

Abstract
Asymmetrically modified nucleosomes contain the two copies of a histone (sister histones) decorated with distinct sets of Post-translational Modifications (PTMs). They are newly identified species with unknown means of establishment and functional implications. Current analytical methods are inadequate to detect the copy-specific occurrence of PTMs on the nucleosomal sister histones. This protocol presents a biochemical method for the in vitro reconstitution of nucleosomes containing differentially isotope-labeled sister histones. The generated complex can be also asymmetrically modified, after including a premodified histone pool during refolding of histone subcomplexes. These asymmetric nucleosome preparations can be readily reacted with histone-modifying enzymes to study modification cross-talk mechanisms imposed by the asymmetrically pre-incorporated PTM using nuclear magnetic resonance (NMR) spectroscopy. Particularly, the modification reactions in real-time can be mapped independently on the two sister histones by performing different types of NMR correlation experiments, tailored for the respective isotope type. This methodology provides the means to study crosstalk mechanisms that contribute to the formation and propagation of asymmetric PTM patterns on nucleosomal complexes.
Keywords: Biochemistry, Issue 121, histones, post-translational modifications, nucleosomes, NMR spectroscopy, epigenetics, chromatin
Introduction
Eukaryotic DNA is tightly packaged within cell nuclei into chromatin. The fundamental building block of chromatin is the nucleosome core particle that contains ~147 bp of DNA wrapped around an octameric complex made up of two copies each of the four core histones (H3, H4, H2A, H2B). Histone proteins harbor a plethora of Post-translational Modifications (PTMs). These covalent substitutions induce alterations in chromatin structure, both directly by affecting the physical chemistry of the system and indirectly by recruiting chromatin-remodeling activities1,2,3. By those means, histone PTMs control chromatin accessibility and, hence, regulate all DNA-based cellular functions4.
PTMs are installed by histone-modifying enzyme systems mainly on the unstructured N-terminal segments (tails) of nucleosome-incorporated core histones. Due to the many modification sites on the relatively short sequence of histone tails, PTMs influence each other by inducing or blocking subsequent modification reactions, an effect known as modification cross-talk5. Because of the overall symmetric architecture of the nucleosome, modification reactions and crosstalk mechanisms were thought to occur similarly onto the two copies of each nucleosomal histone (sister histones). This concept was recently challenged and subsequently disproved. Particularly, in vitro enzymatic assays on free histone H3 tail peptides and on nucleosomes demonstrated that a set of H3 kinases introduced phosphorylation in an asymmetric manner6. Additionally, affinity-purification-based LC-MS/MS analysis revealed the existence of asymmetrically H3-methylated nucleosomes in several types of eukaryotic cells7. Thus, asymmetrically modified nucleosomes constitute novel species, and tools are needed to uncover the mechanisms that control their formation and to analyze the crosstalk effects that this asymmetry might exert.
Commonly, Western Blotting (WB) and Mass Spectrometry (MS) analysis have been used to detect histone PTMs. Despite its easy application, WB suffers from specificity/cross-reactivity problems. On top of that, it is incapable of performing simultaneous multi-PTM analysis and direct quantification of the modification reactions8. On the other hand, MS analysis employs sophisticated instrumentation that requires high-level training, but provides high specificity as well as simultaneous mapping and quantification of multiple PTMs9. However, both methods are disruptive and the nucleosomal complexes are dissociated before analysis, giving rise to a mixture of histones and/or histone-derived peptides. This manipulation removes the ability to distinguish independent modification reactions that occur on each of the two sister histones and to report the copy-specific modification status of the nucleosomal histones.
Nuclear Magnetic Resonance (NMR) spectroscopy evolved as an alternative method to map PTM reactions. NMR is nondisruptive and thus allows the monitoring of PTM events in a real-time manner in reconstituted mixtures, and even in intact cells10,11. The development of routines for fast data acquisition as well as for high-resolution mapping based on 2D hetero-nuclear correlation methods of isotope-labeled (15N and/or 13C) samples12 allowed the simultaneous mapping of different types of PTMs, such as serine/threonine/tyrosine phosphorylation, lysine acetylation/methylation, and arginine methylation13. Depending on the PTM under investigation, 15N- or 13C-labeling protocols can be employed to mark the protein functional group that serves as a modification reporter. Consequently, PTM mapping can be performed by following the characteristic chemical shift displacement of the corresponding functional group 'sensing' the alteration on the chemical environment. In most cases, both N-H and C-H chemical groups can be used to report the evolution of the PTM of interest.
The current protocol describes the generation of nucleosomes containing differentially isotope-labeled sister histones. It combines the flexibility of NMR spectroscopy to map PTMs using both 1H-15N and 1H-13C correlation spectra with the utilization of different protein affinity tags for purification of the selected reconstituted histone complexes. Notably, the protocol employs two different pools of a particular histone for nucleosome reconstitution. These pools are differentially isotope-labeled (one with 15N, the other with 13C), and they are fused to a polyhistidine and a streptavidin affinity tag, respectively. A tandem affinity purification scheme with Ni-NTA and streptavidin-based chromatography initially used by Voigt et al.7 is employed to purify asymmetric species from symmetric counterparts (Figure 1A). Asymmetric histone octamers are used subsequently to reconstitute equivalent nucleosomal complexes (Figure 1B), using the standard salt dialysis method14. Additionally, through the same procedure and by having one of the histone pools pre-modified, a PTM can be incorporated asymmetrically onto the resulting nucleosomes. The reaction of these substrates with histone-modifying enzymes and subsequent NMR-mapping of modification events enable the characterization of crosstalk mechanisms both in-cis (premodified histone copy) and in-trans (unmodified histone copy) (Figure 1C).
