

Abstract
Despite all the technological advances at the light microscopy level, electron microscopy remains the only tool in neuroscience to examine and characterize ultrastructural and morphological details of neurons, such as synaptic contacts. Good preservation of brain tissue for electron microscopy can be obtained by rigorous cryo-fixation methods, but these techniques are rather costly and limit the use of immunolabeling, which is crucial to understand the connectivity of identified neuronal systems. Freeze-substitution methods have been developed to allow the combination of cryo-fixation with immunolabeling. However, the reproducibility of these methodological approaches usually relies on costly freezing devices. Moreover, achieving reliable results with this technique is very time-consuming and skill-challenging. Hence, the traditional chemically fixed brain, particularly with acrolein fixative, remains a time-efficient and low-cost method to combine electron microscopy with immunohistochemistry. Here, we provide a reliable experimental protocol using chemical acrolein fixation that leads to the preservation of primate brain tissue and is compatible with pre-embedding immunohistochemistry and transmission electron microscopic examination.
Keywords: Neuroscience, Issue 122, transmission electron microscopy, monkey, neuroanatomy, immunohistochemistry, antibodies, acrolein, transcardiac perfusion, embedding, ultramicrotome
Introduction
Light microscopy, including confocal and two-photon microscopy, has proven to be an efficient tool for studying in vivo neuronal processes, among other things1,2. Although the typical spatial resolution at the Light Microscopic (LM) level is approximately 200 nm, recent technological advances using different light sources, such as extreme ultraviolet and soft X-ray microscopy, have notably increased this resolution to a nearly 10 nm spatial resolution3,4,5. Other technological advances in imaging include combined magnetic resonance imaging with histology and provide a novel method for measuring the thickness of the myelin sheath in vivo, a parameter that was traditionally measurable only at the Electron Microscopic (EM) level6,7. Although these advances at the LM level provide an excellent tool for studying living processes, a detailed view and characterization of structures, such as synaptic contacts, can only be achieved with EM, which offers a resolution that can reach 0.5 nm. However, observation at the EM level requires the specimens to be dead and altered in some ways, with chemical fixatives and dehydration processes, in order to preserve the cytoarchitecture. Thus, examining biological samples at high resolution can be challenging due to radiation damage from the electron beam, low contrast, structural deviations of membranes, or even the presence of artifacts that can occur following dehydration and epoxy embedding8,9,10.
Preserving specimens in their native form for structural analysis can be achieved by using "Cryo EM of Vitrified Sections" or CEMOVIS, a sectioning approach that involves rapidly freezing and embedding the sample in vitreous ice and examining the sections under the EM at a cryogenic temperature11,12. This procedure allows for the examination of samples while they are still solid and fully hydrated, thus eliminating artifacts caused by dehydration processes13. However, this method involves additional devices for cryo-ultramicrotomy, as well as additional devices on the standard EM, in order to allow this observation at very low temperatures, which generate significant additional costs. In addition, the CEMOVIS approach precludes the use of immunolabeling techniques, since antibodies usually have to be incubated at RT. Alternatively, it is possible to combine ultrastructural analysis with immunohistochemical procedures by using a freeze-substitution approach, during which cryo-fixed specimens are slowly thawed while immerged in cryo-protective chemicals and are then embedded in specialized resins, such as Lowicryls. Post-embedding immunolabeling can then be performed on such material12. However, freeze-substitution and cryo-fixation techniques are time consuming. They require the installation of additional equipment and still require samples to be exposed to organic solvent and chemical fixative that can alter the cytoarchitecture, despite the use of a low temperature14,15. Hence, despite all the technological advances both at the LM and EM level, chemical fixation of brain tissue, particularly with acrolein, remains a low-cost and time-efficient method to combine immunohistochemistry with EM16.
In the last decades, many attempts were made to find a mixture of aldehydes that provide the best tissue preservation. Before the 1960s, the only chemical fixative that gave acceptable results for EM was osmium tetroxide. However, osmium tetroxide is highly toxic and expensive, precluding its use through the vascular system to fix organs such as the brain. Acrolein was introduced in the late 1950s as a reliable method for animal tissue preservation suitable for EM observation of cellular structures17. It penetrates the tissue more deeply and reacts more quickly than other aldehydes when used for fixation by immersion and allows good preservation of cytoplasmic components, with minimal shrinkage of the tissue17. Such a feature gives acrolein fixation a clear advantage over other aldehydes when used in fresh tissue, by allowing a more accurate localization of living molecular compounds, such as enzymes and other proteins18. Indeed, it has been validated through the years as an easy, efficient and low-cost method of fixation for visualization at the EM level in many species, including amphibians and rodents, as it efficiently stabilizes peptides and proteins, retains antigenicity and provides relatively intact ultrastructure when used in combination with another aldehyde fixative16,18,19,20,21. Protocols for acrolein fixation in rodents have since then been standardized and used extensively, particularly by the Pickel group, to implement dual immunolabeling for EM16,22. A few groups have used acrolein fixation in non-human primate brain tissue23. However, to our knowledge, there is only one published protocol efficiently describing chemical fixation with acrolein in non-human primates that is compatible with EM immunolabeling24.
In this article, we provide an easy and reliable method to efficiently chemically fix non-human primate brains with acrolein, allowing for a potentially long-term preservation along with pre-embedding immunolabeling and transmission EM examination.
Protocol
Ethics Statement: All protocols involving animals were approved by the Comité de Protection des Animaux de l'Université Laval and were made in accordance with the Canadian Council on Animal Care's Guide to the Care and Use of Experimental Animals (Ed. 2). The protocol described here was optimized for adult animals of approximately 800 g. The volumes of fixative should be adjusted according to the animal's size.
1. Preparation of Solutions for Transcardiac Perfusion
- Prepare 1 L of a 50 mM sodium Phosphate-buffered Saline (PBS) solution according to the following steps. Prepare the solution at most 24 h before perfusion and keep at 4 °C until use.
- Measure 800 mL of distilled water in a 1 L beaker. Add 5.87 g of dibasic anhydrous sodium phosphate (Na2HPO4), 1.20 g of monobasic monohydrate sodium phosphate (NaH2PO4·H2O), and 9 g of sodium chloride (NaCl). Stir to dissolve.
- Adjust the pH to 7.4 by gradually adding 5 N NaOH. Add distilled water to reach a total volume of 1 L. CAUTION: NaOH is a corrosive chemical compound. Wear the appropriate Personal Protective Equipment (PPE; laboratory coat, gloves, protective goggles, etc.).
- Prepare 2 L of 4% paraformaldehyde (PFA) according to the following steps. This solution should be prepared at most 24 h before surgery and kept at 4 °C until use.
- Measure 1.5 L of distilled water in a 2-L beaker. Add 23.48 g of dibasic anhydrous sodium phosphate (Na2HPO4) and 4.80 g of monobasic monohydrate sodium phosphate (NaH2PO4·H2O). Stir to dissolve. Add distilled water to reach a total volume of 2 L.
- Heat the solution under a venting hood until the temperature reaches approximately 45 - 55 °C. Do not heat to over 60 °C.
- Gradually add 80 g of PFA to the solution and stir until completely dissolved (approximately 30 - 60 min). Keep monitoring the temperature to keep it below 60 °C. CAUTION: PFA is highly volatile in its powder form. It is highly toxic if in contact with eyes or skin and is hazardous in case of inhalation or ingestion. Wear PPE and use with caution under a venting hood.
- Cool the solution down to 4 °C and filter. Store at 4 °C.
- Prepare 1 L of 3% acrolein in 0.1 M Phosphate Buffer (PB) according to the following steps. The PB solution should be prepared at most 24 h prior to perfusion. CAUTION: Acrolein is highly toxic if inhaled and can produce immediate damage. It is also corrosive and highly toxic if absorbed through the skin. It is carcinogen and mutagenic. Wear the appropriate PPE and use under a venting hood.
