

Abstract
The powdery mildew fungi are a group of economically important fungal plant pathogens. Relatively little is known about the molecular biology and genetics of these pathogens, in part due to a lack of well-developed genetic and genomic resources. These organisms have large, repetitive genomes, which have made genome sequencing and assembly prohibitively difficult. Here, we describe methods for the collection, extraction, purification and quality control assessment of high molecular weight genomic DNA from one powdery mildew species, Golovinomyces cichoracearum. The protocol described includes mechanical disruption of spores followed by an optimized phenol/chloroform genomic DNA extraction. A typical yield was 7 µg DNA per 150 mg conidia. The genomic DNA that is isolated using this procedure is suitable for long-read sequencing (i.e., > 48.5 kbp). Quality control measures to ensure the size, yield, and purity of the genomic DNA are also described in this method. Sequencing of the genomic DNA of the quality described here will allow for the assembly and comparison of multiple powdery mildew genomes, which in turn will lead to a better understanding and improved control of this agricultural pathogen.
Keywords: Genetics, Issue 121, fungus, genomic DNA, high molecular weight DNA, obligate pathogen, powdery mildew, sequencing
Introduction
Powdery mildews are a group of obligate biotrophic fungal plant pathogens that, when taken together, are the largest cause of plant disease worldwide1. There are over 900 described species of powdery mildew, which have been taxonomically grouped into five tribes within the family Erisyphaceae2. Due both to their economic importance and the intimate relationship they develop with their hosts, powdery mildew diseases have been the subject of research for > 100 years. Upon infection, powdery mildews elicit drastic changes in the cellular structure, metabolism and molecular biology of their hosts, to benefit this pathogen. However, the study of powdery mildews is particularly challenging due to their obligate lifestyle, and growth of the fungus in pure culture has not yet been described3,4,5,6,7,8. Reliable, stable genetic transformation of powdery mildews has also not yet been accomplished, although transient transformation has been reported in some species9,10.
The sequencing and assembly of powdery mildew genomes has proven difficult due to a number of features of the genome itself. Powdery mildew genomes are large (120 - 180 Mbp) relative to other fungal genomes, and consist of 60 - 90% evenly distributed repetitive elements11. These elements include non-long terminal repeats, as well as uncategorized repetitive elements. Two formae speciales of a single powdery mildew species, Blumeria graminis f. sp. hordei and f. sp. tritici (Bgh and Bgt, respectively) as well as the grape powdery mildew Erysiphe necator, have been sequenced, and draft genomes for several others have been completed12,13,14. The repetitive nature of the genomes has made assembly difficult, and the completed Bgh genome was assembled into 6,989 supercontigs with an L50 of 2 Mb12.
Despite the large genome, the powdery mildews appear to have a small number of protein coding genes, with 5,845 and 6,540 genes predicted in Bgh and Bgt, respectively. The sequenced powdery mildews also appear to lack at least 99 core genes that are essential in other fungi, which is consistent with the dependence of the fungi on their host plant for survival11,12,13,14.
Repetitive sequences near telomers, centromeres, ribosomal RNA gene arrays and regions enriched in transposable elements are poorly assembled from short-read sequencing strategies and are often under-represented in genome assemblies15. Such regions are thought to be responsible for many of the gaps that occur in genome sequences, and this applies to the powdery mildews with their extensive expansion of repetitive elements16. Highly plastic genome regions are often found in such repetitive regions3. They serve as a site of chromosome rearrangements and often encode genes under strong selective pressure, such as the genes encoding effector proteins and the genes encoding enzymes of secondary metabolism. Advances in single-molecule long-read sequencing technologies provide a potential solution for sequencing across repetitive regions of genomes15. For example, Faino et al. (2015) found that including long-sequence read technologies and optical mapping allowed them to produce a gap-less genome sequence for two strains of the fungal plant pathogen Verticillium dahlia, tripling the proportion of repetitive DNA sequences in the genome, increasing the number of gene annotations (and reducing the number of partial or missing gene annotations) and revealing genome rearrangements17.
