

Abstract
The restricted availability of suitable in vitro models that can reliably represent complex human brain development is a significant bottleneck that limits the translation of basic brain research into clinical application. While induced pluripotent stem cells (iPSCs) have replaced the ethically questionable human embryonic stem cells, iPSC-based neuronal differentiation studies remain descriptive at the cellular level but fail to adequately provide the details that could be derived from a complex, 3D human brain tissue.
This gap is now filled through the application of iPSC-derived, 3D brain organoids, "Brains in a dish," that model many features of complex human brain development. Here, a method for generating iPSC-derived, 3D brain organoids is described. The organoids can help with modeling autosomal recessive primary microcephaly (MCPH), a rare human neurodevelopmental disorder. A widely accepted explanation for the brain malformation in MCPH is a depletion of the neural stem cell pool during the early stages of human brain development, a developmental defect that is difficult to recreate or prove in vitro.
To study MCPH, we generated iPSCs from patient-derived fibroblasts carrying a mutation in the centrosomal protein CPAP. By analyzing the ventricular zone of microcephaly 3D brain organoids, we showed the premature differentiation of neural progenitors. These 3D brain organoids are a powerful in vitro system that will be instrumental in modeling congenital brain disorders induced by neurotoxic chemicals, neurotrophic viral infections, or inherited genetic mutations.
Keywords: Developmental Biology, Issue 122, iPSCs, neural progenitor cells, brain organoids, microcephaly, centrosomes, primary cilium
Introduction
Human neurodevelopmental disorders, such as microcephaly, can only be poorly studied in animal models due to the fact that human brains have an extended cortical surface, a unique feature differing from non-human animals.
This aspect makes human brain development a complex process that cannot be sufficiently studied in a 2D, in vitro cell culture system. Emerging 3D culture techniques allow the generation of tissue-like organoids from induced pluripotent stem cells (iPSCs). The in vitro differentiation of pluripotent stem cells in a 3D suspension culture allows the formation of various cell types in a timely and region-specific manner, giving rise to an organized, stratified tissue1,2,3. Thanks to laboratories that pioneered 3D culture technologies and demystified the complexity of organ formation, starting from stem cells, we developed a robust method of generating brain organoids to delineate early events of human brain development and to model microcephaly in vitro1,2,3. It is noteworthy that we adapted the original method developed by Lancaster et al. to generate cerebral organoids1. This method was modified according to our experimental requirements.
The aim of a study from Gabriel et al. was to analyze the cellular and molecular mechanisms of neural stem cell maintenance during brain development. In order to do this, a mechanistic study was performed by analyzing neural progenitor cells (NPCs) in 3D brain organoids derived from a microcephaly patient4. This patient carried a mutation in CPAP, a conserved centrosomal protein required for centrosome biogenesis5. A widely accepted hypothesis is that microcephaly is the result of a depletion of the NPC pool, and this might be due either to cell death or to premature differentiation1,6,7,8,9.
By analyzing the ventricular zones (VZs) of microcephaly brain organoids, it was shown that a significant number of NPCs undergo asymmetrical cell division, unlike brain organoids derived from a healthy donor4. Extensive microscopic and biochemical analyses of microcephalic brain organoids revealed an unexpected role for CPAP in timely cilia disassembly4. Specifically, mutated CPAP is associated with retarded cilium disassembly and delayed cell cycle re-entry, leading to the premature differentiation of NPCs4. These results suggest a role for cilia in microcephaly and their involvement during neurogenesis and brain size control10.
The first part of this protocol is a description of a three-step method to generate homogenous brain organoids. As mentioned before, the original Lancaster protocol was adapted and modified to suit our purpose1. First, human iPSCs are cultured in a defined feeder-free condition on Engelbreth-Holm-Swarm (EHS) matrix. This step avoids the variations of feeder-dependent pluripotent stem cell cultures. In this protocol, the induction of neural differentiation to form neural epithelium starts directly from iPSCs. By skipping the embryoid body (EB) formation step, the neural differentiation proceeds in a more controlled and directed manner. This approach limits the spontaneous and undirected formation of other germ cell layers, such as mesoderm and endoderm. By applying this protocol, neurospheres containing neural rosettes can be harvested on day 5 for EHS matrix embedding and stationary suspension culture. The organoid medium used for the third step of our protocol is supplemented with dorsomorphin and SB431542. Dorsomorphin is a small-molecule inhibitor of bone morphogenic protein (BMP), and SB431542 inhibits the TGFβ/activin/nodal signaling pathway. The combination of these factors could promote neural differentiation more efficiently than retinoic acid alone11,12,13,14.
Altogether, these modifications enable the reproducible generation of brain organoids, with minimal variations across organoids. Importantly, this method was applied to robustly generate microcephalic brain organoids from patient iPSCs, which carry mutations in genes that affect centrosomes and cell-cycle dynamics.
The second part of this protocol gives instructions to prepare brain organoids for the analysis and interpretation of cellular defects in microcephaly. This includes fixation, cryosectioning, immunofluorescent staining, and confocal microscopic analysis. This protocol will provide the reader with a detailed description of expected results and with guidance for interpretation.
