

Abstract
Accurate detection and identification of low frequency mutations can be problematic when assessing residual disease after therapy, screening for emerging resistance mutations during therapy, or when patients have few circulating tumor cells. Wild-type blocking PCR followed by sequencing analysis offers high sensitivity, flexibility, and simplicity as a methodology for detecting these low frequency mutations. By adding a custom designed locked nucleic acid oligonucleotide to a new or previously established conventional PCR based sequencing assay, sensitivities of approximately 1 mutant allele in a background of 1,000 WT alleles can be achieved (1:1,000). Sequencing artifacts associated with deamination events commonly found in formalin fixed paraffin embedded tissues can be partially remedied by the use of uracil DNA glycosylase during extraction steps. The optimized protocol here is specific for detecting MYD88 mutation, but can serve as a template to design any WTB-PCR assay. Advantages of the WTB-PCR assay over other commonly utilized assays for the detection of low frequency mutations including allele specific PCR and real-time quantitative PCR include fewer occurrences of false positives, greater flexibility and ease of implementation, and the ability to detect both known and unknown mutations.
Keywords: Genetics, Issue 121, WTB-PCR, wild-type blocking PCR, blocking oligonucleotide, high sensitivity, sequencing, mutation, FFPE, MYD88, Waldenström's Macroglobulnemia, diffuse large b-cell lymphoma
Introduction
Sanger sequencing has traditionally been the gold standard in testing for both known and unknown somatic mutations. One of the limitations of Sanger sequencing is its limit of detection (~ 10 - 20% mutant allele in a background of WT)1. This level of sensitivity is inappropriate for detecting low level somatic mutations that may be present in samples from premalignant tissues or patients with few circulating tumor cells, or when bone marrow (BM) is patchy. This also makes assessing residual disease after therapy or detecting emerging resistance mutations during therapy difficult by conventional sequencing alone2. By replacing conventional PCR with locked nucleic acid (LNA)-mediated wild-type blocking PCR (WTB-PCR) in Sanger sequencing, sensitivities of up to 0.1% mutant allele in a background of WT can be achieved2,3,4. In WTB-PCR, enrichment for mutant alleles is achieved via the addition of a short (~ 10 - 14 NT) blocking (LNA) oligonucleotide that binds preferentially to WT DNA thereby preventing amplification of WT DNA. The mutant enriched WTB-PCR product can then be sequenced. By blocking WT DNA rather than selecting for specific mutations WTB-PCR allows for enrichment of both known and unknown mutations present in minute cell fractions.
Multiple methods are currently used for detecting mutations in small cells fractions. This includes allele-specific PCR, amplification-refractory mutation system (ARMS), denaturing high performance liquid chromatography (DHPLC), beads, emulsions, amplification, and magnetics (BEAMing), electric field-induced release and measurement (EFIRM), high resolution melting point and others. However, most of these methods are limited by false-positives and the ability to only detect one mutation that the assay was designed for4. WTB-PCR, however, allows the user to visualize sequencing traces which enables the detection of multiple mutation types and can aid in ruling out false positives due to artifacts or deamination events. Next-generation sequencing (NGS) may offer a suitable alternative to conventional sequencing, however, substantially greater costs, complexity, and longer assay time render it an unnecessary option for many disease types with few distinct molecular markers or for monitoring patients on therapy for emerging resistance mutations. Furthermore, high false positive rates when detecting variants with mutant allele frequencies of less than 5% can pose a problem for amplicon-based NGS5,6.
Here we demonstrate the increase in sensitivity achieved by WTB-PCR in screening for mutations in the myeloid differentiation factor 88 gene as described by Albitar et al.3 MYD88 mutations are important diagnostic and prognostic factors in Waldenström's Macroglobulinemia (WM), IgM monoclonal gammopathy of unknown significance (IgM-MGUS), splenic marginal zone lymphoma (SMZL), and diffuse large B-cell lymphoma (DLBCL). MYD88 mutations are found in almost all cases of WM and approximately 50% of patients with Immunoglobulin M (IgM)-secreting MGUS. Contrastingly, MYD88 mutations are found in only 0-6% of patients with SMZL and are absent in multiple myeloma7,8. Because overlapping morphologic, immunophenotypic, cytogenetic, and clinical characteristics between WM and SMZL or IgM-multiple myeloma can often complicate differential diagnoses, the presence of a MYD88 mutation may serve as a useful identifying factor9. MYD88 mutations have also been associated with greater disease burden in patients with DLBCL and poor overall survival following therapy7,10. Additionally, because MYD88 mutations are more frequently found in activated B-cell-like (ABC) DLBCL than germinal center B-cell-like (GCB) DLBCL or primary mediastinal B-cell lymphoma (PMBL), MYD88 mutation status may serve as a surrogate marker for the ABC subtype11,12.