To employ these long-read sequencing technologies, high concentrations of high quality genomic DNA, with minimal sizes > 20 kbp, are needed. Here we describe our methods for conidial collection, purification of high molecular weight DNA from conidia and our quality control assessments using the powdery mildew species Golovinomyces cichoracearum grown on cucumber18. This protocol is based on a protocol developed in the B. Keller laboratory group (University of Zürich, Zürich Switzerland)13,19 and includes several modifications that led to increased DNA yields and a higher proportion of DNA >48.5 kbp in size. The protocol also includes quality control steps based on recommendations from the United States Department of Energy Joint Genome Institute20,21,22.
The function of each reagent included in the lysis buffer, and the rationale for each purification step, are as described in Henry (2008)23. Available published protocols for isolation of high molecular weight genomic DNA from plant and microbial tissues were also consulted during the design of this protocol24,25,26,27,28.
Protocol
1. Preparation of Fungal Material
- Growing Powdery Mildew
- Grow plants in growth chambers with the following conditions: 22 °C day temperature, 20 °C night temperature, 80% relative humidity, 14-h day-length and a light intensity of 125 µE/m2/s (provided by fluorescent lighting).
- Harvesting Powdery Mildew Conidia
- Harvest conidia from infected plants at 7 - 10 days after inoculation using a small vacuum with an in-line filter (11 µm) on which the conidia accumulate. Applying gentle pressure, run the vacuum nozzle along the surface of the leaf to collect the conidia. NOTE: The average yield of G. cichoracearum is 20 mg of conidia from a heavily infected cucumber leaf of approximately 130 cm2 in area.
- Brush about 150 mg of the conidia (approximately 375 µL volume, or conidia from 7-8 infected cucumber leaves) from the filter onto a sheet of clean paper. From here, transfer conidia into 2 mL cryovials with two 5/32-inch diameter pre-chilled (in liquid nitrogen) steel milling balls. Immediately return tubes to liquid nitrogen. Store tubes at -80 °C.
- Breaking Open Conidia by Ball Milling
- Flash freeze aluminum plates of ball mill in liquid nitrogen. Load cryovials into ball-mill rack. Vigorously shake the cryovials with conidia and steel balls in the ball mill for 30 s at 30 cycles/s (at room temperature). Immediately refreeze cryovials in liquid nitrogen. NOTE: Initially, access a sample of conidia by microscopy at 100X magnification for efficiency of cell wall rupture during grinding. If needed, grind for an additional 1 - 2 rounds of 30 s for 30 cycles/s, but keep the number of rounds to a minimum. Routine assessment of conidial breakage by microscopy at this stage is unnecessary once optimal conditions have been established.
2. Genomic DNA Purification
- Conidia Lysis
- Working quickly to avoid thawing conidia, remove the steel balls from cryovials with a magnet. Add 700 µL 65 °C lysis buffer (Table 1) and vortex 5 - 10 s until a slurry is formed.
- Add 300 µL pre-warmed (65 °C) 5% (v/v) sarcosyl and gently invert to mix (1 inversion per 3 s) five times. Incubate tubes for 30 min at 65 °C; invert gently 3 times at 10, 20 and 30 min. Using a wide-bore pipette tip, transfer entire solution to new 2 mL microcentrifuge tube. At all subsequent steps, mix by gentle, slow inversion and transfer the DNA-containing solutions with wide-bore pipette tips.
- DNA Extraction I
- Add 1 volume of chloroform:isoamyl alcohol (24:1 v/v) to the lysis solution and gently invert to mix five times. Incubate for 10 min at room temperature, gently inverting to mix five times at the halfway point and again at the end of the incubation. Centrifuge at room temperature for 15 min at 14,000 x g. Using wide-bore pipette tip, carefully transfer the aqueous layer to new 2 mL microcentrifuge tube; avoid including material from the interface.
- DNA Precipitation I
- Add 1 volume room temperature 100% isopropanol (approximately 750 μL) and gently invert to mix six times. Centrifuge at room temperature for 15 min at 14,000 x g. Carefully remove the supernatant and discard.
- Add 450 μL pre-chilled (-20 °C) 70% ethanol. Centrifuge for 5 min at room temperature at 14,000 x g. Carefully remove the supernatant and discard. Centrifuge at 1,000 x g for 3 s, carefully remove the supernatant with fine pipette tip and discard. Air-dry the pellet for 15 min at room temperature. Do not dry for longer than 15 min.
