

Abstract
Next generation sequencing (NGS) technologies have revolutionized the nature of biological investigation. Of these, RNA Sequencing (RNA-Seq) has emerged as a powerful tool for gene-expression analysis and transcriptome mapping. However, handling RNA-Seq datasets requires sophisticated computational expertise and poses inherent challenges for biology researchers. This bottleneck has been mitigated by the open access Galaxy project that allows users without bioinformatics skills to analyze RNA-Seq data, and the Database for Annotation, Visualization, and Integrated Discovery (DAVID), a Gene Ontology (GO) term analysis suite that helps derive biological meaning from large data sets. However, for first-time users and bioinformatics' amateurs, self-learning and familiarization with these platforms can be time-consuming and daunting. We describe a straightforward workflow that will help C. elegans researchers to isolate worm RNA, conduct an RNA-Seq experiment and analyze the data using Galaxy and DAVID platforms. This protocol provides stepwise instructions for using the various Galaxy modules for accessing raw NGS data, quality-control checks, alignment, and differential gene expression analysis, guiding the user with parameters at every step to generate a gene list that can be screened for enrichment of gene classes or biological processes using DAVID. Overall, we anticipate that this article will provide information to C. elegans researchers undertaking RNA-Seq experiments for the first time as well as frequent users running a small number of samples.
Keywords: Genetics, Issue 122, RNA sequencing, RNA-Seq, Transcriptomics, Gene Expression, Galaxy Project, Tuxedo, Database for Annotation, Visualization and Integrated Discovery (DAVID), C.elegans, Next-Generation Sequencing (NGS), Transcription Profiling, Genomics
Introduction
The first sequencing of the human genome, performed using Fred Sanger's dideoxynucleotide-sequencing method, took 10 years, and cost an estimated US $3 billion1,2. However, in a little over a decade since its inception, Next-Generation Sequencing (NGS) technology has made it possible to sequence the entire human genome within two weeks and for US $1,000. New NGS instruments that allow ever-increasing speeds of sequencing-data collection with incredible efficiency, along with sharp reductions in cost, are revolutionizing modern biology in unimaginable ways as genome sequencing projects are rapidly becoming commonplace. In addition, these developments have galvanized progress in many other areas such as gene-expression analysis through RNA-Sequencing (RNA-Seq), study of genome-wide epigenetic modifications, DNA-protein interactions, and screening for microbial diversity in human hosts. NGS-based RNA-Seq in particular has made it possible to identify and map transcriptomes comprehensively with accuracy and sensitivity, and has replaced microarray technology as the method of choice for expression profiling. While microarray technology has been used extensively, it is limited by its reliance on pre-existing arrays with known genomic information, and other drawbacks such as cross hybridization and restricted range of expression changes that can be measured reliably. RNA-seq, on the other hand, can be used to detect both known and unknown transcripts while producing low background noise due to its unambiguous DNA mapping nature. RNA-Seq, together with the numerous genetic tools offered by model organisms such as yeast, flies, worms, fish and mice, has served as the foundation for many important recent biomedical discoveries. However, significant challenges remain that make NGS inaccessible to the wider scientific community, including limitations of storage, processing, and most of all, meaningful bioinformatics analysis of large volumes of sequencing data.
The rapid advances in sequencing technologies and exponential data accumulation have created a great need for computational platforms that will allow researchers to access, analyze and understand this information. Early systems were heavily dependent upon computer programming knowledge, whereas, genome browsers such as NCBI that allowed non-programmers to access and visualize data did not permit sophisticated analyses. The web-based, open-access platform, Galaxy (https://galaxyproject.org/), has filled this void and proven to be a valuable pipeline that enables researchers to process NGS data and perform a spectrum of simple-to-complex bioinformatics analyses. Galaxy was initially established, and is maintained, by the laboratories of Anton Nekrutenko (Penn State University) and James Taylor (Johns Hopkins University)3. Galaxy offers a wide range of computational tasks making it a 'one-stop shop' for innumerable bioinformatics needs, including all the steps involved in an RNA-Seq study. Itallows users to perform data processing either on its servers or locally on their own machines. Data and workflows can be reproduced and shared. Online tutorials, help section, and a wiki-page (https://wiki.galaxyproject.org/Support) dedicated to the Galaxy Project provide consistent support. However, for first-time users, especially those with no bioinformatics training, the pipeline can appear daunting and the process of self-learning and familiarization can be time consuming. In addition, the biological system studied, and specifics of the experiment and methods used, impact the analytical decisions at several steps, and these can be difficult to navigate without instruction.
The Overall RNA-Seq Galaxy Workflow consists of data upload and quality check followed by analysis using the Tuxedo Suite4,5,6,7,8,9, which is a collective of various tools required for different stages of RNA-Seq data analysis10,11,12,13,14. A typical RNA-Seq experiment consists of the experimental part (sample preparation, mRNA isolation and cDNA library preparation), the NGS and the bioinformatics data analysis. An overview of these sections, and the steps involved in the Galaxy pipeline, are shown in Figure 1.
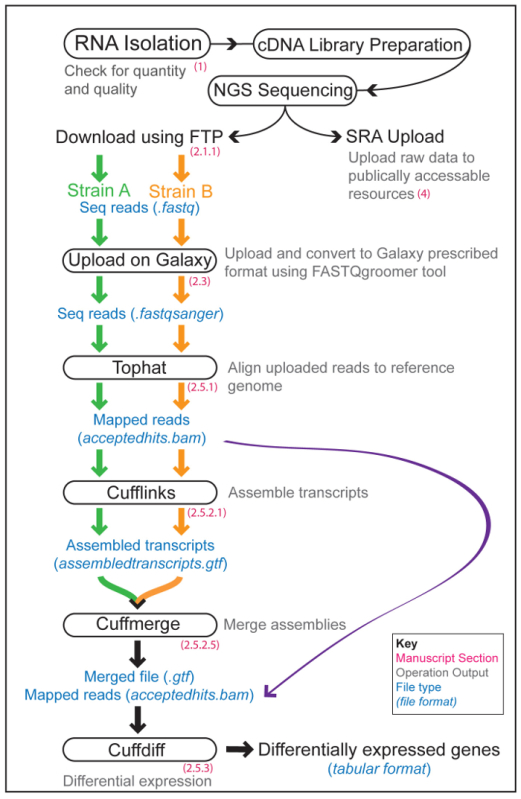 Figure 1: Overview of an RNA-Seq Workflow. Illustration of the experimental and computational steps involved in an RNA-Seq experiment to compare the gene-expression profiles of two worm strains (A and B, orange and green lines and arrows, respectively). The different modules of Galaxy utilized are shown in boxes with the corresponding step in our protocol indicated in red. The outputs of various operations are written in grey with the file formats shown in blue. Please click here to view a larger version of this figure.
Figure 1: Overview of an RNA-Seq Workflow. Illustration of the experimental and computational steps involved in an RNA-Seq experiment to compare the gene-expression profiles of two worm strains (A and B, orange and green lines and arrows, respectively). The different modules of Galaxy utilized are shown in boxes with the corresponding step in our protocol indicated in red. The outputs of various operations are written in grey with the file formats shown in blue. Please click here to view a larger version of this figure.