Protocol
1. Generation of Brain Organoids (23 days)
- Initiation of neuroectoderm (5 days) NOTE: The following points should be considered before the start of differentiation. The reprogramming method (lentiviral-, sendai-virus-, episomal-, or microRNA-based etc.) to obtain human iPSCs should ideally be the same for all patient and control iPSC lines. Various reprogramming kits and instructions based on published protocols are available15,16,17,18. The quality of the human iPSC lines is the key to accomplishing an optimal differentiation. Monitor colony and cell morphology with a microscope and validate pluripotency by testing the expression of markers such as Oct3/4, Nanog, or TRA-1-60.
- Culture human iPSCs under feeder-free conditions in medium A on a dish coated with Engelbreth-Holm-Swarm (EHS) matrix. NOTE: Grow human iPSCs feeder-free and serum-free to maintain a defined culture condition and to avoid an additional step for the removal of mouse embryonic feeder cells (MEFs) before the start of differentiation. The optimal passage number for human iPSCs to start differentiation ranges from passage 15 upon reprogramming to passage 70 in total. When passaging, detach hiPSC colonies as cells aggregate using an appropriate cell detachment solution with low mechanical stress and a 2-mL serological pipette to transfer aggregates to a new dish. Avoid dissociation to single cells, as this might induce differentiation and apoptosis in most iPSC lines. It was reported that long-term, single-cell passaging could increase genomic alterations in human iPSCs19,20. Avoid cell detachment procedures, which require a centrifugation step to remove detachment solution, as this reduces the overall viability of iPSCs. Test all cultures for microbial contaminations, particularly mycoplasma, on a regular basis, because this might alter the quality of the iPSCs and their differentiation capacity.
- Coat a 60 mm tissue culture dish with EHS matrix as per the manufacturer's instructions.
- Thaw an aliquot of 1 x 106 human iPSCs. Seed the iPSCs in a 60-mm tissue culture dish coated in EHS matrix and containing 5 mL of medium A (see the Materials Table). Change the medium daily and passage after 5 to 7 days when the cells reach ~80% confluency. NOTE: Passage the thawed iPSCs at 80% confluency using standard methods, such as the enzyme-free detachment of colonies21, at least once before starting the differentiation. In brief, remove the iPSC medium, wash the cells once with pre-warmed 37 °C Dulbecco's Modified Eagle's Medium: Nutrient Mixture F-12 (DMEM/F12), and incubate the iPSCs following the manufacturer's instructions using reagent A (see the materials table). Don't exceed the recommended incubation time in order to avoid dissociation to single cells. Human iPSCs should be detached and floating as aggregates, not as single cells. They can then be transferred to new EHS matrix-coated dishes; for example, iPSC aggregates from one 60-mm dish can be distributed to 4 new 60-mm dishes.
- Check for mycoplasma with a mycoplasma detection kit according to the manufacturer's instructions. Use only mycoplasma-free iPSCs, as mycoplasma can alter the differentiation capability of iPSCs.
- Dissociate iPSCs (80% confluent) and prepare a single-cell suspension using reagent B.
- Wash the iPSCs once with pre-warmed (37 °C) DMEM/F12.
- Add pre-warmed reagent B (e.g., 1 mL in a 60 mm dish) and incubate the iPSCs for 5 min at 37 °C and 5% CO2.
- Flick 20 times with a fingertip on the side and bottom of the dish to detach the iPSCs. Check for the detachment of cells under the microscope.
- Pipette the cell suspension up and down in the dish 5 times with a 1-mL micropipette.
- Add 3 mL of medium A to dilute 1 mL of reagent B and collect the cell suspension in a 15-mL centrifuge tube.
- Gently spin down the iPSCs (500 x g) for 4 min at room temperature.
- Resuspend the cell pellet in 1 mL of medium B and count the cell number with a hemocytometer. NOTE: Be aware of using only medium B for resuspension. Avoid using medium A, as it contains a too-high concentration of bFGF, which might inhibit the differentiation.
- Dilute the cell suspension to 4.5 x 105 cells per mL in medium B supplemented with 10 µM rho-associated protein kinase inhibitor (Y-27632).
- Add 100 µL per well in a non-adherent, v-bottom, 96-well plate. NOTE: Make sure the cells are equally distributed in the suspension by shaking the tube each time before taking out 100 µL portions. It is important that each well should contain an equivalent cell number in order to obtain neurospheres homogenous in size and shape (round, defined surfaces).
- Gently spin down the plate with the cells at 500 x g and room temperature for 3 min and incubate at 37 °C and 5% CO2.
- Change the medium daily by removing 50 µL and adding 50 µL of fresh medium B into each well for the next 5 days.
2. Embedding Neurospheres in EHS Matrix (4 days)
Prepare neurosphere medium by mixing the following: 1:1 mixture of DMEM/F12 and medium C (v/v), 1:200 (v/v) supplement 1, 1:100 (v/v) supplement 2 without (w/o) Vitamin A, 1:100 L-glutamine, 0.05 mM non-essential amino acids (MEM), 100 U/mL penicillin, 100 µg/mL streptomycin, 1.6 g/L insulin, and 0.05 mM β-mercaptoethanol.
Collect the neurospheres with a 200 µL micropipette using a ~2 mm tip previously cut with sterile scissors.
Place the neurospheres approximately 5 mm away from each other on paraffin film (3 x 3 cm2) in an empty 100 mm dish and carefully remove as much of the remaining medium as possible.
Add a drop (7 µL) of EHS matrix onto each single neurosphere.