- Resuspend the pellet in 300 µL TE (10 mM Tris-Cl, 1 mM ethylene diamine tetra-acetic acid, pH 8.0). Incubate overnight at 4 °C. Gently flick to resuspend any residual material that did not go into solution.
To remove RNA contamination, add 10 µL RNase A (10 mg/mL). Gently invert to mix three times. Centrifuge at 1,000 x g for 3 s. Incubate at 37 °C for 2 h.
- DNA Extraction II
- Add 300 µL phenol:chloroform:isoamyl alcohol (25:24:1 v/v) and gently invert to mix five times. Incubate 10 min at room temperature, gently inverting to mix five times at the halfway mark and again at the end of the incubation. Centrifuge 15 min at room temperature at 14,000 x g. Using a wide-bore pipette tip, transfer the supernatant to new 1.5 mL microcentrifuge tube.
- DNA Precipitation II
- Add 0.01 volume 3 M sodium acetate pH 5.2 (approximately 3 µL), gently invert to mix five times. Add 2.5 volumes pre-chilled (-20 °C) 100% ethanol (approximately 750 μL). Gently invert to mix five times. Incubate overnight at -20° C.
- Centrifuge 30 min at 4 °C at 14,000 x g. Carefully remove the supernatant and discard.
- Add 450 µL pre-chilled (-20 °C) 70% ethanol. Centrifuge for 5 min at 4 °C at 14,000 x g. Carefully remove the supernatant and discard. Centrifuge at 1,000 x g for 3 s. Carefully remove the supernatant with fine pipette tip and discard.
- Air-dry the pellet for 30-60 min at room temperature. Resuspend the pellet in 27.5 µL TE. Incubate overnight at 4 °C. Gently flick to resuspend any residual material that did not go into solution.
- Aliquot 2.5 µL into 22.5 µL TE (final volume 25 μL) for quality control tests (see below). Store DNA samples at 4 °C until submission for sequencing. For long-term storage, store samples at -80 °C and minimize freeze-thaw cycles to prevent shearing. NOTE: Take care when pipetting at this step as the high molecular weight genomic DNA can be difficult to pipette properly, and it is important to ensure that "QC Aliquot" is an accurate representation of genomic DNA sample.
3. Quality Control
- Sample Purity
- Using a TE blank, assess the sample purity of the DNA by loading 1 µL of the QC Aliquot on a small-volume spectrophotometer. Record A260/A280 and A260/A230 readings. Pure DNA will have an A260/A280 ratio of approximately 1.8 and A260/A230 ratio between 1.8 and 2.2.
- Quantification of Genomic DNA
- Perform quantification of QC aliquot using a commercial double-stranded DNA quantification assay kit according to the manufacturer instructions.
- DNA Quality Assessment I
- Load approximately 60 ng genomic DNA and a DNA ladder on an 0.7% agarose gel in 1x TAE (40 mM Tris, 20 mM acetic acid, and 1 mM EDTA, pH 8.0). Run gel apparatus at 70 V for 40 - 45 min until genomic DNA bands are approximately 2 cm from the well and band separation in the ladder is apparent. These conditions may need to be adjusted based on available gel electrophoresis apparatus.
- DNA Quality Assessment II
- Load approximately 60 ng genomic DNA and a high molecular weight DNA ladder (suitable for pulsed-field gel electrophoresis) on a 1% agarose gel in 0.5x TBE (44.5 mM Tris, 44.5 mM boric acid, 1 mM EDTA, pH 8.3) and run in 0.5x TBE running buffer using a 5 - 80 kbp waveform pulsed-field gel electrophoresis protocol for 16 h.
- Assessment of Bacterial, Fungal, and Plant Contaminants
- Perform polymerase chain reaction (PCR) to amplify the appropriate internal transcribed spacers (ITS) regions of the ribosomal genes using primers and amplification conditions listed in Table 2. Load 10 µL of the resultant reaction on a gel. NOTE: A band representing the powdery mildew ITS region should be amplified with the fungal ITS primers. DNA from this amplicon and any amplicon generated with the bacterial, or plant primers should be sequenced to determine the origin of the contamination. Only powdery mildew ITS sequences should be found in the fungal amplicon.