The detailed protocol provided here serves as a template from which new assays can be developed or most existing sequencing assays can be easily adapted to accurately detect low frequency mutations in various sample types. The approach can also be used for monitoring and detecting resistant mutations that may develop in tumors or even bacteria that may develop while patients are being treated with targeted therapy or antibiotics. Furthermore, it addresses and remedies many of the issues associated with mutation enrichment, particularly in formalin-fixed paraffin-embedded (FFPE) tissue.
Protocol
Ethics Statement: All testing of human samples was performed after obtaining Institutional Review Board (IRB) approval.
1. DNA Extraction from FFPE Tissue, Peripheral Blood, and Bone Marrow Aspirate
- For bone marrow FFPE tissue with DNA FFPE extraction kit
- Begin with FFPE tissue from unstained slides (5 - 10 sections at 5 - 10 µm thickness). NOTE: If beginning with tissue shavings, use 3 - 6 sections at 5 - 10 µm thickness and skip to step 1.1.6.
- Place slides in a slide basket and prepare four wash reservoirs (two for Xylene and two for 100% Alcohol). Add a minimum volume of 600 mL of solution to each basket.
- Deparaffinize the slides by doing a 5 min xylene wash in the first tray. Transfer the slides to the second xylene wash reservoir for an additional 5 min.
- After deparaffinization, wash the slides with 100% alcohol for 5 min. Transfer the slides to the second alcohol wash reservoir for an additional 5 min.
- Allow the slides to dry completely under a hood before scraping them with a razor blade into a microcentrifuge tube. NOTE: If an H&E slide with tumor region indicated is available, align the slides and scrape only the tumor region.
- Proceed with instructions from manufacturer handout included in kit.
- For BM aspirate and Peripheral Blood
- Extract according to the manufacturer's instructions handout with these specifications. NOTE: There are numerous commercially available kits and methods for DNA extraction from FFPE tissues and cells. Practically, all can be used. Use a method that is established in the laboratory.
- Use 200 µL peripheral blood (PB) or 100 µL BM + 100 µL PBS.
- Use 4 µL RNase A stock solution.
- Elute with 100 µL elution buffer.
- DNA Quantification
- Measure DNA concentrations using a spectrophotometer ensuring a 260 nm/280 nm ratio of approximately 1.8 (for pure DNA). If the ratio is appreciably lower, it may indicate the presence of protein, phenol, or other contaminants that may interfere with downstream applications.
- Adjust DNA concentrations to approximately 50 - 100 ng/µL with appropriate elution buffer.
2. Wild-Type Blocking PCR
- Primer Design
- Design and obtain primers for genes of interest according to previously described general guidelines of PCR primer design13. Include M13-forward and reverse universal sequencing primers in PCR primers. NOTE: The MYD88 assay was developed to amplify exon 5 of MYD88 (G259 - N291) and 110 nucleotides located in the 5' intronic region to cover the splice site and part of intron 4. The forward and reverse primers were designed with a 5'-M13 sequence (M13-forward: tgt aaa acg acg gcc agt; M13-reverse: cag gaa aca gct atg acc) to allow for annealing of complementary sequencing primers (see Materials Table).
- Locked Nucleic Acid Oligonucleotide Design
- Design the blocking oligonucleotide to be approximately 10 - 15 bases in length and complementary to the WT template where mutant enrichment is desired. NOTE: A shorter oligo will improve mismatch discrimination. To achieve high target specificity, it is important not to use too much blocking nucleotides since this can result in a very "sticky" oligonucleotide. The content and the length and blocking nucleotide should to be optimize according to the specific nucleotide that need to be blocked. It is important to achieve high binding specificity without compromising secondary structure and risking self-complementarity.
- Design the blocking oligo to have a Tm 10 - 15 °C above the extension temperature during thermocycling. Adjust the Tm by adding or removing blocking bases or by substituting blocking bases for DNA while avoiding long stretches (3 - 4 bases) of blocking G or C bases.
- Navigate to the oligo tools website (see Table of Materials) and select the "blocking Oligo Tm Prediction" tool.
- Paste the sequence of the WT template that is desired to be blocked into the "Oligo Sequence" box. Add "+" in front of bases to indicate blocker bases.
- Select the "Calculate" button to determine the approximate Tm of the DNA-Blocker hybrid.
- Design the blocking oligo to avoid secondary structure formation or self-dimers.
- Navigate to the oligo tools website (see Table of Materials) and select the "blocking Oligo Optimizer" tool.