Protocol
1. Reconstitution of Nucleosomes with Differentially Isotope-labeled (and Asymmetrically Modified) Sister Histones
NOTE: The current protocol describes the reconstitution of nucleosomes with differentially isotope-labeled histone H3. To this end, two pools of histone H3 were used; one was 15N-labeled and contained a 6xHis-tag at the N-terminus and the second was 13C-labeled and contained the Strep peptide (WSHPQFEK) fused at the N-terminus. Both tags were separated from the native H3 sequence with a Tobacco Etch Virus (TEV) protease recognition site. To prepare additional asymmetrically modified nucleosomes, one of the two H3 pools is included in a premodified form. Premodification of a histone pool can be performed by reacting the isotope-labeled histone of interest with the respective histone-modifying enzyme in the presence of the necessary for each type of reaction cofactors/PTM donors16. The efficient placement of the respective PTM can be assessed by NMR spectroscopy or mass spectrometry. The amounts of histones/DNA used here are rough recommendations to obtain a nucleosome preparation with an approximate concentration of 10 µM (measured as DNA concentration at 260 nm).This final yield is sufficient to record good quality NMR spectra with relatively short acquisition times.
- Reconstitution of a histone octamer with differentially isotope-labeled (and asymmetrically modified) histone H3 NOTE: Recombinant histone proteins can be produced in mg quantities in BL21(DE3)plysS cells14. To obtain isotope-labeled histones, the same expression/purification protocol is followed with the exception of using for bacterial growth a minimal medium containing 15NH4Cl and/or 13C-Glucose as nitrogen and carbon sources, for 15N- or 13C-labeling respectively. Purified histones are extensively dialyzed against 5 mM β-mercaptoethanol and stored lyophilized before use.
- Dissolve lyophilized histone aliquots, containing ~5 mg of each histone in 1 mL of unfolding buffer (7 M Guanidinium-HCl, 20 mM Tris pH 7.5, 10 mM DTT). Include both types of histone H3. Mix by pipetting and do not vortex. Keep the tubes on ice for 30 min to allow complete unfolding.
- Determine the exact concentration of each histone by measuring the absorbance at 280 nm against unfolding buffer and using extinction coefficients calculated with the ProtParam tool of the ExPASy portal.15
- Mix the core histones to equimolar ratios, keeping in mind to include 50% each of the two histone H3 pools. Start by using the whole content of the less concentrated histone and add all of the others accordingly.
- Adjust final protein concentration to approximately 1 mg/mL using unfolding buffer.
- Transfer the mixture to a 6 kDa-cutoff dialysis membrane and dialyze against 1-2 L of chilled refolding buffer (10 mM Tris pH 7.5, 2 M NaCl, 1 mM EDTA, 2 mM DTT) at 4 °C. Change the buffer at least once after dialysis has proceeded for a minimum of 3 h. Perform a final dialysis O/N at 4 °C.
- Collect dialyzed material and remove any precipitates by centrifugation in a microfuge for 5 min at 4 °C on maximum speed (normally no precipitation should be observed). Determine octamer concentration by measuring the absorbance at 280 nm. In the calculations, use the added extinction coefficients of histones, keeping in mind to double each value.
- Concentrate to a final volume of ~2 mL using a 10 kDa-cutoff centrifugal filter unit. Recalculate octamer concentration and estimate loss. At this point, an octamer concentration between 50 - 100 µM should be obtained.
- Connect the gel filtration column (stated in the Table of Materials) to an FPLC system and equilibrate with 2 column volumes (CV) of 0.22 µm-filtered and degassed refolding buffer at a flow rate of 1 mL/min and a pressure limit of 0.5 MPa. NOTE: The gel filtration run should be performed in the cold room or in a cold cabinet.
- Inject concentrated material using a flow rate of 1 mL/min and collect 1.0 - 1.5 mL fractions.
- Follow the chromatographic profile and observe histone octamer elution at ~65 - 68 mL. NOTE: A small shoulder of the elution peak and a peak at ~80 mL indicates the existence of free H3-H4 tetramers and H2A-H2B dimers, respectively.
- Run 20 µL of each collected fraction in an 18% SDS-PAGE gel. NOTE: Dilute the samples by a factor of at least 2 with water before loading them on the gel to reduce the high salt concentration and thus, avoid distortions. The same applies for subsequent SDS-PAGE analyses of samples in refolding buffer.
- Pool relevant fractions containing pure octamers judged by the equal distribution of the 4 histones on the stained gel and determine concentration as before (step 1.1.7).
- Connect a 1 mL Ni-NTA column (Table of Materials) to an FPLC system and equilibrate with 10 CV of 0.22 µm-filtered and degassed refolding buffer at a flow rate of 1 mL/min. NOTE: The Ni-NTA chromatography should be performed in the cold room or in a cold cabinet.
- Pass purified octamer through the Ni-NTA using a flow rate of 1 mL/min.
- Collect flow-through and keep samples for SDS-PAGE/WB analysis (20 µL and 4 µL, respectively). Subsequently, wash column with 10 CV of refolding buffer or until a baseline signal is reached.
- Elute with 10 CV of refolding buffer containing 250 mM imidazole using a flow rate of 1 mL/min. Keep samples for SDS-PAGE/WB analysis (20 µL and 4 µL, respectively).
- Equilibrate a 1 mL affinity column of a commercial streptavidin (Table of Materials) with 10 CV refolding buffer containing 250 mM imidazole using a batch/gravity flow setup. NOTE: This chromatographic step should be performed in the cold room.
- Pass Ni-NTA elution through the commercial streptavidin affinity column.