Incubate the EHS matrix drops with the neurospheres for 15 min in an incubator.
Wash the neurospheres carefully from the paraffin film by flushing them with neurosphere medium. To flush, use a 1 mL micropipette and a new 100 mm Petri dish containing 10 mL of neurosphere medium.
Incubate the neurospheres for the next 4 days and add 2 mL of fresh neurosphere medium on day 2. NOTE: Make sure that the shelves in the incubator are flat so that the EHS matrix-embedded neurospheres will not clump together on one side of the dish.
3. Organoids in a Rotary Suspension Culture (14 Days)
Prepare brain organoid medium by mixing the following: 1:1 mixture of DMEM/F12 and medium C (v/v), 1:200 (v/v) supplement 1, 1:100 (v/v) supplement 2 w/o Vitamin A, 1:100 L-glutamine, 0.05 mM MEM, 100 U/mL penicillin, 100 µg/mL streptomycin, 1.6 g/L insulin, 0.5 µM dorsomorphin, 5 µM SB431542, and 0.05 mM β-mercaptoethanol.
Add 100 mL of brain organoid medium to each spinner flask through its side arms and place them in an incubator for pre-warming for at least 20 min.
Set up a stirring program at 25 rpm, according to the manufacturer's instructions. NOTE: Before transferring the EHS matrix-embedded neurospheres into spinner flasks, make sure that they are all separated. If two or more are connected through EHS matrix, separate them by cutting the connecting matrix with a scalpel.
Carefully transfer the EHS matrix-embedded neurospheres into spinner flasks containing 100 mL of organoid medium using a 2 mL serological pipette. Use the side arms of the spinner flask to transfer the neurospheres into the flask.
Place the spinner flasks on a magnetic stirring platform in an incubator at 37 °C and 5% CO2; this is day 0 of the organoid culture.
Change the medium once per week (or more often when there is a color change) by removing half of the medium and adding the same amount of fresh medium. NOTE: When taking the spinner flasks out of the incubator, wait 3-5 min to let the organoids sink down to the bottom of the flask. Remove the medium by placing the glass pipette tip (connected to a pump) on the surface of the liquid; aspirate the medium carefully through one side opening/arm of the flask. These manipulations must be done under the laminar hood.
4. Analysis of Brain Organoids
- Fixation of organoids
- Collect the organoids on day 14 of the spinner flask culture with a cut 1 mL micropipette (cut ~5 mm). Put all of them in a 60 mm dish and wash them once with 5 mL of warm DMEM/F12 for 3 min.
- Prepare a 1.5 mL tube with 500 µL of warm 4% paraformaldehyde (PFA). Caution: Wear skin and eye protection and work under a safety hood when handling PFA fixative.
- Place each organoid separately in each tube and fix them for at least 30 min at room temperature. Do not fix the organoids for longer than 60 min. To move the organoids, use an inoculation loop or any other devise that is convenient.
- Remove the PFA and wash the fixed organoids twice for 10 min each with 1 mL of PBS.
- Store the organoids in 1 mL of PBS at 4 °C for up to 7 days, until further use.
- Embedding the organoids for cryosectioning
- Remove the PBS and add 1 mL of 30% sucrose in distilled water solution per tube to dehydrate the organoids before cryofreezing them; after adding sucrose solution, the organoids should be floating at the surface. Store the organoids overnight in sucrose solution at 4 °C; by the next day, the organoids should have sunk down to the bottom of the tube. NOTE: Organoids can be stored for up to 3-5 days at 4 °C in sucrose solution, if necessary.
- Fill a vinyl specimen mold with 400 µL of optimum cutting temperature (OCT) compound and use an inoculation loop to place an organoid at the center of the mold. Label the rim of the mold with the sample name.
- Freeze the organoid-containing mold at -80 °C until cryosectioning.
- Coat glass cryoslides with 0.1% poly-l-lysine solution (PLL) in PBS for 5 min at room temperature and let the slides dry for 3 h. Store the slides at 4 °C and warm them up to room temperate before use. NOTE: PLL-coating is an important step, as it will prevent the organoid slices from floating away. Collect and store PLL solution at 4 °C for up to 3 months. Before reuse, filter it with a 0.22-µm syringe and let it warm to room temperature.
- Section cryofrozen organoids into 20-50 µm-thick slices on PLL-coated glass cryoslides22. Let the slides with the sections dry for 1 h at room temperature. Store the sections at -80 °C until further processing.
5. Immunofluorescent Staining of Organoid Sections
NOTE: For the general characterization of organoids, staining with nestin, a neural progenitor marker, and TUJ1, a pan-neuronal marker, is recommended. As additional examples, immunofluorescent staining with phospho-Vimentin (p-Vim), which labels mitotic apical radial glial cells, and Arl13b, for cilium, are described. To test apoptosis, use the Terminal deoxynucleotidyl transferase (TdT) dUTP Nick-End Labeling (TUNEL) assay. Place the slides in a plastic box during the incubations to protect them from dust, light, and drying out.
Thaw the slides for 30 min at room temperature.
Wash the slides twice with 200 µL of PBS-glycine (0.225 g of glycine in PBS) for 3 min to quench the PFA-induced autofluorescence.
Permeabilize the sections with 200 µL of 0.5% Triton X100/0.1% tween in PBS solution for 10 min at room temperature.