- Measure 800 mL of distilled water. Add 8.66 g of dibasic anhydrous sodium phosphate (Na2HPO4) and 5.38 g of monobasic monohydrate sodium phosphate (NaH2PO4·H2O). Stir until all the salts dissolve. Add distilled water to reach a total volume of 1 L. Keep the solution at 4 °C.
- Before surgery, transfer 900 mL of the PB solution to a 1 L glass container and add 30.94 mL of 97% acrolein solution under a venting hood. Add PB solution to reach a total volume of 1 L and stir. Filter the solution and keep it at 4 °C.
2. Transcardiac Perfusion and Brain Dissection
Keep solutions on ice during the entire surgical procedure. Prepare the pump by placing the tube in the first solution to be used (PBS; 50 mM) and turning the pump on until no air remains in the hose. For the transcardiac perfusion of a macaque monkey, use a 21 G needle and set the outflow at approximately 80 mL/min.
Anesthetize the animal with an intramuscular injection of a mixture of ketamine (20 mg/kg), xylazine (4 mg/kg), and acepromazine (0.5 mg/kg). Maintain the animal under isoflurane (3%) sedation.
Attach the animal's limbs to a venting table.
With a scalpel, remove the skin up to the armpits. Cut the abdominal muscles. Use heavy-duty surgical scissors to cut the ribs laterally by carefully avoiding the vital organs.
Cut the diaphragm with surgical scissors and raise the rib cage to expose the heart. Once the diaphragm is cut, proceed quickly, since the heart will stop beating within minutes.
- Remove the pericardium with a scalpel blade and insert the needle into the left ventricle. With a scalpel, carefully make a small excision to the right atrium. Quickly start the pump at 72 mL/min and hold the needle in place. If possible, tilt the animal in order to have its head on a lower level than the heart. NOTE: Be careful not to pierce the interventricular septum when inserting the needle.
- Rinse the blood with approximately 300 mL of PBS until the lungs are white and no blood comes out of the right atrium.
Stop the pump, quickly transfer the hose/tube to the 3% acrolein solution, and start the pump again. Gradually increase the pumping speed to 80 mL/min once the heart has stopped beating. Perfuse approximately 500 mL of the acrolein solution.
Stop the pump, quickly transfer the hose to the 4% PFA solution, and start the pump again. This step requires approximately 1 L of PFA. NOTE: The fixation is complete when the forelimbs are rigid and the neck is stiff.
Cut the head and carefully dissect the brain out of the skull. Take care to not damage the brain with the surgical instruments. The perfusion is optimal when the brain is pale (no trace of blood) and rigid (Figure 1A).
Immerse the intact brain in 4% PFA for 1 h at 4 °C.
Serially cut the brain with a cooling vibratome (4 °C) in the desired plane into 50 µm thick sections and collect them in PBS (0.1 M) (Figure 1B). NOTE: This step can be carried out in many ways according to the desired protocol. Hemispheres can be separated prior to vibratome cutting, or kept whole. If the brain is too large for the vibratome platform, it can be cut into smaller blocks. After this step, sections can be stored for a long period of time at -30 °C in an antifreeze solution made of 40% PB (50 mM), 30% glycerol, and 30% ethylene glycol.
3. Pre-embedding Immunohistochemistry (Figure 1C)
- Prepare a 4 L stock solution of Tris-buffered Saline (TBS, 50 mM, pH 7.6) as follows.
- Measure 2L of distilled water in a 4 L beaker, add 24.23 g of trihydroxymethyl aminomethane (THAM; C4H11NO3), and stir to dissolve.
- Adjust the pH to 7.6 with approximately 148 mL of 1 N HCl. The acid should be added with caution to avoid reaching a pH below 7.6. The total volume should be 4 L. CAUTION: HCl is highly corrosive. Wear the appropriate PPE.
Select sections (from step 2.11) containing the region of interest to be processed for EM immunohistochemistry.
Wash the free-floating sections 3x in PBS (0.1 M, pH 7.4) for 5 min at RT to rinse the antifreeze solution.
- Prepare a 0.5% solution of NaBH4 diluted in PBS.
- Weigh 0.05 g of NaBH4 and dilute it in 10 mL of PBS. Do not cover. Prepare this solution just before use.
Incubate the sections in the freshly prepared NaBH4 solution for 30 min at RT. Rock gently. Do not cover.
Wash 3x in PBS for 10 min at RT, rocking vigorously until none of the reaction gas remains.
Prepare a blocking solution for EM with 2% appropriate normal serum and 0.5% cold fish gelatin diluted in PBS. NOTE: The quantity should be calculated in order to have enough for the following three steps. Use a serum made from the same animal species hosting the secondary antibody. Avoid the use of antigen retrieval methods or addition of small amounts of triton (to increase the penetration of antibodies), as these methods significantly compromise the quality of the tissue.
Incubate the sections in the blocking solution for 1 h at RT. Rock gently.
Prepare primary antibody solution diluted in the blocking solution. NOTE: The concentration of the primary antibody is usually the same as for LM immunohistochemistry, but consider conducting tests with different antibody concentrations beforehand, as some primary antibodies may not work with acrolein. If so, it is possible to use a mix of glutaraldehyde (0.1 - 2%) and 4% PFA for transcardiac perfusion and obtain similar results. Optimize the incubation time and temperature as well.
Incubate the sections in primary antibody solution overnight at RT with gentle rocking. Use a previously tested incubation time and temperature.
Wash the sections 3x in PBS for 5 min at RT with gentle rocking.
Prepare a 1:1,000 solution of biotinylated secondary antibody diluted in the blocking solution. NOTE: The secondary antibody must be raised against the host species used to generate the primary antibody.
Incubate the sections in secondary antibody solution for 1.5 h at RT. Rock gently.
- Prepare an avidin-biotin-peroxidase (ABC) solution at least 60 min prior to the end of the secondary antibody incubation.
- Use a calibrated pipette to measure 8.80 µL/mL of solutions A and B and dilute them in PBS.
- Rock mildly for at least 60 min at RT to allow the complete binding between the avidin and biotin molecules.
After the incubation in secondary antibody solution, wash 3x in PBS for 10 min at RT.
Incubate the sections in the ABC solution for 1 h at RT. Rock gently.
Wash once in PBS and twice in TBS for 10 min at RT with gentle rocking.
- Prepare a fresh solution of 0.05% 3,3'-diaminobenzidine (DAB) with 0.005% H2O2 diluted in TBS.
- Weigh 12.5 mg of DAB and dilute it in 25 mL of cold TBS. Protect from light. CAUTION: The DAB powder is highly volatile and harmful if inhaled. It is carcinogenic and teratogenic. Thus, pregnant or nursing women should not manipulate this product, even when diluted. Use a N95 mask when manipulating and wear PPE.
- Filter the solution and add 4.5 µL of 30% H2O2 just before use.
Incubate sections in the DAB solution for 3 to 7 min at RT. Rock gently. NOTE: The brown precipitate should not be too dark in order to avoid a high level of background staining. The incubation time should be optimized accordingly.
Stop the reaction by quickly washing twice in cold TBS, then twice for 10 min in cold TBS at RT, followed by twice for 10 min in PB, with mild rocking. NOTE: The use of PB (and not PBS) is critical in order to eliminate any traces of NaCl, as it would react with osmium and form crystals.
4. Osmification and Embedding for Electron Microscopic Observation
Prepare a solution of 1% osmium tetroxide (OsO4) diluted in PB. Protect from light. CAUTION: Osmium is highly toxic and should not come in contact with the skin, eyes, or mouth and should not be inhaled. It can cause death if ingested. It should only be used under a venting hood and with appropriate PPE.
