2–11) |
| Primary indication for intravenous paracetamol [n (%)] | ||
| Postoperative analgesia | 19 (54) | 33 (55) |
| Cardiac surgery | 19 (54) | 15 (25) |
| Thoracic surgery | 0 (0) | 11 (18) |
| Abdominal surgery | 0 (0) | 6 (10) |
| Other | 0 (0) | 1 (2) |
| Medical conditions | 16 (46) | 27 (45) |
| Alprostadil administration | 0 (0) | 8 (13) |
| Procedural/respiratory | 16 (46) | 8 (13) |
| Traumatic pain | 0 (0) | 5 (8) |
| Fever | 0 (0) | 3 (5) |
| Other | 0 (0) | 3 (5) |
| Gestational status [n (%)] | ||
| Extreme preterm (<28 weeks’ GA) | 10 (29) | 5 (8) |
| Preterm (<37 weeks’ GA) | 17 (49) | 28 (47) |
| Full-term (37–42 weeks’ GA) | 18 (51) | 32 (53) |
| Current body weighta (kg) by gestational age subgroup [median (range)] | ||
| Extreme preterm (<28 weeks’ GA) | 0.69 (0.55–1.30) | 0.90 (0.61–1.41) |
| Preterm (<37 weeks’ GA) | 0.96 (0.46–2.80) | 2.08 (0.61–3.66) |
| Full-term (37–42 weeks’ GA) | 3.16 (2.70–4.20) | 3.22 (1.80–4.30) |
| Postnatal agea (days) by gestational age subgroup [median (range)] | ||
| Extreme preterm (<28 weeks’ GA) | 10 (1–26) | 17 (6–24) |
| Preterm (<37 weeks’ GA) | 9 (1–26) | 6 (1–27) |
| Full-term (37–42 weeks’ GA) | 6 (2–12) | 2 (1–10) |
GA gestational age, HPLC high-performance liquid chromatography, MS/MS tandem mass spectrometry, NCT National Clinical Trial, PARA-NEO Paracetamol in Neonates, UV ultraviolet detection
On the day of the first paracetamol dose
Demographic characteristics of the model-building study population are summarized in Tables 2 and 3 for continuous and categorical variables, respectively. The median gestational age was 37 weeks (range 23–41), and most study subjects (66 %) received the first paracetamol dose within 1 week after birth. Median body weight at the time of the first paracetamol dose was 2.80 kg (range 0.46–4.20). Body length was missing from one patient record, which precluded calculation of BMI and estimated GFR for that subject. Total bilirubin measurements were not obtained within the requisite timeframe for 13 subjects. Additionally, two subjects did not have serum creatinine measurements obtained within the requisite timeframe, and three subjects had serum creatinine measurements that were excluded because they were considered to reflect maternal renal function (postnatal age ≤3 days).
Table 2.
Subject demographics for continuous covariates tested in the model-building dataset
| Characteristic | N | Mean | SD | CV | Median | Range |
|---|---|---|---|---|---|---|
| Current body weighta (kg) | 35 | 2.30 | 1.22 | 0.530 | 2.80 | 0.46–4.20 |
| Current body lengtha (cm) | 34 | 43.4 | 9.15 | 0.211 | 47.5 | 25.0–56.0 |
| BMI (kg/m2) | 34 | 11.1 | 2.91 | 0.263 | 12.0 | 6.04–16.7 |
| Gestational age (weeks) | 35 | 33.6 | 6.57 | 0.196 | 37 | 23–41 |
| Postnatal agea (days) | 35 | 7.49 | 5.73 | 0.766 | 6 | 1–26 |
| Postmenstrual agea (weeks) | 35 | 34.6 | 6.28 | 0.181 | 37.6 | 23.1–41.6 |
| Total bilirubin (mg/dL) | 22 | 6.65 | 4.88 | 0.734 | 4.8 | 0.9–17.5 |
| Serum creatinineb (mg/dL) | 30 | 0.707 | 0.242 | 0.342 | 0.7 | 0.3–1.1 |
| Estimated GFRc (mL/min/1.73 m2) | 29 | 30.1 | 16.6 | 0.551 | 24.1 | 12.6–70.9 |
BMI body mass index, CV coefficient of variation, GFR glomerular filtration rate, SD standard deviation
On the day of the first paracetamol dose
Serum creatinine concentrations obtained at ≤3 days postnatal age were considered to reflect maternal renal function and were excluded from the analysis
Estimated GFR was calculated using the updated Schwartz formula (Eq. 2) [26]
Table 3.
Subject demographics for categorical covariates tested in the model-building dataset
| Characteristic | N (%) |
|---|---|
| Sex | |
| Male | 20 (57) |
| Female | 15 (43) |
| Race | |
| White/Caucasian | 16 (46) |
| Black/African American | 14 (40) |
| American Indian/Alaska Native | 1 (3) |
| Asian | 1 (3) |
| Declined to respond | 3 (9) |
| Ethnicity | |
| Non-Hispanic | 24 (69) |
| Hispanic | 8 (23) |
| Declined to respond | 3 (9) |
The model-building dataset was best described by a one-compartment structural model with first-order elimination. A two-compartment model had a higher BIC than a comparable one-compartment model (847.6 vs. 841.7) and provided no visual improvement in standard diagnostic plots relative to a one-compartment model. Additionally, condition numbers of 14 and 161,905 were obtained, respectively, for comparable one- and two-compartment models, which suggested that the two-compartment model was an over-parameterized representation of the data [23]. When additive, proportional, and combined additive and proportional error functions were tested for characterization of RUV, the proportional function produced the lowest OFV and was selected for inclusion in the model (Eq. 3):
| (3) |
where Yij is the observed paracetamol concentration for the ith individual at time j, Ymij is the model-predicted paracetamol concentration, and εij is a normally distributed random error with a mean of zero and a variance of σ2.