Wash them twice with 200 µL of PBS-glycine solution for 3 min.
Incubate them with 200 µL of 0.5% fish gelatin/0.1% Triton X100 in PBS for 1 h at room temperature or overnight at 4 °C to block unspecific antigen binding. NOTE: If a TUNEL assay is required, perform the assay as per the manufacturer's protocol. Start with the TUNEL assay before performing the immunostaining, as it might interfere with secondary antibodies and quench fluorophores when used afterwards.
Dilute the antibodies in blocking solution at the following concentrations: nestin, 1:200; p-Vim, 1:500; TUJ1, 1:200; Arl13b, 1:20; and secondary antibodies, 1:1,000.
Incubate with 200 µL of the first primary antibody (e.g., nestin) for 1-2 h at room temperature or overnight at 4 °C.
Wash 3 x 3 min with 200 µL of blocking solution.
Dilute the first (anti-mouse 488) secondary antibody 1:1,000 in blocking solution and incubate the slide in 200 µL for 1-2 h at room temperature. From now on, always protect the slides from light.
Wash 3 x 3 min with 200 µL of blocking solution.
Incubate with 200 µL of the next primary antibody (e.g., TUJ1) for 1-2 h at room temperature or overnight at 4 °C.
Wash 3 x 3 min with 200 µL of blocking solution.
Add 200 µL of the next secondary antibody (anti-rabbit 647) for 1-2 h at room temperature.
Wash 3 x 3 min. with 200 µL of blocking solution.
Add 200 µL of 4',6-diamidino-2-phenylindole (DAPI) at a 30 nM concentration in PBS for 15 min at room temperature for nuclear staining.
Wash 2 x 3 min with 200 µL of blocking solution.
Wash 1 x 1 min with 200 µL of distilled water and let the sections dry for 10-20 min, until no obvious water drop is visible any more.
Mount the sections with embedding medium. Store them protected from light at 4 °C for up to several weeks.
Proceed with microscopic analysis to image an overview of the organoid, ventricular zone, primitive cortical plate, and other areas of interest.
Use a confocal microscope with a 63X oil objective and fluorescent filters, chosen according to the fluorescent dye-tagged secondary antibodies used1,10.
Representative Results
The generation of brain organoids requires at least three weeks of continuous culturing (Figure 1A). To accomplish reproducible results, we recommend that the researcher documents every step and, importantly, avoids any alterations regarding medium components, time points, and cell handling. Here, we give a summary of how to evaluate if critical milestones are reached in order to obtain organoids of sufficient quality at the end of the experiment. The formation of neurospheres in a 96-well plate should be clearly visible from day 4. Neurospheres can be recognized as well-defined spheres on the bottom of each well (Figure 1B, step 1).
On day 5, neurospheres should be ~500 µm in diameter and exhibit a smooth surface, with a bright rim surrounding a dark center. It might be possible to already observe neural rosette-like extended structures within this bright rim (Figure 1C, start of step 2). If the neurospheres fall apart or appear more like cell aggregates, differentiation should not be continued further. Finally, organoids should increase and exhibit a similar size during the differentiation process in spinner flasks (Figure 1D, step 3). See the troubleshooting table (Table 1) for further information.
The quality of day-14 organoids should be verified by light microscopy. VZs are composed of thick layers of nestin-positive NPCs/radial glial cells with a palisade nuclear shape at the apical side. On the other hand, the primitive cortical plate will contain abundant TUJ1-positive neurons on the basal side, spatially distinct from the apical side (Figure 1E). Importantly, TUJ1-positive neurons should not be seen at the apical side of the VZ.
To analyze the division plane of apical radial glial cells (aRGs), we recommend using phospho-vimentin (p-Vim) and Arl13b staining. Arl13b labels the primary cilia of the innermost aRGs lining the lumen of the VZ. Thus, the Arl13b-positive ciliated region serves as an orientation guide to locate and to analyze the division plane of p-Vim-positive aRGs in anaphase. Typically, the VZ of a 14-day-old control organoid displays a significant number of aRGs, whose division planes are horizontally oriented. A horizontally oriented division plane is essential for the early symmetric expansion of aRGs (Figure 1F and G)23,24. When sufficient aRG expansion is reached, neurogenesis begins, the dividing cells change their orientation towards the lumen, and the division plane switches from horizontal (symmetric) to vertical (asymmetric)4,12,24.