The first tool in the Tuxedo Suite is an alignment program called 'Tophat'. It breaks down the NGS input reads into smaller fragments and then maps them to a reference genome. This two-step process ensures that reads spanning intronic regions whose alignment can otherwise be disrupted or missed are accounted for and mapped. This increases coverage and facilitates the identification of novel splice junctions. Tophat output is reported as two files, a BED file (with information about splice junctions that include genomic location) and a BAM file (with mapping details of each read). Next, the BAM file is aligned against a reference genome to estimate the abundance of individual transcripts within each sample using the subsequent tool in the Tuxedo Suite called 'Cufflinks'. Cufflinks functions by scanning the alignment to report full-length transcript fragments or 'transfrags' that span all the possible splice variants in the input data for every gene. Based on this, it generates a 'transcriptome' (assembly of all the transcripts generated per gene for every gene) for each sample being sequenced. These Cufflinks assemblies are then collapsed or merged together along with the reference genome to produce a single annotation file for downstream differential analysis using the next tool, 'Cuffmerge'. Finally, the 'Cuffdiff' tool measures differential gene expression between samples by comparing the TopHat outputs of each of the samples to the final Cuffmerge output file (Figure 1). Cufflinks uses FPKM/RPKM (Fragments/Reads Per Kilobase of transcript per Million mapped reads) values to report transcript abundances. These values reflect the normalization of the raw NGS data for depth (average number of reads from a sample that align to the reference genome) and gene length (genes have different lengths, so counts have to be normalized for length of a gene to compare levels between genes). FPKM and RPKM are essentially the same with RPKM being used for single-end RNA-Seq where every read corresponds to a single fragment, whereas, FPKM is used for paired-end RNA-Seq, as it accounts for the fact that two reads can correspond to the same fragment. Ultimately, the outcome of these analyses is a list of genes differentially expressed between the conditions and/or strains tested.
Once a successful Galaxy run is completed and a 'gene list' is generated, the next logical step requires more bioinformatics analyses to deduce meaningful knowledge from the datasets. Many software packages have emerged to cater to this need, including publicly-available web-based computational packages such as DAVID (the Database for Annotation, Visualization and Integrated discovery)15. DAVID facilitates assigning biological meaning to large gene lists from high-throughput studies by comparing the uploaded gene list to its integrated biological knowledgebase and revealing the biological annotations associated with the gene list. This is followed by Enrichment Analysis, i.e., tests to identify if any biological process or gene class is overrepresented in the gene list(s) in a statistically significant manner. It has become a popular choice because of a combination of a wide, integrated knowledge-base and powerful analytical algorithms that enable researchers to detect biological themes enriched within genomics-derived 'gene lists'10,16. Additional advantages include its ability to process gene lists created on any sequencing platform and a highly user-friendly interface.
The nematode Caenorhabditis elegans is a genetic model system, well known for its many advantages such as small size, transparent body, simple body plan, ease of culture and great amenability to genetic and molecular dissection. Worms have a small, simple and well-annotated genome that includes up to 40% conserved genes with known human homologs17. Indeed, C. elegans was the first metazoan whose genome was completely sequenced18, and one of the first species where RNA-Seq was used to map an organism's transcriptome19,20. Early worm studies involved experimentation with different methods for high-throughput RNA capture, library preparation and sequencing as well as bioinformatics pipelines that contributed to the advancement of the technology21,22. In recent years, RNA-Seq-based experimentation in worms has become commonplace. But, for traditional worm biologists the challenges posed by computational analysis of RNA-Seq data remain a major impediment for greater and better utilization of the technique.
In this article, we describe a protocol for using the Galaxy platform to analyze high-throughput RNA-Seq data generated from C. elegans. For many first-time and small-scale users, the most cost-efficient and straightforward way to undertake an RNA-Seq experiment is to isolate RNA in the lab and utilize a commercial (or in-house) NGS facility for preparation of sequencing cDNA libraries and the NGS itself. Hence, we have first detailed the steps involved in isolation, quantification and quality assessment of C. elegans RNA samples for RNA-Seq. Next, we provide step-by-step instructions for using the Galaxy interface for analyses of the NGS data, beginning with tests for post-sequencing quality-control checks followed by alignment, assembly, and differential quantification of gene expression. In addition, we have included directions to scrutinize the gene lists resulting from Galaxy for biological enrichment studies using DAVID. As a final step in the workflow, we provide instructions for uploading RNA-Seq data on to public servers such as the Sequence Read Archive (SRA) on NCBI (http://www.ncbi.nlm.nih.gov/sra) to make it freely accessible to the scientific community. Overall, we anticipate that this article will provide comprehensive and sufficient information to worm biologists undertaking RNA-Seq experiments for the first time as well as frequent users running a small number of samples.
Protocol
1. RNA Isolation
- Precautionary measures
- Wipe down the entire working surface, instruments and pipettes using a commercially-available RNase spray to eliminate any RNases present.
- Wear gloves at all times, regularly changing them with fresh ones during the different steps of the protocol.
- Use only filter tips and keep all samples on ice as much as possible to avoid RNA degradation. NOTE: In order to obtain the best data from NGS platforms, it is critical to begin with high-quality RNA. RNA isolation and preparation methods vary depending on sample origin, method of sequencing and investigator preference. Several commercially available kits can be used for this purpose or RNA can also be isolated using a standard phenol-chloroform method of RNA extraction. With either methodology, the precautionary measures listed above should be followed throughout the process to minimize contamination and obtain pristine RNA samples.
- Harvesting Worms
- Synchronize the worm population by hypochlorite bleaching treatment23 to obtain 1,000-1,500 age-matched C. elegans adult worms per strain.
- Wash the worms off plates using M9 buffer solution and spin at 325 x g on a table top centrifuge for 30 s. Aspirate out the M9 buffer leaving behind a pellet of worms. Repeat this step at least thrice to eliminate bacterial carryover.
- To the worm pellet, add ~ 500 µL of lysis buffer (if using a commercial kit) or Trizol (a mono-phasic solution of phenol and guanidine isothiocyanate; if phenol:chloroform extraction described in 1.3.3 is undertaken) to disrupt worm tissues, deactivate RNases and stabilize nucleic acids. NOTE: The protocol can be paused here by flash freezing the samples in liquid nitrogen followed by storage at -80 °C.
- RNA Isolation
- Sonicate worm samples at 45% amplitude in cycles of 20 s. 'ON' and 40 s. 'OFF' (8-12 cycles per strain). Keep samples on ice at all times. NOTE: Ensure that the sonicator probe is immersed in the buffer and is kept at a constant level throughout. Avoid frothing of the sample and clean the probe thoroughly in-between samples. Sonication cycles may vary depending on the type of sonicator used. It is recommended that sonication conditions are first optimized on a test sample before starting an experiment.
- If using a commercially available kit, proceed with RNA Isolation as per the prescribed protocol. For RNA isolation using a phenol-chloroform method, perform the following steps.