During testing of potential covariates, subjects with missing information for a covariate undergoing evaluation were excluded from both the base and covariate models being tested. Current body weight was found to have a strong influence on paracetamol pharmacokinetics. Current body length and BMI were also tested as markers of body size, but their performance was inferior to that of weight. Postnatal age was identified as a significant covariate on clearance during the forward selection process but it failed to meet the criterion for inclusion as a covariate during backward elimination. Additionally, the final forward selection step showed that total bilirubin was a significant covariate on paracetamol clearance and volume of distribution: decreases in OFV ranged from 4.2 to 7.3, depending on the pharmacokinetic parameter and functional form of the covariate. However, total bilirubin was not included in the model due to the fact that these laboratory values were unavailable for 37 % of the study subjects. None of the other potential covariates met the criterion for inclusion at the final forward selection step. Based on OFV, a power function of mean-centered weight best described the relationship between weight and paracetamol pharmacokinetic parameters (Eq. 4):
| (4) |
where θi is the individual model-predicted pharmacokinetic parameter (e.g. clearance) for an individual with current body weight of WTi (kg), θpop is the population mean for the pharmacokinetic parameter θ when current body weight is equal to the mean current body weight of the study population, 2.30 is the mean current body weight of the study population (kg), θcov is the covariate effect, and ηi is the between-subject random effect for the ith individual on the pharmacokinetic parameter θ. In the final backward elimination step, removal of weight on clearance and volume of distribution increased the OFV by 65.5 and 81.8, respectively. The θcov exponent in Eq. 4 was first estimated to be 1.07 and 0.892 for clearance and volume of distribution, respectively. To simplify the final model equations, these values were rounded and fixed at 1.1 and 0.9, which caused a trivial change in OFV (increase of 0.2).
Parameter estimates derived from the final covariate model are provided in Table 4. Overall, model parameters were estimated with reasonably good precision, and bootstrap estimates agreed well with those obtained from the final covariate model. Only one of the 1000 bootstrap datasets (0.1 %) failed to minimize during parameter estimation. Standard diagnostic plots are also provided to illustrate the fit of the final covariate model. Plots of observed versus population- and individual-predicted paracetamol concentrations are provided in Fig. 1, and conditional weighted residuals versus time and population-predicted paracetamol concentrations are shown in Fig. 2.
Table 4.
Parameter estimates for the final covariate model
| Parameter | Model fit
|
Bootstrap 95 % CIa
|
||
|---|---|---|---|---|
| Estimated value (%RSE) |
BSV (ω2), as % CV (% RSE) |
Estimated value | BSV (ω2), as % CV | |
| CL (L/h)b | 0.348 (5.5) | 30.8 (19.9) | 0.311–0.387 | 23.9–36.2 |
| Effect of weightc | 1.1 fixed | 1.1 fixed | ||
| Vd (L)b | 2.46 (3.5) | 14.3 (51.2) | 2.29–2.64 | 0.1–19.9 |
| Effect of weightc | 0.9 fixed | 0.9 fixed | ||
| Proportional residual unexplained variability (σ2) | 0.0576 (20.8) | 0.0373–0.0844 | ||
BSV between-subject variability, CI confidence interval, CL total body clearance, % CV percent coefficient of variation, % RSE percent relative standard error, Vd volume of distribution
Bootstrap success rate was 99.9 % (n = 1000)
At the mean current body weight of the study population (2.30 kg)
Exponent on mean-centered weight, i.e. the covariate effect (θcov) in Eq. 4
Fig. 1.
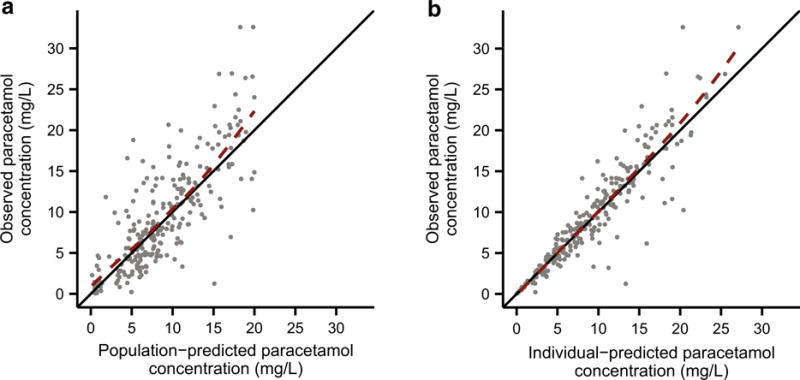
Diagnostic plots for the final covariate model. Observed versus a population-predicted and b individual-predicted paracetamol plasma concentrations. The solid black lines depict the lines of identity (y = x), and the dashed red lines depict the LOESS fits of the data
Fig. 2.