- Collect flow through and keep samples for SDS-PAGE/WB analysis (20 µL and 4 µL, respectively). Subsequently, wash with 10 CV of refolding buffer.
- Elute with 10 CV of refolding buffer containing 2.5 mM desthiobiotin. Keep samples for SDS-PAGE/WB analysis (20 and 4 µL respectively).
- Measure octamer concentration, add 10 times less TEV protease and let reaction proceed overnight at 4 °C in a standard plastic centrifuge tube. NOTE: The necessary amount of TEV to obtain complete digestion (removal of the affinity tags) depends on the origin (construct) and on the activity (freshly made, recombinant TEV is more active compared to the one stored for prolonged periods at - 80 °C). Try initially adding 10 times less TEV compared to the octamer (estimated as protein concentrations) and adjust accordingly.
- Check for the efficient digestion by running 20 µL of the mixture prior and after TEV addition on an 18% SDS-PAGE gel. If digestion is not complete, add more TEV protease and continue incubation for another 3-4 h.
- Dialyze sample against refolding buffer using a 50 kDa-cutoff dialysis membrane to remove TEV protease and adjust buffer composition.
- Concentrate the purified asymmetric octamer using a 10 kDa-cutoff centrifugal filter unit (the same filter unit from step 1.1.7 can be also used) to a volume of 1.0-2.0 mL. Measure the final concentration. Either use soon after for nucleosome reconstitution or adjust to 50% (v/v) glycerol to store at -20 °C for longer periods. At this point and expecting at least 70% loss during the previous steps, the octamer concentration should be in the range of 20 - 50 µM.
- Nucleosome reconstitution
- If octamer was stored in 50% glycerol, dialyze overnight at 4 °C against refolding buffer before proceeding with nucleosome reconstitution.
- Adjust DNA (containing a nucleosome-positioning sequence) salt concentration to 2 M NaCl by using a 5 M NaCl stock solution. NOTE: The purified DNA either isolated after purification of a plasmid that contains multiple copies of the desired sequence or synthesized by PCR amplification should be stored concentrated to ~50 µM in water or a standard Tris-EDTA buffer.
- Mix octamer and DNA using the optimum ratio determined from a small-scale test. Use refolding buffer to adjust the final volume to ~3.0 mL. Always add the octamer last to prevent any possibility of mixing the octamer and the DNA at <2 M salt concentrations.
- Perform initial small-scale reconstitutions to determine the octamer:DNA ratio that leads to optimum nucleosome yields. For this, assemble three reactions of 0.9:1.0, 1.0:1.0, and 1.1:1.0 DNA:octamer ratios in a volume between 50-100 µL. Typically, use a concentration of 5 µM DNA.
- Proceed with reconstitution as described below (steps 1.2.4-1.2.8) and at the end, estimate the amount of soluble and good quality reconstituted nucleosome by measuring the DNA concentration at 260 nm and by running a 6% native-PAGE DNA gel, stained with ethidium bromide, respectively (Figure 5A).
- Transfer the mix to a 6 kDa-cutoff dialysis membrane and dialyze against 1 L of refolding buffer overnight at 4 °C.
- Reduce the salt concentration of the dialysis buffer in a step-wise manner from 2, to 0.85, 0.64, and 0.2 M NaCl respectively. Keep sample in each buffer for at least 3 h. Sometimes precipitate is formed after the last transition. In this case continue dialysis as planned and remove precipitate by microfuge centrifugation at the end of the next step.
- Transfer dialysis membrane in a beaker with 1 L of assay buffer (25 mM NaH2PO4/Na2HPO4 pH 6.8, 25 mM NaCl, 2 mM DTT) and continue dialysis O/N at 4 °C.
- Collect sample, remove any precipitate formed at step 1.2.5 by centrifugation in a microfuge for 5 min at 4 °C on maximum speed and concentrate using a 30 kDa-cutoff centrifugal filter unit to a final volume of ~300 µL.
- Measure DNA concentration at 260 nm, run an 18% SDS-PAGE protein gel (4 µL of sample) and a 6% native-PAGE DNA gel (0.2 µL of sample) and store sample at 4 °C for several weeks. The final concentration should be in the range of 10-20 µM.
2. NMR Analysis of Modification Reactions on the Two Sister Histones
NOTE: Assignment of 2D 1H-15N correlation spectra of nucleosomal histone tails can be found at references16,17,18. Additionally, assignments of CHx groups of lysines, serines and threonines on 2D 1H-13C correlation spectra can be found at reference16.
- NMR setup and recording of reference spectra NOTE: Enzymatic assays were performed using 140 µL of the nucleosome sample in assay buffer into a 3-mm NMR tube at 300 K. Different NMR tubes with other sample volumes can be also used. The aforementioned temperature is appropriate to obtain good quality 1H-15N correlation spectra of nucleosomal histone tails and good enzymatic activities. 2D-SOFAST-HMQC 1H-15N and 2D-HSQC 1H-13C spectra were recorded respectively with a setup that confers similar acquisition times. Typical acquisition parameters are 128 transients with 1.024 (1H) x 128 (15N) complex points and 32 transients with 1.024 (1H) x 64 (13C) complex points for the two different types of correlation spectra. For both 2D spectra, the acquisition time was ~30 min.
- Set 300 K as the sample temperature in the spectrometer. Add 10% D2O (v/v) to the sample to be used for the spectrometer frequency lock.
- Place the sample in the spectrometer and perform basic operations (locking, tuning, shimming). Additionally, determine optimal pulse lengths and general acquisition parameters.
- Record 1D and 2D 1H-15N and 1H-13C reference spectra.
- Process reference spectra and ensure that the signals to be used for mapping the modification reactions are well resolved and have good intensities.