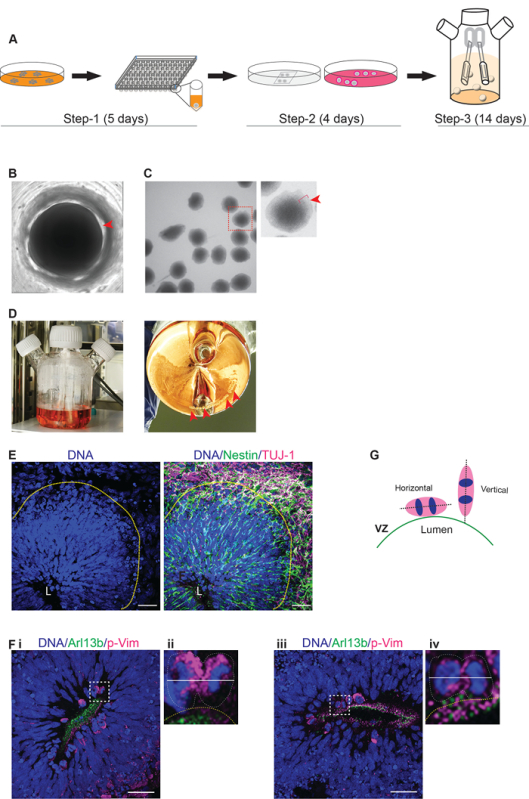 Figure 1:Generation of Brain Organoids from Human iPS Cells. (A) Workflow of the differentiation protocol. Step 1: Start of differentiation. During this phase, the formation of neurospheres from human iPSCs in a 96-well plate occurs. The time duration is 5 days. Step 2: Embedding the neurospheres in EHS matrix droplets. A stationary suspension culture of EHS matrix-embedded neurospheres has a time duration of 4 days. Step 3: Organoids in spinner flasks. The transfer of EHS matrix-embedded neurospheres to spinner flasks takes place. The time duration is 14 days. (B) A neurosphere in a multi-well plate. Representative image of a neurosphere on day 4 in a 96-well plate during step 1 is shown. The sphere should be clearly visible on the bottom of the well, with a smooth, round surface (red arrow). (C) Morphology of neurospheres prior to EHS matrix embedding. Neurospheres collected on day 5 of step 1 should be uniform in size and display a smooth surface with a bright rim (bracket and arrow). (D) Organoids in spinner flasks. A spinner flask with brain organoids during step 3 (red arrows) is shown here. (E) Immunofluorescent imaging of cryosectioned organoids displaying a typical VZ and primitive cortical plate. The left panel shows the nuclear staining of cells at the VZ. The VZ spans from the apical side (lumen L) to the basal side (yellow line). Note that cells within the VZ display palisade-like nuclei, suggesting that they are radial glial cells. The right panel shows the immunofluorescent staining of nestin-positive NPCs (green) within the VZ and TUJ1-positive neurons (magenta) in the primitive cortical plate. Scale bar = 50 µm. (F) Immunofluorescent imaging of cryosectioned organoids. Two examples of VZs stained with p-Vim and Arl13b (i and iii). The regions marked by white squares are magnified at the right for each image inset (ii and iv). The division plane of anaphase apical radial glial cells (aRGs). Dividing apical radial glial cells at the apical side of the VZ are p-Vim-positive (magenta). Examples for symmetrically dividing aRGs are shown (white squares and insets). The division plane is given as a white line. The division plane of an anaphase cell is horizontal to the lumen surface line (yellow dotted line) and is marked by Arl13b staining (green). Scale bar = 50 µm. (G) This schematic provides the vertical or horizontal orientation of aRGs in anaphase relative to the lumen. The dividing cells of control organoids on day 14 are mostly horizontally oriented (0-30°) relative to the lumen surface of the VZ. The apical side of the lumen is lined by the primary cilia of aRGs, which specify the lumen surface of the VZ (green line). In contrast, most of the radial glial cells of microcephaly organoids display a vertically oriented (60-90°) division plane. The white line shows the axis of the division plane. Please click here to view a larger version of this figure.
Figure 1:Generation of Brain Organoids from Human iPS Cells. (A) Workflow of the differentiation protocol. Step 1: Start of differentiation. During this phase, the formation of neurospheres from human iPSCs in a 96-well plate occurs. The time duration is 5 days. Step 2: Embedding the neurospheres in EHS matrix droplets. A stationary suspension culture of EHS matrix-embedded neurospheres has a time duration of 4 days. Step 3: Organoids in spinner flasks. The transfer of EHS matrix-embedded neurospheres to spinner flasks takes place. The time duration is 14 days. (B) A neurosphere in a multi-well plate. Representative image of a neurosphere on day 4 in a 96-well plate during step 1 is shown. The sphere should be clearly visible on the bottom of the well, with a smooth, round surface (red arrow). (C) Morphology of neurospheres prior to EHS matrix embedding. Neurospheres collected on day 5 of step 1 should be uniform in size and display a smooth surface with a bright rim (bracket and arrow). (D) Organoids in spinner flasks. A spinner flask with brain organoids during step 3 (red arrows) is shown here. (E) Immunofluorescent imaging of cryosectioned organoids displaying a typical VZ and primitive cortical plate. The left panel shows the nuclear staining of cells at the VZ. The VZ spans from the apical side (lumen L) to the basal side (yellow line). Note that cells within the VZ display palisade-like nuclei, suggesting that they are radial glial cells. The right panel shows the immunofluorescent staining of nestin-positive NPCs (green) within the VZ and TUJ1-positive neurons (magenta) in the primitive cortical plate. Scale bar = 50 µm. (F) Immunofluorescent imaging of cryosectioned organoids. Two examples of VZs stained with p-Vim and Arl13b (i and iii). The regions marked by white squares are magnified at the right for each image inset (ii and iv). The division plane of anaphase apical radial glial cells (aRGs). Dividing apical radial glial cells at the apical side of the VZ are p-Vim-positive (magenta). Examples for symmetrically dividing aRGs are shown (white squares and insets). The division plane is given as a white line. The division plane of an anaphase cell is horizontal to the lumen surface line (yellow dotted line) and is marked by Arl13b staining (green). Scale bar = 50 µm. (G) This schematic provides the vertical or horizontal orientation of aRGs in anaphase relative to the lumen. The dividing cells of control organoids on day 14 are mostly horizontally oriented (0-30°) relative to the lumen surface of the VZ. The apical side of the lumen is lined by the primary cilia of aRGs, which specify the lumen surface of the VZ (green line). In contrast, most of the radial glial cells of microcephaly organoids display a vertically oriented (60-90°) division plane. The white line shows the axis of the division plane. Please click here to view a larger version of this figure.