- Centrifuge sonicated samples at 16,000 x g for 10 min. at 4° C.
- Transfer supernatant into a 1.5 mL RNase-free microfuge tube and add 100 µL of chloroform (1/5th the volume of RNA/DNA isolation reagent). Caution: Chloroform is toxic. To minimize exposure and avoid inhalation, work in a chemical hood when handling this substance.
- Vortex the samples thoroughly for 30 - 60 s. and let the samples sit at room temperature for 3 min.
- Centrifuge at 11,750 x g for 15 min. at 4 °C. Transfer only the top aqueous layer to a new RNase-free microfuge tube taking care not to aspirate the DNA-containing white interface. Repeat steps 1.3.4 through 1.3.6.
- Add 250 µL (70% of aqueous phase or 1/2 RNA/DNA isolation reagent volume) of 2-propanol and invert the tube to mix. Let tubes sit at room temperature for 10 min or leave overnight at -80 °C.
- Centrifuge samples at 11,750 x g for 10 min. at 4 °C. Decant the supernatant very carefully, leaving behind a few µL at the bottom of the tube so that the pellet is not disturbed.
- Wash pellet with 500 µL of 75% ethanol (made using RNase-free water) and spin down at 16,000 x g for 5 min. at 4 °C.
- Remove as much supernatant as possible without disturbing the pellet. Air dry the pellet in a hood for a few minutes.
- Add 30 µL of RNase-free water and help dissolve the RNA pellet by heating for 10 min. at 60 °C.
- Check RNA quality and quantity using a bioanalyzer. NOTE: Bioanalyzer generates an RNA Integrity Number (RIN) as a measure of RNA quality. An RIN of at least 8 is the recommended threshold for RNA-Seq samples (higher is better). RNA quantity and quality can also be checked spectrophotometrically but should also be followed by visual assessment of RNA integrity. To do this, run the samples on a 1.2% agarose gel long enough to obtain suitable separation of the 28s and 18s ribosomal RNA bands. The presence of two distinct bands (1.75 kb for 18s rRNA and 3.5 kb for 28s rRNA in the case of C. elegans) is an acceptable measure of RNA quality.
- Use ~100 ng/µL RNA to ship to the vendor/NGS facility for preparation of sequencing libraries. NOTE: RNA samples should be shipped on dry ice to the sequencing service provider. Most providers conduct an independent RNA quality-control test before library preparation.
2. RNA-Seq Data Analysis
- Download of Raw Sequencing Data
- Download the compressed raw fastq sequencing data encoded in the fastq.gz format from the NGS provider using a "file transfer protocol" (ftp).
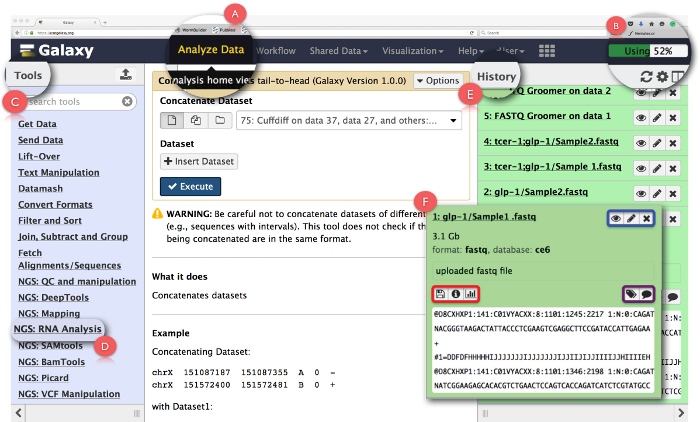 Figure 2: Layout of the Galaxy User Interface Panel and Key RNA-Seq Functions. Key features of the page are expanded and highlighted. (A) highlights the 'Analyze data' function in the webpage header used to access Analysis Home View. (B) is the 'Progress bar' that indicates the space on the Galaxy server utilized by the operation. (C) is the 'Tools Section' that lists all the tools that can be run on the Galaxy interface. (D) shows the 'NGS: RNA Analysis' tool section used for RNA-Seq analysis. (E) depicts the 'History' panel that lists all the files generated using Galaxy. (F) shows an example of the dialogue box that opens up when clicking on any file in the History section. Within (F), the blue box highlights icons that can be used to view, editthe attributes or delete the dataset, the purple box highlights icons that can be used to 'edit' the dataset tags or annotation, and, the red box indicates icons to download the data, view details of the task performed or rerun the operation. Please click here to view a larger version of this figure.
Figure 2: Layout of the Galaxy User Interface Panel and Key RNA-Seq Functions. Key features of the page are expanded and highlighted. (A) highlights the 'Analyze data' function in the webpage header used to access Analysis Home View. (B) is the 'Progress bar' that indicates the space on the Galaxy server utilized by the operation. (C) is the 'Tools Section' that lists all the tools that can be run on the Galaxy interface. (D) shows the 'NGS: RNA Analysis' tool section used for RNA-Seq analysis. (E) depicts the 'History' panel that lists all the files generated using Galaxy. (F) shows an example of the dialogue box that opens up when clicking on any file in the History section. Within (F), the blue box highlights icons that can be used to view, editthe attributes or delete the dataset, the purple box highlights icons that can be used to 'edit' the dataset tags or annotation, and, the red box indicates icons to download the data, view details of the task performed or rerun the operation. Please click here to view a larger version of this figure.
- Getting Started with Galaxy NOTE: Galaxy can be run on a free public server using a web-based platform providing cloud access and free limited storage. It can also be downloaded and run locally on the user's machine or computational clusters hosted by institutions but local processing, may be constrained by data-storage limits and processing power limitations of user machines. Details on downloading and installation can be accessed at https://wiki.galaxyproject.org/Admin/GetGalaxy. In this protocol we describe the web-based usage of the Galaxy pipeline.
- After downloading and storing the NGS data on the user's machine, access Galaxy at https://usegalaxy.org/.
- Register a user account by clicking on 'User' in the header of the page, login and begin by getting acquainted with the user interface panel. NOTE: It is recommended that first time users utilize the 'Start here' tutorial provided on the home page to get familiarized with the basic set up of Galaxy (https://github.com/nekrut/galaxy/wiki/Galaxy101-1).
- Click on 'Analyze Data' (Figure 2A) in the header panel to access the 'Analysis Home View' which is also the startup screen on Galaxy. NOTE: The header also houses other links whose details can be seen by hovering the mouse pointer over them. The upper right-hand corner of the header has a progress bar that monitors space utilized for the tasks (Figure 2B).
- Click on 'NGS: RNA Analysis' task in the 'Tools Menu' on the left panel (Figure 2C) to access all the tools required for RNA-seq data analysis. NOTE: The 'Tools Menu' catalogs all the operations that Galaxy offers. This menu is split based on tasks and clicking on any one will open up a list of all the tools needed to accomplish that task.