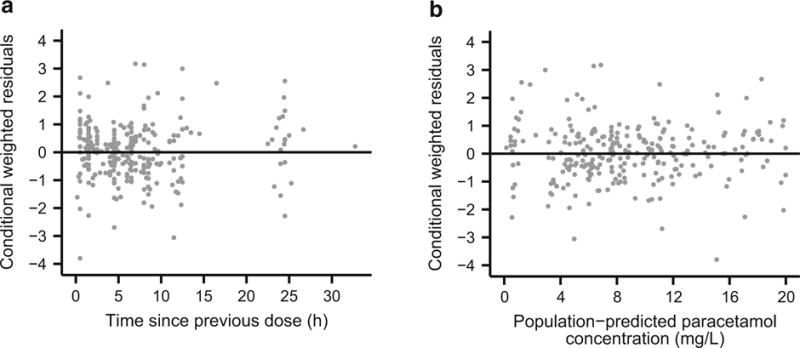
Diagnostic plots for the final covariate model. Conditional weighted residuals of paracetamol plasma concentrations versus a time since previous dose and b population-predicted paracetamol concentrations. The solid black lines depict y = 0
Simulation-based visualizations of model appropriateness were generated with NPDEs and a visual predictive check. The mean NPDE from the model-building dataset was 0.0485, and the variance was 0.935. These values were not significantly different from the expected mean of 0 (Wilcoxon signed-rank test, p = 0.231) and variance of 1 (Fisher variance test, p = 0.476) (Fig. 3a). Additionally, there were no visible trends in NPDEs when plotted against time since previous dose (Fig. 3c), population-predicted paracetamol concentration (Fig. 3e), and current body weight (Fig. 3g). The visual predictive check demonstrated good agreement between observed paracetamol concentrations and model-based simulations (Fig. 4a), and a numerical predictive check determined that 92.7 % of the observations fell within the simulation-based 90 % prediction interval.
Fig. 3.
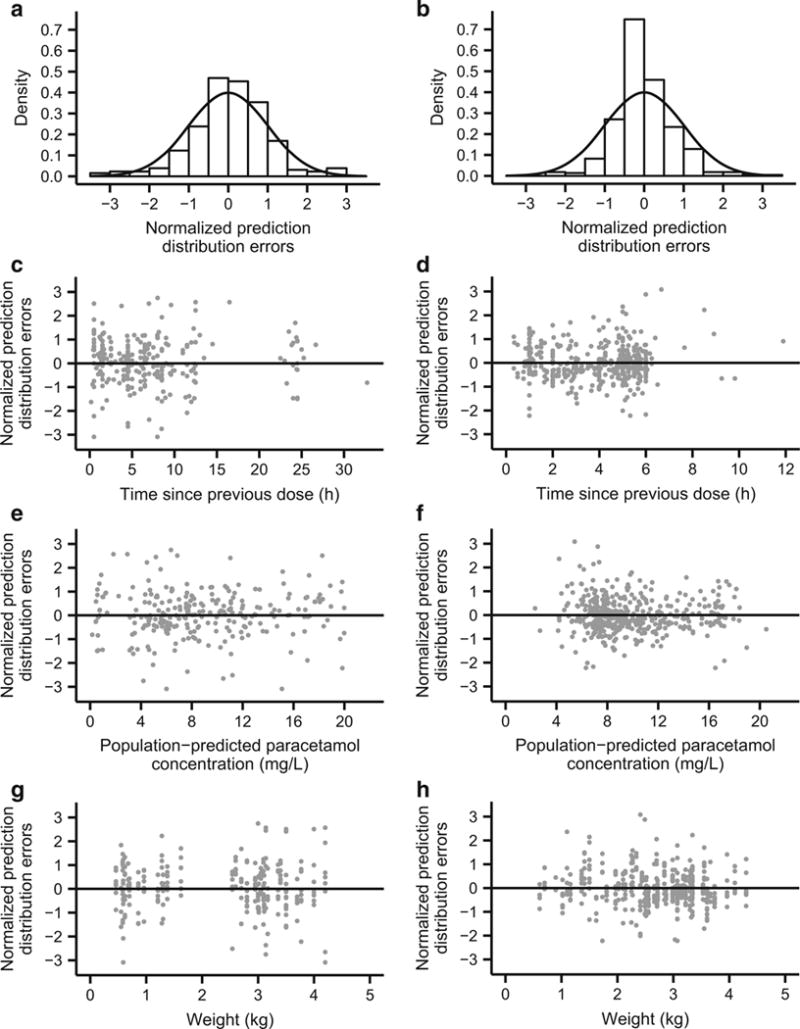
NPDEs of paracetamol plasma concentrations from the model-building dataset (a, c, e, g) and the external evaluation dataset (b, d, f, h). Density histograms of NPDEs (a, b) with overlaid solid black curves depicting standard normal distributions for comparison. NPDEs versus time since previous dose (c, d), population-predicted paracetamol concentration (e, f), and current body weight (g, h). NPDEs normalized prediction distribution errors
Fig. 4.
