

Abstract
Obesity promotes a chronic inflammatory state that is largely mediated by tissue-resident macrophages as well as monocyte-derived macrophages. Diet-induced obesity (DIO) is a valuable model in studying the role of macrophage heterogeneity; however, adequate macrophage isolations are difficult to acquire from inflamed tissues. In this protocol, we outline the isolation steps and necessary troubleshooting guidelines derived from our studies for obtaining a suitable population of tissue-resident macrophages from mice following 18 weeks of high-fat (HFD) or high-fat/high-cholesterol (HFHCD) diet intervention. This protocol focuses on three hallmark tissues studied in obesity and atherosclerosis including the liver, white adipose tissues (WAT), and the aorta. We highlight how dualistic usage of flow cytometry can achieve a new dimension of isolation and characterization of tissue-resident macrophages. A fundamental section of this protocol addresses the intricacies underlying tissue-specific enzymatic digestions and macrophage isolation, and subsequent cell-surface antibody staining for flow cytometric analysis. This protocol addresses existing complexities underlying fluorescent-activated cell sorting (FACS) and presents clarifications to these complexities so as to obtain broad range characterization from adequately sorted cell populations. Alternate enrichment methods are included for sorting cells, such as the dense liver, allowing for flexibility and time management when working with FACS. In brief, this protocol aids the researcher to evaluate macrophage heterogeneity from a multitude of inflamed tissues in a given study and provides insightful troubleshooting tips that have been successful for favorable cellular isolation and characterization of immune cells in DIO-mediated inflammation.
Keywords: Immunology, Issue 122, obesity, diet-induced inflammation, macrophages, enzymatic tissue digestion, cell sorting, single cell suspension, monoclonal antibodies, FACS, liver perfusion, aorta digestion, adipose tissue digestion
Introduction
Mouse models have been extensively used to study the dynamics of human diseases. Proper isolation of tissue resident cells from mice in a diseased state can provide a platform for understanding the molecular and cellular contributions to the pathogenesis of a disease1. One disorder that is of critical importance is obesity. The incidence of obesity continues to rise worldwide in parallel with insulin resistance and type 2 diabetes mellitus, cardiovascular disease, and fatty liver disease2,3. Excessive nutrient consumption is further skewed by decreased physical activity triggering altered signals emanating from adipose tissue, which can alter the cellular milieu of other peripheral tissues such as the aorta and liver4. Such disruption of metabolic homeostasis results in a chronic low grade systemic inflammation5.
Classical activation of macrophages resident to the aorta and liver as well as their recruitment to white adipose tissue (WAT) has been shown to not only initiate dysregulation of metabolic signals but also sustain inflammation6,7. The phenotypic and functional heterogeneity of macrophages is strongly associated with the pathogenesis of obesity related co-morbidities7. The dynamic plasticity in macrophage polarization allows for these cells to exhibit a range of activated phenotypes that coordinate the progression and resolution of inflammation8. While classically activated (M1) macrophages are implicated in the propagation of inflammation, alternatively activated (M2) macrophages have been associated with resolution and tissue repair9,10.
As the body undergoes metabolic stress, white adipose tissue abnormally accumulates. The expanded adipose tissue attracts and retains inflammatory cells that profoundly alter normal adipocyte function to promote insulin resistance, hyperglycemia and ultimately type 2 diabetes mellitus, insulin resistance or hyperglycemia11,12. In parallel, white adipose tissue remodels in response to inflammatory signals released by infiltrated classically activated (M1) adipose tissue macrophages (ATMs)13,14. This multi-cellular organ exerts a cascade of signals that derails the normal function of other body organs such as the aorta and liver4.
The liver is a metabolic powerhouse that adapts in response to stimuli originating from nearby dysregulated WAT15. Hepatic macrophages or Kupffer cells, in response to metabolic changes, secrete inflammatory cytokines that transform both parenchymal and non-parenchymal cell phenotype and promote tissue remodeling. Hepatic lipid accumulation, inflammation, excessive extracellular matrix deposits, necrosis and eventual function loss follows the inflammatory insults contributing to the wide spectrum of liver damage associated with non-alcoholic fatty liver disease16,17,18.
In parallel to compromised WAT and liver function, large arteries accumulate lipids within the arterial wall as the body undergoes chronic metabolic stress19. Arterial lipid accumulation triggers the secretion of chemokines by activated endothelial cells and subsequent recruitment of monocytes20. Once recruited, monocytes proliferate, differentiate, ingest lipoproteins and become foam cells. Atherogenesis is initiated and sustained by the pro-inflammatory activity of recruited and tissue resident lipid-laden macrophages. Succumbing to the extracellular and intracellular stress signals relayed in this atherogenic microenvironment, these macrophages then engage in an apoptotic signaling cascade. As these foam cells die, they contribute their lipid filled contents to the necrotic core of the lesion, which then leads to plaque rupture, myocardial infarction, and stroke.
Collectively, the heterogeneity of macrophage phenotypes in part orchestrates the obesity induced by inflammatory changes observed in dysregulated tissues such as WAT, liver and aorta8,21. Characterization of recruited and tissue resident macrophages could provide insight into potential molecular targets that manipulate macrophage phenotype1. To effectively characterize macrophages from obesity-induced inflamed tissues, a single-cell suspension can be obtained through enzymatic digestion. Such dissociation protocols must be effective in sufficiently degrading connective tissue while minimizing immune cell death and providing optimal cell yield. The enzyme mixture is dependent on the type of tissue and its structural make up. Resilient tissues such as the aorta requires stronger enzymatic activity, as compared to the liver and WAT, to achieve tissue dissociation. From the single cell suspension, tissue resident macrophages can be unambiguously characterized or isolated for further downstream analyses such as transcriptional profiling.
Here a tissue-specific protocol is described that uses collagenase-dependent tissue digestion and polychromatic flow cytometry to effectively isolate and characterize tissue-resident macrophages obtained from traditional diet induced obesity, atherosclerosis, simple steatosis and steatohepatitis mouse models. Simultaneous staining of cell surface markers with antibodies against leukocyte- (CD45 and/or CD11b) and macrophage- (F4/80) specific antigens is often used to identify macrophage populations22. Fluorescence-activated cell sorting (FACS) is a powerful strategy used to sort these identified populations at high purity. The sorted population can then be evaluated for phenotype specific gene profiles using downstream molecular analysis (such as quantitative real time polymerase chain reaction)23. Although standard flow cytometry and flow cytometry-based cell sorting are powerful tools in distinguishing macrophages within a vastly heterogeneous cell suspension, the former protocols must be optimized to ensure successful output. In this study, protocols that effectively isolate and characterize viable tissue specific macrophages are described; more importantly, this study provides crucial insight into technical issues that often arise, as well as proactive and trouble-shooting strategies to prevent and/or resolve them.
Protocol
All experimental protocols (Sections 1, 1.2, and 1.3) were approved by the Institutional Animal Care and Use Committee (IACUC) at Pennsylvania State University.
| Tissue | Dissociation Buffers Preparation | Final Volume | Storage |
| White Adipose Tissue (WAT) | Dissociation Buffer: 2.5% HEPES, 10 mg/mL Bovine serum albumin (BSA), 3 mg/mL (0.3%) Collagenase Type II in Dulbecco’s Modified Eagle Medium (DMEM) with 4.5 g/L glucose without L-glutamine and sodium pyruvate | 500 mL | minus 80°C (10 mL aliquots) |
| Liver | 25x Perfusion Buffer Concentrate (PBC): 3.55 M NaCl, 168 mM KCl, 240 mM HEPES, 150 mM NaOH in distilled deionized H2O | 500 mL | minus 20°C (40 mL aliquots) |
| Preservation Buffer (PRB): 1% BSA in 1x Perfusion Buffer | 1 L | 4 °C | |
| Dissociation Buffer: 1x Perfusion Buffer supplemented with 4.76 mM CaCl2 and 72 U/mL Collagenase Type IV | 50 mL (per mouse) | Prepare immediately prior to use | |
| Aorta | Dissociation Buffer: 125 U/mL Collagenase Type XI, 60 U/mL Hyaluronidase type I, 60 U/mL DNase I, 450 U/mL Collagenase Type I, 20 mM HEPES in 1x Phosphate Buffered Saline (PBS) | 500 mL | minus 80°C (10 mL aliquots) |
Table 1: Tissue specific perfusion buffer recipes.