- Create new analysis history by clicking on the gear icon at the top of the 'History' panel on the right (Figure 2E). Choose 'Create New' option from the pop-up menu. Give this 'History' a suitable name to identify the analysis. NOTE: The 'History' panel shows all the files uploaded for analysis as well as all the output files that are generated by running tasks on Galaxy. Clicking on a file name in this panel opens up a dialogue box with detailed information about the task performed and a snippet of the dataset (Figure 2F). Icons in this box enable the user to 'view', 'edit the attributes' or 'delete' the dataset (Figure 2F, highlighted in blue). Additionally, the user can also 'edit' dataset tags or annotation (Figure 2F, highlighted in purple), 'download' the data, 'view details' of the task, 'rerun' the task or even 'visualize' the dataset from this dialogue box (Figure 2F, highlighted in red).
- Click the 'Upload File' function under 'Get Data' in the 'ToolsMenu' to upload raw fastq files. NOTE: Clicking on this or any other tool opens up a short description of the operation, and the test itself, in the middle 'Analysis Interface' panel. This panel laces together the 'Tools' from the left panel and the 'Input Files' from the right 'History' panel (Figure 2E). Here, input files from 'History' are selected and other parameters defined to run a given task. The resultant output dataset from every test is saved back in 'History'. Included with the test in the 'Analysis Interface" panel are explanations for all the parameters available for running a given tool along with a detailed list of all the output files the tool generates.
- After the task opens in the 'Analysis Interface', click on 'Choose Local File' or 'Choose FTP File' (faster upload), navigate to the folder containing the sequencing files and select the appropriate dataset to be uploaded.
- Allow Galaxy to 'Auto-detect' the uploaded file type (default setting). Select 'C. elegans' in the pull down menu for the genome.
- Click on 'Start' to initiate data upload. Once the file is uploaded, it will be saved in the 'History' panel and can be accessed from there.
- If multiple sequencing data files are produced for a single sample, combine them using the 'Concatenate' tool. To do this, open up the 'Text Manipulation' option in the 'Tools Menu'.
- Click on the 'Concatenate' tool, choose the files that need to be combined from the drop-down box in the middle of 'Analysis interface' and click 'Execute'. NOTE: Output files produced using this task are generated in the fastq format. The mapping program has a limit of 16,000,000 sequences per fastq file and when that limit is reached a new fastq file is generated for the remaining sequences. The 'Concatenate' tool is needed in such instances to combine the datasets.
- Convert the uploaded fastq format files to the required fastqsanger format for Galaxy RNA-Seq analysis by using the 'fastq groomer' tool found under the 'NGS: QC and manipulation' section (see supplemental file).
- Choose the appropriate fastq dataset under the 'File to Groom' option and run the tool using default parameters. NOTE: Output files produced using this task are generated in the fastqsanger format.
- fastqsanger Data Quality-Control Tests
- Check the quality of the uploaded fastqsanger reads using the 'FastQC' tool located under 'NGS: QC and manipulation' in the 'Tools' Menu.
- Choose the groomed fastqsanger data file from the dropdown menu for 'Short read data from the current library' and run the tool using default parameters. NOTE: Pay special attention to the quality of the reads and presence of any adapter sequences. Adapters are usually removed as part of the post RNA-Seq data processing by NGS providers but in some instances, may be left behind. For explanation of quality standards go to http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Check with the NGS provider and if adapters are present, trim them using the 'Clip' tool from the 'NGS: QC and manipulation' task menu. NOTE: Output files produced using this task are generated in the raw txt format as well as in html that can be opened on any web browser.
- Data Analysis with Tuxedo Suite
- TopHat
- Download the latest version of C. elegans reference genome fasta and gtf (Gene Transfer Format) files from Upload file' as described above in 2.2.6.
- Open the 'NGS: RNA Analysis' section and click on the 'TopHat' tool to map the sequencing reads to the downloaded reference genome.
- Select the appropriate answer from the dropdown menu to the question 'Is this single-end or paired-end data?'
- Choose the appropriate fastq file.
- Select 'Use a genome from history' in the next dropdown menu and choose reference genome downloaded in step 2.4.1.1.
- Select 'Default' for the other parameters and click 'Execute'. NOTE: Among the output files produced using this task, the 'Accepted Hits' file is used for subsequent steps.
- Cufflinks and Cuffmerge
- Select the 'Cufflinks' tool in the 'NGS: RNA Analysis' section to assemble the transcripts, estimate their abundance and test for differential expression.
- In the first dropdown menu, choose the mapped 'Accepted hits (BAM format)' file obtained from TopHat analysis.
- In the second dropdown menu, set reference annotation to the gtf file downloaded in step 2.4.1.1.
- Select 'Yes' for the 'Perform bias correction' option and run the task using the default settings for all other parameters. NOTE: Among the output files produced using this task, the 'Accepted Transcripts' file is used for subsequent steps.
- Open 'Cuffmerge' tool in the 'NGS: RNA Analysis' to merge the 'Assembled Transcripts' produced for all the RNA-Seq samples. NOTE: The first box in the tool self-populates and lists all the gtf files produced by Cufflinks.
- Select the 'Assembled Transcripts' file for all the strains/conditions tested, including biological replicates of the same strain/condition (See discussion for biological replicates).
- Select 'Yes' for 'Use Reference Annotation' and choose the gtf file downloaded in step 2.4.1.1.
- In the following box, again select 'Yes' for the 'Use Sequence Data' option and choose the whole genome fasta file downloaded in step 2.4.1.1.
- Keeping the other parameters as default, click 'Execute'. NOTE: Cuffmerge generates a single gtf output file.
- Cuffdiff
- Navigate to the 'Cuffdiff' tool in the 'NGS: RNA Analysis' section. In the 'Transcripts' menu, select the merged output file from Cuffmerge.
- Label conditions 1 and 2 with the two strains/condition names. NOTE: Cuffdiff can perform comparisons between more than two strains or conditions as well as time course experiments. Simply use the 'Add new conditions' option to add each new strains/condition, as needed.
- For each strain/condition, under 'Replicates' select individual 'Accepted Hits' output files from TopHat that correspond to the different biological replicates of that strain/condition. Hold down the 'cmd' key, if using a Macintosh computer, and 'ctrl' key, if using a PC, to select multiple files.
- Leave all other options as default parameters. Click 'Execute' to run the task. NOTE: Cuffdiff generates numerous output files in a tabular format as the final readout of the RNA-Seq analysis. These include files with FPKM tracking for transcripts, genes (combined FPKM values of transcripts sharing a gene identity), primary transcripts and coding sequences. All data files generated can be viewed on any spreadsheet application and contain similar attributes such as gene name, locus, fold change (in log2 scale) as well as statistical data on comparisons between strains/conditions, including p value and q values. The data in these files can be sorted based on statistical significance of differences or fold change in gene expression (magnitude and direction of change, as in up- or down- regulated genes) and manipulated as per the users' requirements. If conversion between different gene identifiers is needed (e.g., Wormbase gene ID vs. cosmid number), tools available on Biomart (http://www.biomart.org/) can be utilized.