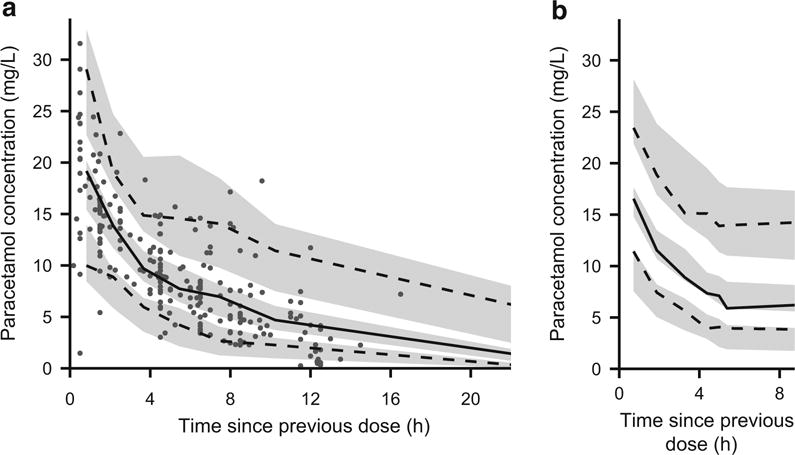
Visual predictive checks of the final covariate model for a the model-building dataset and b the external evaluation dataset. The solid black lines depict the observed 50th percentiles, and the dashed black lines depict the observed 5th and 95th percentiles. The shaded gray regions depict the 95 % confidence intervals surrounding the predicted 5th, 50th, and 95th percentiles. Individual observations are depicted as gray dots. Individual observations were omitted from b because the density of points would obscure the percentile lines and prediction intervals
Key subject characteristics for the external dataset are summarized in Tables 1 and 5. Compared with the model-building dataset, the external dataset had a lower proportion of extremely preterm neonates and tended toward younger postnatal ages. Additionally, whereas the model-building dataset was obtained from a US-based study with fairly even representation of White/Caucasian and Black/ African American races, the external dataset was derived from a Belgian study with predominantly White/Caucasian subjects [6].
Table 5.
Key subject demographics from the external evaluation dataset
| Characteristic | N | Mean | SD | CV | Median | Range |
|---|---|---|---|---|---|---|
| Current body weight (kg) | 60 | 2.62 | 0.894 | 0.341 | 2.71 | 0.61–4.30 |
| Gestational age (weeks) | 60 | 35.6 | 4.34 | 0.122 | 37 | 24–41 |
| Postnatal age (days) | 60 | 6.08 | 6.85 | 1.126 | 3 | 1–27 |
| Postmenstrual age (weeks) | 60 | 36.5 | 3.89 | 0.106 | 37.4 | 26.4–42.0 |
CV coefficient of variation, SD standard deviation
Bias (prediction error) and precision (absolute prediction error) were quantitated by applying the final covariate model to the external dataset. Population-predicted concentrations from the model tended to be slightly higher than observed values (Table 6). The mean NPDE from the external dataset was −0.00772, which was not significantly different from the expected mean of 0 (Wilcoxon signed-rank test, p = 0.178), but the NPDE variance from the external dataset was 0.516, which was lower than the expected variance of 1 (Fisher variance test, p = 2.09 × 10−18). Thus, the final covariate model overpredicted the amount of variability in the external dataset (Fig. 3b). However, the NPDE showed no bias when plotted against time since previous dose (Fig. 3d), population-predicted paracetamol concentration (Fig. 3f), and current body weight (Fig. 3h). Additionally, a visual predictive check demonstrated reasonably good agreement between paracetamol concentration observations from the external dataset and model-based simulations (Fig. 4b). Finally, the numerical predictive check determined that 95.9 % of the observations fell within the 90 % prediction interval, which also indicated that the model overestimated the amount of variability in the external dataset.
Table 6.
Predictive performance of the final covariate model when applied to the external dataset
| Concentration (mg/L)
|
Percentage of observed concentration
|
|||
|---|---|---|---|---|
| Median | 95 % CI | Median | 95 % CI | |
| Prediction error | 0.911 | 0.495–1.33 | 10.1 | 6.13–14.3 |
| Absolute prediction error | 2.22 | 2.07–2.35 | 25.3 | 23.1–28.1 |
CI confidence interval
4 Discussion
Previous neonatal studies of intravenous paracetamol and propacetamol, a prodrug that is rapidly hydrolyzed by plasma esterases to form paracetamol, have used one- [22, 35, 36] and two-compartment [6, 8] models. In the present study, the model-building dataset was best described by a one-compartment model. At the mean current body weight of the study population, the final covariate model gave parameter estimates of 0.151 L/h/kg (0.348 L/h/2.30 kg) and 1.07 L/kg (2.46 L/2.30 kg) for clearance and volume of distribution, respectively. Clearance estimates from earlier neonatal studies ranged from 0.090–0.21 L/h/kg, depending in part on gestational, postmenstrual, or postconceptional age [6, 8, 22, 35, 36]. Previously reported volume of distribution estimates were slightly lower than those observed here, with values ranging from 0.56–0.76 L/kg [8, 35].