| Problem: | Possible reasons: | Suggestions: |
| Step 1.1.1.2 | Poor efficiency of reprogramming | Check hiPSCs for pluripotency markers: if quality is low, manually pick undifferentiated colonies for few passages to enrich pluripotent colonies |
| Human iPSCs differentiate before start of differentiation | ||
| Stress due to early passaging or passaging as single cells | Do not passage before 80% confluency is reached. Passage as aggregates, not single cells | |
| Contamination with mycoplasma | Test for mycoplasma contamination | |
| Step 2 | Poor quality of hiPSCs | Improve quality of hiPSCs (see above) |
| Neurospheres do not form at all or fall apart at day 5 after start of differentiation | Starting number of cells per well is too low or too high | Accurately count the number of cells and distribute them equally in each well |
| hiPSC medium was used instead of neural differentiation medium | Use neural differentiation medium | |
| Centrifugation step was too harsh or to mild | Spin 96-well plate 500 x g for 3 min and check if cells accumulated centrally at the bottom of each well | |
| No Y-27632 at start of differentiation | Use Y-27632 to enhance the cell survival | |
| Cells attached and grew at the bottom of the plate | Make sure that a non-adherent 96-well v-bottom plate is used. | |
| Step 2 | No daily medium change | Change half amount of medium daily |
| Neurospheres are not homogenous in size | Starting number of cells per well was not equal | Mix the tube with single cell suspension each time before taking out 100 µL cell suspension |
| Medium change was not done for each well daily | Make sure every well gets medium change daily | |
| Step 2 | hiPSC were not dissociated into single cells before the start of differentiation | Check under the microscope if hiPSC are dissociated to single cells after accutase treatment. If there are still aggregates, pipette cells 10 times up and down with 100 µL micropipette and check again or repeat accutase treatment |
| Neurospheres do not display a bright rim or round surface; they form large cysts at the surface | Starting number of cells per well was too high | Accurately count the number of cells and distribute them equally in each well |
| Medium change was not done daily for each well | Change half the medium daily | |
| Step 2.7 | Poor quality of hiPSCs | Improve quality of hiPSCs (see above) |
| EHS matrix embedded neurospheres stick and clump together | Not enough medium (volume) | Provide a minimum of 10 mL medium in a 100 mm petridish |
| Shelf of incubator not even | Place dish on an even surface | |
| After placing into incubator, move the dish so that neurospheres are distributed evenly |
Table 1: Troubleshooting Table.
Discussion
MCPH is a complex human neurodevelopmental disorder that cannot be recapitulated in animal models in vivo or in simple human cell culture approaches in vitro. The clinical manifestation of MCPH begins to appear during the first trimester, when early neurogenesis begins. Thus, 3D brain organoids represent a reliable experimental system to model MCPH development. In addition, 3D human brain organoids are an ideal approach since i) they allow for the adaptation of a spectrum of patient samples with various genetic backgrounds, ii) they display organized tissue containing different neural cell types, and, importantly, iii) various differentiation stages of neurodevelopment in this in vitro approach are linked to their in vivo counterparts25,26,27. For example, the neural rosette is an equivalent structure to the developing neural tube.
Lancaster et al. used twice the number of patient iPSCs compared to control iPSCs in order to succeed in generating patient organoids. Of note, with the protocol described here, by modifying the existing methods, we could generate control and microcephaly brain organoids starting from the same cell numbers4. This was possible due to the defined culture conditions of iPSCs and due to a more directed differentiation. In this protocol, organoid generation was improved by replacing the commonly performed EB formation step by initiating neural differentiation directly from iPSCs. Secondly, in step 3, dorsomorphin and SB431542 were supplemented into the culture medium instead of retinoic acid alone. Retinoic acid is readily isomerized when applied to cell culture medium28. Therefore, its biological activity is less defined than that of more stable compounds such as dorsomorphin. Dorsomorphin inhibits the BMP4 signaling pathway, and SB431542 is an inhibitor of the activin/nodal signaling pathway (TGF-β1 ALK inhibitor). A combination of both compounds leads to the inhibition of BMP and the activin/nodal signaling pathway, induces neural differentiation, and reduces the differentiation into other lineages in various cell lines from human ES or human iPS cells with a similar efficiency29. Compound stability and universal cell response to a compound are important points for establishing a protocol that can be applied to different patient cell lines to model a disease in vitro. Thus, a robust and efficient differentiation results in homogenous brain organoid cultures and, eventually, in more reproducible data.