Incubate the sections in OsO4 solution for 30 min at RT under the venting hood (without agitation) and cover them with aluminum foil to protect them from light. Completely flatten the sections prior to adding the OsO4 solution. NOTE: The sections become very dark and rigid and should be manipulated with care following this step.
- Prepare water-repellent epoxy resin during the osmification.
- Add appropriate amounts of each component of the epoxy resin mix (20 g of epoxy resin, 20 g of hardener, 0.6 g of accelerator and 0.4 g of plasticizer) to a large plastic cup. Stir with a wooden stick or plastic pipette until a homogeneous brown color is obtained. NOTE: It is critical to use the exact proportion of each component.
- Transfer equal quantities to aluminum cups of appropriate sizes, depending on the number of sections to be processed. Allow it to rest.
Wash the osmificated sections 3x in PB for 10 min at RT with low-speed rocking.
Dehydrate the sections in the following series of graded ethanol for 2 min each: 2 times in 35% ethanol; 1 time each in 50, 70, 80, 90, and 95% ethanol; and 3x in 100% ethanol.
Transfer the sections to glass vials to complete the dehydration process by incubating the sections 3 times for 2 min in propylene oxide. CAUTION: Propylene oxide is a highly volatile and toxic organic solvent. It can cause serious damage in the eyes or skin in case of contact or if inhaled or ingested. It has been classified as a grade 2 carcinogenic substance. It should only be used under a venting hood and with PPE. It is also highly flammable and should be kept away from any heat source. NOTE: Prior to this step, sections should be carefully transferred in glass vials, since propylene oxide is an organic solvent and is incompatible with plastic. At this point, sections are very fragile and should be manipulated with care. Sections can be alternatively transferred to glass vials prior to step 4.5.
Transfer sections carefully, one by one, in the aluminum cups and avoid contact with air as much as possible. Flat-embed the sections in previously mixed water-repellent epoxy resin and incubate them overnight under the venting hood at RT. NOTE: At this step, the sections are completely dehydrated and very fragile and should be manipulated with care.
Using mineral oil, prepare grease-coated glass slides along with greased plastic coverslips.
Soften the resin by incubating aluminum cups at 60 °C for 12-15 min at most. Carefully flatten the sections on the greased side of the glass slide. Place the greased coverslip and carefully push out any remaining air.
Incubate the slides at 60 °C for 48 h. NOTE: It is critical to not exceed 48 h incubation time, as the resin will become too hard.
Remove the plastic coverslip.
5. Sample Preparation for Ultrathin Sectioning and Observation Using a Transmission Electron Microscope
Use binoculars to find the region of interest and cut a small quadrangular piece of approximately 1 mm2 with a scalpel.
File the tip of a resin block and glue the quadrangular piece onto it (Figure 1C). Allow the glue to dry for at least 1 h or overnight prior to sectioning.
- Using an ultramicrotome, cut the quadrangular piece into 80 µm-thick sections (Figure 1D).
- Place the resin block in an ultramicrotome deck in the vertical position and, using a sharp razor blade, gradually cut each side of the resin block to form a trapezoid with smooth sides.
- Put the deck in its horizontal position and rotate the block until the longest side of the trapezoid is facing downward.
- Use a diamond trimming tool or a glass knife to trim the surface of the quadrangular piece. Set the ultramicrotome to cut 300 µm thick sections at 1 mm/s. Adjust the knife to be vertically parallel to the quadrangular piece and to display a very small horizontal angle of approximately 1°. NOTE: This angle will allow the user to approach the tissue almost parallel to the surface of the block, where immunolabeled elements are more likely to be found. When using these parameters with the diamond trimming tool, the resin displays a shiny white color. When the tissue is being cut, it changes to a purple or greenish color.
- Use an ultra 45° diamond knife equipped with a boat filled with distilled water to cut 80 µm thick sections, smoothen sections by passing over them with a piece of absorbing paper tipped in xylene, and collect serial sections on formvar-coated nickel slot grids or bare 150 mesh copper grids (Figure 1E).
Place the grids in a grid storage box.
- Stain the grids with lead citrate.
- Use a 5 mL syringe and a 0.2 µm syringe filter to prepare a 1:1 solution of filtered lead citrate stock solution and filtered distilled water. Protect it from light. NOTE: The stock solution of lead citrate should be made fresh every month to avoid the formation of solid deposits. See the material data sheet for the stock recipe. In addition, if many series of grids need to be stained, change the diluted solution when it becomes milky.
- Place each grid onto a drop of the diluted solution, with the section in contact with the solution. Incubate for 3 min.
- Use small tweezers to hold the grid, and thoroughly rinse it in two beakers containing distilled water.
- Remove excess water by gently using absorbing paper. Store the grids in a grid box. Wait 30 min before examining the sections by Transmission Electron Microscopy (TEM; Figure 1F).
Representative Results
In this section, we present representative results that were obtained following the observation, at the transmission EM level, of immunostained primate brain tissue chemically fixed with a mixture of 3% acrolein and 4% PFA. We achieved good preservation of the ultrastructure, as indicated by the relatively intact myelin sheath and the neat visualization of double membranes (Figure 2A). Synaptic contacts, along with neuronal elements from the microenvironment, can easily be identified (Figure 2B). Neuronal elements labeled with diaminobenzidine (DAB) immunoprecipitate are recognized at the EM level by their filled cytoplasm or axoplasm. The plasma membrane and the outer surface of organelles are also typically lined with the electron-dense precipitate (Figure 3).
In this particular experiment, we used antibodies against the serotonin transporter (SERT), choline acetyltransferase (ChAT), or Tyrosine Hydroxylase (TH) to visualize immunolabeled neuronal elements in the external (GPe) or internal (GPi) segment of the squirrel monkey globus pallidus (Figure 3). In order to do so, we used a combination of fixative chemicals that preserve antigenicity as well as ultrastructure, allowing a detailed morphological investigation. Although many antibodies can be used with the transcardiac perfusion protocol described above, we recommend that users perform optimization concentration tests beforehand, since some primary antibodies are known to not provide optimal immunolabeling with acrolein fixation. Alternatively, when antibodies do not provide optimal immunolabeling with acrolein fixation, a dilution of 0.1 - 2% glutaraldehyde in 4% PFA can be used for transcardiac perfusion. It provides tissue quality relatively equivalent to acrolein-fixed brain tissue with preserved antigenicity for many antibodies (Figure 4).
Finally, we provide typical examples of EM photomicrographs obtained following inappropriate manipulations. A poor fixation results in altered myelin sheaths (Figure 5A, B) and difficulties in visualization of the double membranes of neurites (Figure 5C, D), preventing a reliable identification and analysis of labeled and unlabeled neuronal elements. An excessive incubation time in the DAB solution creates excessive background and non-specific staining that can potentially generate false-positive results. Background or non-specific staining sometimes appears as incomplete staining of neuronal elements (Figure 6A, B), but more often as numerous and closely located stained neuronal elements (Figure 6C, D). The use of detergent in the blocking solution significantly alters the quality of the tissue. It may lead to missing organelles in labeled elements (Figure 7A) or degradation of myelin sheaths (Figure 7B) and cellular membranes (Figure 7C), rendering any acute interpretation of the microenvironment difficult. Finally, a misstep in the osmification process, such as using rinsing solution containing sodium chloride, produces unreliable results where the cellular structures are difficult to distinguish (Figure 7D).