To compare the neonatal pharmacokinetic parameter estimates from the present study with adult values, estimates can be extrapolated to a standard 70 kg adult using allometric scaling. If allometric exponents of 0.75 and 1 are applied to the clearance and volume of distribution terms, respectively [37], this yields values of 4.51 L/h/70 kg for clearance and 74.9 L/70 kg for volume of distribution. These values are consistent with allometrically standardized neonatal estimates from prior studies of both intravenous [6, 22, 36] and enteral [13, 38] paracetamol. In neonates, paracetamol clearance values are approximately one-quarter to one-third of typical adult values [39], and the relatively low neonatal clearance can be attributed to incomplete maturation of hepatic drug metabolism pathways [14, 40–42].
Current body weight was the only covariate that met criteria for inclusion in the final model. Previous population pharmacokinetic studies found weight, as a correlate of body size, was the major covariate influencing intravenous paracetamol pharmacokinetics in neonates [6, 22, 36]. Postmenstrual age [6, 36], postconceptional age [22], and unconjugated bilirubin [6, 36] have also been shown to have minor effects on neonatal clearance of intravenous paracetamol but these characteristics were not identified as significant covariates in the present study. The current study was expected to have reasonably good power to detect an effect of postmenstrual age on clearance because it had a good representation of extreme preterm, preterm, and full-term neonates (Table 1) and was obtained from subjects with a fairly wide range of postmenstrual ages (23.1–41.6 weeks). Given that prior studies in neonates have found only minor increases in paracetamol clearance with increasing postmenstrual or postconceptional age, the failure to identify postmenstrual age as a significant covariate was not surprising. Indeed, maturation of paracetamol clearance appears to be fairly slow up until a postmenstrual age of 40 weeks and then occurs more rapidly during the first year of life [42]; therefore, the ability of this study to detect any age-related covariate effects was likely limited by the fact that the postnatal age of most study subjects (66 %) did not exceed 7 days.
During forward covariate selection, total bilirubin was inversely correlated with clearance, which agrees with previous findings that high unconjugated bilirubin was associated with reduced clearance [6, 36]. Physiologically, these observations can be attributed to the fact that both paracetamol and bilirubin undergo substantial clearance via glucuronidation, and concentrations of paracetamol and unconjugated or total bilirubin are therefore expected to be correlated. Because the association between total bilirubin and paracetamol pharmacokinetic parameters was fairly weak, and these laboratory values were unavailable for 37 % of the study subjects, it was decided that omission of this covariate from the final model was preferable to the data imputation that would be required if the covariate was retained.
Perhaps the most comprehensive study to date on the pharmacokinetics of intravenous paracetamol in neonates is the pooled analysis performed by Allegaert et al. [6]. A subset of that pooled data was used to externally evaluate the final covariate model reported herein. This external dataset from the PARANEO trial was selected because the study design and subject demographics were relatively consistent with those of the study from which the model-building dataset was obtained. The final covariate model demonstrated acceptable bias and precision when applied to the external dataset (Table 6). The most substantial difference between model predictions and external data observations was an overestimation of variability, which was particularly evident in the NPDEs and numerical and visual predictive checks. This discrepancy could be attributable to differences in patient demographics, study design or execution, or analytical drug quantification. However, given the gestational age distributions from the two studies, larger variability might be expected in the model-building dataset, based on the higher proportion of extremely preterm subjects. Overall, the external evaluation indicated that the final covariate model performed adequately despite notable study differences in the proportion of extremely preterm neonates, postnatal age, racial composition, and geographic location.
One major strength of the present study was the inclusion of a large number of extremely preterm neonates. Additionally, the final covariate model was successfully evaluated against a dataset obtained from a similar, independent clinical trial—a validation procedure that is rarely performed in neonatal clinical research. This study demonstrated that the pharmacokinetics of intravenous paracetamol can be predicted using body weight in neo-nates ranging from extreme preterm to full-term gestational status. This finding reinforces previous work that supported the use of a simplified neonatal dosing regimen in which maturational changes in paracetamol pharmacokinetics could be accommodated using only equivalent per kilogram dosing, without requiring different doses or dosing intervals dependent on gestational or postmenstrual age [6]. The results of the present study suggest that extension of such a parsimonious dosing regimen to extremely preterm neonates may be valid; however, these findings should be interpreted with caution based on the limitations outlined below.
Although the number and proportion of extremely pre-term neonates in this study was substantially higher than in previous reports, the sample size was still relatively small, as is often the case for neonatal trials. Additionally, BSV in the final covariate model remained fairly large, particularly for clearance (30.8 % CV), and it is possible that other unmeasured factors could be incorporated as covariates to further reduce the unexplained BSV. Another important limitation is related to hepatotoxicity, the principal safety concern that accompanies use of the drug. Paracetamol-induced hepatotoxicity is not associated with exposure to the parent drug per se but rather depends on the amount of exposure to a relatively minor toxic metabolite, N-acetyl-p-benzoquinone imine (NAPQI) [43]. In humans, cyto-chrome P450 (CYP) 2E1 is predominantly responsible for the conversion of paracetamol to NAPQI [43]. Hepatic expression of CYP2E1 increases during the neonatal period, approaching adult values by approximately 90 days postnatal age [44]. Additionally, glucuronidation accounts for the majority of paracetamol clearance in adults, but glucuronidation capacity is immature in neonates [14, 45]. Thus, maturational changes in hepatotoxicity risk may not be reflected in the pharmacokinetics of the parent drug, and the pharmacokinetics of paracetamol metabolites should be studied to address this safety concern more thoroughly. Finally, although this study provides critical information regarding the pharmacokinetics of intravenous paracetamol in neonates, it should be noted that pharmacodynamic data for paracetamol are still quite limited in this patient population [46]. Further studies are needed to determine appropriate pharmacodynamic targets, which may vary by indication (e.g. analgesia, antipyresis, or patent ductus arteriosus closure) [47, 48].