- Recover sample and transfer it to a microfuge tube.
- Setup of the enzymatic reaction, NMR monitoring and quantitative analysis
- Copy and arrange a series of interleaved 2D 1H-15N and 1H-13C spectra. Aim for a total acquisition time of several hours and adjust accordingly in a new run based on the enzymatic rates obtained from the first reaction.
- Add to the sample the necessary cofactors for the respective type of modification reaction (1 mM ATP/2 mM MgCl2 for phosphorylation, 1 mM acetyl-CoA for acetylation, 1 mM SAM for methylation, etc.).
- Add the enzyme of interest, mix by pipetting, transfer the sample back in the NMR tube and subsequently in the spectrometer (perform these operations quickly). NOTE: If there is no data about the expected enzymatic activity, adjust the substrate-to-enzyme molar ratio to 10:1. Perform a first trial run and adjust accordingly for the real run.
- Perform a fast automatic shimming and use the rest of parameters from the reference spectra.
- Initiate the series of the interleaved 2D 1H-15N and 1H-13C spectra.
- Follow the progression of the reaction in real-time and stop acquisition when the reaction reaches completion or a desired level.
- Process spectra as before with exactly the same parameters.
- Repeat the whole run using a different acquisition order for the two types of correlation spectra. If in the first experiment a 1H-15N correlation was first in the series, start now with a 1H-13C correlation. By this and having same acquisition time for both spectra, it is possible to plot and compare PTM reactions on both differentially isotope-labeled histones on the same time scale. Additionally, it is possible to calculate averages and plot error bars. NOTE: A thorough description of the methodologies for NMR signal analysis and subsequent quantification of enzymatic reactions can be found here19.
- Select NMR signals from each time-course NMR spectrum that report the progression of the reaction and calculate their relative signal intensities using a visualization and analysis software for NMR spectra. Do this for signals that report the unmodified as well as the modified state. Extract modification levels.
- Plot the calculated values of modification levels against the reaction time.
Representative Results
Properly refolded octameric species are isolated after a run of the reconstitution mixture through a gel filtration column (Figure 2). The reconstituted octameric pool containing the three different types of octamers is subjected to the tandem affinity purification scheme. Samples are collected from all the steps and analyzed by SDS-PAGE and subsequently WB. Verification of the correct execution of the protocol that results in the isolation of asymmetric species is achieved by following the presence of the two affinity tags (His- and Strep-) in the different samples (Figure 3). Subsequently, the affinity tags are removed after TEV protease digestion (Figure 4). Asymmetric octamers disposed of affinity tags are used to reconstitute asymmetric nucleosomes. Initially, a small test-scale reconstitution is used to determine the optimum molar ratio of DNA:octamer that results in a high yield formation of a monodisperse complex. Subsequently, the optimized mixing ratio is employed to reconstitute nucleosomes in a large-scale. Nucleosome reconstitutions are analyzed by protein/DNA gels and NMR spectroscopy for the proper assembly and the presence of the differentially isotope-labeled sister H3 histones respectively (Figure 5). An application example is depicted in Figure 6. Particularly, differentially isotope-labeled and asymmetrically phosphorylated on H3S10 nucleosomes are reacted with Gcn5 acetyltransferase, and acetylation of H3K14 on both H3 copies is followed in real-time by NMR spectroscopy. The analysis reveals that H3S10 phosphorylation stimulates H3K14 acetylation by Gcn5 in cis compared to in trans.
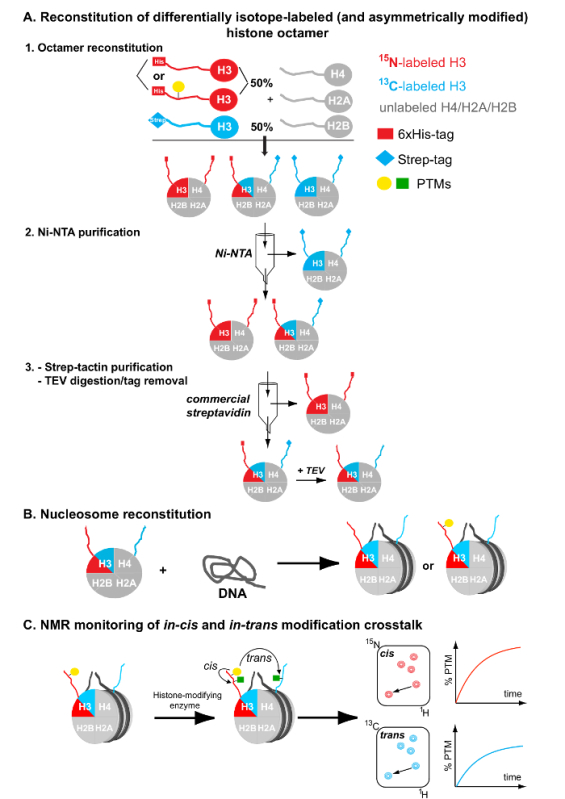 Figure 1: Flowchart for the Preparation of Differentially Isotope-labeled (and Asymmetrically Modified) Nucleosomes. (A) Reconstitution and tandem affinity purification of differentially isotope-labeled (and asymmetrically modified) histone octamers. (B) Nucleosome reconstitution by combining the purified asymmetric octamers and a DNA nucleosome-positioning sequence. (C) NMR monitoring of in-cis and in-trans modification crosstalk after mixing the asymmetrically modified and differentially isotope-labeled nucleosomes with histone-modifying enzymes. Please click here to view a larger version of this figure.