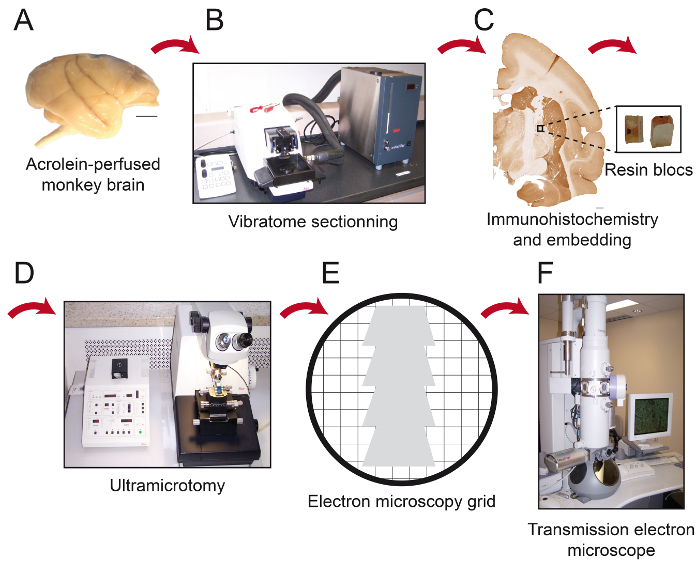 Figure 1: Schematics of Essential Steps of the Protocol. The monkey brain (A) is cut into serial sections with a cooling vibratome (B). It is then processed for pre-embedding immunohistochemistry and electron microscopy, after which the region of interest is placed on the tip of a resin block (C) and cut into 80 nm thick sections with an ultramicrotome (D). Ultrathin sections are then collected on bare 150 mesh copper grids or formvar-coated nickel grids (E), stained with lead citrate and ready to be observed under transmission electron microscope (F). Scale bars: 1 mm. Please click here to view a larger version of this figure.
Figure 1: Schematics of Essential Steps of the Protocol. The monkey brain (A) is cut into serial sections with a cooling vibratome (B). It is then processed for pre-embedding immunohistochemistry and electron microscopy, after which the region of interest is placed on the tip of a resin block (C) and cut into 80 nm thick sections with an ultramicrotome (D). Ultrathin sections are then collected on bare 150 mesh copper grids or formvar-coated nickel grids (E), stained with lead citrate and ready to be observed under transmission electron microscope (F). Scale bars: 1 mm. Please click here to view a larger version of this figure.
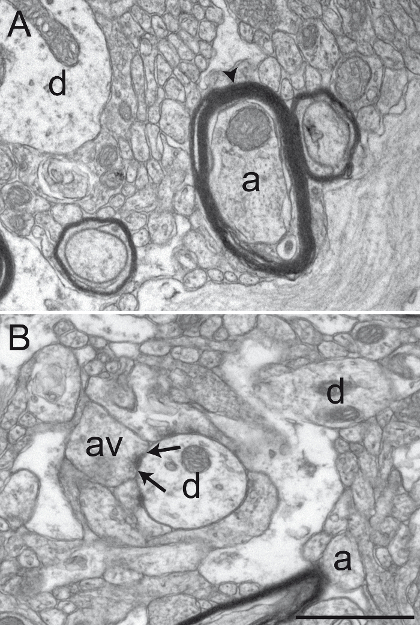 Figure 2: Well-preserved Primate Brain Tissue after Acrolein-PFA Transcardiac Perfusion. Electron micrographs of squirrel monkey (Saimiri sciureus) brain tissue of the GPi (A) and GPe (B) showing representative well-preserved material after performing the acrolein-PFA transcardiac perfusion and the immunoperoxidase-diaminobenzidine technique. The myelin sheath of axons (a) is relatively intact (see arrowhead in A), and the general ultrastructure is well preserved in A and B. Dendritic profiles (d), small unmyelinated axons (a), and axon varicosities (av) can easily be identified. An example of an axon varicosity establishing a symmetrical synaptic contact (between arrows) with a dendritic profile (d) is shown in B. Scale bar: 1 µm. Please click here to view a larger version of this figure.
Figure 2: Well-preserved Primate Brain Tissue after Acrolein-PFA Transcardiac Perfusion. Electron micrographs of squirrel monkey (Saimiri sciureus) brain tissue of the GPi (A) and GPe (B) showing representative well-preserved material after performing the acrolein-PFA transcardiac perfusion and the immunoperoxidase-diaminobenzidine technique. The myelin sheath of axons (a) is relatively intact (see arrowhead in A), and the general ultrastructure is well preserved in A and B. Dendritic profiles (d), small unmyelinated axons (a), and axon varicosities (av) can easily be identified. An example of an axon varicosity establishing a symmetrical synaptic contact (between arrows) with a dendritic profile (d) is shown in B. Scale bar: 1 µm. Please click here to view a larger version of this figure.
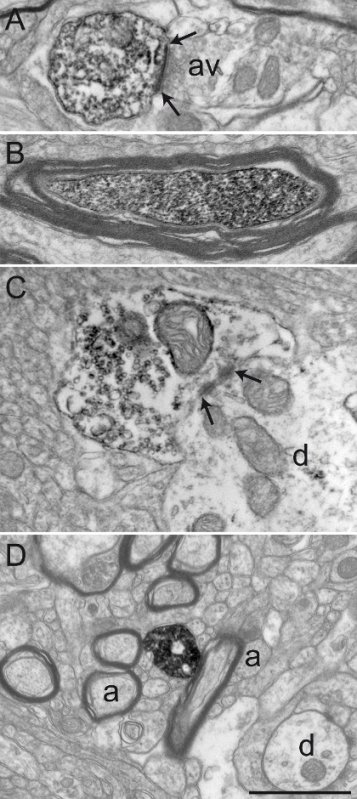 Figure 3: Sections from Squirrel Monkey GPe and GPi Immunolabeled for Choline Acetyltransferase (ChAT), Serotonin Transporter (SERT), and Tyrosine Hydroxylase (TH) by using the Immunoperoxidase-diaminobenzidine Technique. Immunolabeled elements can easily be identified by their cytoplasm or axoplasm filled with electron-dense DAB precipitate. Labeled dendritic profiles are recognized by filled microfilaments, as seen in A, where a ChAT-immunostained dendrite in the GPi receives a synaptic contact (between arrows) from an unlabeled axon varicosity (av). The electron micrograph in B shows a myelinated axon in the GPe with a relatively intact myelin sheath whose axoplasm is immunolabeled for TH. The example in C shows an axon varicosity in the GPi immunolabeled for SERT and is seen to establish a symmetrical synaptic contact (between arrows) with a dendrite (d). In this example, the DAB precipitate lines the plasma membrane and the outer surface of organelles (mitochondrion and synaptic vesicles). The axon varicosity shown in D was observed in the GPe and immunolabeled for TH and represents an example of DAB precipitate entirely filling the axoplasm, with synaptic vesicles being visible but more difficult to delineate. Elements of the microenvironment can easily be identified, as exemplified by myelinated and unmyelinated axons (a) and occasional dendrites (d) surrounding the labeled axon varicosity. Electron micrographs are modified from 25,26,27. Scale bar: 1 µm. Please click here to view a larger version of this figure.
Figure 3: Sections from Squirrel Monkey GPe and GPi Immunolabeled for Choline Acetyltransferase (ChAT), Serotonin Transporter (SERT), and Tyrosine Hydroxylase (TH) by using the Immunoperoxidase-diaminobenzidine Technique. Immunolabeled elements can easily be identified by their cytoplasm or axoplasm filled with electron-dense DAB precipitate. Labeled dendritic profiles are recognized by filled microfilaments, as seen in A, where a ChAT-immunostained dendrite in the GPi receives a synaptic contact (between arrows) from an unlabeled axon varicosity (av). The electron micrograph in B shows a myelinated axon in the GPe with a relatively intact myelin sheath whose axoplasm is immunolabeled for TH. The example in C shows an axon varicosity in the GPi immunolabeled for SERT and is seen to establish a symmetrical synaptic contact (between arrows) with a dendrite (d). In this example, the DAB precipitate lines the plasma membrane and the outer surface of organelles (mitochondrion and synaptic vesicles). The axon varicosity shown in D was observed in the GPe and immunolabeled for TH and represents an example of DAB precipitate entirely filling the axoplasm, with synaptic vesicles being visible but more difficult to delineate. Elements of the microenvironment can easily be identified, as exemplified by myelinated and unmyelinated axons (a) and occasional dendrites (d) surrounding the labeled axon varicosity. Electron micrographs are modified from 25,26,27. Scale bar: 1 µm. Please click here to view a larger version of this figure.