1. Tissue Isolation and Dissociation
- WAT Isolation and Dissociation
- Prepare the tissue-specific buffers as per Table 1, and store them as described.
- Prepare the following reagents.
- Prepare the digestion buffer by thawing the appropriate volume of WAT dissociation buffer and store at 4 °C until use. Immediately before use, warm buffer to 37 °C. Prepare the FACS buffer by preparing 1x phosphate-buffered saline (PBS) containing 2% fetal bovine serum (FBS).
- WAT Isolation
- Euthanize a mouse in a carbon dioxide (CO2) filled chamber. Check for effectiveness prior to continuing to the dissection stage.
- Dip the euthanized mouse briefly in a beaker containing 70% ethanol until thoroughly soaked and coated with ethanol. Place the mouse ventral surface up on the dissection stage and fasten the mouse fore and hind paws to the dissection board using 21 G needles.
- Use medium point tip forceps to grasp the abdominal skin anterior to the urethral opening, then use sharp dissecting scissors to make a nick in the grasped abdominal skin.
- Insert the lower blade of the scissors into the small incision between the skin and peritoneum and make a lateral incision from abdomen to the rib cage.
- Gently pull back the skin to expose the intact peritoneal cavity and make a lateral incision through the peritoneum and either fold back the transparent membrane or excise the tissue to expose the abdominal WAT.
- Collect the perigonadal fat pads as described in Mann et al.24
- The perigonadal fat pads in male mice are loosely bound to the epididymis and testes. First locate the testes and then with medium point forceps, grasp hold of the epididymis head and pull gently.
- Using sharp dissecting scissors, excise each epididymal fat pad by cutting along the surface of epididymis (head, body and tail) and the testes.
- With the forceps, gently pull on the fat pad while cutting through the connective tissue directly bound to the epididymis structure.
- Using the forceps, firmly grip the end of fat pad proximal to the epididymis and gently peel the fat pad away from the gonads
- In female mice, the perigonadal fat pads are loosely bond to the uterus body and uterine horn.
- Using medium point forceps, grip the perigonadal fat tissue and gently pull the tissue away from the uterus body and uterine horn.
- Excise each fat pad by cutting along the uterus body and uterine horn using sharp dissecting scissors.
- Place the fat pads in a petri-dish filled with PBS and keep on ice to keep tissue moist.
- Dissociation of WAT into Single Cell Suspensions
- Remove excess PBS from the petri dish and use a single edge razor blade to mince the abdominal WAT into small pieces.
- With the sharp edge of the razor blade, gently scrape the minced abdominal WAT into a labeled 15 mL polypropylene collection tube containing 2 mL WAT dissociation buffer.
- Incubate this tissue-digestion buffer mixture under slow continuous rotation at 37 °C for 45 min.
- Filter the digested tissue through a 70 µm cell strainer into a 50 mL collection tube while moving the rubber piston of a syringe plunger in a circular motion and continue to sieve the cell suspension through the filter.
- Pipet 2 mL of Dulbecco's Modified Eagle Medium (DMEM) to a 15 mL collection tube, gently pipet up and down and wash the filter with the single-cell suspension.
- Wash the filter two additional times with cold DMEM and place the single cell suspension on ice.
- Centrifuge the cell suspension at 300 x g at 4 °C for 8 min.
- Use a vacuum aspirator to carefully remove floating adipocytes (first) and remaining supernatant. Make sure not to disturb the pelleted stromal vascular fraction (SVF).
- With a 1,000 µL pipette, gently re-suspend the pellet in ammonium chloride-potassium (ACK) buffer to lyse erythrocytes according to the manufacturer's instructions.
- Dilute the above cell suspension with DMEM and centrifuge the cell suspension at 300 g at 4 °C for 8 min.
- Re-suspend cells in cold PBS containing 2% FBS (FACS Buffer) and count the cells in a Burker chamber/hemocytometer.
- Transfer either 1 x 106 cells (for basic flow cytometry) or the entire cell suspension (for FACS) to labeled 5 mL round-bottom polystyrene tubes. Continue to Section 2 for flow cytometry staining protocol.
- Liver Tissue Isolation and Dissociation NOTE: This protocol was adapted from Smedsrød25.
- Prepare the tissue-specific buffers as per Table 1 and store them as described.
- Prepare the following reagents.
- Prepare 1x Perfusion Buffer (PB), by diluting 40 mL of PBC into 960 mL ultrapure H2O. Pre-warm a syringe filled with 50 mL of PB at 37 °C, just before perfusion starts. Eliminate any present air bubbles.
- To eliminate air bubbles, invert the syringe where the leur lock tip is pointing upward. Tap or flick the syringe until air bubbles are to the top pf the syringe. Gently push on the plunger to expel the air
- Prepare the digestion buffer by diluting 0.5 mL of 476 mM CaCl2 solutionto 49.5 mL 1x Perfusion buffer. Add 3600U of Collagenase Type IV to the 4.76 mM CaCl2 – PB solution. Pre-warm a syringe filled with 50 mL of digestion buffer at 37 °C just before perfusion starts. Eliminate any present air bubbles as described in Step 1.2.2.1.1.
- Prepare the preservation Buffer (PRB) by dissolving 2.5 g bovine serum albumin (BSA) in 250 mL 1x perfusion buffer. Store at 4 °C or keep on ice until use.
- Prepare FACS Buffer with 1x PBS and 2% FBS.
- Liver Tissue Isolation
- Euthanize a mouse by CO2 asphyxiation, saturate mouse with 70% ethanol to prevent contamination of intraabdominal organs with fur.
- Position the mouse ventral side up on a polystyrene foam dissecting board and secure the mouse fore and hind paws to the dissection board using 21 G needles.
- To expose the thoracic wall and peritoneal cavity, grasp the abdominal skin near the urethral opening using a medium point tip forceps. Then use a sharp dissecting scissors to create a small incision to the grasped abdominal skin.
- Insert the lower blade of the scissors into the small incision between the skin and peritoneum and make a lateral incision from groin to chin.
- Gently pull back the skin to expose the intact peritoneal cavity and thoracic wall, make a lateral incision through the peritoneum and excise the membrane.
- Lift the sternum and carefully incise the diaphragm, be sure not to disturb the inferior vena cava (IVC). Move the gastrointestinal organs to the side and locate the subhepatic IVC. Use hemostatic forceps to clamp the suprahepatic IVC to maintain localized perfusion. Note: Excessive accumulation of visceral white adipose tissue can often mask the IVC. To better visualize this vessel, a small area of adipose tissue next to the IVC can be excised. The incised window within the pliable fat tissue can then be positioned over the IVC to expose the vessel for cannulation.
- Attach the syringe filled with pre-warmed PB to the 23 G blood collection and infusion set. Gently push on the plunger until the tubing and needle are filled with PB.
- Insert the 23 G needle, parallel to the level of the subhepatic IVC. Secure the needle in place using a 4 cm hemostatic clamp. NOTE: Cannulation of the subhepatic IVC is preferred in diet-fed mice in that the IVC can be better visualized and cannulated within the fat-filled cavity.