Figure 1: Flowchart for the Preparation of Differentially Isotope-labeled (and Asymmetrically Modified) Nucleosomes. (A) Reconstitution and tandem affinity purification of differentially isotope-labeled (and asymmetrically modified) histone octamers. (B) Nucleosome reconstitution by combining the purified asymmetric octamers and a DNA nucleosome-positioning sequence. (C) NMR monitoring of in-cis and in-trans modification crosstalk after mixing the asymmetrically modified and differentially isotope-labeled nucleosomes with histone-modifying enzymes. Please click here to view a larger version of this figure.
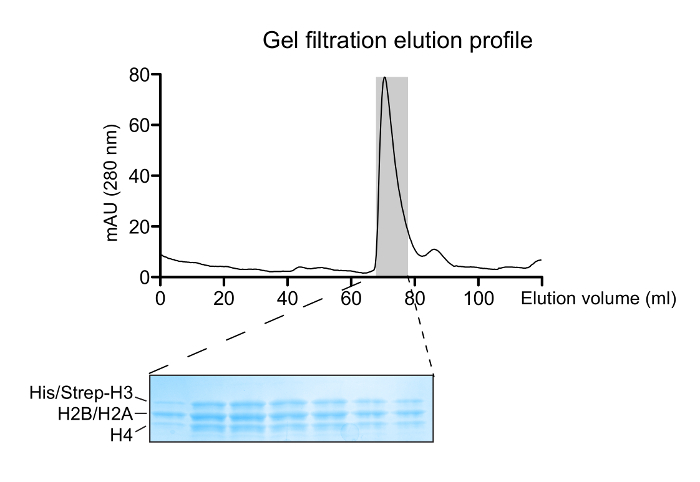 Figure 2: Reconstitution of Histone Octamers. A gel filtration elution profile of the refolded histone mixture. The major peak at ~70 mL corresponds to the histone octamers. A 14 - 20% gradient SDS-PAGE gel (Coomassie Blue staining) of eluted fractions corresponding to the octamer peak shows the correct histone stoichiometry. Please click here to view a larger version of this figure.
Figure 2: Reconstitution of Histone Octamers. A gel filtration elution profile of the refolded histone mixture. The major peak at ~70 mL corresponds to the histone octamers. A 14 - 20% gradient SDS-PAGE gel (Coomassie Blue staining) of eluted fractions corresponding to the octamer peak shows the correct histone stoichiometry. Please click here to view a larger version of this figure.
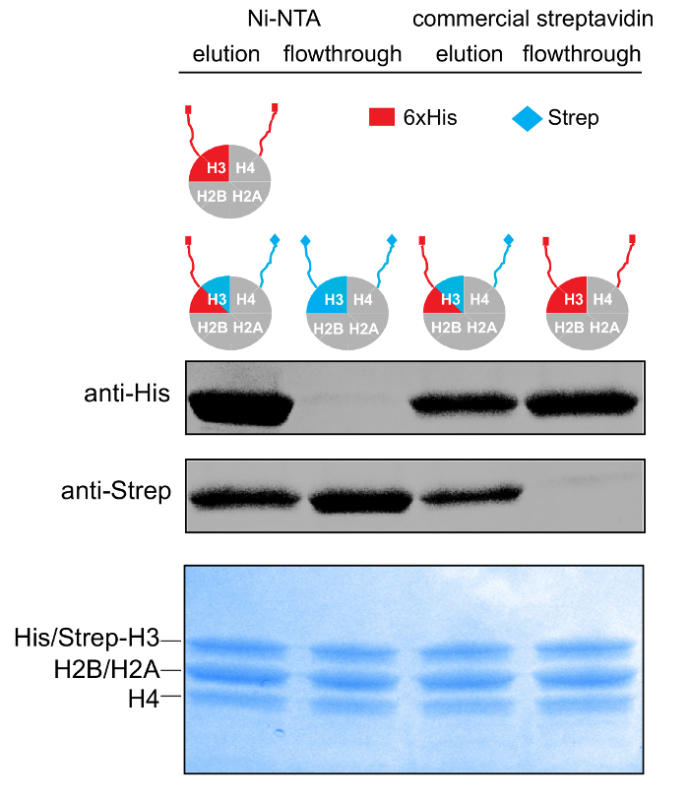 Figure 3: Purification of Asymmetric Histone Octamers. 14-20% gradient SDS-PAGE gel (Coomassie staining) and WB against the His- and Strep- affinity tags of samples from the different stages of the tandem affinity purification scheme. Please click here to view a larger version of this figure.
Figure 3: Purification of Asymmetric Histone Octamers. 14-20% gradient SDS-PAGE gel (Coomassie staining) and WB against the His- and Strep- affinity tags of samples from the different stages of the tandem affinity purification scheme. Please click here to view a larger version of this figure.