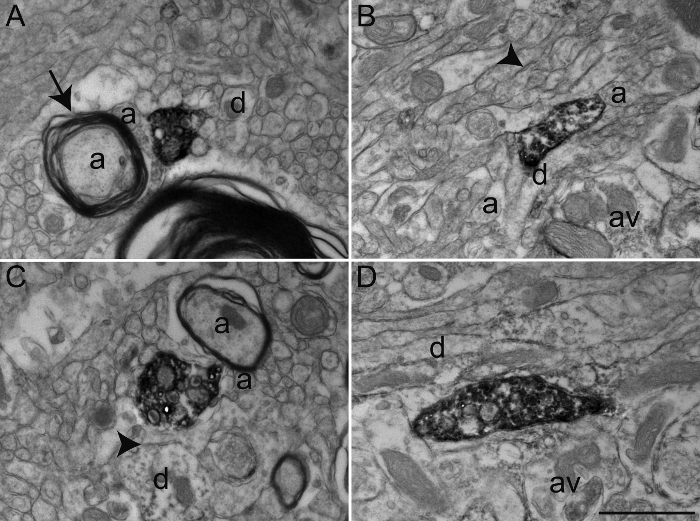 Figure 4: Examples of Primate Brain Tissue after Glutaraldehyde-PFA Transcardiac Perfusion. Representative electron micrographs of macaque monkey (Macaca fascicularis) brain tissue of the GPe (A) and GPi (B-D) after performing transcardiac perfusion with 0.2% glutaraldehyde mixed with 4% PFA and immunoperoxidase-diaminobenzidine technique with an antibody against the serotonin transporter (SERT). As in Figure 2, immunolabeled elements can be identified by their cytoplasm and axoplasm filled with electron-dense DAB precipitate. General ultrastructure is relatively intact and elements of the microenvironment can easily be identified, as shown by myelinated and unmyelinated axons (a) and occasional dendrites (d) and axon varicosities (av) surrounding the labeled axon varicosities, as described in Figure 2. However, note the inconsistency in the quality of ultrastructure, denoted by well-defined plasma membranes (arrowheads), but relatively damaged myelin sheath (arrow). Scale bar: 1 µm. Please click here to view a larger version of this figure.
Figure 4: Examples of Primate Brain Tissue after Glutaraldehyde-PFA Transcardiac Perfusion. Representative electron micrographs of macaque monkey (Macaca fascicularis) brain tissue of the GPe (A) and GPi (B-D) after performing transcardiac perfusion with 0.2% glutaraldehyde mixed with 4% PFA and immunoperoxidase-diaminobenzidine technique with an antibody against the serotonin transporter (SERT). As in Figure 2, immunolabeled elements can be identified by their cytoplasm and axoplasm filled with electron-dense DAB precipitate. General ultrastructure is relatively intact and elements of the microenvironment can easily be identified, as shown by myelinated and unmyelinated axons (a) and occasional dendrites (d) and axon varicosities (av) surrounding the labeled axon varicosities, as described in Figure 2. However, note the inconsistency in the quality of ultrastructure, denoted by well-defined plasma membranes (arrowheads), but relatively damaged myelin sheath (arrow). Scale bar: 1 µm. Please click here to view a larger version of this figure.
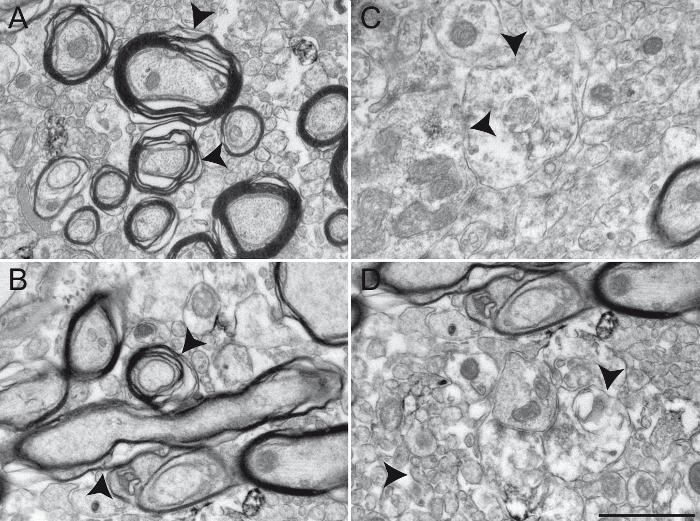 Figure 5: Examples of Primate Brain Tissue Obtained following an Unsuccessful Transcardiac Perfusion. Results from an unsuccessful chemical fixation are shown here in the squirrel monkey GPi (A - B) and GPe (C - D) transcardially perfused with 0.9% NaCl rinsing solution and a mixture of ice-cold 4% PFA and 15% picric acid diluted in 0.1 M PB (pH 7.4). Brains were post-fixed 1 h at 4 °C in 4% PFA and 30% sucrose and cut into 60 µm thick sagittal sections with a cooling vibratome. Inappropriately fixed brain tissue can be recognized by a damaged myelin sheath (A and B, arrowheads), as well as by blurry or undefined plasma membranes (C and D, see arrowheads for examples). Different neuronal elements are difficult to identify, rendering any interpretation of the ultrastructure unreliable. Scale bar: 1 µm. Please click here to view a larger version of this figure.
Figure 5: Examples of Primate Brain Tissue Obtained following an Unsuccessful Transcardiac Perfusion. Results from an unsuccessful chemical fixation are shown here in the squirrel monkey GPi (A - B) and GPe (C - D) transcardially perfused with 0.9% NaCl rinsing solution and a mixture of ice-cold 4% PFA and 15% picric acid diluted in 0.1 M PB (pH 7.4). Brains were post-fixed 1 h at 4 °C in 4% PFA and 30% sucrose and cut into 60 µm thick sagittal sections with a cooling vibratome. Inappropriately fixed brain tissue can be recognized by a damaged myelin sheath (A and B, arrowheads), as well as by blurry or undefined plasma membranes (C and D, see arrowheads for examples). Different neuronal elements are difficult to identify, rendering any interpretation of the ultrastructure unreliable. Scale bar: 1 µm. Please click here to view a larger version of this figure.
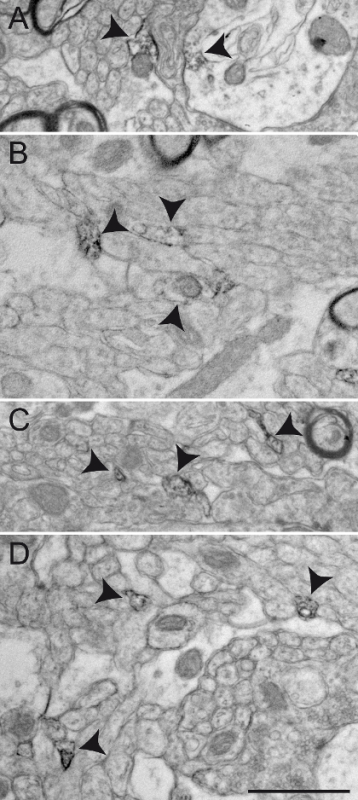 Figure 6: Examples of Non-specific ChAT Immunolabeling in the Squirrel Monkey GPe. Background staining or non-specific immunolabeling sometimes appears under the EM as partial staining of large cellular elements, as illustrated in A - B (arrowheads). Such unspecific staining appears more often at the surface of immunostained sections. Other examples of non-specific immunolabeling include the frequent observation of very small elements evoking small, unmyelinated axons partially or completely filled with DAB and located very close to one another, as demonstrated by arrowheads in C and D. Scale bar: 1 µm. Please click here to view a larger version of this figure.