- Gently push on the plunger to begin perfusion, color will rapidly flush from the liver (right lobe first) if the needle is appropriately positioned in the vessel. Enlargement of the lobes is also visible if the needle is properly inserted into the subhepatic IVC.
- Promptly cut the portal vein to allow the PB to flow freely through the liver.
- Perfuse the liver until blood is no longer visible. On occasion gently massage liver lobes between fore-finger and thumb to facilitate perfusion. NOTE: It is important to use non-serrated blunt edged tools to handle the liver to prevent tissue damage.
- When 5 mL of PB remains within the syringe, change to the syringe filled with pre-warmed dissociation buffer. Do not disturb the position of the secured needle during this time. Note: Enlarged livers significantly exceeding 1.5 g should be perfused with a greater volume of pre-warmed digestion buffer to guarantee successful liver dissociation.
- Perfuse the liver until fully digested. Gently massage liver lobes to facilitate perfusion. NOTE: Separation of Glisson's capsule from parenchyma is observable in a successfully digested liver. To assess this, use the blunt edge of a pair of forceps, to gently press on the left lateral lobe. An impression should appear on the surface and the indentation should fill slowly once the forceps is removed.
- Remove the needle from the subhepatic IVC. Do not remove the hemostatic forceps clamping the suprahepatic IVC.
- Excise the whole liver by carefully cutting through connecting ligaments using a short straight blade dissecting scissors and remove the gallbladder.
- Place the whole liver in a 10-cm petri-dish containing 10 mL ice-cold Preservation Buffer and store at 4 оC until ready to liberate cells.
- Liver Cell Dissociation
- Transfer the liver to a 10 cm dish containing 10 mL ice-cold DMEM.
- Grip the severed suprahepatic IVC attached to the excised liver using a toothed forceps. Use medium point forceps to release the cells from the Glisson's capsule by applying rapid stroking motion to each lobe.
- Pass the saturated DMEM through a 70 µm cell strainer attached to a 50 mL collection tube.
- Pipet 10 mL ice-cold DMEM to the 10 cm petri dish and repeat steps 1.2.4.1 to 1.2.4.3 until media saturation is no longer observed. Place the cell suspension on ice or at 4 °C.
- Gently mix the cell suspension by inverting the collection tube and then centrifuge at 54 x g for 2 min at 4 °C.
- Collect the supernatant in a clean 50 mL collection tube. Then centrifuge the cell suspension at 54 x g for 2 min at 4 °C.
- Repeat Step 1.2.4.6 two additional times before proceeding to Step 1.2.4.8.
- Transfer the supernatant to a clean 50 mL-collection tube and centrifuge the cell suspension at 300 x g for 10 min at 4 °C.
- Re-suspend the non-parenchymal cell pellet in FACS buffer and count them in a Burker chamber/hemocytometer.
- Transfer 1 x 106 cells (for basic flow cytometry) or 15 x 106 - 20 x 106 cells (for FACS) to labeled 5-mL round-bottom polystyrene tubes. Continue to Section 2 for flow cytometry staining protocol.
- Aorta Isolation and Dissociation NOTE: This protocol was adapted from Butcher et al.26
- Prepare the tissue-specific buffers based upon the recipes provided in Table 1 and store them as described.
- Prepare the following reagents.
- Prepare the digestion buffer by thawing the appropriate volume of WAT dissociation buffer and store at 4 °C until use. Immediately before use, warm buffer to 37 °C.
- Prepare a 10x heparin solution by dissolving heparin sodium salt in 1x PBS at a concentration of 200 U/mL. Dilute the 10x heparin stock solution with 1x PBS to prepare a 1x Heparin solution. Store the 10x and 1x Heparin solutions at 4 °C until use.
- Prepare the FACS Buffer with 1x PBS containing 2% FBS.
- Aorta Dissection
- Euthanize a mouse in a carbon dioxide filled chamber and rinse with 70% ethanol.
- Place the mouse ventral side up on the dissection stage, spread the mouse fore and hind paws and fasten them to the dissection board using 21 G needles.
- Immediately after euthanizing the mouse, perform cardiac puncture. Refer to Steps 1.3.3.4 to 1.3.3.9, if necessary.
- Locate the xiphoid process at the lower end of the sternum
- To do so, place a finger midpoint along the width of the animal's neck. Trace along the ventral midline from neck to the caudal end of the sternum until a cartilaginous extension is felt.
- Use medium point forceps to grasp the cartilaginous extension, and if necessary mark the location of the xiphoid process by removing the patch of hair or skin at the area.
- Partially insert a 26 G needle under the xiphoid process at 20-30° angle.
- Gently apply negative pressure to the syringe by slowly withdrawing the plunger, then insert the needle further into the cavity until blood flows.
- If blood flow stops, slowly rotate the needle or move slightly in and out.
- Use the pair of forceps to grab hold of the abdominal skin anterior to urethral opening, and use a pair of sharp dissecting scissors to cut along the ventral midline through the peritoneum from groin to chin.
- Pull back the skin to expose abdominal organs and rib cage with fingers or blunt forceps gently move abdominal organs to the side.
- Use the forceps to grab hold of the xiphoid process, lift the sternum, and incise the diaphragm.
- Using the dissecting scissors, cut through the lateral ribs on both sides. Gripping the sternum, flip the rib cage upwards and cut through the connecting base. Remove the rib cage exposing the underlying thoracic organs.
- Perfuse the aorta with a 26-gauge needle attached to a 10 mL-syringe filled with 1x heparin solution by injecting 1-2 mL of solution into the left ventricle (LV) of the heart. NOTE: Be sure to perfuse at a slow rate with little pressure to ensure intact atherosclerotic aortic lesions.
- Excise the thymus, lungs and connective tissue. Refer to Steps 1.3.3.16 and 1.3.3.17, if necessary.
- Anterior to the heart locate the white bi-lobed (or butterfly shaped) organ and firmly grab hold of the thymus with the forceps. Pull both lobes upward away from the cavity and cut at the base with the scissors.
- To remove the lungs, pinch a lung lobe with the forceps, pull the tissue away from the thoracic cavity and cut at the base. Repeat for each remaining lobe.
- Use a dissection microscope and micro-dissecting tools, and collect the aortic arch (including the innominate artery, left common carotid artery and left subclavian artery), the ascending, descending, thoracic and abdominal aorta.
- Locate the ascending aorta at the left ventricle of the heart.
- With a pair of curved 0.07 x 0.04 mm-tip forceps gently grasp the portion of the ascending aorta emerging from the left ventricle. NOTE: Compared to the surrounding yellow-tinged fat tissue, the aorta will be a bright white vessel originating from the LV when sufficiently flushed with the heparin solution.
- If the aorta was not properly flushed by cardiac perfusion, flush the aorta again while the aorta remains connected to the cavity. To do so, cut near the heart-aorta junction, remove the heart, and then cut horizontally through the lower-end of the abdominal aorta. Insert a 26 G needle in the opening of the ascending aorta and gently flush the aorta with the 1x heparin solution.
- Gently tug on the ascending aorta to better discriminate between the fat tissue and embedded aorta.
- Still gently pulling on the ascending aorta, use the tip of closed spring dissecting scissors to clear the fat that encapsulate the ascending aorta and aortic arch. To do so, stroke the fat tissue with the tip of closed scissor blades to tear away the connecting fat. NOTE: Refrain from excising any fat tissue by cutting until the ascending aorta and arch can be clearly visualized. This is to prevent cutting the aorta.
- If the ascending aorta and arch can be clearly delineated from the surround fat tissue, use the spring dissecting scissors to excise excess fat.