Representative Results
A representative example of 60ng purified genomic DNA from G. cichoracearum run on an agarose gel using gel electrophoresis and using pulsed-field gel electrophoresis are shown in Figures 1 and 2, respectively. Genomic DNA preparations that pass, marginally pass and fail quality control are represented. Genomic DNA preparations that fully pass quality control are ideal for sequencing using long-read genome sequencing approaches. Preparations that marginally pass quality control are acceptable, but preparations that fully pass are preferred. Preparations that fail quality control are not acceptable for long-read genome sequencing approaches.
 Figure 1: Agarose Gel Electrophoresis of Purified Genomic DNA. Lane 1: A DNA ladder, with bands ranging from 20 kbp to 75 bp; Lane 2: genomic DNA that passed quality control; Lane 3: genomic DNA that was considered marginal; and Lane 4: genomic DNA that failed quality control. Acceptable and marginal genomic DNA samples have a single band running above 20 kbp with minimal smearing at < 20 kbp. Genomic DNA samples that fail quality control have smearing below 20 kbp, and may include low molecular weight fragments at approximately 100 bp (*). Pulsed-field gel electrophoresis is required to distinguish between acceptable and marginal DNA samples. Ethidium bromide was added to the gel prior to electrophoresis to visualize the genomic DNA. Samples were electrophoresed at 60 V for 50 min. The gel was imaged under UV light using a gel documentation system.
Figure 1: Agarose Gel Electrophoresis of Purified Genomic DNA. Lane 1: A DNA ladder, with bands ranging from 20 kbp to 75 bp; Lane 2: genomic DNA that passed quality control; Lane 3: genomic DNA that was considered marginal; and Lane 4: genomic DNA that failed quality control. Acceptable and marginal genomic DNA samples have a single band running above 20 kbp with minimal smearing at < 20 kbp. Genomic DNA samples that fail quality control have smearing below 20 kbp, and may include low molecular weight fragments at approximately 100 bp (*). Pulsed-field gel electrophoresis is required to distinguish between acceptable and marginal DNA samples. Ethidium bromide was added to the gel prior to electrophoresis to visualize the genomic DNA. Samples were electrophoresed at 60 V for 50 min. The gel was imaged under UV light using a gel documentation system.
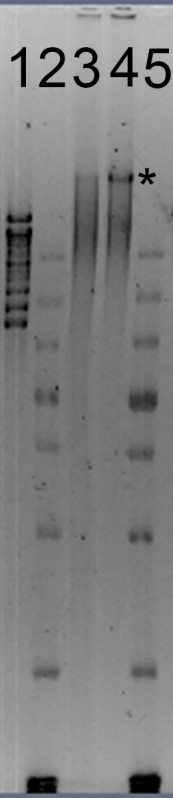 Figure 2: Pulsed-Field Gel Electrophoresis (PFGE) of Purified Powdery Mildew Genomic DNA. Lane 1: molecular weight standards, with the highest band at 48.5 kbp and the lowest at 8.1 kbp. Lanes 2 and 5: molecular weight standards showing bands of 20, 10, 7, 5, 4, 3, 2 and 1.5 kbp (at the bottom of the gel image). Lane 3: a marginal DNA sample, with the majority of the DNA between 20 and 48.5 kbp in size. Lane 4: an acceptable DNA sample, with the majority of the DNA > 48.5 kbp (*). Samples were run at 75 V for 15 h using a 5-80 kbp PFGE waveform program. DNA-staining dye was added to the gel after electrophoresis and the gel was imaged under UV light using a gel documentation system.
Figure 2: Pulsed-Field Gel Electrophoresis (PFGE) of Purified Powdery Mildew Genomic DNA. Lane 1: molecular weight standards, with the highest band at 48.5 kbp and the lowest at 8.1 kbp. Lanes 2 and 5: molecular weight standards showing bands of 20, 10, 7, 5, 4, 3, 2 and 1.5 kbp (at the bottom of the gel image). Lane 3: a marginal DNA sample, with the majority of the DNA between 20 and 48.5 kbp in size. Lane 4: an acceptable DNA sample, with the majority of the DNA > 48.5 kbp (*). Samples were run at 75 V for 15 h using a 5-80 kbp PFGE waveform program. DNA-staining dye was added to the gel after electrophoresis and the gel was imaged under UV light using a gel documentation system.