3. Gene Ontology (GO) Term Analysis using DAVID
Access DAVID from the website https://david.ncifcrf.gov/. Click on 'Start Analysis' in the header of the webpage. In 'Step 1', copy and paste the list of genes obtained from Galaxy into box A. In 'Step 2', select 'Wormbase Gene ID' as the identifier for the input genes. NOTE: DAVID recognizes most publicly available annotation categories, so other gene identifiers (such as Entrez gene ID or gene symbol) can also be used.
In 'Step 3', choose 'Gene List' (genes to be analyzed) under 'List Type' and then click on the 'Submit List' icon. NOTE: 'Analysis Wizard', will open up to list all the hyperlinked DAVID tools that can be run on the uploaded gene list (Figure 3). Click on these links to access relevant corresponding modules as per the user's requirement. To identify the tools appropriate for a given task, click on 'Which DAVID tools to use?' link on the 'Analysis Wizard' page. Click on the 'Start Analysis' link in the header to return to the 'Analysis Wizard' home page at any point during the analysis.
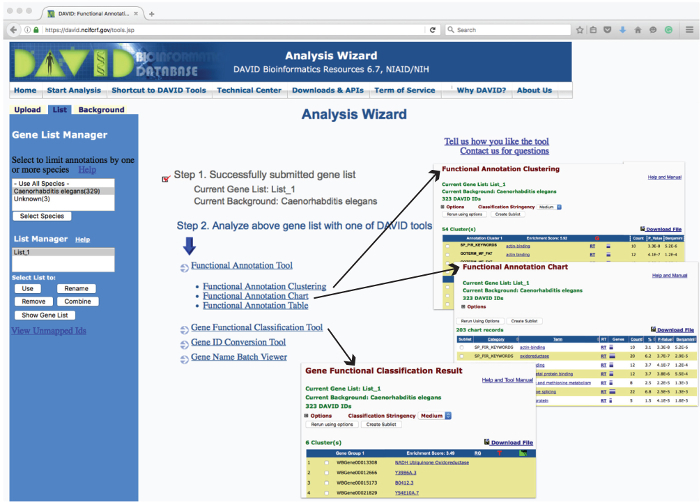 Figure 3: Layout of the DAVID Analysis Wizard Webpage and Examples of Operation Outputs. The 'Analysis Wizard' web user-interface lists the tools used to analyze uploaded gene list for enrichment based on various parameters. Clicking on these tools reports the analyzed data in a new web page. Examples of the tabular reports generated from 'Gene Functional Classification', 'Functional Annotation Chart' and 'Functional Annotation Clustering' are shown as insets (arrows). Please click here to view a larger version of this figure.
Figure 3: Layout of the DAVID Analysis Wizard Webpage and Examples of Operation Outputs. The 'Analysis Wizard' web user-interface lists the tools used to analyze uploaded gene list for enrichment based on various parameters. Clicking on these tools reports the analyzed data in a new web page. Examples of the tabular reports generated from 'Gene Functional Classification', 'Functional Annotation Chart' and 'Functional Annotation Clustering' are shown as insets (arrows). Please click here to view a larger version of this figure.
- Functional Annotation Tool 1: Functional Annotation Clustering
- Click on 'Functional Annotation Clustering' module to go to the summary page. Keep the default annotation categories and click on 'Functional Annotation Clustering' to generate clusters of similar annotation terms ranked by their enrichment score.
- Click on the hyperlinked name of each term to read details about it and 'RT' (related terms) to list other similar terms related to the category.
- Click on the purple bar to list the genes associated with a term and the red 'G' to list all the genes associated with all the terms within a cluster.
- Click on the green icon to see a two-dimensional view of all the genes and terms in a cluster. NOTE: The last three columns list the analytic and statistical results for each term. The results for this and all other analytics can be downloaded in a .txt format by clicking the 'Download File' link.
- Functional Annotation Tool 2: Functional Annotation Chart
- Return to the summary page and click on 'Functional Annotation Chart' to identify significantly overrepresented biological terms (e.g. transcription factor activity or kinase activity) associated with the gene list.
- Click on term name to get more detailed information and 'RT' (related terms) to list other related terms.
- Click on the purple bar to list all associated genes of corresponding individual category. NOTE: The last two columns list the statistical-tests' results for each category.
- Functional Annotation Tool 3: Functional Annotation Table
- Return to the summary page and click on 'Functional Annotation Table' to see a list of all the annotations associated with the genes on a list without any statistical calculations. NOTE: This tool can be useful for gene-by-gene analysis of a list or to look at specific, highly interesting genes.
- Gene Functional Classification Tool
- Return to 'Analysis Wizard' and click on 'Gene Functional Classification' module to segregate the input gene list into functionally-related groups of genes ranked as per their 'Enrichment Score', a measure of overall enrichment of the gene group in the list.
- Click on term name to get more detailed information and 'RG' to reveal functionally related genes of the gene group
- Click on the red 'T' (term reports) to list associated biology and the green icon to see a two-dimensional view of all the genes and terms.
- Gene-name Batch Viewer
- Return to 'Analysis Wizard' and click on 'Gene-name Batch Viewer' to translate 'Wormbase Gene IDs' into their corresponding gene names. (WBGene00022855 = tcer-1).
- Click on gene name to obtain more gene-specific information.
- Click on the 'RG' (related genes) link next to each gene to reveal genes predicted to be functionally related to the gene of interest.
4. Uploading RAW Data onto the NCBI Sequence Read Archive (SRA)
Access the SRA webpage at Sign in to NCBI' link or register a new account.
Click on 'Bioproject'.
Click on 'Submission' under the 'Using Bioproject' heading on the left.
Select the option 'New Submission'. Update details of the submitter. Continue through the remaining seven tabs, filling in the details of the experiment and data being uploaded. Click 'Submit' when completed. NOTE: In the fifth 'Biosample' tab, leave the slot for 'Biosample' empty.
Refresh the resulting page by clicking on the 'My Submissions' link. The submitted data will be listed with an assigned submission number, brief description and upload status.
Click on 'Biosample' at the top of this page, in the 'start a new submission' box and create a 'new submission'. Submit separate submissions for each sample.
As in the case with 'Bioproject' in 4.4, update the details of the submitter and continue through the rest of the tabs filling in the details of each tab. Once completed review and click 'Submit'.
Navigate to http://www.ncbi.nlm.nih.gov/sra to create the final ‘Sequence Read Archive (SRA)’ submission.
Click on 'Login to SRA' under 'Getting Started'.
On the next page click on the 'NCBI PDA' link. An 'Update Preferences' link will open up. Complete the form and click 'Save Preferences'.
On the resulting page, click on the 'Create New Submission' link. Enter a suitable name under 'Alias' and click 'Save'. A table with the submission ID and other details will be created.
Click on 'New Experiment' and register at least one unique sequencing library for each 'BioSample'.
Designate and link the previously created 'BioProject' and 'BioSample' submission ID's. A 'New Experiment' will be created.
Click on 'New Run' at the bottom of the page after the SRA Experiment has been made and identify the data files that need to be linked to it.