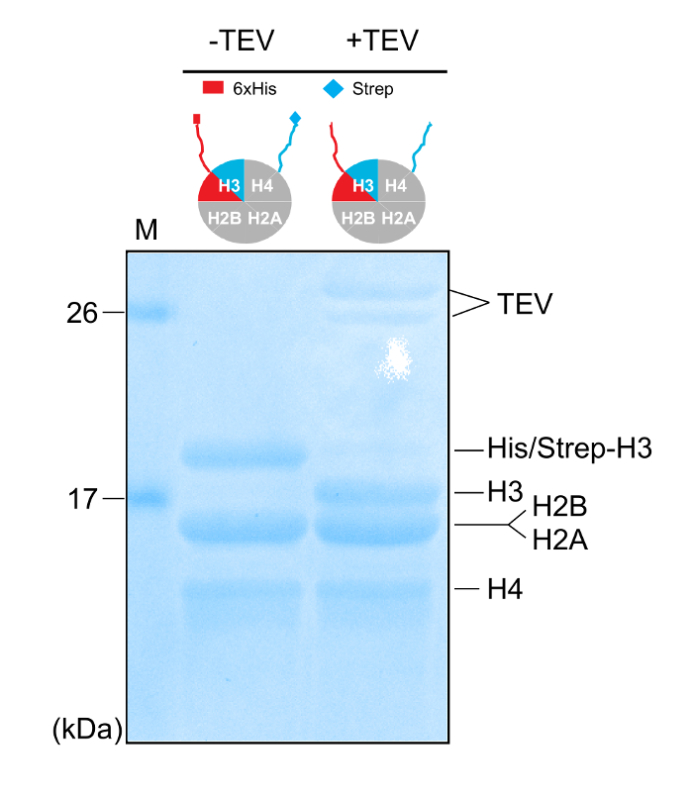 Figure 4: Removal of the Affinity Tags. 18% SDS-PAGE gel (Coomassie staining) of purified asymmetric histone octamers before and after mixing with the TEV protease. Please click here to view a larger version of this figure.
Figure 4: Removal of the Affinity Tags. 18% SDS-PAGE gel (Coomassie staining) of purified asymmetric histone octamers before and after mixing with the TEV protease. Please click here to view a larger version of this figure.
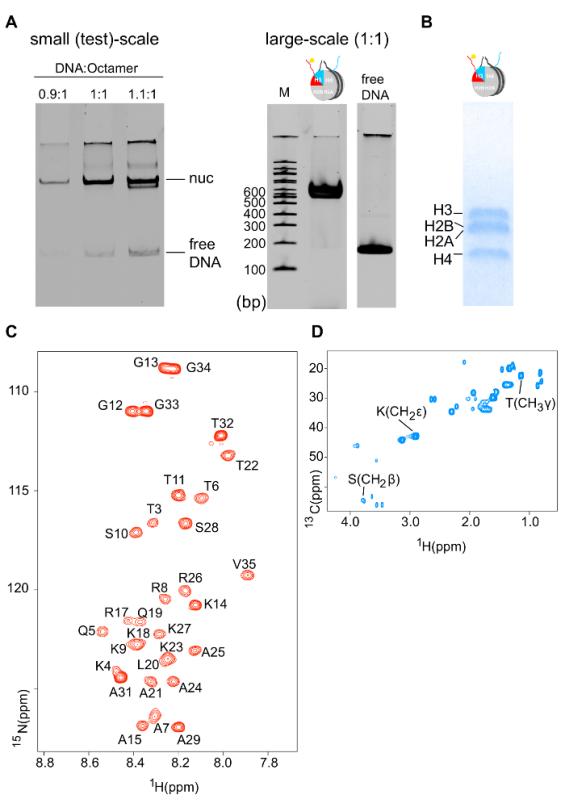 Figure 5: Biochemical and NMR Characterization of Differentially Isotope-labeled Nucleosomes. (A) 6% native-PAGE DNA gel (ethidium bromide staining) of a small test-scale nucleosome reconstitution using different molar ratios of DNA:octamer (left). 6% native-PAGE DNA gel (ethidium bromide staining) of a large-scale nucleosome reconstitution using the optimized DNA:octamer molar ratio (right). (B) 18% SDS-PAGE protein gel (Coomassie staining) of reconstituted nucleosomes shows the correct stoichiometry of the four core histones. (C) 1H-15N SOFAST-HMQC spectrum of the 15N-labeled H3 copy of differentially isotope-labeled nucleosomes (only H3 tail residues are visible – the protein core is invisible due to the slow tumbling rate). (D) 1H-13C HSQC spectrum of the 13C-labeled H3 copy of differentially isotope-labeled nucleosomes. Please click here to view a larger version of this figure.
Figure 5: Biochemical and NMR Characterization of Differentially Isotope-labeled Nucleosomes. (A) 6% native-PAGE DNA gel (ethidium bromide staining) of a small test-scale nucleosome reconstitution using different molar ratios of DNA:octamer (left). 6% native-PAGE DNA gel (ethidium bromide staining) of a large-scale nucleosome reconstitution using the optimized DNA:octamer molar ratio (right). (B) 18% SDS-PAGE protein gel (Coomassie staining) of reconstituted nucleosomes shows the correct stoichiometry of the four core histones. (C) 1H-15N SOFAST-HMQC spectrum of the 15N-labeled H3 copy of differentially isotope-labeled nucleosomes (only H3 tail residues are visible – the protein core is invisible due to the slow tumbling rate). (D) 1H-13C HSQC spectrum of the 13C-labeled H3 copy of differentially isotope-labeled nucleosomes. Please click here to view a larger version of this figure.