| Reagent | Final Concentration |
| Potassium metabisulfite (K2S2O5) | 0.25 M |
| Tris buffer pH 7.5 | 0.2 M |
| Ethylenediaminetetraacetic acid (EDTA) | 50 mM |
| Sodium chloride (NaCl) | 2 M |
| Cetyltrimethyl ammonium bromide (CTAB) | 2% v/v |
Table 1: Preparation of Lysis Buffer. Bring to a volume of 5 mL with dH2O.
| Target | Primer Name | Primer Sequence | Tm (°C) | Expected Product Size (bp) |
| Fungal ITS2 | FW (ITS9) | GAACGCAGCRAAIIGYGA | 56-61.3 | 336 |
| RV (ITS4) | TCCTCCGCTTATTGATATGC | 52.1 | ||
| Bacterial 16S V4 | FW (515F) | GTGCCAGCMGCCGCGGTAA | 63.8-66.5 | 331 |
| RV (805R) | GGACTACHVGGGTWTCTAAT | 46.9-53.8 | ||
| Bacterial 16S V4-V5 | FW (515F-Y) | GTGYCAGCMGCCGCGGTAA | 60.8-66.5 | 450 |
| RV (926R) | CCGYCAATTYMTTTRAGTTT | 43.9-53.8 | ||
| Fungal 18S V4 | FW | CCAGCASCYGCGGTAATTCC | 58.7-61.5 | 431 |
| RV | ACTTTCGTTCTTGATYRA | 43.2-48.3 | ||
| Cucumis sativus PDS (LOC101204524) | CsPDS F | GTGAGTAAGGTTACCGTTTGGGGCTTATCCCAAT | 63.8 | 750 |
| CsPDS R | GCGTGAGCTCGGTACTCTCATCCACTCTTGCAC | 66.6 |
Table 2: Primers for Contamination Assessment.
Discussion
In order to obtain pure, high molecular weight genomic DNA from the obligate biotrophic powdery mildew fungi, a modified version of previously described methods was developed30. The average yield using this optimized protocol is 7 µg DNA per 150 mg conidia, a doubling of the yield obtained with a prior protocol. Also, the average size increased from approximately 20 kbp to over 48.5 kbp. This protocol was optimized in the cucumber-G. cichoracearum system. In order to obtain the best quality and highest purity DNA in other powdery mildew or obligate biotrophic fungi, additional modification of this protocol may be required. In the future, this protocol may be used to obtain high molecular weight DNA suitable for long-read DNA sequencing from other obligate biotrophic fungi, though further modification may be required.
Balancing high DNA yield and high molecular-weight of extracted DNA was a concern during the optimization of this protocol. Steps including vortexing were eliminated in order to minimize shearing of the DNA, and were replaced by gently mixing by inversion. This change increased the quality of the extracted DNA without significantly reducing the yield. Additionally, removal of the steel balls at the beginning the cell lysis step significantly increased DNA yield when compared to leaving the balls in the solution until the end of the lysis step.
No G. cichoracearum conidia remained intact after the ball milling procedure in step 1.3. To obtain good yields of high molecular weight genomic DNA, it is crucial that the fungal cell wall is disrupted during this step. Conidial integrity can be assessed microscopically, and additional ball milling or alternative methods of disruption may be required for other fungal species.
Some fungal conidia, notably the Blumeria sp., contain pigments that can contaminate DNA extraction and purification attempts. In these cases, we recommend including the DNA filtration step as outlined in the Bourras et al. (2015) protocol19 and an additional wash step with TE after the filtration step. The addition of the filtration step resulted in a small but significant increase in DNA degradation, and should only be included if excessive pigmentation remains after the second DNA precipitation step. These pigments, if allowed to remain in solution, may interfere with later analysis of quality and quantity of DNA and could potentially interfere with sequencing reactions.
This protocol includes both a traditional agarose gel electrophoresis step and a pulsed-field gel electrophoresis step. The first is an initial assessment of the fraction of DNA that is > 20 kbp and the fraction that runs as a smear at < 20 kbp. For the purpose of the downstream sequencing reactions required for powdery mildew genomes, the fraction of DNA < 20 kbp should be minimal (see Figure 1, lane 2 versus lane 4). The pulsed-field gel electrophoresis assessment provides a better estimate of the size as DNA > 20 kbp can be separated by size. DNA of > 48.5 kbp is desirable for use in subsequent sequencing reactions (see Figure 2, lane 4).