Compute the MD5 sum of each data file. To do this on a MacIntosh terminal, navigate to Applications/Utilities/Terminal. In terminal, type in 'md5' (without the quotes) followed by a space. Drag and drop the files that need to be uploaded into terminal from finder and click 'Enter'.
Terminal will return an alphanumeric MD5 sum. Enter this as part of the submission process for the file upload. Use the username and password provided by the system to upload files using FTP.
Representative Results
In C. elegans, elimination of the germline stem cells (GSCs) extends lifespan, enhances stress resilience, and elevates body fat24,28. Loss of GSCs, either brought about by laser-ablation or by mutations such as glp-1, causes lifespan extension through activation of a network of transcription factors29. One such factor, TCER-1, encodes the worm homolog of the human transcription elongation and splicing factor, TCERG130. The following representative results illustrate how RNA-Seq was used to identify genes whose expression is modulated by TCER-1/TCERG1 following germline loss in our recently published study31. The transcriptomes of age-matched, day 2 adults of glp-1 and tcer-1;glp-1 mutants were compared. For each strain, mRNA was isolated from two biological replicates (four samples totally) using the protocol described in section 1. RNA samples were shipped to a commercial service provider that prepared cDNA libraries from the four samples and performed 50 bp single end sequencing. The raw NGS data was downloaded as described in section 2.1.
Post sequencing data evaluation
Table 1 is a compilation of test results to assess the quality of raw sequencing reads. 'FASTQ' quality check analysis highlights the number of sequences read with no 'poor quality' reads along with 48-49% GC content and a constant sequence read length of 51 bp. This step also checks the sequencing data for many other features such as Kmer content and is collectively made up of 11 tests in total. The C. elegans genome is ~100 Mbp. Based on the number of sequencing reads from each sample that mapped to the genome, the genome coverage (last column) was estimated using the Lander/Waterman equation 'C = LN/G', wherein, C stands for coverage, G is the haploid genome length, L is the read length and N is the number of reads. We used default parameters for all the steps and obtained 48 - 49% GC content in all the samples. As can be seen, genome coverage was between 9x to 11x in the samples.
Identification of TCER-1/TCERG-1-regulated Genes by Differential Gene Expression Analysis on Galaxy
Through the steps detailed in sections 2.2 to 2.4, the Galaxy pipeline3 was used to obtain a list of genes differentially expressed between glp-1 and tcer-1;glp-1 mutants. Galaxy enabled us to combine the NGS data from the two replicates for each strain and performed differential analysis to generate tabular files highlighting the genome wide expression profile. Using a threshold of at least one-fold change in magnitude and P value of at least 0.05, a list of 835 genes that were differentially expressed between the two strains was generated31. The list was divided based on whether expression of the genes was down-regulated in tcer-1;glp-1 mutants (359 UP genes whose transcription is likely enhanced by TCER-1/TCERG1) or up-regulated (476 DOWN genes whose transcription is likely repressed by TCER-1/TCERG1) as compared to glp-1 (Figure 4).
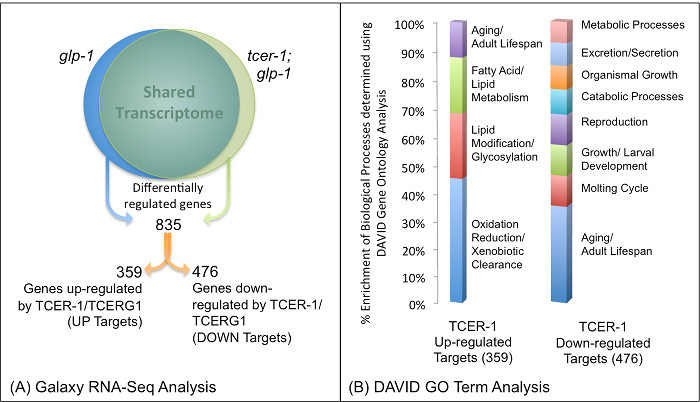 Figure 4: Identification of TCER-1/TCERG1-regulated Genes in Germline-less C. elegans Mutants using RNA-Seq: Results of Galaxy (A) and DAVID (B) Analyses.(A) Differential gene expression analysis of RNA-Seq data comparing the transcriptomes of glp-1 and tcer-1;glp-1 yielded a total of 835 genes, of which 359 were identified as being up-regulated by TCER-1/TCERG1 (UP) and 476 as down-regulated by TCER-1/TCERG1 (DOWN). (B) Results of 'Functional Annotation Clustering' analysis of genes identified as TCER-1/TCERG1 targets using DAVID.Percentage enrichment of Biological Processes for both the Up-regulated (UP) and Down-regulated (DOWN) Classes of TCER-1/TCERG1 targets. The graphic shown here is obtained by plotting the enriched gene groups (X-axis) and their respective percent enrichment (Y-axis) obtained as the output of DAVID analysis. Figure modified from Amrit et al.31 and reproduced with permission. Please click here to view a larger version of this figure.
Figure 4: Identification of TCER-1/TCERG1-regulated Genes in Germline-less C. elegans Mutants using RNA-Seq: Results of Galaxy (A) and DAVID (B) Analyses.(A) Differential gene expression analysis of RNA-Seq data comparing the transcriptomes of glp-1 and tcer-1;glp-1 yielded a total of 835 genes, of which 359 were identified as being up-regulated by TCER-1/TCERG1 (UP) and 476 as down-regulated by TCER-1/TCERG1 (DOWN). (B) Results of 'Functional Annotation Clustering' analysis of genes identified as TCER-1/TCERG1 targets using DAVID.Percentage enrichment of Biological Processes for both the Up-regulated (UP) and Down-regulated (DOWN) Classes of TCER-1/TCERG1 targets. The graphic shown here is obtained by plotting the enriched gene groups (X-axis) and their respective percent enrichment (Y-axis) obtained as the output of DAVID analysis. Figure modified from Amrit et al.31 and reproduced with permission. Please click here to view a larger version of this figure.
Gene Ontology Enrichment Analysis
To obtain an overview of the gene classes enriched in TCER-1/TCERG1 targets, we carried out gene ontology (GO) term analysis using DAVID. The TCER-1/TCERG1-regulated UP and DOWN gene lists were uploaded independently onto DAVID and analyzed as described in section 3. Little was known about the genes and cellular processes targeted by TCER-1/TCERG1 previously30, so we found the DAVID analysis to be especially revealing and helpful. Functional Annotation analysis of the UP genes revealed five Annotation Clusters with an Enrichment Score of >1.3, the highest including Cytochrome P450 enzyme-encoding genes and xenobiotic response genes, followed by genes implicated in lipid modifications. This was reinforced by the results of the Gene Functional Classification analysis that identified groups attributed with similar molecular activities with significant enrichment scores. Using spreadsheet, the identified groups were plotted against their respective enrichment scores (Figure 4). Our previous data suggested that TCER-1/TCERG1 functioned with the conserved longevity transcription factor, DAF-16/FOXO3A, to promote the longevity of GSC-less adults30. DAF-16/FOXO3A, in turn, has been implicated in modulating lipid metabolism in recent studies27,32,33. Based on this evidence, and the identification of lipid-metabolic genes and pathways as potential TCER-1/TCERG1 targets in the DAVID analyses, we focused on the fat metabolism genes identified in the RNA-Seq study for detailed mechanistic studies. Following this lead, and through subsequent molecular genetic, biochemical, and functional experimentation, we demonstrated that TCER-1/TCERG1 along with DAF-16/FOXO3A coordinately enhanced both lipid catabolic and anabolic processes in response to germline loss31. Similarly, Functional Annotation Clustering of the DOWN TCER-1/TCERG1 targets identified Annotation Clusters enriched for cytoskeletal functions, positive regulation of growth, reproduction and aging (Figure 4). These observations, and our supporting experimental evidences, suggest that upon germline loss, TCER-1/TCERG1 also represses growth and reproductive physiology in somatic cells as well as the expression of anti-longevity genes31.