- Gently grab hold of the aortic arch with the forceps. Again using the spring scissor's tip, stroke at the fat along the length of the descending, thoracic and abdominal aorta to the caudal end of the abdominal aorta, do so on the right side of the sleeve of fat.
- Continue to grasp along the aorta when tearing away the fat. Be sure to do so gently to prevent damaging the aorta.
- Gently pull the aorta upward toward the microscope so that the fat is now positioned behind the aorta.
- Insert the closed scissor blades between the fat-aorta interface near the anterior end of the descending aorta, severe the fat tissue's attachment to the aorta's surface by bluntly using a sweeping motion. NOTE: Thoroughly removing excess fat and connecting tissue while the aorta is still within the cavity is recommended.
- Excise the heart and cut at the caudal end of the abdominal aorta transversely. Remove the whole aorta.
- Place the aorta in a 35 mm dish and tease away any excess fat on the aorta using two 21-gauge needles. Place the excised aorta in 1.7 mL microcentrifuge tube on ice.
- Dissociation of the Aorta into Single Cell Suspensions
- Pipet 0.2 mL aorta dissociation buffer to the tube containing the aorta. Insert the spring scissor blades toward the tapered end of the tube and rapidly mince the aorta to facilitate enzymatic digestion.
- Transfer the tissue-solution mixture to a 15 mL collection tube and pipette 1.8 mL aorta dissociation buffer for a total volume of 2 mL. NOTE: For easy transfer of the tissue suspension, cut the 1 cm off the tapered end of a standard 1,000 µL pipette tip. Use the shortened tip to transfer the suspension with a micropipette.
- Incubate the minced aorta in the aorta dissociation buffer for 55 min at 37 °C, shaking at 220 rpm (0.514 x g).
- Pass the suspension through 50-µm cell strainer into a 15 mL polypropylene collection tube. Use the rubber piston of a syringe plunger to facilitate filtering.
- Rinse the 15 mL collection tube with 1 mL FACS buffer, collect the suspension and pass through the cell strainer.
- Wash the 50 µm cell strainer two additional times with 1 mL FACS buffer.
- Pellet the cells by centrifugation at 300 x g for 5 min at 4 °C. Re-suspend cells in cold FACS Buffer. Count the cells in a Burker chamber/hemocytometer.
- Transfer either 1 x 106 (for basic flow cytometry) cells or the entire cell suspension (for FACS) to a labeled 5 mL round-bottom polystyrene tubes. Continue to Section 2 for flow cytometry staining protocol.
2. Flow Cytometry and FACS Staining
| Fluorophore | R-Phycoerythrin (PE) | PE/Cyanine (Cy) 7 | PE/Cyanine (Cy) 5 | Pacific Blue (PB) |
| Laser (nm) | Blue (488 nm) / Yellow (561-570 nm) - Green (532 nm) | Blue (488 nm) / Yellow (561-570 nm) - Green (532 nm) | Blue (488 nm) / Yellow (561-570 nm) - Green (532 nm) | Violet (405 nm) |
| ExcitationMAX (nm) | 496 | 496 | 496 | 401 |
| EmissionMAX (nm) | 578 | 785 | 667 | 455 |
| Phenotypic Marker | F4/80 | CD11c | CD11b | CD45 |
| EMR1, Ly71 | αX integrin, integrin αX chain, CR4, p150, ITGAX | αM integrin, Mac-1, Mo1, CR3, Ly-40, C3biR, ITGAM | T200, Ly-5, LCA | |
| Targeted Cell Type | Tissue Resident Macrophages | Classically Activated (M1) Macrophages | Monocytes / macrophages | Leukocytes (Macrophages / monocytes, lymphocytes, and granulocytes) |
| Subset of Dendritic cells | Dendritic cells, NK cells, Activated T cells, and a subset of intestinal intraepithelial lymphocytes (IEL) | Granulocytes, dendritic cells, NK cells, and subsets of T and B cells | ||
| Dilution Factor | 1:50 | 1:100 | 1:100 | 1:100 |
| Isotype Controls | PE Rat IgG2a | PE/Cy7 Armenian Hamster IgG | PE/Cy5 Rat IgG2b | Pacific Blue Rat IgG2c |
Table 2: List of fluorophore tagged antibodies specific for discriminating tissue resident macrophages.
Collect and prepare the following items: FACS Buffer 5 mL round-bottom polystyrene tubes, anti-mouse CD16/32 Fc receptor blocking antibody, fluorophore conjugated or unconjugated primary antibodies, fluorophore-conjugated secondary antibodies (if necessary), 1x phosphate-buffered saline (PBS). NOTE: For staining large amounts of cells, antibody concentration is most critical rather than cell number. For example, if staining 10 x 106 cells the staining buffer volume and antibody concentration should be the same as if staining 1.0 x 106 cells in 100 µL buffer volume. For staining 1.0 x 108 cells, increasing the antibody amount 5-fold is recommended.
Add additional ice-cold FACS Buffer to each FACS tube if necessary, pellet cells by centrifugation at 751 x g for 5 min (4 °C). Aspirate the supernatant following centrifugation.
Add anti-mouse CD16/CD32 Fc receptor blocking antibody to all samples according to the manufacturer's instructions. Incubate for 15 min at 4 °C or on ice.
- Add fluorophore-conjugated primary antibody cocktail directly to the Fc receptor blocking antibody-containing cell suspension and incubate samples at 4 °C for 30 min. Protect samples from light to minimize bleaching of fluorophore-conjugated antibodies.
- In the event unconjugated primary antibodies were used, follow these steps subsequent to completing Step 2.4.
- Wash primary antibody stained samples by centrifugation at 751 x g for 5 min (4 °C).
- Remove supernatant by decanting and re-suspend pellet in 100 µL FACS buffer.
- Add conjugated secondary antibody to all necessary samples and incubate for 30 min at 4 °C. Proceed to Step 2.5.
Following antibody incubation, add 2 mL of ice cold FACS buffer then pellet cells at 751 x g for 5 minutes (4 °C). Decant supernatant and be sure not to disturb pellet.
Mix cells gently and then re-suspend with 200-400 µL of FACS buffer, then keep this in the dark at 4 °C until standard flow cytometry analysis or cell sorting.
For short term fixation of stained cells, proceed to Steps 2.8 to 2.10. Use fixation for standard flow cytometry only.
Immediately following Step 2.5, re-suspend cells in 0.5 mL 2-4% paraformaldehyde (PFA) for 15-30 min in 4 °C. NOTE: Caution: PFA is a known irritant with carcinogenic and toxic effects. When using PFA, handle with extreme care. Be sure to use the appropriate personal protective equipment and exhaust ventilation.
Following fixation, wash cells by adding 2 mL FACS buffer to all samples and centrifuge at 751 x g in 4 °C for 5 min. Remove supernatant following centrifugation.
Resuspend fixed cells in 200 µL of ice cold FACS buffer and store at 4 °C. Store fix cells up to 1 week at 4 °C, ideally perform flow cytometry acquisition within 48 hours to minimize autofluorescence.
Acquire flow cytometry data according to the flow cytometer manufacturer's instructions or perform fluorescent activated cell sorting as described by Basu et al.23
Representative Results
When using apolipoprotein E deficient (ApoE KO) C57BL/6 (BL6) mice maintained on a high fat high cholesterol diet (HCHFD or HCD) for 18 weeks, 1 x 104 - 2 x 104 CD45+F4/80+ aortic macrophages can be isolated when two samples are pooled. Livers dissected from HFHCD-fed ApoE KO mice, produced greater than 5 x 105 sorted Kupffer cells (which depends on available sorting time). When using high fat diet (HFD) fed wild type (WT) C57BL/6 mice, 5 x 105 to 1 x 106 resident adipose tissue macrophages (ATMs) can be sorted from the stromal vascular fraction (SVF). The mentioned total number of macrophages that can be sorted from a given tissue was adequate for performing gene expression analysis using quantitative real time polymerase chain reaction (qPCR). For tissues where fewer numbers of macrophage populations are recovered, such as the aorta, it is recommended to use co-precipitants (for example glycogen) and overnight precipitation when isolating RNA is needed for these analyses.