The fungal material used in these experiments was propagated in growth chambers and contamination was limited using strict isolation protocols and personnel controls. Because of this, contamination with other, non-powdery mildew pathogens was minimal, and sequencing of the ITS region amplified using fungi-specific primers recovered only G. cichoracearum sequences. This may not be the case for powdery mildews collected from field sites or greenhouses lacking the same isolation controls, and contamination assessment in these cases will be critical for the assembly of high-quality genomes from the extracted DNA.
No one modification of the various fungal DNA isolation protocols was uniquely important. Rather slight modifications at many steps resulted in the relatively high yields of high molecular weight powdery mildew DNA reported here.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This protocol was developed in support of a Joint Genome Institute-Community Sequencing Project (#1657). The work was supported in part by the Philomathia Foundation, the National Science Foundation (#0929226) and the Energy Biosciences Institute to S. Somerville; and by Swiss National Science Foundation (#310030_163260) to B. Keller. We would like to thank our colleagues, M. Figureroa and M. Miller (University of Minnesota), R. Panstruga (RWTH Aachen University), C. Pedersen (University of Copenhagen), P. Spanu (Imperial College), J. Taneja (University of California Berkeley), M. Wildermuth (University of California Berkeley), R. Wise (Iowa State University) and S. Xiao (University of Maryland) for their generous advice and for volunteering their protocols as we developed the protocol described here.
References
- Agrios GN. Plant Pathology. 4th. Academic Press; 1969. [Google Scholar]
- Braun U, Cook RT. Taxonomic manual of the Erysiphales (Powdery Mildews) The Netherlands: CBS-KNAW Fungal Biodiversity Centre Utrecht; 2012. [Google Scholar]
- Both M, Csukai M, Stumpf MPH, Spanu PD. Gene expression profiles of Blumeria graminis indicate dynamic changes to primary metabolism during development of an obligate biotrophic pathogen. Plant Cell. 2005;17(7):2107–2122. doi: 10.1105/tpc.105.032631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glawe DA. The powdery mildews: A review of the world's most familiar (yet poorly known) plant pathogens. Annu Rev Phytopathol. 2008;46:27–51. doi: 10.1146/annurev.phyto.46.081407.104740. [DOI] [PubMed] [Google Scholar]
- Fabro G, et al. Genome-wide expression profiling Arabidopsis at the stage of Golovinomyces cichoracearum haustorium formation. Plant Physiol. 2008;146(3):1421–1439. doi: 10.1104/pp.107.111286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hückelhoven R, Panstruga R. Cell biology of the plant-powdery mildew interaction. Curr Opin Plant Biol. 2011;14(6):738–746. doi: 10.1016/j.pbi.2011.08.002. [DOI] [PubMed] [Google Scholar]
- Micali CO, Neumann U, Grunewald D, Panstruga R, O'Connell R. Biogenesis of a specialized plant-fungal interface during host cell internalization of Golovinomyces orontii haustoria. Cell Microbiol. 2011;13(2):210–226. doi: 10.1111/j.1462-5822.2010.01530.x. [DOI] [PubMed] [Google Scholar]
- Chandran D, Inada N, Hather G, Kleindt CK, Wildermuth MC. Laser microdissection of Arabidopsis cells at the powdery mildew infection site reveals site-specific processes and regulators. Proc Natl Acad Sci. 2010;107(1):460–465. doi: 10.1073/pnas.0912492107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanu PD, Panstruga R. Powdery mildew genomes in the crosshairs. New Phytol. 2012;195(1):20–22. doi: 10.1111/j.1469-8137.2012.04173.x. [DOI] [PubMed] [Google Scholar]
- Vela-Corcía D, Romero D, Torés JA, De Vicente A, Pérez-García A. Transient transformation of Podosphaera xanthii by electroporation of conidia. BMC Microbiol. 2015;15(20) doi: 10.1186/s12866-014-0338-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacquard S. In: Advances in Botanical Research. Francis MM, editor. Vol. 70. Academic Press; 2014. pp. 109–142. [Google Scholar]