Figure 6: Examples of Non-specific ChAT Immunolabeling in the Squirrel Monkey GPe. Background staining or non-specific immunolabeling sometimes appears under the EM as partial staining of large cellular elements, as illustrated in A - B (arrowheads). Such unspecific staining appears more often at the surface of immunostained sections. Other examples of non-specific immunolabeling include the frequent observation of very small elements evoking small, unmyelinated axons partially or completely filled with DAB and located very close to one another, as demonstrated by arrowheads in C and D. Scale bar: 1 µm. Please click here to view a larger version of this figure.
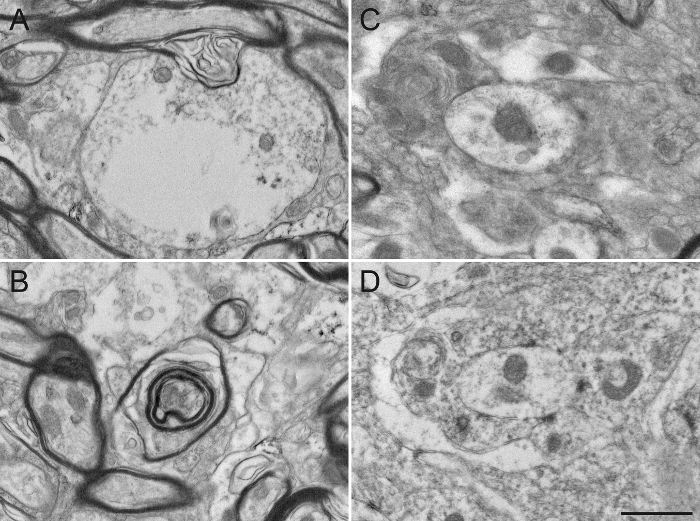 Figure 7: Damaged Squirrel Monkey GPe Tissue after Missteps in Sample Preparation for Electron Microscopy. The use of detergent, such as Triton X-100, in the blocking solution, even at a low concentration of 0.02%, substantially alters the integrity of the ultrastructure by damaging the cytoplasm of dendrites (A) or the myelin sheath of axons (B). The different neuronal elements are also difficult to distinguish from one another (C), since the plasma membranes are damaged and difficult to delineate. The osmification process is also an important step in sample preparation. The use of sodium chloride in rinsing solutions (D) alters the fixation of the tissue, rendering subsequent analysis difficult. Scale bar: 800 nm. Please click here to view a larger version of this figure.
Figure 7: Damaged Squirrel Monkey GPe Tissue after Missteps in Sample Preparation for Electron Microscopy. The use of detergent, such as Triton X-100, in the blocking solution, even at a low concentration of 0.02%, substantially alters the integrity of the ultrastructure by damaging the cytoplasm of dendrites (A) or the myelin sheath of axons (B). The different neuronal elements are also difficult to distinguish from one another (C), since the plasma membranes are damaged and difficult to delineate. The osmification process is also an important step in sample preparation. The use of sodium chloride in rinsing solutions (D) alters the fixation of the tissue, rendering subsequent analysis difficult. Scale bar: 800 nm. Please click here to view a larger version of this figure.
Discussion
In this article, we present a reliable protocol for transcardiac perfusion of non-human primates and pre-embedding immunohistochemistry suitable for EM sample examination. Although typical cryo-EM, such as CEMOVIS, provides a good preservation of brain ultrastructure, it also limits the use of immunohistochemistry12. Other techniques, including cryo-substitution and Tokuyaso technique, allow post-embedding immunohistochemistry, but these techniques are expensive due to additional devices needed during the process and can be time-consuming and skill-challenging12,14,15. Moreover, in order to efficiently use the cryo-fixation method, the sample has to be relatively small (up to 200 µm in thickness when using high-pressure freezing28 and 10 µm at atmospheric pressure29). Ideally, to get good results with cryo-fixation of primate brain tissue, the sample has to be taken from a biopsy, causing problems in finding the exact location of the region of interest. This problem must be circumvented by using stereotaxic coordinates. The chemical fixation with acrolein and pre-embedding technique proposed above provides an easy, low-cost, time-efficient, and reliable method for sample preparation of primate brain tissue and immunolabeling for EM. By following these steps, one will obtain a well-preserved ultrastructure along with antigenicity to allow for the immunolabeling of most proteins. However, chemical fixation for EM also has its disadvantages. First, while fixative solutions such as acrolein preserve the morphological details of the brain tissue, it is possible that some morphological changes occur during the chemical fixation process and alter the results compared to those that would be obtained with CEMOVIS or cryo-substitution techniques. Second, the fixation process and subsequent dehydration needed for resin embedding remove most of the extracellular fluids and squeeze cellular components together, causing shrinkage of tissue that significantly modifies their size and shape compared to cryo-fixed cells21,30. Nonetheless, aldehyde fixation has been used successfully in many laboratories around the world and is widely accepted in the literature as a reliable method for studying the ultrastructural features of neurons and glial cells, despite previously mentioned concerns21,30,31,32.
By comparison with the method described above, the postembedding immunohistochemistry that is needed for cryo-fixed samples embedded in methacrylate resin, is less sensitive and the detection of central nervous system antigens is limited14,33. However, aldehyde fixation also has its limitations in regards to antigenicity. Therefore, it is important to test the specificity of the antibodies for a given chemical fixation protocol at the LM level before starting the EM preparation. The quality of immunohistochemistry on acrolein-fixed brain tissue also depends on previously breaking the strong aldehyde bonds created by the chemical fixation. This step can be achieved by incubating the sections before immunohistochemistry with sodium borohydride (see steps 3.3 - 3.5). Omitting this step would definitely result in suboptimal immunostaining34. If the antibodies to be used do not give optimal staining on brain sections fixed with acrolein, it is possible to alternatively use glutaraldehyde (0.1 - 2%) diluted in 4% PFA. This has been proven to well-preserve brain ultrastructure while sufficiently maintaining antigenicity for many antibodies and to provide brain tissue suitable for long-term storage with minimal alterations20,21,35,36,37. It is also possible to achieve relatively good fixation and antigenicity suitable for EM with PFA alone by significantly elevating the pH of the solution, but a combination of more than one fixative during perfusion has provided better results34, and studies support that PFA fixation alone generally produce poorly preserved tissue for EM examination38,39. However, in some rare cases, antigenicity is very difficult to maintain, and PFA fixation alone remains the only viable option for EM examination.
Many steps in this protocol should be followed carefully in order to obtain the optimal results. For instance, the preparation of the 4% PFA solution must be performed at temperatures above 45 °C in order to allow the PFA powder to dissolve, but it is imperative that the temperature remains below 60 °C. Otherwise, the PFA solution depolymerizes into formaldehyde and formic acid, which fixes the tissue differently and forms an acidic solution that could significantly alter tissue quality40,41. Additionally, it is critical that the perfusion steps be carried out rapidly once the diaphragm has been cut, since hypoxia and hypercapnia will produce irreversible physiological changes to the brain that could alter the quality and integrity of the tissue31. Defective fixation could be detrimental to the preservation of brain tissue and permanently alter the ultrastructure of the tissue, such as plasma membranes, mitochondria and synapses. Thus, solution preparation and perfusion steps need to be carried out with care. Successful EM sample preparation also highly depends upon a good post-fixation in osmium tetroxide. Indeed, direct perfusion with osmium tetroxide has been described as producing reasonably intact tissue for EM observation21,39. However, many differences in terms of the amount of the extracellular space and the appearance of the plasma membrane between osmium-perfused and aldehyde-perfused animals have been noticed. Furthermore, the blackening and hardening of the tissues after osmium perfusion rendered the removal of the brain from the skull, the subsequent dissection, and the differentiation between white and grey matter more difficult, favoring aldehyde perfusion for better tissue preservation and easier manipulation21. Aldehyde fixation alone appears to reduce the extracellular space, which alters the integrity of the tissue and gives the impression of tight junctions where extracellular space should be seen, thus failing to show acceptable electron micrographs even after lead citrate staining42. However, postfixation in osmium tetroxide reversed this situation by allowing a most definite separation between tissue elements, which is necessary for their identification42, thus clearly justifying the importance of carrying out the osmification steps (steps 4.1 - 4.2) with caution.