With these modifications, critical steps within the protocol are minimized to the initiation of differentiation in step 1. At this point, it is important to gently dissociate the iPSCs into a single-cell suspension and to distribute them accurately and evenly to each well of the 96-well plate. Rho-associated protein kinase inhibitor (Y-27632) must be added to medium B during the first 24 h, as it supports the survival of iPSCs upon their dissociation to single cells. It is important to bring cells in close contact with each other by spinning them down to the bottom of the wells. Once neurospheres are formed at the end of step 1, the successful completion of further experimental procedures can be expected. In case neurospheres do not form, the pluripotency of the human iPSCs, the non-adherence of the cells in the 96-well plate, and the centrifuging conditions must be checked. The whole procedure on day 0 of step 1 should take no longer than 1 h due to the sensitivity of human iPSCs. Medium changes during the following days must be done carefully, avoiding sheer forces, in order not to destroy freshly formed cell contacts between the cells.
It is noteworthy that centrosomal mutants have defective cell proliferation and altered cell-cycle dynamics. Thus, modeling MCPH requires a powerful protocol that can withstand the compromised cellular functions. Hence, the current protocol allows for the generation of homogenous organoids of high quality and serves as a unique tool to study stem cell homeostasis at the VZ. In contrast to other protocols, this protocol enables the generation of organoids from control and patient iPSCs, starting with equal cell numbers. For studies based on in vitro differentiation, identical culture conditions for different iPSCs are a basic requirement to identify disease-related alterations and to avoid culture-condition artifacts. Besides modeling microcephaly of genetic origin, this protocol can also be applied to microcephaly of non-genetic origin, including from neurotrophic viral infections, chemicals, or radiation.30 In addition, modern molecular biology tools, such as CRISPR/Cas9 genome-editing technologies, can be applied to 3D organoids to dissect specific aspects of the human brain development paradigm in vitro31,32,33.
On the other hand, the generation of brain organoids to date has still not gone beyond the first and early second trimester of human development34. Surpassing this limitation and enabling the generation of mature brain organoids in vitro would open new avenues to model neurodegenerative disorders that manifest at later stages, such as Parkinson's or Alzheimer's disease. For future applications, protocols directing differentiation to more specific regions, like the forebrain or midbrain, might be of interest to study complex neurological disorders like autism and schizophrenia35,36,37,38.
Importantly, certain aspects should be taken into consideration when drawing conclusions from organoid studies. The first limitation is that there is no obvious vascularization in organoids. Thus, gaseous exchange and nutrient supply in vitro only approximate in vivo conditions. Second, since brain organoids are not connected to complex organ systems, the organoid model lacks a complete immune, metabolic, and hormonal system. Nevertheless, the absence of these aspects sometimes provides an advantage. For example, the organoids described here could replace immunosuppressed in vivo models when addressing the impact of the immune system in the pathogenesis of brain disorders.
With the use of spinner flasks, a large number of organoids can be generated for biochemical experiments, whole transcriptome sequencing analysis, and high-throughput drug screenings. Taken together, by using this current protocol, one can generate brain organoids from human iPSCs cells, which can then be utilized in a broad range of applications, from disease modeling to drug testing platforms, in order to reliably replace animal trials in the future.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
This work was supported by the Fritz Thyssen Foundation (Az.10.14.2.152). We are grateful to the tissue embedding facility and the microscope core facility of CMMC. We are grateful for the discussions and technical support provided by the members of the Laboratory for Centrosome and Cytoskeleton Biology. We thank Li Ming Gooi for proofreading the manuscript.
References
- Lancaster MA, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiraku M, Sasai Y. Mouse embryonic stem cell culture for generation of three-dimensional retinal and cortical tissues. Nat Protoc. 2012;7:69–79. doi: 10.1038/nprot.2011.429. [DOI] [PubMed] [Google Scholar]
- Nakano T, et al. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell. 2012;10:771–785. doi: 10.1016/j.stem.2012.05.009. [DOI] [PubMed] [Google Scholar]
- Gabriel E, et al. CPAP promotes timely cilium disassembly to maintain neural progenitor pool. EMBO J. 2016;35(8):803–819. doi: 10.15252/embj.201593679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Dosari MS, Shaheen R, Colak D, Alkuraya FS. Novel CENPJ mutation causes Seckel syndrome. J Med Genet. 2010;47:411–414. doi: 10.1136/jmg.2009.076646. [DOI] [PubMed] [Google Scholar]
- Wollnik B. A common mechanism for microcephaly. Nat Genet. 2010;42:923–924. doi: 10.1038/ng1110-923. [DOI] [PubMed] [Google Scholar]
- Thornton GK, Woods CG. Primary microcephaly: do all roads lead to Rome? Trends Genet. 2009;25:501–510. doi: 10.1016/j.tig.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbelanne M, Tsang WY. Molecular and cellular basis of autosomal recessive primary microcephaly. Biomed Res Int. 2014;2014:547986. doi: 10.1155/2014/547986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homem CC, Repic M, Knoblich JA. Proliferation control in neural stem and progenitor cells. Nat. Rev. Neurosci. 2015;16:647–659. doi: 10.1038/nrn4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel E, et al. CPAP promotes timely cilium disassembly to maintain neural progenitor pool. EMBO J. 2016;35:803–819. doi: 10.15252/embj.201593679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak SK, et al. Small molecules greatly improve conversion of human-induced pluripotent stem cells to the neuronal lineage. Stem Cells Int. 2012. [DOI] [PMC free article] [PubMed]