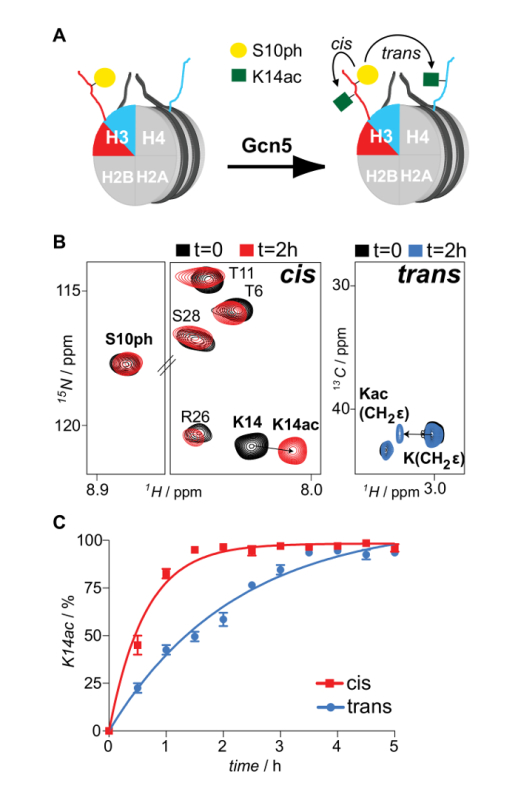 Figure 6: NMR Monitoring of In cis and In trans Modification Cross-talk Imposed by Asymmetric Phosphorylation of H3S10 on the H3K14 Acetylation Activity of Gcn5 Acetyltransferase. (A) Schematic representation of differentially isotope-labeled and asymmetrically phosphorylated on H3S10 nucleosomes reacted with Gcn5 acetyltransferase and H3K14 acetylation mapping, in cis and in trans. (B) 1H-15N SOFAST-HMQC and 1H-13C HSQC spectra (selected regions) of the asymmetric nucleosomes before and after reaction with Gcn5. (C) Time-resolved NMR monitoring of H3K14 acetylation by Gcn5 on asymmetrically H3S10 phosphorylated nucleosomes. The averages and variation (error bars) from two independent experiments are shown. Reproduced with permission16. Please click here to view a larger version of this figure.
Figure 6: NMR Monitoring of In cis and In trans Modification Cross-talk Imposed by Asymmetric Phosphorylation of H3S10 on the H3K14 Acetylation Activity of Gcn5 Acetyltransferase. (A) Schematic representation of differentially isotope-labeled and asymmetrically phosphorylated on H3S10 nucleosomes reacted with Gcn5 acetyltransferase and H3K14 acetylation mapping, in cis and in trans. (B) 1H-15N SOFAST-HMQC and 1H-13C HSQC spectra (selected regions) of the asymmetric nucleosomes before and after reaction with Gcn5. (C) Time-resolved NMR monitoring of H3K14 acetylation by Gcn5 on asymmetrically H3S10 phosphorylated nucleosomes. The averages and variation (error bars) from two independent experiments are shown. Reproduced with permission16. Please click here to view a larger version of this figure.
Discussion
For nucleosome reconstitution, the current protocol utilizes a 165 bp long DNA template containing the 601-Widom nucleosome positioning sequence20, but similar performance is expected using various lengths of DNA templates. The protocol was designed and employed using asymmetric types of histone H3. With the same principle, the method can be applied also for the other core histones and additionally can be used to reconstitute complexes carrying two distinct core histones with different isotope labels. The protocol can be also slightly modified to initially reconstitute histone sub-complexes and not directly histone octamers. Following this modification, asymmetric H3-H4 tetramers can be reconstituted by employing the same tandem purification scheme. The latter are mixed with separately reconstituted H2A-H2B dimers and DNA to assemble nucleosomes16. The final yields and the overall performance of the modified protocol are similar to the original one.
The protocol is strongly dependent on the ability of the two affinity columns to efficiently bind the relevant species and thus to yield at the end highly pure asymmetric complexes. It is cumbersome to provide direct and concrete proof that the final purified complex is indeed and only asymmetric. However, WB analysis of samples from all the purification steps with the relevant antibodies provides reliable indications for the success of the tandem purification scheme (Figure 3). The same results, and hence additional reassurance, were obtained by analyzing the same samples for the presence of the affinity tags by NMR methods16. Still, if WB analysis indicates contamination of asymmetric species with symmetric ones, a small variation can be adopted at the first purification step (Ni-NTA) that can improve the outcome. Particularly, instead of an isocratic elution, an imidazole gradient elution (10-250 mM) can be employed and then, only the first elution fractions that are highly enriched in the asymmetric species are pooled and run through the affinity column. This can be assessed by WB analysis of the elution fractions for the presence of the strep affinity tag.
The selection of the isotope-label in relation to the PTM of interest is rather flexible because for most cases, NMR is capable of mapping the same PTM using both 1H-15N and 1H-13Ccorrelation spectra. However, for certain amino acids, like lysines, the chemical shift dispersion for the side-chain 1H-13C resonances is rather limited. Additionally, when the target sites of a lysine-modifying enzyme are unknown, site-specific read-out of lysine PTMs is not straightforward. In these cases, alternative methods employ site-selective 13C-labeling, combined with semi-synthetic histone production21 or the use of newly established NMR pulse sequences that enables specific detection of modification states through selective excitation routines22. NMR spectroscopy confers rather low sensitivity. In this regard, relatively high amounts of nucleosomes (µM range) are needed to perform the enzymatic reactions. This prevents the evolution of the method to a high-throughput setup.
Concurrently, a new chemical method has been introduced for the synthesis of chemically pure, asymmetrically modified nucleosomes, with high yields, carrying defined PTMs23. This approach has been used, in combination with fluorography, to study PTM crosstalk mechanisms. Due to the low-resolution capability of fluorographic methods in detecting PTMs, conclusions were derived indirectly by comparing overall enzymatic activities on a set of nucleosomes carrying different combinations of symmetric and asymmetric PTMs, rather than by direct observation of PTM reactions on the corresponding sites of each sister histone. However, incorporation of isotopically labeled histones and the use of NMR spectroscopy for PTM readout could constitute the aforementioned method an additional tool for high-resolution PTM analysis of asymmetrically modified nucleosomes.
In connection with the identification of new types of asymmetrically modified nucleosomes in the future, the current method aims to constitute a high-resolution tool for analyzing in cis and in trans PTM crosstalk reactions on nucleosomal substrates. Particularly, of high importance is the decoding of crosstalk mechanisms that give rise to bivalent nucleosomes24 that contain asymmetrically, both transcriptionally active and repressive PTMs.