5 Conclusions
A one-compartment model successfully characterized the pharmacokinetics of intravenous paracetamol in preterm and term neonates. Clearance and volume of distribution increased with body weight, and weight was the principal predictor of intravenous paracetamol pharmacokinetics in extremely preterm to full-term neonates. An external evaluation supported the generalizability of the final covariate model to other similar patient populations.
Key Points.
In neonates ranging from extreme preterm to full-term gestational status, body weight is the principal predictor of intravenous paracetamol pharmacokinetics.
A parsimonious regimen based only on equivalent per kilogram doses may be sufficient to accommodate maturational changes in paracetamol pharmacokinetics, even for extremely preterm neonates; however, additional studies are needed to ensure that such a simplified dosing regimen does not increase the risk of paracetamol-induced hepatotoxicity in any neonatal subpopulations.
Acknowledgments
The authors would like to thank Syamala Mankala of the Division of Clinical Pharmacology at the Children’s National Health System (Washington, DC, USA) for administrative support.
Funding This work was supported by National Institutes of Health grants from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (R01HD060543, to John N. van den Anker) and the National Center for Advancing Translational Sciences (UL1TR000075, to the Children’s National Health System), and by a contract for analytical laboratory services from McNeil Consumer Healthcare (Division of McNEIL-PPC, Inc., Fort Washington, PA, USA, to Diana G. Wilkins). Karel Allegaert was supported by a Fundamental Clinical Investigatorship (1800214N) from the Fund for Scientific Research—Flanders (FWO-Vlaanderen, Belgium). Sarah F. Cook received stipend support from the Howard Hughes Medical Institute (Med into Grad Initiative); Sarah F. Cook and Chris Stock-mann were supported by pre-doctoral fellowships from the American Foundation for Pharmaceutical Education; and Jessica K. Roberts was supported by a Pharmacotherapy Subspecialty Award from the Primary Children’s Hospital Foundation (Salt Lake City, UT, USA).
Footnotes
Compliance with Ethical Standards
Conflicts of interest Sarah Cook, Jessica Roberts, Samira Samiee-Zafarghandy, Chris Stockmann, Amber King, Nina Deutsch, Elaine Williams, Karel Allegaert, Diana Wilkins, Catherine Sherwin, and John van den Anker have no potential conflicts of interest to declare.
Ethical approval All human studies were approved by the appropriate Ethics Committees and were carried out in concordance with ICH Guidelines for Good Clinical Practice [19]. Informed consent was obtained prior to study inclusion.
References
- 1.Hall RW, Anand KJ. Pain management in newborns. Clin Perinatol. 2014;41(4):895–924. doi: 10.1016/j.clp.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allegaert K, Tibboel D, van den Anker J. Pharmacological treatment of neonatal pain: in search of a new equipoise. Semin Fetal Neonatal Med. 2013;18(1):42–7. doi: 10.1016/j.siny.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pacifici GM, Allegaert K. Clinical pharmacology of paracetamol in neonates: a review. Curr Ther Res Clin Exp. 2015;77:24–30. doi: 10.1016/j.curtheres.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duggan ST, Scott LJ. Intravenous paracetamol (acetaminophen) Drugs. 2009;69(1):101–13. doi: 10.2165/00003495-200969010-00007. [DOI] [PubMed] [Google Scholar]
- 5.US FDA. Drug approval package: Ofirmev (acetaminophen) injection, 10mg/mL. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/022450_ofirmev_toc.cfm Accessed 24Mar 2015.