Here, the effects of diet-induced metabolic disorders on macrophage phenotype using basic flow cytometry, fluorescence-activated cell sorting (FACS), and downstream post sort analyses are shown. These findings corroborate previously published observations that mice fed a HFD or HFHCD exhibit increased infiltration of classically activated (M1) macrophages in affected tissues such as the aorta (Figure 1B). Taking advantage of flow cytometry, FACS, and gene expression profiling (via quantitative polymerase chain reaction (qPCR), the predominating phenotype for Ron receptor tyrosine kinase-expressing CD45+ F4/80+ macrophages derived from tissues isolated from diet-fed mice was observed. Ron receptor-expressing aortic macrophages which demonstrated an anti-inflammatory phenotype were decreased in HFHCD-fed mice (Figure 1C and D). Sorted Ron receptor-expressing macrophages derived from digested aortas demonstrated increased arginase 1 (Arg1) gene expression, which is a well-established M2 macrophage marker (Figure 1E). Pro-inflammatory macrophages which were characterized as CD45+ F4/80+ CD11c+ were increased in aortas isolated from HFHCD-fed mice (Figure 1B and D). Characterizing liver-resident macrophages further elucidated the prevailing phenotype of Ron receptor-expressing subpopulations (Figure 2A). CD11c+ pro-inflammatory macrophage populations demonstrated a decrease expression of genes that are strongly associated with an anti-inflammatory (M2) phenotype such as Arg1 and Ron (Figure 2B). Similar trends were observed in macrophage populations isolated from digested white adipose tissue (Figure 3). Macrophage population with a pro-inflammatory signature showed decreased surface expression of the Ron receptor (Figure 3). Combining the approaches, basic flow cytometry and FACS, resulted in conclusive data that further validates the Ron receptor as a regulator of alternative (M2) activation in macrophages32. Such bias toward an anti-inflammatory (M2) phenotype has been shown to play a protective role in the development and progression of obesity, atherosclerosis and steatohepatitis31.
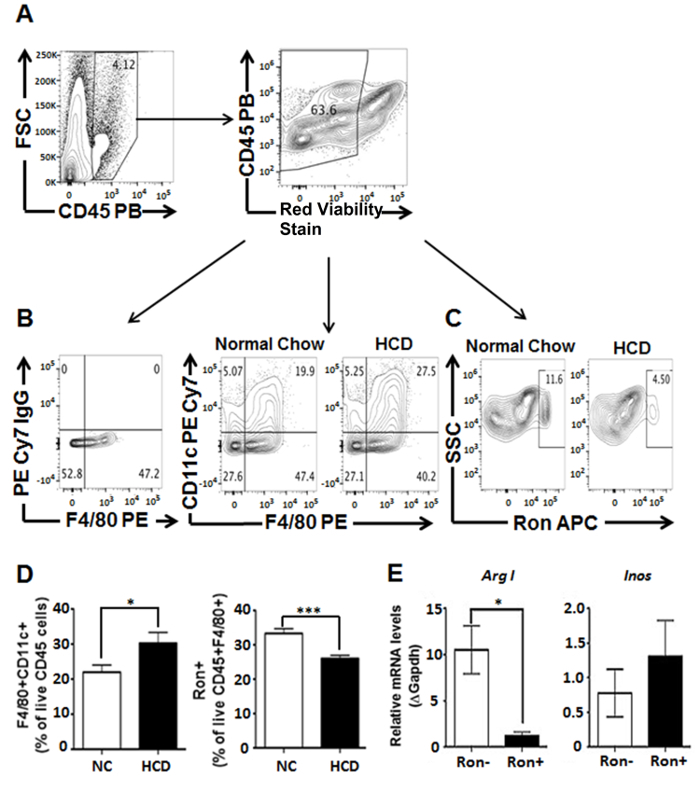 Figure 1: Characterization of macrophages isolated from dissociated aorta removed from ApoE KO mice maintained on a HCD for 18 weeks. (A) Cells were first gated on CD45+ leukocytes, excluding cellular debris. (B) CD45 staining in conjunction with F4/80 is used to delineate double positive populations presumed to be macrophages. Additional gating was used to demonstrate the percentage of CD11c+CD45+F4/80+ (M1) and (C) Ron+CD45+F4/80+ (potential M2) macrophages in aortas derived from mice fed on either a normal chow or high cholesterol diet for 18 weeks. (D) Increased percentage of CD11c+CD45+F4/80+ (M1) macrophages, as well as a decreased percentage of Ron+CD45+F4/80+ (potential M2) macrophages were observed in aortas derived from mice fed HCD compared to mice fed a normal chow diet. (E) Gene expression analysis of sorted Ron+CD45+F4/80+ macrophages demonstrated increased expression of arginase I (a well-known murine M2 marker). Values were obtained using Student's t-test analyses performed using statistical analysis software and represented as mean ± SEM. P < 0.05 was considered statistically significant (*P < 0.05, ***P < 0.001). Figure has been modified from Yu et al. (2016)31. Please click here to view a larger version of this figure.
Figure 1: Characterization of macrophages isolated from dissociated aorta removed from ApoE KO mice maintained on a HCD for 18 weeks. (A) Cells were first gated on CD45+ leukocytes, excluding cellular debris. (B) CD45 staining in conjunction with F4/80 is used to delineate double positive populations presumed to be macrophages. Additional gating was used to demonstrate the percentage of CD11c+CD45+F4/80+ (M1) and (C) Ron+CD45+F4/80+ (potential M2) macrophages in aortas derived from mice fed on either a normal chow or high cholesterol diet for 18 weeks. (D) Increased percentage of CD11c+CD45+F4/80+ (M1) macrophages, as well as a decreased percentage of Ron+CD45+F4/80+ (potential M2) macrophages were observed in aortas derived from mice fed HCD compared to mice fed a normal chow diet. (E) Gene expression analysis of sorted Ron+CD45+F4/80+ macrophages demonstrated increased expression of arginase I (a well-known murine M2 marker). Values were obtained using Student's t-test analyses performed using statistical analysis software and represented as mean ± SEM. P < 0.05 was considered statistically significant (*P < 0.05, ***P < 0.001). Figure has been modified from Yu et al. (2016)31. Please click here to view a larger version of this figure.
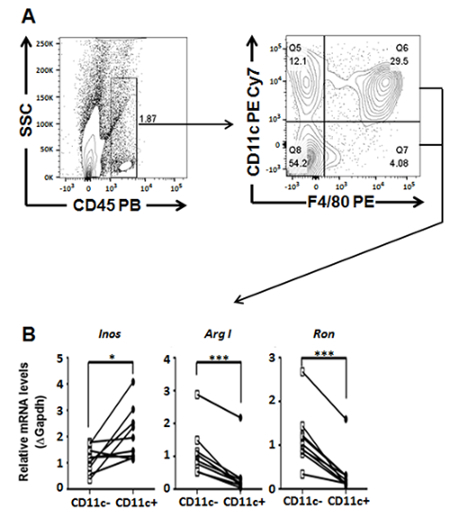 Figure 2: Gene transcript profiling of Kupffer cell populations sorted from digested livers dissected from ApoE KO mice maintained on a HCD for 18 weeks. (A) General gating scheme for characterizing and sorting Kupffer cell populations. (B) Gene expression analysis of sorted Ron expressing (Ron+) and non-expressing (Ron-) CD45+F4/80+ macrophages by quantitative RT PCR. Student's t-test analyses were performed using statistical software and values were represented as mean ± SEM. P < 0.05 was considered statistically significant (*P < 0.05, ***P < 0.001). Figure has been modified from Yu et al. (2016)31.