- Paste the sequence of the WT template that is desired to be blocked into the box. Add "+" in front of bases to indicate blocking bases.
- Select the two boxes for "Secondary Structure" and "Self Only". Press the Analyze button to review the oligo for potentially troublesome secondary structures or self-dimers. NOTE: The scores represent very rough estimates of the Tm's of the secondary structures and self-dimers. Lower scores are therefore optimal and can be achieved by limiting blocker-blocker pairing. The blocking oligonucleotide for MYD88 [MYD88blocker (TCAGA+AG+C+G+A+C+T+G+A+T+CC/invdT/)] was designed to cover amino acids Q262-I266 and features a 3'-inverted dT to inhibit both extension by DNA polymerase and degradation by 3'-exonuclease. This specific blocking oligo is a 17mer with 10 blocking bases which are denoted as "+N". The remaining 7 bases are ordinary DNA nucleotides.
- WTB-PCR Setup and Thermocycling
- Prepare a WTB-PCR master mix (MMX) using 2.5 µL PCR reaction buffer 10x w/ 20 mM MgCl2, 250 µM dNTPs, 0.2 µM forward and reverse primer, 1.2 µM MYD88blocker, and DNAse, RNAse-free, ultra-pure H2O to create a final solution volume of 21.75 µL per reaction. NOTE: Working concentrations are for the final reaction volume of 25 µL (after addition of DNA template and polymerase). Conventional PCR MMX is prepared by simply omitting the addition of blocker. All protocol steps remain identical for both WTB and conventional PCR. This can be used in validation and determining enrichment achieved by addition of blocker.
- When calculating the amount of MMX to prepare make sure to account for 3 additional reactions (positive and negative controls and a non-template control to check for contamination) and at least 1 additional reaction for pipetting error.
- Vortex MMX thoroughly. The MMX can be stored at -80 °C for up to a year.
- Add 0.25 µL Taq DNA polymerase per reaction to the MMX and invert to mix. Once the polymerase has been added to the MMX, keep it on ice.
- Put a new 96 well PCR plate on a cold plate and pipette 22 µL of MMX with polymerase to each reaction well.
- Add 3 µL genomic DNA (50 - 100 ng/µL to each of the wells containing the MMX with polymerase.
- Seal the plate and centrifuge briefly to ensure the solution reaches the bottom of each well.
- Load the PCR plate on a thermocycler.
- Set the thermocycling conditions for the WTB-PCR reaction as follows: initial denaturation at 95 °C for 6 min; 40 cycles of denaturation at 95 °C for 30 s, primer annealing at 56 °C for 30 s, and extension at 72 °C for 1 min 20 s; final extension at 72 °C for 10 min. NOTE: Best practice involves completing the remainder of the protocol in a physically separate area to avoid amplicon contamination in future setups.
Perform gel electrophoresis according to previously described protocols14 in order to determine if WTB-PCR was successful and to confirm a lack of amplification in the non-template control.
- Purification of PCR Product by magnetic beads
- Remove magnetic beads from 4 °C and bring to room temperature.
- Transfer 10 µL PCR product to a new PCR plate.
- Vortex the magnetic beads vigorously to fully resuspend the magnetic particles.
- Add 18 µL magnetic beads to each well on the new plate. Pipette mix 10 times.
- Incubate at room temperature for 5 min.
- Place the PCR plate onto the side-skirted magnet plate for 2 min to separate beads from solution.
- Aspirate the supernatant with a multichannel pipette, avoiding the bead pellet.
- Dispense 150 µL 70% ethanol into each well and incubate at room temperature for at least 30 s. Aspirate the ethanol with a multichannel pipette and discard tips. Repeat this wash procedure once more.
- Using a 20 µL multichannel pipette aspirate the remaining ethanol from each well and discard tips.
- Once wells are dry (~ 10 min), remove from magnet plate and add 40 µL nuclease free water to each well and pipette mix 15 times or vortex gently.
- Incubate at room temperature for 2 min.
- Place the PCR plate onto magnet plate for 1 min to separate beads from solution.
- Transfer 35 µL of purified product to a new PCR plate. This is purified PCR product is now ready to proceed with bi-directional sequencing or can be stored at -20 °C until needed. NOTE: When developing a new assay, quantification of purified PCR product may be required. 1 - 3 ng/µL amplicon DNA is optimal for bi-directional sequencing. If concentration is consistently low, increase PCR product and magnetic beads proportionally (1:1.8). If concentration is consistently high, elute with a greater volume of water.
3. Sequencing of WTB-PCR Product
- Bi-Directional Sequencing NOTE: The following protocol is a modified form of the manufacturer's instructions that has been optimized to use fewer reagents.