- 6.Allegaert K, Palmer GM, Anderson BJ. The pharmacokinetics of intravenous paracetamol in neonates: size matters most. Arch Dis Child. 2011;96(6):575–80. doi: 10.1136/adc.2010.204552. [DOI] [PubMed] [Google Scholar]
- 7.Zuppa AF, Hammer GB, Barrett JS, Kenney BF, Kassir N, Mouksassi S, et al. Safety and population pharmacokinetic analysis of intravenous acetaminophen in neonates, infants, children, and adolescents with pain or fever. J Pediatr Pharmacol Ther. 2011;16(4):246–61. doi: 10.5863/1551-6776-16.4.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Ganzewinkel C, Derijks L, Anand KJ, van Lingen RA, Neef C, Kramer BW, et al. Multiple intravenous doses of paracetamol result in a predictable pharmacokinetic profile in very preterm infants. Acta Paediatr. 2014;103(6):612–7. doi: 10.1111/apa.12638. [DOI] [PubMed] [Google Scholar]
- 9.Allegaert K, Murat I, Anderson BJ. Not all intravenous paracetamol formulations are created equal. Paediatr Anaesth. 2007;17(8):811–2. doi: 10.1111/j.1460-9592.2007.02227.x. [DOI] [PubMed] [Google Scholar]
- 10.Bartocci M, Lundeberg S. Intravenous paracetamol: the ‘Stockholm protocol’ for postoperative analgesia of term and preterm neonates. Paediatr Anaesth. 2007;17(11):1120–1. doi: 10.1111/j.1460-9592.2007.02322.x. [DOI] [PubMed] [Google Scholar]
- 11.van den Anker JN, Tibboel D. Pain relief in neonates: when to use intravenous paracetamol. Arch Dis Child. 2011;96(6):573–4. doi: 10.1136/adc.2011.211060. [DOI] [PubMed] [Google Scholar]
- 12.Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology: drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349(12):1157–67. doi: 10.1056/NEJMra035092. [DOI] [PubMed] [Google Scholar]
- 13.Anderson BJ, van Lingen RA, Hansen TG, Lin YC, Holford NH. Acetaminophen developmental pharmacokinetics in premature neonates and infants: a pooled population analysis. Anesthesiol-ogy. 2002;96(6):1336–45. doi: 10.1097/00000542-200206000-00012. [DOI] [PubMed] [Google Scholar]
- 14.Allegaert K, Vanhaesebrouck S, Verbesselt R, van den Anker JN. In vivo glucuronidation activity of drugs in neonates: extensive interindividual variability despite their young age. Ther Drug Monit. 2009;31(4):411–5. doi: 10.1097/FTD.0b013e3181a8cc0a. [DOI] [PubMed] [Google Scholar]
- 15.De Cock RF, Piana C, Krekels EH, Danhof M, Allegaert K, Knibbe CA. The role of population PK-PD modelling in paedi-atric clinical research. Eur J Clin Pharmacol. 2011;67(Suppl 1):5–16. doi: 10.1007/s00228-009-0782-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knibbe CA, Danhof M. Individualized dosing regimens in children based on population PKPD modelling: are we ready for it? Int J Pharm. 2011;415(1–2):9–14. doi: 10.1016/j.ijpharm.2011.02.056. [DOI] [PubMed] [Google Scholar]
- 17.Tod M, Jullien V, Pons G. Facilitation of drug evaluation in children by population methods and modelling. Clin Pharma-cokinet. 2008;47(4):231–43. doi: 10.2165/00003088-200847040-00002. [DOI] [PubMed] [Google Scholar]
- 18.Krekels EH, van Hasselt JG, Tibboel D, Danhof M, Knibbe CA. Systematic evaluation of the descriptive and predictive performance of paediatric morphine population models. Pharm Res. 2011;28(4):797–811. doi: 10.1007/s11095-010-0333-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) doi: 10.1111/j.1365-2125.1994.tb05705.x. Available at: http://www.ich.org/ Accessed 15 Apr 2015. [DOI] [PMC free article] [PubMed]
- 20.van den Anker J. Metabolism and toxicity of acetaminophen. Available at: https://clinicaltrials.gov/show/NCT01328808 Accessed 1 Mar 2015.
- 21.Universitaire Ziekenhuizen Leuven. Pharmacokinetics, -dynamics and safety of intravenous paracetamol in neonates (PARANEO) Available at: https://clinicaltrials.gov/show/NCT00969176 Accessed 1 Mar 2015.
- 22.Allegaert K, Anderson BJ, Naulaers G, de Hoon J, Verbesselt R, Debeer A, et al. Intravenous paracetamol (propacetamol) pharmacokinetics in term and preterm neonates. Eur J Clin Pharmacol. 2004;60(3):191–7. doi: 10.1007/s00228-004-0756-x. [DOI] [PubMed] [Google Scholar]
- 23.Bonate PL. Pharmacokinetic-pharmacodynamic modeling and simulation. New York: Springer Science+Business Media, Inc.; 2006. [Google Scholar]
- 24.Ludden TM, Beal SL, Sheiner LB. Comparison of the akaike information criterion, the Schwarz criterion and the F test as guides to model selection. J Pharmacokinet Biopharm. 1994;22(5):431–45. doi: 10.1007/BF02353864. [DOI] [PubMed] [Google Scholar]
- 25.Mould DR, Upton RN. Basic concepts in population modeling, simulation, and model-based drug development—part 2: introduction to pharmacokinetic modeling methods. CPT Pharma-comet Syst Pharmacol. 2013;2(4):e38. doi: 10.1038/psp.2013.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwartz GJ, Munoz A, Schneider MF, Mak RH, Kaskel F, Warady BA, et al. New equations to estimate GFR in children with CKD. J Am Soc Nephrol. 2009;20(3):629–37. doi: 10.1681/ASN.2008030287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McLeay SC, Morrish GA, Kirkpatrick CM, Green B. The relationship between drug clearance and body size: systematic review and meta-analysis of the literature published from 2000 to 2007. Clin Pharmacokinet. 2012;51(5):319–30. doi: 10.2165/11598930-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 28.Owen JS, Fiedler-Kelly J. Introduction to population pharmacokinetic/pharmacodynamic analysis with nonlinear mixed effects models. London: Wiley; 2014. [Google Scholar]