Please click here to view a larger version of this figure.
Figure 2: Gene transcript profiling of Kupffer cell populations sorted from digested livers dissected from ApoE KO mice maintained on a HCD for 18 weeks. (A) General gating scheme for characterizing and sorting Kupffer cell populations. (B) Gene expression analysis of sorted Ron expressing (Ron+) and non-expressing (Ron-) CD45+F4/80+ macrophages by quantitative RT PCR. Student's t-test analyses were performed using statistical software and values were represented as mean ± SEM. P < 0.05 was considered statistically significant (*P < 0.05, ***P < 0.001). Figure has been modified from Yu et al. (2016)31.
Please click here to view a larger version of this figure.
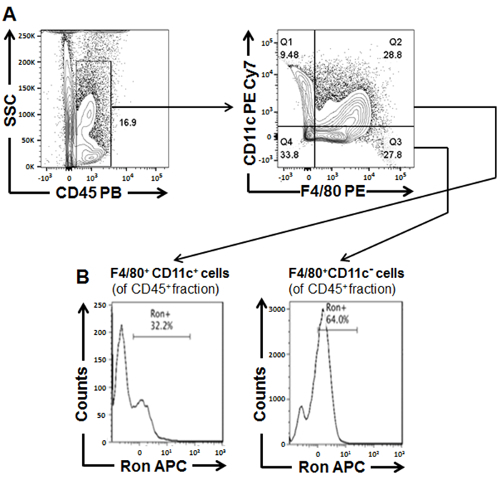 Figure 3:Characterization of ATMs isolated from white adipose tissue dissected from WT BL6 mice fed a HFD for 18 weeks. (A) General gating scheme for characterizing and sorting adipose tissue derived macrophage populations (B) Percentage of Ron+ cells within CD45+F4/80+CD11c- and CD45+F4/80+CD11c+ macrophage populations sorted from WAT. Figure has been modified from Yu et al. (2016)31.
Please click here to view a larger version of this figure.
Figure 3:Characterization of ATMs isolated from white adipose tissue dissected from WT BL6 mice fed a HFD for 18 weeks. (A) General gating scheme for characterizing and sorting adipose tissue derived macrophage populations (B) Percentage of Ron+ cells within CD45+F4/80+CD11c- and CD45+F4/80+CD11c+ macrophage populations sorted from WAT. Figure has been modified from Yu et al. (2016)31.
Please click here to view a larger version of this figure.
Discussion
Diet-induced metabolic disorder models that mimic co-morbidities such as atherosclerosis, simple steatosis, steatohepatitis and type 2 diabetes are extensively used to better understand the underlying molecular mechanisms of disease progression. Collagenase dependent digestion is often used to dissociate tissues to liberate cells from the extracellular matrix (ECM)16,27. Enzymes such as collagenase disrupt collagen which provides structural support for neighboring cells. The tissues' structural composition dictates the tissue matrix stiffness (resistance to deformation) and which crude collagenase product is most efficient in ensuring successful ECM disruption28. WAT, which is composed of "soft matrix" is often digested with Collagenase Type II to liberate adipocytes and resident immune cells while simultaneously maintaining the integrity of cell surface insulin receptors29. The microarchitecture of the fibril components of the aorta contributes to the "stiff matrix", for which effective aortic digestion with collagenase type XI (which has the highest collagenase activity) is used in combination with additional enzymes. Unlike the aorta, the liver digestion uses a collagenase with a weaker enzymatic activity, Collagenase Type IV, to disrupt the matrix and liberate viable parenchymal and non-parenchymal cells30. Chronically inflamed tissues derived from diet-fed wild type or apolipoprotein E deficient (ApoE KO) mice on a C57BL6 background often undergo remodeling that can prevent proper enzymatic digestion. This section will discuss strategies to minimize and/or eliminate the possibility of improper tissue dissociation and low cell recovery.
A common feature of tissues derived from diet-fed mouse models that mimic obesity, atherosclerosis, and/or non-alcoholic fatty liver disease, is abnormal tissue remodeling. In white adipose tissue, the aorta, and liver, ECM composition is altered, often resulting in excessive deposition of fibrillar components such as collagens. Wild type C57BL6 mice maintained on a high fat diet for eighteen weeks, experience tissue-specific abnormalities such as expanded white adipose tissue and enlarged fatty liver (simple steatosis). Often times these metabolic features are not troublesome obstacles to overcome during the tissue dissociation process. On the other hand, in other diet induced models that mirror more exacerbated phenotypes, the extensive remodeling of the tissue can pose a problem during tissue digestion. HFHCD fed ApoE KO mice are often used to model atherosclerosis and steatohepatitis. A common feature associated with advanced steatohepatitis is the excessive deposition of ECM in the liver (or fibrosis). Fibrotic liver has been shown to be quite problematic during enzymatic tissue dissociation and often produces low cell yield31. To improve cell yield, it is critical that the digestion protocols be followed without deviation. Although normal livers can be minced and submerged in digestion buffer to achieve dissociation, perfusion with digestion buffer maximizes contact between the extracellular matrix of the liver and the collagenase solution; and therefore this approach is highly recommended. Additionally, 20-30 mL of digestion buffer is often an adequate volume for successful dissociation of normal livers; however, in regards to mouse models that develop enlarged and/or fibrotic livers, perfusion with 50 mL of digestion buffer is preferred to ensure proper digestion while minimizing cell death. For livers with weights that significantly exceed 1.5 g, increasing the volume of digestion buffer is recommended to guarantee successful digestion.