Disclosures
The author has nothing to disclose.
Acknowledgments
The author thanks Dr. Philipp Selenko (FMP-Berlin) for providing wet-lab space and infrastructure to perform experiments and Deutsche Forschungsgemeinschaft (DFG) for funding the work through a research grant (LI 2402/2-1).
References
- Hansen JC. Conformational Dynamics of the Chromatin Fiber in Solution: Determinants, Mechanisms, and Functions. Annu. Rev. Biophys. Biomol. Struct. 2002;31:361–392. doi: 10.1146/annurev.biophys.31.101101.140858. [DOI] [PubMed] [Google Scholar]
- Clapier CR, Cairns BR. The Biology of Chromatin Remodeling Complexes. Annu. Rev. Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- Musselman CA, Lalonde M, Cote J, Kutateladze TG. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 2012;19(12):1218–1227. doi: 10.1038/nsmb.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suganuma T, Workman JL. Signals and Combinatorial Functions of Histone Modifications. Annu. Rev. Biochem. 2011;80:473–499. doi: 10.1146/annurev-biochem-061809-175347. [DOI] [PubMed] [Google Scholar]
- Lee JS, Smith E, Shilatifard A. The Language of Histone Crosstalk. Cell. 2010;142(5):682–685. doi: 10.1016/j.cell.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liokatis S, et al. Phosphorylation of histone H3 Ser10 establishes a hierarchy for subsequent intramolecular modification events. Nat. Struct. Mol. Biol. 2012;19(8):819–823. doi: 10.1038/nsmb.2310. [DOI] [PubMed] [Google Scholar]
- Voigt P, et al. Asymmetrically Modified Nucleosomes. Cell. 2012;151(1):181–193. doi: 10.1016/j.cell.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelhofer TA, et al. An assessment of histone-modification antibody quality. Nat. Struct. Mol. Biol. 2011;18(1):91–93. doi: 10.1038/nsmb.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Lin S, Garcia BA, Zhao Y. Quantitative Proteomic Analysis of Histone Modifications. Chem. Rev. 2015;115(6):2376–2418. doi: 10.1021/cr500491u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liokatis S, Dose A, Schwarzer D, Selenko P. Simultaneous Detection, of Protein Phosphorylation and Acetylation by High-Resolution NMR Spectroscopy. J. Am. Chem. Soc. 2010;132(42):14704–14705. doi: 10.1021/ja106764y. [DOI] [PubMed] [Google Scholar]
- Binolfi A, et al. Intracellular repair of oxidation-damaged α-synuclein fails to target C-terminal modification sites. Nat. Commun. 2016;7:10251. doi: 10.1038/ncomms10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schanda P, Kupce E, Brutscher B. SOFAST-HMQC experiments for recording two-dimensional heteronuclear correlation spectra of proteins within a few seconds. J. Biomol. NMR. 2005;33(4):199–211. doi: 10.1007/s10858-005-4425-x. [DOI] [PubMed] [Google Scholar]
- Theillet FX, et al. Cell signaling, post-translational protein modifications and NMR spectroscopy. J. Biomol. NMR. 2012;54(3):217–236. doi: 10.1007/s10858-012-9674-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer PN, et al. Reconstitution of Nucleosome Core Particles from Recombinant Histones and DNA. Methods Enzymol. 2004;375:23–43. doi: 10.1016/s0076-6879(03)75002-2. [DOI] [PubMed] [Google Scholar]
- Artimo P, et al. ExPASy :SIB bioinformatics resource portal. Nucleic Acids Res. 2012;40(W1):597–603. doi: 10.1093/nar/gks400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liokatis S, klingberg R, Tan S, Schwarzer D. Differentially Isotope-Labeled Nucleosomes to Study Asymmetric Histone Modification Crosstalk by Time-Resolved NMR Spectroscopy. Angew. Chem. Int. Ed. 2016;55(29):8262–8265. doi: 10.1002/anie.201601938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stützer A, et al. Modulations of DNA Contacts by Linker Histones and Post-translational Modifications Determine the Mobility and Modifiability of Nucleosomal H3 Tails. Mol. Cell. 2016;61(2):247–259. doi: 10.1016/j.molcel.2015.12.015. [DOI] [PubMed] [Google Scholar]
- Zhou BR, et al. Histone H4 K16Q Mutation, an Acetylation Mimic, Causes Structural Disorder of Its N-terminal Basic Patch in the Nucleosome. J. Mol. Biol. 2012;421(1):30–37. doi: 10.1016/j.jmb.2012.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theillet FX, et al. Site-specific NMR mapping and time-resolved monitoring of serine and threonine phosphorylation in reconstituted kinase reactions and mammalian cell extracts. Nat. Protoc. 2013;8(7):1416–1432. doi: 10.1038/nprot.2013.083. [DOI] [PubMed] [Google Scholar]
- Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998;276(1):19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- Hackenberger CP, Schwarzer D. Chemoselective ligation and modification strategies for peptides and proteins. Angew. Chem. Int. Ed. 2008;47(52):10030–10074. doi: 10.1002/anie.200801313. [DOI] [PubMed] [Google Scholar]
- Theillet FX, et al. Site-specific mapping and time-resolved monitoring of lysine methylation by high-resolution NMR spectroscopy. J. Am. Chem. Soc. 2012;134(18):7616–7619. doi: 10.1021/ja301895f. [DOI] [PubMed] [Google Scholar]
- Lechner CC, Agashe ND, Fierz B. Traceless Synthesis of Asymmetrically Modified Bivalent Nucleosomes. Angew. Chem. Int. Ed. 2016;55(8):2903–2906. doi: 10.1002/anie.201510996. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125(2):315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]