- 29.Ette EI. Stability and performance of a population pharmacokinetic model. J Clin Pharmacol. 1997;37(6):486–95. doi: 10.1002/j.1552-4604.1997.tb04326.x. [DOI] [PubMed] [Google Scholar]
- 30.Brendel K, Comets E, Laffont C, Mentre F. Evaluation of different tests based on observations for external model evaluation of population analyses. J Pharmacokinet Pharmacodyn. 2010;37(1):49–65. doi: 10.1007/s10928-009-9143-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karlsson MO, Savic RM. Diagnosing model diagnostics. Clin Pharmacol Ther. 2007;82(1):17–20. doi: 10.1038/sj.clpt.6100241. [DOI] [PubMed] [Google Scholar]
- 32.Keizer RJ, Karlsson MO, Hooker A. Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT Pharmacometrics Syst Pharmacol. 2013;2:e50. doi: 10.1038/psp.2013.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13(2):143–51. doi: 10.1208/s12248-011-9255-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sheiner LB, Beal SL. Some suggestions for measuring predictive performance. J Pharmacokinet Biopharm. 1981;9(4):503–12. doi: 10.1007/BF01060893. [DOI] [PubMed] [Google Scholar]
- 35.Allegaert K, Van der Marel CD, Debeer A, Pluim MA, Van Lingen RA, Vanhole C, et al. Pharmacokinetics of single dose intravenous propacetamol in neonates: effect of gestational age. Arch Dis Childhood Fetal Neonatal Ed. 2004;89(1):F25–8. doi: 10.1136/fn.89.1.F25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palmer GM, Atkins M, Anderson BJ, Smith KR, Culnane TJ, McNally CM, et al. I.V. acetaminophen pharmacokinetics in neonates after multiple doses. Br J Anaesth. 2008;101(4):523–30. doi: 10.1093/bja/aen208. [DOI] [PubMed] [Google Scholar]
- 37.Anderson BJ, Holford NH. Mechanism-based concepts of size and maturity in pharmacokinetics. Ann Rev Pharmacol Toxicol. 2008;48:303–32. doi: 10.1146/annurev.pharmtox.48.113006.094708. [DOI] [PubMed] [Google Scholar]
- 38.Anderson BJ, Woollard GA, Holford NH. A model for size and age changes in the pharmacokinetics of paracetamol in neonates, infants and children. Br J Clin Pharmacol. 2000;50(2):125–34. doi: 10.1046/j.1365-2125.2000.00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Allegaert K, Olkkola KT, Owens KH, Van de Velde M, de Maat MM, Anderson BJ. Covariates of intravenous paracetamol pharmacokinetics in adults. BMC Anesthesiol. 2014;14:77. doi: 10.1186/1471-2253-14-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levy G, Khanna NN, Soda DM, Tsuzuki O, Stern L. Pharmacokinetics of acetaminophen in the human neonate: formation of acetaminophen glucuronide and sulfate in relation to plasma bilirubin concentration and D-glucaric acid excretion. Pediatrics. 1975;55(6):818–25. [PubMed] [Google Scholar]
- 41.Miller RP, Roberts RJ, Fischer LJ. Acetaminophen elimination kinetics in neonates, children, and adults. Clin Pharmacol Ther. 1976;19(3):284–94. doi: 10.1002/cpt1976193284. [DOI] [PubMed] [Google Scholar]
- 42.Anderson BJ, Holford NH. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab Pharmacokinet. 2009;24(1):25–36. doi: 10.2133/dmpk.24.25. [DOI] [PubMed] [Google Scholar]
- 43.McGill MR, Jaeschke H. Metabolism and disposition of acetaminophen: recent advances in relation to hepatotoxicity and diagnosis. Pharm Res. 2013;30(9):2174–87. doi: 10.1007/s11095-013-1007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnsrud EK, Koukouritaki SB, Divakaran K, Brunengraber LL, Hines RN, McCarver DG. Human hepatic CYP2E1 expression during development. J Pharmacol Exp Ther. 2003;307(1):402–7. doi: 10.1124/jpet.102.053124. [DOI] [PubMed] [Google Scholar]
- 45.Krekels EH, Danhof M, Tibboel D, Knibbe CA. Ontogeny of hepatic glucuronidation; methods and results. Curr Drug Metab. 2012;13(6):728–43. doi: 10.2174/138920012800840455. [DOI] [PubMed] [Google Scholar]
- 46.Allegaert K, Naulaers G, Vanhaesebrouck S, Anderson BJ. The paracetamol concentration-effect relation in neonates. Paediatr Anaesth. 2013;23(1):45–50. doi: 10.1111/pan.12076. [DOI] [PubMed] [Google Scholar]
- 47.Gibb IA, Anderson BJ. Paracetamol (acetaminophen) pharmacodynamics: interpreting the plasma concentration. Arch Dis Child. 2008;93(3):241–7. doi: 10.1136/adc.2007.126896. [DOI] [PubMed] [Google Scholar]
- 48.Ohlsson A, Shah PS. Paracetamol (acetaminophen) for patent ductus arteriosus in preterm or low-birth-weight infants. Cochrane Database Syst Rev. 2015;3:CD010061. doi: 10.1002/14651858.CD010061.pub2. [DOI] [PubMed] [Google Scholar]