| Tissue | Problem | Possible Reason | Solution |
| White Adipose Tissue (WAT) | Poor cell dissociation | Poor Digestion | Make sure digestion buffer is at 37°C |
| Cell Death | Excessive collagenase digestion | Reduce time of digestion | |
| Reduce volume of digestion buffer | |||
| Aorta | Poor cell dissociation | Poor Digestion | Make sure digestion buffer is at 37 °C |
| Aorta pieces in digestion buffer were too large | Make sure aorta is cut into approximately 1 mm pieces | ||
| Aorta pieces were not shaking in dissociation buffer during incubation | Be sure aorta pieces are shaking in the digestion solution | ||
| Cell Death | Excessive collagenase digestion | Reduce time of digestion | |
| Reduce volume of digestion buffer | |||
| Liver | Poor cell dissociation | Poor Digestion | Make sure digestion buffer is at 37 °C |
| Increase volume of digestion buffer | |||
| Improper cannulation of IVC (swelling of tissue surrounding IVC occurs) | Be sure needle is properly inserted into the IVC | ||
| Ruptured IVC | Properly secure needle in IVC prior to perfusion | ||
| Reduce perfusion speed | |||
| Ruptured tissue | Reduce perfusion speed | ||
| Cell Death | Improper release of cells from the Glisson’s capsule | Be sure to dissociate cells from capsule using previously discussed stroking method | |
| Excessive collagenase digestion | Reduce time of digestion | ||
| Reduce volume of digestion buffer |
Table 3: Troubleshooting unsuccessful tissue dissociation. Flow cytometry based cell sorting or FACS is a powerful technique for isolating cell populations where high purity is a necessity. When purifying cells by cell sorting, achieving high cell yield but also a high purity sort requires using the proper sorting strategies. In this section, methods for improving multi-fluorophore flow cytometry-based cell sorting of tissue resident macrophage derived from diet-fed mice are described. For distinct tissue resident macrophage isolation, surface marker selection is a critical step. Macrophages derived from WAT, aorta, and liver are often distinguished using a CD45+, CD11b+, F4/80+ gating strategy. Additional flow cytometric panels can be used to identify tissue resident macrophages. These panels include antibodies which probe for the surface expression of macrophage specific glycoproteins (CD64, CD68, and CD14), major histocompatibility complexes (MHCII) and apoptotic cell tyrosine kinase receptors (MerTK)32,33. Specific macrophage phenotype can then be delineated by probing for the selective surface expression of M2 (CD163, CD209, and CD206) or M1 markers (CD38, CD40, CD80, and CD86)34,35. Auto-fluorescence generated by lipid-laden macrophages can present some issues when gating populations. Using antibodies tagged with fluorochromes that are excited by the yellow-green laser (such as PE, PE/Cy5, PE/Cy7) or red laser (such as APC, APC Cy7) results in emitted fluorescence that is significantly brighter than the auto-fluorescence and thus can improve results36. When selecting fluorophore conjugates with such high staining index and potential emission spectra overlap, inclusion of appropriate controls is critical. When identifying gating boundaries, inclusion of single stain (SS) controls and isotype controls allow for the delineation of positive/negative populations and the measurement of non-specific background signal caused by primary antibodies, respectively37. For circumstances where cells are scarce, we recommend using compensation particles for SS and isotype controls. Compensation particles typically emit brighter signals than biological controls and also have less variance in background fluorescence. Additionally, fluorescence minus one (FMO) controls are ideal for delineating gating boundaries. By including FMO controls, the maximum fluorescence expected for a staining subset is revealed in a given channel when the fluorochrome-tagged antibody specific for that particular fluorescence channel is excluded38. In our experience, in addition to single stained compensation particle controls, an unstained biological comparison control should be included for setting more precise positive/negative boundaries.
Excluding cellular debris and cell aggregates in the gating strategy is also an additional approach used to minimize auto-fluorescence. To distinguish cellular debris from the viable cell population, using forward scatter (FSC) and side scatter (SSC) is the most common gating strategy. For isolated cells that will be used for post sort analyses and require higher viability for DNA/RNA extractions, it is recommended that viability stains not be used with any fixation and/or permeabilization procedures. Formaldehyde fixing of cells can compromise nucleic acid integrity due to nucleic acid-protein crosslinking and thus limit isolation efficiency, detection, and accurate quantification. Cell aggregates as mentioned before cannot only contribute to emitted auto-fluorescent signals, but also result in "coincidence aborts". Such action occurs to maintain high purity in a sort but if too frequent, it reduces the sorted cell yield. Sorting buffer (FACS buffer) supplemented with DNAse and MgCl2 during cell staining can minimize cell aggregation. It is recommended to not use EDTA in combination with DNAse as it inhibits the enzyme's activity. Filtering the single cell suspension through a 70 µm cell strainer prior to sorting can also liberate cells from aggregates. It is imperative to note that to minimize aggregation, cell suspensions should be at a concentration of 5 million cells per mL in a minimum volume of 0.4 mL. Cells isolated from lipid laden tissues tend to be more prone to aggregation and this can result in coincidence aborts and low sort yield. It is recommended that samples are further diluted if aggregation persists. Sorted macrophages can be collected in polypropylene round-bottom collection tubes containing fetal bovine rich medium to maximize cell recovery. An initial high FBS concentration ensures cell recovery since the concentration eventually becomes diluted with each sorted droplet. For collecting cells that will be used for DNA or RNA analysis, cells can be sorted directly into the appropriate extraction reagent (e.g. TriZol) to prevent RNAse contamination. When sorting high volumes, sorting cells first into culture media supplemented with lower concentrations of FBS, is recommended. Immediately following sorting, cells should then be pelleted and lysed for DNA/RNA extraction. The isolated genetic material can then be profiled for altered gene expression. Pro-inflammatory genes including TNFα, IL1-β, IL-6, IL-12, 1L-23, IFNγ, Nos2, and MCP1 (CCL2) are often upregulated in macrophages exhibiting a classically activated (M1) phenotype39. On the other hand, alternatively activated (M2) phenotype in macrophages is often marked by induction of genes encoding Chi3l3 (Ym1), Fizz1, Arginase 1, CD206, CD163, CD209 1L-10, and TGFβ34,40. Recently, CD38, Fpr2, and Gpr18 have been validated as M1-specific genes and c-Myc and Egr2 as M2-specific genes34.
Although multi-color flow cytometry-based cell sorting is a valuable tool for isolating macrophages at high purity, this approach can be costly. The powerful advantages of FACS mediated sorting are dependent on operations personnel that can maneuver a cell sorter in addition to the high cost of cell sorter maintenance reagents. Alternative approaches can be used in substitute of costly flow cytometry based cell sorting. They include magnetic activated cell sorting (MACS) or density gradient centrifugation. The first alternative cell sorting method mentioned encompasses magnetic and/or microbead column isolation kits to separate cells of interest from blood or solid tissues41. The second cell sorting approach separates a heterogeneous cell suspension based on density and force of centrifugation. Unfortunately, density mediated centrifugation is not practical for isolating macrophages from aorta- or WAT-derived single cell suspensions. Oftentimes, the product obtained from differential centrifugation is contaminated, and of low yield. Consequently, smaller tissues (such as the aorta or WAT) that result in few cells initially following enzymatic digestion are not ideal candidates for differential centrifugation. On the other hand, cell suspensions derived from dissociated livers can produce an adequate number of sorted macrophages that can be used in post sort experiments and analyses such as culture stimulations, qPCR, or western blotting analysis. These alternative approaches can also be used to enrich populations prior to FACS, allowing for cleaner sorts. Of note, tissue resident macrophages make up a small percentage of the entire cell population in WAT, liver and the aorta. Enrichment of cells prior to FACS sorting has been an approach used when isolating populations of cell that are less frequent. One issue that is common in isolating small populations is that large cell numbers must be processed to obtain enough cells for subsequent analysis. Enrichment or pre-sorting can be used to resolve such an issue. This method aids in obtaining a more concise population of cells through positive and negative selection but it also allows conservation of time as FACS sorting can be an enduring process for dense tissue sources such as the liver.
Recent advances in inflammation biology highlight the importance of phenotypic and functional characterization of macrophage heterogeneity to further the understanding of the complex role of these immune cells in regulating chronic inflammation. In brief, this comprehensive protocol provides a multi-dimensional approach to characterizing tissue resident macrophages from three hallmark tissues studied in established diet induced obesity and inflammation models. More importantly this protocol takes into account the difficulty and the measures necessary to isolate clean single cell suspensions from dysregulated inflamed tissues such as WAT, aortic plaques and fatty livers. The protocol allows the researcher to apply flow cytometry and FACS sorting tools in an innovative dimension to characterize tissue resident macrophages, key regulators of inflammation in obesity. In-depth characterization of macrophage population dynamics can provide insight into monocyte trafficking in inflamed tissues and allow for continued mechanistic evaluations through a multitude of experimental approaches on sorted macrophages. Further characterization of macrophage populations can provide a pivotal insight into the biological underpinnings that regulate macrophage heterogeneity in health and disease.
Disclosures
Authors have no conflict of interests to disclose.
Acknowledgments
We would like to thank the Flow Cytometry Core Facility at The Pennsylvania State University Millennium Science Complex.