- Prepare forward and reverse sequencing reaction mixes with 0.25 µL Ready Reaction mix, 1.88 µL Sequencing Buffer, 1.78 µM M13-F or M13-R sequencing primers and add and DNAse, RNAse-free, ultra-pure H2O to create a final solution volume of 9 µL per reaction. This sequencing reaction mix can be stored at -20 °C for up to a year.
- Pipette 9 µL of the forward sequencing reaction mix into each well of a new 96 well PCR plate for the forward sequencing reaction. Repeat on a separate plate for the reverse sequencing reaction.
- Add 1 µL of the purified WTB-PCR product to each well on both the forward and reverse plates.
- Seal the plate and centrifuge briefly to ensure the solution reaches the bottom of each well.
- Load the PCR plate on a thermocycler.
- Set the thermocycling conditions for the sequencing reaction as follows: 96 °C for 1 min; 30 cycles of 96 °C for 10 s, 50 °C for 5 s, 60 °C for 4 min; hold at 4 °C until ready for purification.
- Purify Sequencing Products
- Prepare a fresh 1:25 solution of 3 M sodium acetate (pH 5.2) and 100% ethanol.
- Prepare a fresh 70% ethanol solution.
- Add 30 µL of sodium acetate/100% ethanol solution to each well of both the forward and reverse sequencing plates and pipette mix 5 times.
- Reseal the plate and incubate in the dark at room temperature for 20 min.
- Centrifuge the plate at 2,250 x g for 15 min.
- Remove the plate sealer and invert over a waste container. NOTE: Invert only once or the pellet may loosen from the well bottoms.
- Place the inverted plate on a clean paper towel and centrifuge at 150 x g for 1 min.
- Add 150 µL 70% ethanol to each well.
- Reseal the plate and spin at 2,250 x g for 5 min.
- Repeat steps 3.2.6 and 3.2.7.
- If the wells are not completely dry, allow them to air dry at room temperature. Make sure the samples are protected from light while drying.
- Add 10 µL Formamide to each well and pipette mix 10 times. Reseal the plate.
- Denature on thermocycler at 95 °C for 3 min followed by 4 °C for 5 min.
- After denaturing, replace the plate sealer with a septa and sequence on sequencing platform according to manufacturer's instructions15.
4. Analysis of Sequencing Results
Representative Results
A conceptual overview of WTB-PCR during extension is presented in Figure 1. Because a single nucleotide mismatch in the blocker-DNA hybrid greatly decreases its melting temperature (ΔTm=20 - 30 °C), amplification of the WT allele is blocked while mutant template DNA is free to complete extension17. In this manner, mutant DNA is amplified exponentially while WT DNA is amplified linearly.
A demonstration of the mutant enrichment achieved by WTB-PCR is presented in Figure 2. Genomic DNA from patients with and without mutations were tested by both conventional and WTB-PCR and then sequenced to demonstrate the typical enrichment achievable by WTB-PCR and a lack of false positives in WT DNA. The working concentration of blocker used in WTB-PCR MMX should be determined by titration experiments and should achieve the desired level of mutant enrichment while not resulting in false positives or blocking amplification of WT DNA entirely. Sequencing analysis of WTB-PCR product demonstrates enrichment for the mutant allele and a limit of detection in excess of 0.5% mutant allele in a background of WT compared with 16% in conventional PCR3.
The effects of this increase in sensitivity in clinical testing may vary depending on the relative quantity of neoplastic cells in the samples tested. In a methods comparison study, the WTB-PCR assay described here has demonstrated that 64% of MYD88 mutations would be missed by conventional testing of patients with WM or MGUS3.
A characteristic drop-off in signal intensity (Figure 3) is often seen if too high a concentration of blocker is used or if post-PCR purification failed to remove blocker prior to bi-directional sequencing. The latter is demonstrated when magnetic bead purification is substituted for enzymatic purification. Though enzymatic purification is an attractive option when working with greater sample numbers, it is inappropriate for application with WTB-PCR as it fails to remove blocker from solution prior to sequencing.
An example of sequence artifacts frequently found in FFPE-derived DNA as a result of cytosine deamination (C:G>T:A) are presented in Figure 43,18,19,20. Though the actual causes of cytosine deamination are poorly understood, any PCR based assay that enriches for mutant alleles will detect these low frequency artifacts21,22. False positives due to deamination are best avoided by starting with high quality template material; in the many cases where this is not possible, treatment with uracil DNA glycosylase (UDG) during extraction can aid in limiting the frequency and intensity of deamination artifacts. UDG treatment of FFPE tissue during extraction (as part of the DNA FFPE kit) excises deaminated cytosine residues thereby preventing artificially induced C:G>T:A mutations. However, 5-methylcytosine residues that frequently occur at CpG dinucleotides are deaminated to thymine, which cannot be excised by UDG. The resulting sequencing artifacts are fairly recognizable and often appear as tandem mutations as seen in Figure 4. If samples have already been extracted, UDG treatment can be implemented in a secondary extraction with relatively low DNA loss.