| Sample | Total Sequences | Length | %GC | Total Reads (Galaxy) | Mapped Reads (Galaxy) | Genome Coverage |
| glp-1 | 4000000 | 51 | 49 | 20700539 | ~16,000,000 | 11x |
| glp-1; tcer-1 | 4000000 | 51 | 49 | 18055444 | ~13,000,000 | 9x |
| glp-1 | 4000000 | 51 | 48 | 18947463 | ~14,000,000 | 10x |
| glp-1; tcer-1 | 4000000 | 51 | 48 | 13829643 | ~10,000,000 | 7x |
Table 1: RNA-Seq Sample Details. Compilation of raw data attributes evaluated post-sequencing to confirm the success of the sequencing run. Sequencing data from the representative experiment consists of two biological conditions, a control strain (glp-1) and a mutant strain (tcer-1;glp-1) with two biological replicates sequenced for each. 'FastQC' quality check analysis highlights the number of sequences read with no "poor quality" reads, 48 - 49% GC content and a constant sequence read length of 51bp. Modified and reproduced with permission from Amrit et al.31.
Supplemental File: Command chain in brief for the tools run on the Galaxy pipeline for RNA-Seq data analysis. Please click here to download this file.
Discussion
Significance of the Galaxy Sequencing Platform in Modern Biology
The Galaxy Project has become instrumental in helping biologists without bioinformatics training to process and analyze high-throughput sequencing data in a fast and efficient manner. Once considered a herculean task, this publicly-available platform has made running complex bioinformatics algorithms to analyze NGS data a straightforward, reliable, and easy process. Apart from hosting a wide range of bioinformatics tools, the key to success for Galaxy is also the simplicity of its user interface that laces together the various aspects of complex sequencing analysis in an intuitive and seamless manner. Owing to these features, the Galaxy pipeline has acquired wide usage amongst biologists, including C. elegans researchers. In addition to familiarizing the user with the RNA-Seq Analysis pipeline, Galaxy also helps lay the foundation for basic biologists to grasp the concept of data analysis and understand the tools involved. This knowledge primes the user to perhaps further pursue more complex bioinformatics platforms such as 'R' and 'Python'. Besides Galaxy, other tools and packages are available commercially and as open-source solutions, which can be used for RNA-Seq analysis. The commercial options are often stand-alone software packages that are user-friendly but can be expensive for individual researchers who do not use NGS often. Alternatively, open source platforms such as BioWadrobe34 and ArrayExpressHTS35 require working knowledge of the command line and running scripts, which poses significant challenges for non-bioinformaticians. Hence, Galaxy remains a popular and indispensable resource.
Critical steps within the protocol
The effortless advantages of Galaxy and DAVID notwithstanding, a successful RNA-Seq experiment still relies fundamentally on careful design and execution of the experimental step. For instance, it is critical to ensure genetic homogeneity before comparing two strains by RNA-Seq, and to determine if there are differences in developmental rates. Isolating RNA from age-matched strains is critical as well. Similarly, to account for variability of gene expression within the same strain, it is important to run two or more 'biological replicates' of each strain. This essentially means growing and harvesting worms from the strains being sequenced in at least twoindependent experiments, although three biological replicates is the recommended standard. Galaxy unifies the data from multiple biological replicates so that the reported gene-expression differences between strains are not simply a consequence of 'within-sample' variability.
A critical design decision is about the use of single-end vs. paired-end sequencing. With single-end sequencing, each fragment is sequenced uni-directionally so the process is faster, cheaper and suited for transcriptional profiling. In paired-end sequencing, once the fragment is sequenced from one end to the other, a second round of sequencing is resumed in the opposite direction. It provides more in-depth data and additional positioning information of the genome, so is more suited for de novo genome assembly, new SNP identification and for identifying epigenetic modifications, deletions, insertions, and inversions. Similarly, the total number of reads and extent of genome coverage required for adequate differential expression studies is context dependent. For small genomes, such as bacteria and fungi, ~5 million reads is sufficient, whereas, in worms and flies ~10 million reads provide adequate coverage. For organisms with large genomes such as mice and humans, 15-25 million reads is the required range. In addition, to the read number and coverage, it is also important that most of the NGS reads align to the reference genome. An alignment of <70% reads is indicative of poor NGS or the presence of contaminants. Overall, for C. elegans RNA-Seq studies, three biological replicates sequenced with 50 bp unidirectional sequencing resulting in ~10-15 million reads and ~5-10X genome coverage for each sample is an ideal aim.
Despite the ease of using Galaxy, there are a few points to remember in order to ensure a smooth and glitch-free data analysis experience. It is necessary for the user to have a basic understanding of the purpose and functioning of the various tools used. Each Galaxy tool requires selection of parameters and understanding the tool will help the user optimize settings based on the requirement of the experiment. The Galaxy help pages explain every parameter and it is recommended that the user peruse these details to decide on test variables.
The gene list obtained post RNA-Seq analysis is merely a list of genes until it is mined for biologically relevant data using DAVID. This is a crucial exercise that converts individual gene-based data into biological-process based results. Exploring the RNA-Seq gene list using the various analytics DAVID provides is therefore an integral and important part of the protocol.
Modifications, troubleshooting and limitations
A common glitch with NGS data analysis is tasks or tests that fail, especially at the quality-control stages. Of the tests that FastQC runs on a sample, a few could come up as failed. However, this does not inevitably mean the sample does not meet the fastq quality standards. The failure could have an alternative explanation that should be explored carefully.
For instance, if the 'Per base sequence content' test fails (suggesting that there is a greater than 10% difference between bases in any position), check the method for the oligodt library preparation. Previous work has shown that Illumina NGS libraries may have a propensity for the 13th base being sequenced to have a bias for certain bases causing the sample to fail the test. Similarly, a failure of the 'Kmer content' test can sometimes be attributed to the fact that libraries derived from random priming will nearly always show Kmer bias at the start due to an incomplete sampling of the random primers. Therefore, it is important to consider these and other impediments in the analysis pipeline before determining the fate of the experiment.