References
- Davies LC, Taylor PR. Tissue-resident macrophages: Then and Now. Immunology. 2015;144(4):541–548. doi: 10.1111/imm.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444(12):840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444(12):875–880. doi: 10.1038/nature05487. [DOI] [PubMed] [Google Scholar]
- Jung U, Choi M-S. Obesity and Its Metabolic Complications: The Role of Adipokines and the Relationship between Obesity, Inflammation, Insulin Resistance, Dyslipidemia and Nonalcoholic Fatty Liver Disease. Int J Mol Sci. 2014;15(4):6184–6223. doi: 10.3390/ijms15046184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuela F, Grazia M, Marco DR, Maria Paola L, Giorgio F, Marco B. Inflammation as a link between obesity and metabolic syndrome. Nutr Metab. 2012;2012:1–7. doi: 10.1155/2012/476380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira-Potter VJ. Inflammation and macrophage modulation in adipose tissues. Cell Microbiol. 2014;16(10):1484–1492. doi: 10.1111/cmi.12336. [DOI] [PubMed] [Google Scholar]
- Chawla A, Nguyen KD, Goh YPS. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol. 2011;11(11):738–749. doi: 10.1038/nri3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey A, Allen J, Hankey-giblin PA. Ontogeny and polarization of macrophages in inflammation : blood monocytes versus tissue macrophages. Front Immunol. 2015;5(1):1–15. doi: 10.3389/fimmu.2014.00683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills CD, Lenz LL, Ley K. Macrophages at the fork in the road to health or disease. Front Immunol. 2015;6(2):1–6. doi: 10.3389/fimmu.2015.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: Phenotypical vs. functional differentiation. Front Immunol. 2014;5(8):1–22. doi: 10.3389/fimmu.2014.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilherme A, Virbasius JV, Vishwajeet P, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9(5):367–377. doi: 10.1038/nrm2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TD, Holden CR, et al. Metabolic remodeling of white adipose tissue in obesity. Am J Physiol Endocrinol Metab. 2014;307(3):262–277. doi: 10.1152/ajpendo.00271.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisaka S, Usui I, et al. Regulatory Mechanisms for Adipose Tissue M1 and M2 Macrophages in Diet Induced Obese Mice. Diabetes. 2009;58(9):2574–2582. doi: 10.2337/db08-1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotipic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi K, Abrams GA. Metabolic liver disease of obesity and role of adipose tissue in the pathogenesis of nonalcoholic fatty liver disease. World J Gastroenterol. 2007;13(26):3540–3553. doi: 10.3748/wjg.v13.i26.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedossa P, Paradis V. Liver extracellular matrix in health and disease. J Pathol. 2003;200(4):504–515. doi: 10.1002/path.1397. [DOI] [PubMed] [Google Scholar]
- Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology. 2010;52(5):1836–1846. doi: 10.1002/hep.24001. [DOI] [PubMed] [Google Scholar]
- Milic S, Lulic D, Stimac D. Non-alcoholic fatty liver disease and obesity: Biochemical, metabolic and clinical presentations. World J Gastroenterol. 2014;20(28):9330–9337. doi: 10.3748/wjg.v20.i28.9330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams IL, Wheatcroft SB, Shah AM, Kearney MT. Obesity, atherosclerosis and the vascular endothelium: mechanisms of reduced nitric oxide bioavailability in obese humans. Int J Obesity. 2002;26(12):754–764. doi: 10.1038/sj.ijo.0801995. [DOI] [PubMed] [Google Scholar]
- Mestas J, Ley K. Monocyte-Endothelial Cell Interactions in the Development of Atherosclerosis. Trends Cardiovasc Med. 2008;18(6):228–232. doi: 10.1016/j.tcm.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Rev Immunol. 2013;14(10):986–995. doi: 10.1038/ni.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S, Campbell HM, Dittel BN, Ray A. Purification of specific cell population by fluorescence activated cell sorting (FACS) J Vis Exp. 2010. p. e1546. [DOI] [PMC free article] [PubMed]
- Mann A, Thompson A, Robbins N, Blomkalns AL. Localization, Identification, and Excision of Murine Adipose Depots. J Vis Exp. 2014. p. e52174. [DOI] [PMC free article] [PubMed]
- Smedsrød B. Protocol for preparation of mouse liver Kupffer cells and liver sinusoidal endothelial cells. 2012;1:1–10. Available from: http://munin.uit.no/handle/10037/4575. [Google Scholar]
- Butcher MJ, Herre M, Ley K, Galkina E. Flow cytometry analysis of immune cells within murine aortas. J Vis Exp. 2011. p. e2848. [DOI] [PMC free article] [PubMed]
- Salamone M, Saladino S, Pampalone M. Tissue Dissociation and Primary Cells Isolation Using Recombinant Collagenases Class I and II. Chemical Engineering Transactions. 2014;38:247–252. Matsushita 1994. [Google Scholar]
- Wells RG. The role of matrix stiffness in regulating cell behavior. Hepatology. 2008;47(4):1394–1400. doi: 10.1002/hep.22193. [DOI] [PubMed] [Google Scholar]
- Fain JN. Isolation of Free Brown and White Fat Cells. Diabetologia. 1968;375(1964):555–561. doi: 10.1016/0076-6879(75)35184-7. [DOI] [PubMed] [Google Scholar]
- Seglen P. Preparation of isolated rat liver cells. Method Cell Biol. 1976;13:29–83. doi: 10.1016/s0091-679x(08)61797-5. [DOI] [PubMed] [Google Scholar]
- Yu S, Allen JN, et al. The Ron Receptor Tyrosine Kinase Regulates Macrophage Heterogeneity and Plays a Protective Role in Diet-Induced Obesity, Atherosclerosis, and Hepatosteatosis. J Immunol. 2016;197(1):256–265. doi: 10.4049/jimmunol.1600450. [DOI] [PubMed] [Google Scholar]
- Yu YRA, O'Koren EG, et al. A protocol for the comprehensive flow cytometric analysis of immune cells in normal and inflamed murine non-lymphoid tissues. PLoS ONE. 2016;11(3):1–23. doi: 10.1371/journal.pone.0150606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zizzo G, Hilliard BA, Monestier M, Cohen PL. Efficient clearance of early apoptotic cells by human macrophages requires "M2c" polarization and MerTK induction. J Immunol. 2012;100(2):130–134. doi: 10.4049/jimmunol.1200662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonski KA, Amici SA, et al. Novel markers to delineate murine M1 and M2 macrophages. PLoS ONE. 2015;10(12):5–11. doi: 10.1371/journal.pone.0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rőszer T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediators of Inflamm. 2015;2015:1–16. doi: 10.1155/2015/816460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho KW, Morris DL, Lumeng CN. Flow Cytometry Analysis of Adipose Tissue Macrophages. Methods Enzymol. 2011;4(164):297–314. doi: 10.1016/B978-0-12-411619-1.00016-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y-J, Brox T, Feiden W, Weickert J. Technical Note: Flow Cytometry Controls, Instrument Setup, and the Determination of Positivity. Cytometry A. 2007;71(1):8–15. [Google Scholar]
- Tung JW, Heydari K, et al. Modern Flow Cytometry: A Practical Approach. Clin Lab Med. 2007;27(3):453–468. doi: 10.1016/j.cll.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000. 2014;6(3):13. doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcheray F, Viaud S, et al. Macrophage activation switching: An asset for the resolution of inflammation. Clin Exp Immunol. 2005;142(3):481–489. doi: 10.1111/j.1365-2249.2005.02934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plouffe BD, Murthy SK, Lewis LH. Fundamentals and Application of Magnetic Particles in Cell Isolation and Enrichment. Rep Prog Phys. 2010;1(78):1–6. doi: 10.1088/0034-4885/78/1/016601. [DOI] [PMC free article] [PubMed] [Google Scholar]