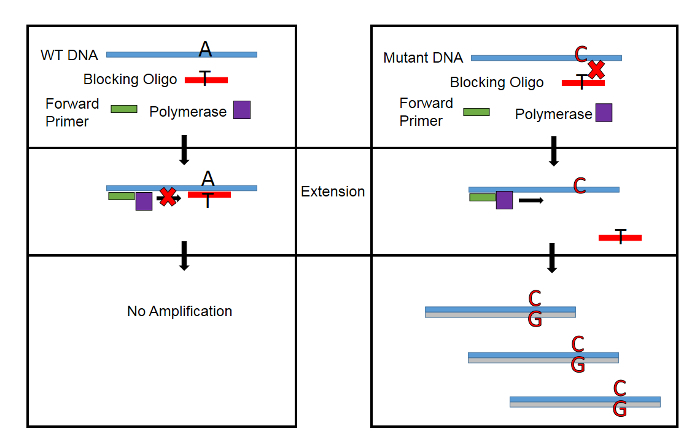 Figure 1: Conceptual Overview of WTB-PCR. A single nucleotide mismatch in the Blocker-DNA hybrid decreases Tm by up to 30 °C. By designing the blocking oligonucleotide to have a Tm of 10 - 15 °C above the temperature during extension, amplification of WT DNA is blocked while allowing amplification of mutant DNA. Please click here to view a larger version of this figure.
Figure 1: Conceptual Overview of WTB-PCR. A single nucleotide mismatch in the Blocker-DNA hybrid decreases Tm by up to 30 °C. By designing the blocking oligonucleotide to have a Tm of 10 - 15 °C above the temperature during extension, amplification of WT DNA is blocked while allowing amplification of mutant DNA. Please click here to view a larger version of this figure.
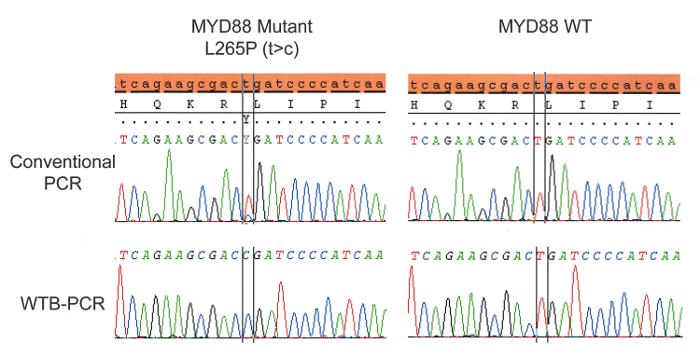 Figure 2: Genomic DNA from patients with and without mutations were tested by both conventional and WTB-PCR and then sequenced to demonstrate the typical enrichment achievable by WTB-PCR. The final concentration of blocker used to achieve WTB-PCR was selected to achieve maximum mutant enrichment while not causing false positives in WT DNA or blocking amplification of WT DNA entirely. Please click here to view a larger version of this figure.
Figure 2: Genomic DNA from patients with and without mutations were tested by both conventional and WTB-PCR and then sequenced to demonstrate the typical enrichment achievable by WTB-PCR. The final concentration of blocker used to achieve WTB-PCR was selected to achieve maximum mutant enrichment while not causing false positives in WT DNA or blocking amplification of WT DNA entirely. Please click here to view a larger version of this figure.
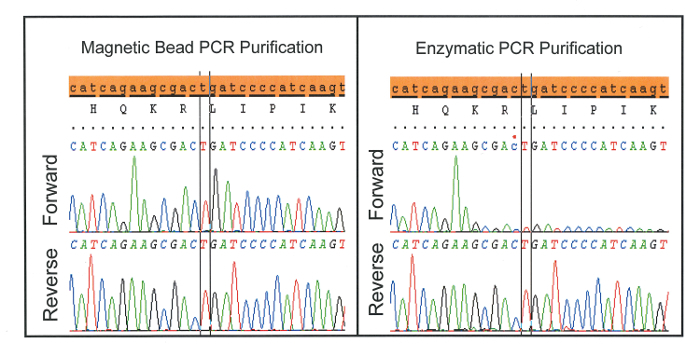 Figure 3: Characteristic drop-off in signal intensity seen when enzymatic PCR purification is used instead of magnetic beads. This is likely because enzymatic purification fails to remove blocker prior to bi-directional sequencing. Please click here to view a larger version of this figure.