The conditions and concentrations under which these steps are carried have been tested in our laboratory, specifically for pre-embedding immunohistochemistry using DAB as the precipitate on primate brain sections. Although skill-challenging, double-immunohistochemistry is possible through post-embedding immunohistochemistry with gold particles43. The electron-dense DAB precipitate obtained after the immunoperoxidase-diaminobenzidine technique proposed here is very easy to identify at the EM level, since it outlines plasma membranes and outer surfaces of organelles, while still allowing the identification of sub-cellular components, such as mitochondria and synaptic contacts. Additionally, once the present technique is mastered for single immunohistochemistry, it is possible to perform double immunohistochemistry by combining DAB precipitate to gold particles, which are more easily discerned from one another than gold particles of different sizes. We would recommend doing rigorous testing of antibody specificity, osmium concentrations, and incubation time and temperature before processing brain sections for EM. Protocols for pre-embedding and double immunohistochemistry for EM have been previously published and can be adapted for primate brain tissue16.
In conclusion, the transcardiac perfusion protocol for non-human primates presented above allows for the long-term storage of brain sections that can subsequently be used for EM pre-embedding immunohistochemistry. Sections obtained are also suitable for neuroanatomical studies at the LM level. Hence, by using a protocol that is suitable for both EM and LM, it is possible to reduce the number of animals used, a cost-efficient and ethical consideration. It also allows a direct comparison between results obtained at the LM and EM levels on the same animal and allows the use of Correlative Light and Electron Microscopic (CLEM) studies. CLEM studies have mostly focused on the use of genetically engineered mice expressing fluorescent proteins, such as EGFP, which could later be labeled with electron-dense precipitate and observed at the EM level44. Alternatively, to circumvent the problem of immunohistochemistry, cryo-fixation can be combined with an intraneuronal injection of electron-dense markers, such as quantum dots45, prior to freezing, allowing visualization at the EM level46. However, quantum dots are not biocompatible, and their use is costly and time-consuming due to the additional freezing apparatus needed for subsequent tissue preparation. Although these techniques still favor cryo-fixation and freeze-substitution methods, there are some developments for the use of chemical tags on semi-thin (300 nm) sections to label different cellular proteins with synthetic fluorophores, which can be correlated with EM analysis47. Techniques allowing, for instance, the correlation between one specific axon localization using fluorescence and its synaptic relationship with neurons of a given target structure using EM, are rather promising for the understanding of the organization of the nervous system, specifically in primates. In this case, acrolein fixation should be favored over glutaraldehyde, as the latter will produce autofluorescence that could alter the proper visualization of brain structures at the LM level. However, very few studies of this type have been undertaken in primates, mostly due to technical challenges and the high cost that such experiments impose. Thus, new developments are needed for successful and low-cost CLEM studies in primates, such as improving fixation methods or using chemical tags visible at both the LM and EM levels.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This study was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC, 401848-2011 to M.P.). M.P received a career award from the Fonds de recherche du Québec-Santé (FRQ-S). L.E. was the recipient of a doctoral fellowship from the FRQ-S (FRQ-S 14D 29441). We thank Marie-Josée Wallman for technical assistance.
References
- Pozzi P, Gandolfi D, et al. High-throughput spatial light modulation two-photon microscopy for fast functional imaging. Neurophotonics. 2015;2(1):015005. doi: 10.1117/1.NPh.2.1.015005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, et al. A comparison study of detecting gold nanorods in living cells with confocal reflectance microscopy and two-photon fluorescence microscopy. J. Microsc. 2010;237(2):200–207. doi: 10.1111/j.1365-2818.2009.03324.x. [DOI] [PubMed] [Google Scholar]
- Chao W, Kim J, Rekawa S, Fischer P, Anderson EH. Demonstration of 12 nm resolution Fresnel zone plate lens based soft X-ray microscopy. Opt. Express. 2009;17(20):17669–17677. doi: 10.1364/OE.17.017669. [DOI] [PubMed] [Google Scholar]
- Wachulak P, Bartnik A, Fiedorowicz H. A 50 nm spatial resolution EUV imaging-resolution dependence on object thickness and illumination bandwidth. Opt Express. 2011. [DOI] [PubMed]
- Wachulak P, et al. A compact "water window" microscope with 60 nm spatial resolution for applications in biology and nanotechnology. Microsc. Microanal. 2015. pp. 1–10. [DOI] [PubMed]
- Stikov N, et al. In vivo histology of the myelin g-ratio with magnetic resonance imaging. Neuroimage. 2015;118:397–405. doi: 10.1016/j.neuroimage.2015.05.023. [DOI] [PubMed] [Google Scholar]
- Stikov N, et al. Quantitative analysis of the myelin g-ratio from electron microscopy images of the macaque corpus callosum. Data Brief. 2015;4:368–373. doi: 10.1016/j.dib.2015.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollenhauer HH. Artifacts caused by dehydration and epoxy embedding in transmission electron microscopy. Microsc. Res. Tech. 1993;26(6):496–512. doi: 10.1002/jemt.1070260604. [DOI] [PubMed] [Google Scholar]
- Henderson R. Realizing the potential of electron cryo-microscopy. Q. Rev. Biophys. 2004;37(1):3–13. doi: 10.1017/s0033583504003920. [DOI] [PubMed] [Google Scholar]
- Sander B, Golas MM. Visualization of bionanostructures using transmission electron microscopical techniques. Microsc. Res. Tech. 2011;74(7):642–663. doi: 10.1002/jemt.20963. [DOI] [PubMed] [Google Scholar]
- Ren G, Rudenko G, Ludtke SJ, Deisenhofer J, Chiu W, Pownall HJ. Model of human low-density lipoprotein and bound receptor based on cryoEM. Proc. Natl. Acad. Sci. U.S.A. 2010;107(3):1059–1064. doi: 10.1073/pnas.0908004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster P, Schwarz H, Griffiths G. Preparation of cells and tissues for immuno EM. Methods Cell Biol. 2008;88:45–58. doi: 10.1016/S0091-679X(08)00403-2. [DOI] [PubMed] [Google Scholar]
- Kürner J, Medalia O, Linaroudis AA, Baumeister W. New insights into the structural organization of eukaryotic and prokaryotic cytoskeletons using cryo-electron tomography. Exp. Cell Res. 2004;301(1):38–42. doi: 10.1016/j.yexcr.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Humbel BM. Freeze-substitution. Handbook of cryo-preparation methods for electron microscopy. 2009;13:319–341. [Google Scholar]
- Stierhof Y, Humbel BM, van Donselaar E, Schwarz H. Cryo-fixation, freeze-substitution rehydration and Tokuyaso cryo-sectioning. Handbook of cryo-preparation methods for electron microscopy. 2009;14:344–365. [Google Scholar]
- Leranth C, Pickel MV. Electron microscopic pre-embedding double-immunohistochemical methods. Neuroanatomical tract-tracing methods 2. 1989. pp. 129–172.