- Morizane A, Doi D, Kikuchi T, Nishimura K, Takahashi J. Small-molecule inhibitors of bone morphogenic protein and activin/nodal signals promote highly efficient neural induction from human pluripotent stem cells. J. Neurosci. Res. 2011;89:117–126. doi: 10.1002/jnr.22547. [DOI] [PubMed] [Google Scholar]
- Shi Y, Kirwan P, Livesey FJ. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat Protoc. 2012;7:1836–1846. doi: 10.1038/nprot.2012.116. [DOI] [PubMed] [Google Scholar]
- Zhou J, et al. High-efficiency induction of neural conversion in human ESCs and human induced pluripotent stem cells with a single chemical inhibitor of transforming growth factor beta superfamily receptors. Stem Cells. 2010;28:1741–1750. doi: 10.1002/stem.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Okita K, et al. A more efficient method to generate integration-free human iPS cells. Nat Methods. 2011;8:409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- Yu J, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324:797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieu PT, Fontes A, Vemuri MC, Macarthur CC. Generation of induced pluripotent stem cells with CytoTune, a non-integrating Sendai virus. Methods Mol. Biol. 2013;997:45–56. doi: 10.1007/978-1-62703-348-0_5. [DOI] [PubMed] [Google Scholar]
- Bai Q, et al. Temporal analysis of genome alterations induced by single-cell passaging in human embryonic stem cells. Stem Cells Dev. 2015;24:653–662. doi: 10.1089/scd.2014.0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garitaonandia I, et al. Increased risk of genetic and epigenetic instability in human embryonic stem cells associated with specific culture conditions. PLoS One. 2015;10:e0118307. doi: 10.1371/journal.pone.0118307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnuma K, et al. Enzyme-free passage of human pluripotent stem cells by controlling divalent cations. Sci. Rep. 2014;4:4646. doi: 10.1038/srep04646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currle DS, Monuki ES. Flash freezing and cryosectioning E12.5 mouse brain. J Vis Exp. 2007. [DOI] [PMC free article] [PubMed]
- Yingling J, et al. Neuroepithelial stem cell proliferation requires LIS1 for precise spindle orientation and symmetric division. Cell. 2008;132:474–486. doi: 10.1016/j.cell.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P. A small step for the cell, a giant leap for mankind: a hypothesis of neocortical expansion during evolution. Trends Neurosci. 1995;18:383–388. doi: 10.1016/0166-2236(95)93934-p. [DOI] [PubMed] [Google Scholar]
- Hu BY, et al. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc. Natl. Acad. Sci. U. S. A. 2010;107:4335–4340. doi: 10.1073/pnas.0910012107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson PG, Stice SS. Development and differentiation of neural rosettes derived from human embryonic stem cells. Stem Cell Reviews. 2006;2:67–77. doi: 10.1007/s12015-006-0011-1. [DOI] [PubMed] [Google Scholar]
- Dhara SK, Stice SL. Neural differentiation of human embryonic stem cells. J. Cell Biochem. 2008;105:633–640. doi: 10.1002/jcb.21891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie VB, et al. Synthesis and evaluation of synthetic retinoid derivatives as inducers of stem cell differentiation. Org Biomol Chem. 2008;6:3497–3507. doi: 10.1039/b808574a. [DOI] [PubMed] [Google Scholar]
- Kim DS, et al. Robust enhancement of neural differentiation from human ES and iPS cells regardless of their innate difference in differentiation propensity. Stem Cell Reviews. 2010;6:270–281. doi: 10.1007/s12015-010-9138-1. [DOI] [PubMed] [Google Scholar]
- Gabriel E, et al. Recent Zika Virus Isolates Induce Premature Differentiation of Neural Progenitors in Human Brain Organoids. Cell stem cell. 2017. [DOI] [PubMed]
- Wu J, Hunt SD, Xue H, Liu Y, Darabi R. Generation and Characterization of a MYF5 Reporter Human iPS Cell Line Using CRISPR/Cas9 Mediated Homologous Recombination. Sci. Rep. 2016;6:18759. doi: 10.1038/srep18759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HL, Gee P, Ishida K, Hotta A. Efficient genomic correction methods in human iPS cells using CRISPR-Cas9 system. Methods. 2016;101:27–35. doi: 10.1016/j.ymeth.2015.10.015. [DOI] [PubMed] [Google Scholar]
- Grobarczyk B, Franco B, Hanon K, Malgrange B. Generation of Isogenic Human iPS Cell Line Precisely Corrected by Genome Editing Using the CRISPR/Cas9 System. Stem Cell Reviews. 2015;11:774–787. doi: 10.1007/s12015-015-9600-1. [DOI] [PubMed] [Google Scholar]
- Camp JG, et al. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc. Natl. Acad. Sci. U. S. A. 2015;112:15672–15677. doi: 10.1073/pnas.1520760112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell. 2016;165:1238–1254. doi: 10.1016/j.cell.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo J, et al. Midbrain-like Organoids from Human Pluripotent Stem Cells Contain Functional Dopaminergic and Neuromelanin-Producing Neurons. Cell Stem Cell. 2016;19:248–257. doi: 10.1016/j.stem.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani J, et al. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell. 2015;162:375–390. doi: 10.1016/j.cell.2015.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Avanzo C, et al. Alzheimer's in 3D culture: challenges and perspectives. Bioessays. 2015;37:1139–1148. doi: 10.1002/bies.201500063. [DOI] [PMC free article] [PubMed] [Google Scholar]