Figure 3: Characteristic drop-off in signal intensity seen when enzymatic PCR purification is used instead of magnetic beads. This is likely because enzymatic purification fails to remove blocker prior to bi-directional sequencing. Please click here to view a larger version of this figure.
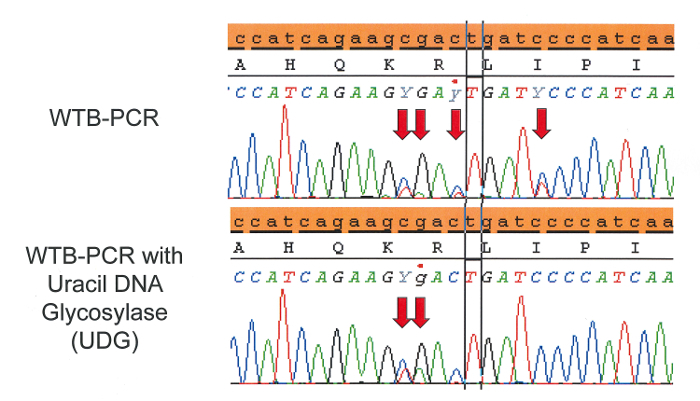 Figure 4: C:G>T:A sequencing artifacts arise in FFPE tissue when cytosine or methylated cytosine are deaminated via formalin fixation to uracil or thymine, respectively. Uracil DNA glycosylase (UDG) can excise uracil prior to WTB-PCR helping to reduce sequencing artifacts. However, thymine resulting from deaminated 5-methylcytosine, which frequently occurs at CpG islands, cannot be excised by UDG. Decreasing the concentration of blocker used in WTB-PCR may help to reduce the occurrence of sequencing artifacts that are not remedied by UDG treatment. Please click here to view a larger version of this figure.
Figure 4: C:G>T:A sequencing artifacts arise in FFPE tissue when cytosine or methylated cytosine are deaminated via formalin fixation to uracil or thymine, respectively. Uracil DNA glycosylase (UDG) can excise uracil prior to WTB-PCR helping to reduce sequencing artifacts. However, thymine resulting from deaminated 5-methylcytosine, which frequently occurs at CpG islands, cannot be excised by UDG. Decreasing the concentration of blocker used in WTB-PCR may help to reduce the occurrence of sequencing artifacts that are not remedied by UDG treatment. Please click here to view a larger version of this figure.
Discussion
The WTB-PCR assay described here uses a generic set of primers with a blocking oligo designed to block amplification of WT DNA during extension (Figure 1). The WTB-PCR product is then sequenced for mutational analysis. The utility of WTB-PCR/Sanger lies in its simplicity, high-sensitivity, and high-throughput. Using the guidelines described here, most existing Sanger based assays can be simply modified via the addition of a blocking oligonucleotide to greatly increase sensitivity. In the example assay presented here the addition of a single blocking oligonucleotide to PCR increased the limit of detection from approximately 16% mutant allele in a background of WT for the conventional assay to >0.5% in the WTB-PCR assay3. The effect of which is a 64% reduction in false negatives seen in clinical testing3. Confirming the presence of mutations in MYD88 has significant diagnostic and prognostic implications; falsely reporting a case as negative may have serious consequences on overall therapy and patient management. Testing with WTB-PCR is therefore of great importance, particularly in patients with relatively low abnormal cellularity.
WTB-PCR techniques vary considerably and are sometimes referred to interchangeably with LNA, BNA, or PNA-mediated PCR clamping or PCR-clamp-probe assays2,4,23,24. Some variations utilizing WTB-PCR involve a qPCR assay which necessitates designing an additional fluorescent probe for each specific mutation. A significant challenge associated with this technique includes the need to develop competitively binding probes that accurately discriminate between WT and mutant alleles. Furthermore, because a mutation specific probe is required, the qPCR approach lacks the ability to detect unknown mutations. Another variation of WTB-PCR blocks WT amplification in an allele-specific manner. Instead of using mutation-specific primers however, a blocking probe specific for the WT allele inhibits complete primer binding. While this approach does not require a mutation-specific probe like qPCR, it fails to discriminate between mutations and polymerase induced errors can lead to increased false positives. Exponential amplification of those polymerase induced errors through PCR may obscure the detection of rare mutational events. Any approach that uses PCR amplification to enrich for mutations has its accuracy limited by the frequency of PCR errors25,26,27. A fundamental advantage of WTB-PCR/Sanger over many other high-sensitivity methodologies is that it prevents amplification of both WT template and mutant template whose mutations occur outside the gene region targeted by the blocking oligonucleotide. Therefore, PCR errors introduced by polymerases are effectively filtered out along with WT DNA. Contrary to AS-PCR or qPCR however, enrichment is retained for unknown mutations that occur within the region covered by the blocking oligo. In the case of MYD88, WTB-PCR allowed the detection of both L265P and R264* mutations3. Others have similarly reported the detection of multiple, low-frequency mutations with the use of WTB-PCR4,28. This makes WTB-PCR ideal for both research and clinical purposes.