Another important feature that may impact RNA-Seq data analysis is the rapid and exponential advancements that are occurring in NGS methods and analytic software. Ideally, one expects an identical gene list to result from analyzing a sample NGS data on two pipelines or two versions of the same pipeline. However, while constantly improving algorithms are lowering aberrations in RNA-Seq analysis and producing gene lists of greater accuracy, this often leads to disparities. For instance, analyzing a sample NGS data using an older vs. newer version of the same toolset may produce significantly different gene lists. A modest variation is expected but users need to be aware that large discrepancies may be reflective of weaknesses in the design or performance of the experiment.
Collectively, the Galaxy Project and DAVID analytical tools have transformed the way NGS data can be harnessed to extract biologically relevant information. This has opened entirely new levels of independence and investigation to the scientific community, including C. elegans researchers. For instance, the constantly reducing cost of sequencing coupled with better and faster sequencing technology are ushering in an era of transcriptomics at the level of single worms, individual worm tissues and even few select worm cells. These endeavors involve dramatic increases in NGS data being generated. Keeping up with the analytical end of this workflow will be a challenge, but due to its versatility, Galaxy is likely to be instrumental in empowering the transition from whole organism transcriptomics to RNA-Seq at single cell level in C. elegans. The resulting advances in knowledge are likely to provide extraordinary insights into fundamental biology.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors would like to express their gratitude to the laboratories, groups and individuals who have developed Galaxy and DAVID, and thus made NGS widely accessible for the scientific community. The help and advice provided by colleagues at the University of Pittsburgh during our bioinformatics training is acknowledged. This work was supported by an Ellison Medical Foundation New Scholar in Aging award (AG-NS-0879-12) and a grant from the National Institutes of Health (R01AG051659) to AG.
References
- Venter JC, et al. The sequence of the human genome. Science. 2001;291(5507):1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Afgan E, et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016;44(W1):W3–W10. doi: 10.1093/nar/gkw343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011;12(3):R22. doi: 10.1186/gb-2011-12-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A, Pimentel H, Trapnell C, Pachter L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 2011;27(17):2325–2329. doi: 10.1093/bioinformatics/btr355. [DOI] [PubMed] [Google Scholar]
- Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31(1):46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Giardine B, et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15(10):1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Gao S, Muegge K, Zhang W, Zhou B. Advanced Applications of RNA Sequencing and Challenges. Bioinform Biol Insights. 2015;9(1):29–46. doi: 10.4137/BBI.S28991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER. Next-generation sequencing platforms. Annu Rev Anal Chem (Palo Alto Calif) 2013;6:287–303. doi: 10.1146/annurev-anchem-062012-092628. [DOI] [PubMed] [Google Scholar]
- Yang IS, Kim S. Analysis of Whole Transcriptome Sequencing Data: Workflow and Software. Genomics Inform. 2015;13(4):119–125. doi: 10.5808/GI.2015.13.4.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri P, Draghici S. Ontological analysis of gene expression data: current tools, limitations, and open problems. Bioinformatics. 2005;21(18):3587–3595. doi: 10.1093/bioinformatics/bti565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaye DD, Greenwald I. OrthoList: a compendium of C. elegans genes with human orthologs. PLoS One. 2011;6(5):e20085. doi: 10.1371/journal.pone.0020085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium CeS. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 1998;282(5396):2012–2018. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- Agarwal A, et al. Comparison and calibration of transcriptome data from RNA-Seq and tiling arrays. BMC Genomics. 2010;11:383. doi: 10.1186/1471-2164-11-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, et al. Scaffolding a Caenorhabditis nematode genome with RNA-seq. Genome Res. 2010;20(12):1740–1747. doi: 10.1101/gr.111021.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnert R, Ratsch G. rQuant.web: a tool for RNA-Seq-based transcript quantitation. Nucleic Acids Res. 2010;38:W348–W351. doi: 10.1093/nar/gkq448. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamm AT, Stadler MR, Zhang H, Gent JI, Fire AZ. Multimodal RNA-seq using single-strand, double-strand, and CircLigase-based capture yields a refined and extended description of the C. elegans transcriptome. Genome Res. 2011;21(2):265–275. doi: 10.1101/gr.108845.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrit FR, Ratnappan R, Keith SA, Ghazi A. The C. elegans lifespan assay toolkit. Methods. 2014;68(3):465–475. doi: 10.1016/j.ymeth.2014.04.002. [DOI] [PubMed] [Google Scholar]
- Hsin H, Kenyon C. Signals from the reproductive system regulate the lifespan of C. elegans. Nature. 1999;399(6734):362–366. doi: 10.1038/20694. [DOI] [PubMed] [Google Scholar]
- Alper S, et al. The Caenorhabditis elegans germ line regulates distinct signaling pathways to control lifespan and innate immunity. J Biol Chem. 2010;285(3):1822–1828. doi: 10.1074/jbc.M109.057323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbaugh MJ, et al. Lipid-mediated regulation of SKN-1/Nrf in response to germ cell absence. Elife. 2015;4 doi: 10.7554/eLife.07836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapierre LR, Gelino S, Melendez A, Hansen M. Autophagy and lipid metabolism coordinately modulate life span in germline-less. C. elegans. Curr Biol. 2011;21(18):1507–1514. doi: 10.1016/j.cub.2011.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rourke EJ, Soukas AA, Carr CE, Ruvkun G. C. elegans major fats are stored in vesicles distinct from lysosome-related organelles. Cell Metab. 2009;10(5):430–435. doi: 10.1016/j.cmet.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazi A. Transcriptional networks that mediate signals from reproductive tissues to influence lifespan. Genesis. 2013;51(1):1–15. doi: 10.1002/dvg.22345. [DOI] [PubMed] [Google Scholar]
- Ghazi A, Henis-Korenblit S, Kenyon C. A transcription elongation factor that links signals from the reproductive system to lifespan extension in Caenorhabditis elegans. PLoS Genet. 2009;5(9):e1000639. doi: 10.1371/journal.pgen.1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrit FR, et al. DAF-16 and TCER-1 Facilitate Adaptation to Germline Loss by Restoring Lipid Homeostasis and Repressing Reproductive Physiology in C. elegans. PLoS Genet. 2016;12(2):e1005788. doi: 10.1371/journal.pgen.1005788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MC, O'Rourke EJ, Ruvkun G. Fat metabolism links germline stem cells and longevity in C. elegans. Science. 2008;322(5903):957–960. doi: 10.1126/science.1162011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick M, Chen K, Ramaswamy P, Kenyon C. New genes that extend Caenorhabditis elegans' lifespan in response to reproductive signals. Aging Cell. 2012;11(2):192–202. doi: 10.1111/j.1474-9726.2011.00768.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartashov AV, Barski A. BioWardrobe: an integrated platform for analysis of epigenomics and transcriptomics data. Genome Biol. 2015;16:158. doi: 10.1186/s13059-015-0720-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves A, Tikhonov A, Brazma A, Kapushesky M. A pipeline for RNA-seq data processing and quality assessment. Bioinformatics. 2011;27(6):867–869. doi: 10.1093/bioinformatics/btr012. [DOI] [PMC free article] [PubMed] [Google Scholar]