- Luft JH. The use of acrolein as a fixative for light and electron microscopy. Anat. Rec. 1959;133:305–305. [Google Scholar]
- Saito T, Keino H. Acrolein as a fixative for enzyme cytochemistry. J. Histochem. Cytochem. 1976;24(12):1258–1269. doi: 10.1177/24.12.187691. [DOI] [PubMed] [Google Scholar]
- King JC, Lechan RM, Kugel G, Anthony EL. Acrolein: a fixative for immunocytochemical localization of peptides in the central nervous system. J. Histochem. Cytochem. 1983;31(1):62–68. doi: 10.1177/31.1.6187805. [DOI] [PubMed] [Google Scholar]
- Sabatini DD, Bensch K, Barrnett RJ. Cytochemistry and electron microscopy. The preservation of cellular ultrastructure and enzymatic activity by aldehyde fixation. J. Cell Biol. 1963;17:19–58. doi: 10.1083/jcb.17.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson U, Schultz R. Fixation of the central nervous system for electron microscopy by aldehyde perfusion: I. Preservation with aldehyde perfusates versus direct perfusion with osmium tetroxide with special reference to membranes and the extracellular space. J. Ultrastruct. Res. 1965;12:160–186. doi: 10.1016/s0022-5320(65)80014-4. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Pickel VM. Dual ultrastructural localization of enkephalin and tyrosine hydroxylase immunoreactivity in the rat ventral tegmental area: multiple substrates for opiate-dopamine interactions. J. Neurosci. 1992;12(4):1335–1350. doi: 10.1523/JNEUROSCI.12-04-01335.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathai A, Ma Y, Paré J, Villalba RM, Wichmann T, Smith Y. Reduced cortical innervation of the subthalamic nucleus in MPTP-treated parkinsonian monkeys. Brain. 2015;138(4):946–962. doi: 10.1093/brain/awv018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba RM, Paré J, Smith Y. Three-dimensional electron microscopy imaging of spines in non-human primates. Transmission electron microscopy methods for understanding the brain. 2016;115:81–103. [Google Scholar]
- Eid L, Champigny M, Parent A, Parent M. Quantitative and ultrastructural study of serotonin innervation of the globus pallidus in squirrel monkeys. Eur. J. Neurosci. 2013;37(10):1659–1668. doi: 10.1111/ejn.12164. [DOI] [PubMed] [Google Scholar]
- Eid L, Parent A, Parent M. Asynaptic feature and heterogeneous distribution of the cholinergic innervation of the globus pallidus in primates. Brain Struct. Funct. 2016;221:1139–1155. doi: 10.1007/s00429-014-0960-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid L, Parent M. Morphological evidence for dopamine interactions with pallidal neurons in primates. Front. Neuroanat. 2015;9:111–114. doi: 10.3389/fnana.2015.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald K. High-pressure freezing for preservation of high resolution fine structure and antigenicity for immunolabeling. Methods Mol. Biol. 1999;117:77–97. doi: 10.1385/1-59259-201-5:77. [DOI] [PubMed] [Google Scholar]
- Gilkey J, Staehelin L. Advances in ultrarapid freezing for the preservation of cellular ultrastructure. J. Electron Microsc. Tech. 1986;3:177–210. [Google Scholar]
- Korogod N, Petersen CCH, Knott GW. Ultrastructural analysis of adult mouse neocortex comparing aldehyde perfusion with cryo fixation. eLife. 2015;4 doi: 10.7554/eLife.05793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson U, Schultz R. Fixation of the central nervous system for electron microscopy by aldehyde perfusion: III. Structural changes after exsanguination and delayed perfusion. J. Ultrastruct. Res. 1966;14:47–63. [Google Scholar]
- Schultz RL, Maynard EA, Pease DC. Electron microscopy of neurons and neuroglia of cerebral cortex and corpus callosum. Am. J. Anat. 1957;100(3):369–407. doi: 10.1002/aja.1001000305. [DOI] [PubMed] [Google Scholar]
- Gocht A. Use of LR white resin for post-embedding immunolabelling of brain tissue. Acta Anat. (Basel) 1992;145(4):327–339. doi: 10.1159/000147385. [DOI] [PubMed] [Google Scholar]
- Eldred WD, Zucker C, Karten HJ, Yazulla S. Comparison of fixation and penetration enhancement techniques for use in ultrastructural immunocytochemistry. J. Histochem. Cytochem. 1983;31(2):285–292. doi: 10.1177/31.2.6339606. [DOI] [PubMed] [Google Scholar]
- Manocha SL. Effect of glutaraldehyde fixation on the localization of various oxidative and hydrolytic enzymes in the brain of rhesus monkey, Macaca mulatta. Histochem. J. 1970;2(3):249–260. doi: 10.1007/BF01003474. [DOI] [PubMed] [Google Scholar]
- Mrini A, Moukhles H, Jacomy H, Bosler O, Doucet G. Efficient immunodetection of various protein antigens in glutaraldehyde-fixed brain tissue. J. Histochem. Cytochem. 1995;43(12):1285–1291. doi: 10.1177/43.12.8537644. [DOI] [PubMed] [Google Scholar]
- Storm-Mathisen J, Ottersen OP. Immunocytochemistry of glutamate at the synaptic level. J. Histochem. Cytochem. 1990;38(12):1733–1743. doi: 10.1177/38.12.1979340. [DOI] [PubMed] [Google Scholar]
- Hwang SJ, Rustioni A, Valtschanoff JG. Kainate receptors in primary afferents to the rat gracile nucleus. Neurosci. Lett. 2001;312(3):137–140. doi: 10.1016/s0304-3940(01)02204-2. [DOI] [PubMed] [Google Scholar]
- Palay SL, McGee-Russeel SM, Gordon S, Jr, Grillo MA. Fixation of neural tissues for electron microscopy by perfusion with solutions of osmium tetroxide. J. Cell Biol. 1962;12:385–410. doi: 10.1083/jcb.12.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corthell J. Basic Molecular Protocols in Neuroscience: Tips, Tricks, and Pitfalls. 1st ed. Academic Press; 2014. Chapter 9, Perfusion and Immersion Fixation; pp. 83–90. [Google Scholar]
- Helander KG. Formaldehyde prepared from paraformaldehyde is stable. Biotech. Histochem. 2000;75(1):19–22. doi: 10.3109/10520290009047980. [DOI] [PubMed] [Google Scholar]
- van Harreveld A, Khattab FI. Perfusion fixation with glutaraldehyde and post-fixation with osmium tetroxide for electron microscopy. J. Cell. Sci. 1968;3(4):579–594. doi: 10.1242/jcs.3.4.579. [DOI] [PubMed] [Google Scholar]
- Renno WM. Post-embedding double-gold labeling immunoelectron microscopic co-localization of neurotransmitters in the rat brain. Med. Sci. Monit. 2001;7(2):188–200. [PubMed] [Google Scholar]
- Ellisman MH, Deerinck TJ, Shu X, Sosinsky GE. Picking Faces out of a Crowd: Genetic Labels for Identification of Proteins in Correlated Light and Electron Microscopy Imaging. Methods Cell Biol. 2012;111:139–155. doi: 10.1016/B978-0-12-416026-2.00008-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrecque S, et al. Hyperspectral multiplex single-particle tracking of different receptor subtypes labeled with quantum dots in live neurons. J. Biomed. Opt. 2016;21(4):046008. doi: 10.1117/1.JBO.21.4.046008. [DOI] [PubMed] [Google Scholar]
- Bailey R, Smith A, Nie S. Quantum dots in biology and medicine. Physica E Low Dimens. Syst. Nanostruct. 2004;25(1):1–12. [Google Scholar]
- Perkovic M, et al. Correlative light- and electron microscopy with chemical tags. J. Struct. Biol. 2014;186(2):205–213. doi: 10.1016/j.jsb.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]