Along with high sensitivity and inherent internal controls for eliminating false positives, WTB-PCR's flexibility for adapting an existing sequencing assay with very few additional steps with blocker design make it attractive option for many labs with established assays. The same set of PCR primers used in a conventional assay can typically be used in the WTB-PCR variation. Other protocol changes that are highly recommended for WTB-PCR-including UDG treatment of FFPE tissue and appropriate post-PCR purification methodologies-are equally transferrable. Blocker design, therefore is the critical element in implementing a WTB-PCR assay. Though the guidelines presented in this protocol represent the most relevant factors in that design, various blocking oligonucleotides should be tested to find one that blocks WT amplification efficiently without secondary effects to PCR. This includes adding or removing blocker bases to adjust Tm, shifting the position of the blocking oligo relative to the WT template, and changing the overall length of the oligo. Blocker titration experiments should also be employed to establish a balance between acceptable occurrences of sequencing artifacts and limit of detection.
Selecting an appropriate methodology for detecting mutations that are present in minute cell fractions depends greatly on the application and disease/mutation types. If mutant quantification is desired, qPCR or digital PCR may offer more viable solutions than WTB-PCR/Sanger. Though WTB-PCR is primarily a qualitative assay, it is possible to determine a rough estimate of the mutant allele frequency by testing a sample with conventional and WTB-PCR in parallel. Because the limit of detection for the conventional assay is ~ 10 - 20% mutant allele in a background of WT, it is appropriate to conclude that mutations detected by the WTB-PCR assay but not the conventional are present at a concentration less than the limit of detection for the conventional assay. Few assays offer the versatility, simplicity, and robustness of WTB-PCR. The low cost and short turnaround time makes it ideal for assessing residual disease after therapy or monitoring emerging resistance mutations during therapy. Additionally, the ability to detect previously undescribed mutations make WTB-PCR ideal for research purposes.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors have no acknowledgements.
References
- Vogelstein B, Kinzler KW. Digital PCR. Proc Natl Acad of Sci U S A. 1999;96(16):9236–9241. doi: 10.1073/pnas.96.16.9236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milbury CA, Li J, Makrigiorgos GM. PCR-based methods for the enrichment of minority alleles and mutations. Clin Chem. 2009;55(4):632–640. doi: 10.1373/clinchem.2008.113035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albitar A, Ma W, DeDios I, Estella J, Agersborg S, Albitar M. Positive selection and high sensitivity test for MYD88 mutations using locked nucleic acid. Int J Lab Hematol. 2016;38(2):133–140. doi: 10.1111/ijlh.12456. [DOI] [PubMed] [Google Scholar]
- Dominguez PL, Kolodney MS. Wild-type blocking polymerase chain reaction for detection of single nucleotide minority mutations from clinical specimens. Oncogene. 2005;24(45):6830–6834. doi: 10.1038/sj.onc.1208832. [DOI] [PubMed] [Google Scholar]
- Gray PN, Dunlop CLM, Elliott AM. Not All Next Generation Sequencing Diagnostics are Created Equal: Understanding the Nuances of Solid Tumor Assay Design for Somatic Mutation Detection. Cancers. 2015;7(3):1313–1332. doi: 10.3390/cancers7030837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EN, et al. Biased estimates of clonal evolution and subclonal heterogeneity can arise from PCR duplicates in deep sequencing experiments. Genome Biol. 2014;15(8):420. doi: 10.1186/s13059-014-0420-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varettoni M, et al. Prevalence and clinical significance of the MYD88 (L265P) somatic mutation in Waldenström's macroglobulinemia and related lymphoid neoplasms. Blood. 2013;121(13):2522–2528. doi: 10.1182/blood-2012-09-457101. [DOI] [PubMed] [Google Scholar]
- Xu L, et al. MYD88 L265P in Waldenström macroglobulinemia, immunoglobulin M monoclonal gammopathy, and other B-cell lymphoproliferative disorders using conventional and quantitative allele-specific polymerase chain reaction. Blood. 2013;121(11):2051–2058. doi: 10.1182/blood-2012-09-454355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz MA. Waldenström macroglobulinemia: 2012 update on diagnosis, risk stratification, and management. Amer J Hematol. 2012;87(5):503–510. doi: 10.1002/ajh.23192. [DOI] [PubMed] [Google Scholar]
- Salar A, et al. MYD88 (L265P) Mutation Confers Very Poor Response and Outcome after Second-Line Therapy in Patients with Diffuse Large B-Cell Lymphoma (DLBCL) Blood. 2014;124(21):1690–1690. [Google Scholar]
- Ngo VN, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470(7332):115–119. doi: 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011;43(9):830–837. doi: 10.1038/ng.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieffenbach C, Lowe T, Dveksler G. General concepts for PCR primer design. PCR Methods Appl. 1993;3(3):S30–S37. doi: 10.1101/gr.3.3.s30. [DOI] [PubMed] [Google Scholar]
- Lee PY, Costumbrado J, Hsu CY, Kim YH. Agarose Gel Electrophoresis for the Separation of DNA Fragments. J Vis Exp. 2012. p. e3923. [DOI] [PMC free article] [PubMed]
- Applied Biosystems. User Guide for Applied Biosystems 3730/3730xl DNA Analyzer. 2014.