- Spanu PD, et al. Genome expansion and gene loss in powdery mildew fungi reveal tradeoffs in extreme parasitism. Science. 2010;330(6010):1543–1546. doi: 10.1126/science.1194573. [DOI] [PubMed] [Google Scholar]
- Wicker T, et al. The wheat powdery mildew genome shows the unique evolution of an obligate biotroph. Nat Genet. 2013;45(9):1092. doi: 10.1038/ng.2704. [DOI] [PubMed] [Google Scholar]
- Jones L, et al. Adaptive genomic structural variation in the grape powdery mildew pathogen, Erysiphe necator. BMC Genomics. 2014;15:1081. doi: 10.1186/1471-2164-15-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomma BPHJ, et al. Mind the gap; seven reasons to close fragmented genome assemblies. Fungal Genet Biol. 2016;90:24–30. doi: 10.1016/j.fgb.2015.08.010. [DOI] [PubMed] [Google Scholar]
- Parlange F, et al. A major invasion of transposable elements accounts for the large size of the Blumeria graminis f.sp. tritici genome. Funct Integr Genomics. 2011;11:671–677. doi: 10.1007/s10142-011-0240-5. [DOI] [PubMed] [Google Scholar]
- Faino L, et al. Single-molecule real-time sequencing combined with optical mapping yields completely finished fungal genome. mBio. 2015;6 doi: 10.1128/mBio.00936-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam L, et al. Comparison of Erysiphe cichoracearum and E. cruciferarum and a survey of 360 Arabidopsis thaliana accessions for resistance to these two powdery mildew pathogens. Mol Plant Microbe In. 1999;12(12):1031–1043. doi: 10.1094/MPMI.1999.12.12.1031. [DOI] [PubMed] [Google Scholar]
- Bourras S, et al. Multiple avirulence loci and allele-specific effector recognition control the PM3 race-specific resistance of wheat to powdery mildew. Plant Cell. 2015;27(10):2991–3012. doi: 10.1105/tpc.15.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joint Genome Institute DNA sample submission guidelines. 2016. http://1ofdmq2n8tc36m6i46scovo2e.wpengine.netdna-cdn.com/wp-content/uploads/2016/04/DNA-Preparation-Requirements-1.pdf.
- Chovatia M, Sharma A. Genomic DNA sample QC version 4.0. 2014. http://1ofdmq2n8tc36m6i46scovo2e.wpengine.netdna-cdn.com/wp-content/uploads/2013/11/Genomic-DNA-Sample-QC.pdf.
- Daum C. iTag sample amplification QC version 1.3. 2016. http://1ofdmq2n8tc36m6i46scovo2e.wpengine.netdna-cdn.com/wp-content/uploads/2016/04/iTag-Sample-Amplification-QC-v1.3.pdf.
- Henry RJ, editor. Plant genotyping II: SNP technology. Oxfordshire, UK: CABI; 2008. [Google Scholar]
- Healey A, Furtado A, Cooper T, Henry RJ. Protocol: a simple method for extracting next-generation sequencing quality genomic DNA from recalcitrant plant species. Plant Methods. 2014;10(21) doi: 10.1186/1746-4811-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G, Hammar S, Grumet R. A quick and inexpensive method for removing polysaccharides from plant genomic DNA. Biotechniques. 1992;13(1):52–54. [PubMed] [Google Scholar]
- Jordon-Thaden IE, Chanderbali AS, Gitzendanner MA, Soltis DE. Modified CTAB and TRIzol protocols improve RNA extraction from chemically complex Embryophyta. Appl Plant Sci. 2015;3(5):1400105. doi: 10.3732/apps.1400105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray MG, Thompson WF. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980;8(19):4321–4325. doi: 10.1093/nar/8.19.4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco M, Sáez CA, Brown MT, Bitonti MB. A simple and effective method for high quality co-extraction of genomic DNA and total RNA from low biomass Ectocarpus siliculosus, the model brown alga. PLoS One. 2014;9(5):e96470. doi: 10.1371/journal.pone.0096470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson IW, Schiff CL, Hughes DE, Somerville SC. Quantitative trait loci analysis of powdery mildew disease resistance in the Arabidopsis thaliana accession Kashmir-1. Genetics. 2001;158(3):1301–1309. doi: 10.1093/genetics/158.3.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- User Manual: Pippin Pulse Electrophoresis Power Supply. Sage Science; 2008. http://www.sagescience.com/wp-content/uploads/2014/01/Pippin-Pulse-User-Manual-RevH.pdf. [Google Scholar]