- Applied Biosystems. User Guide for SeqScape Software 3. 2012.
- Mouritzen P, Nielsen AT, Pfundheller HM, Choleva Y, Kongsbak L, Møller S. Single nucleotide polymorphism genotyping using locked nucleic acid (LNA) Expert Rev Mol Diagn. 2003;3(1):27–38. doi: 10.1586/14737159.3.1.27. [DOI] [PubMed] [Google Scholar]
- Quach N, Goodman MF, Shibata D. In vitro mutation artifacts after formalin fixation and error prone translesion synthesis during PCR. BMC Clin Pathol. 2004;4(1) doi: 10.1186/1472-6890-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallegos Ruiz MI, Floor K, Rijmen F, Grünberg K, Rodriguez JA, Giaccone G. EGFR and K-ras mutation analysis in non-small cell lung cancer: comparison of paraffin embedded versus frozen specimens. Anal Cell Pathol. 2007;29(3):257–264. doi: 10.1155/2007/568205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solassol J, et al. KRAS mutation detection in paired frozen and formalin-fixed paraffin-embedded (FFPE) colorectal cancer tissues. Int J Mol Sci. 2011;12(5):3191–3204. doi: 10.3390/ijms12053191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost SE, et al. Identification of high-confidence somatic mutations in whole genome sequence of formalin-fixed breast cancer specimens. Nucleic Acids Res. 2012;40(14):e107–e107. doi: 10.1093/nar/gks299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do H, Wong SQ, Li J, Dobrovic A. Reducing sequence artifacts in amplicon-based massively parallel sequencing of formalin-fixed paraffin-embedded DNA by enzymatic depletion of uracil-containing templates. Clin Chem. 2013;59(9):1376–1383. doi: 10.1373/clinchem.2012.202390. [DOI] [PubMed] [Google Scholar]
- Oldenburg RP, Liu MS, Kolodney MS. Selective amplification of rare mutations using locked nucleic acid oligonucleotides that competitively inhibit primer binding to wild-type DNA. J Invest Dermatol. 2008;128(2):398–402. doi: 10.1038/sj.jid.5700920. [DOI] [PubMed] [Google Scholar]
- Mancini M, et al. Two novel methods for rapid detection and quantification of DNMT3A R882 mutations in acute myeloid leukemia. J Mol Diagn. 2015;17(2):179–184. doi: 10.1016/j.jmoldx.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Parsons BL, Heflich RH. Genotypic selection methods for the direct analysis of point mutations. Mutat Res Rev Muta Res. 1997;387(2):97–121. doi: 10.1016/s1383-5742(97)00026-4. [DOI] [PubMed] [Google Scholar]
- Liu Q, Swiderski P, Sommer SS. Truncated amplification: a method for high-fidelity template-driven nucleic acid amplification. BioTechniques. 2002;33(1):129–139. doi: 10.2144/02331rr04. [DOI] [PubMed] [Google Scholar]
- Kaur M, Zhang Y, Liu WH, Tetradis S, Price BD, Makrigiorgos GM. Ligation of a primer at a mutation: a method to detect low level mutations in DNA. Mutagenesis. 2002;17(5):365–374. doi: 10.1093/mutage/17.5.365. [DOI] [PubMed] [Google Scholar]
- Albitar A, et al. High Sensitivity Testing Shows Multiclonal Mutations in Patients with CLL Treated with BTK Inhibitor and Lack of Mutations in Ibrutinib-Naive Patients. Blood. 2015;126(23):716. [Google Scholar]