

Abstract
The Golgi-Cox method of neuron staining has been employed for more than two hundred years to advance our understanding of neuron morphology within histological brain samples. While it is preferable from a practical perspective to prepare brain sections at the greatest thickness possible, in order to increase the probability of identifying stained neurons that are fully contained within single sections, this approach is limited from a technical perspective by the working distance of high-magnification microscope objectives. We report here a protocol to stain neurons using the Golgi-Cox method in mouse brain sections that are cut at 500 μm thickness, and to visualize neurons throughout the depth of these sections using an upright microscope fitted with a high-resolution 30X 1.05 N.A. silicone oil-immersion objective that has an 800 μm working distance. We also report two useful variants of this protocol that may be employed to counterstain the surface of mounted brain sections with the cresyl violet Nissl stain, or to freeze whole brains for long-term storage prior to sectioning and final processing. The main protocol and its two variants produce stained thick brain sections, throughout which full neuron dendritic trees and dendrite spines may be reliably visualized and quantified.
Keywords: Neuroscience, Issue 122, neuron morphology, histology, Golgi-Cox staining, cerebral cortex, hippocampus, dendrite, spines, pyramidal neuron
Introduction
The visualization of individual neurons within tissue samples allows for the in situ analysis of neuron morphological characteristics, which has significantly advanced our understanding of the brain and how it may be influenced by endogenous disease or exogenous environmental factors. The Golgi-Cox staining method is a cost-effective, relatively simple means of staining a random sample of neurons within the brain. First developed by Golgi1 and modified by Cox2 in the 1800s, researchers have further refined this technique over the years to produce clear, well-stained neurons that can be used to visualize and quantify both dendritic tree morphology and spine density3,4,5,6,7,8,9.
A major technical consideration for the visualization of stained neurons within brain sections is the maximum slice thickness, which is limited by the working distance of available high-magnification/high-resolution microscope objectives. Common oil-immersion objectives in the 60 - 100X range provide excellent resolution, but are limited by their working distances that are typically no greater than 200 μm. Brain sections cut at the 200 μm range may be adequate to visualize certain neuron types that can be contained within this slice thickness, for example pyramidal neurons in shallow layers of the cerebral cortex10,11,12, pyramidal neurons in the CA1 region of the hippocampus13,14, and granule cells in the dentate gyrus of the hippocampus15. Neurons with relatively longer dendritic trees, such as pyramidal neurons within deep layers of the cerebral cortex that for mouse can extend more than 800 μm from the cell body16, provide a greater challenge because brains would need to be sectioned at a perfect angle to contain the entire dendrite tree within 200 μm slices. This may not even be feasible if a dendrite or any of its branches extend in the rostral or caudal direction. While it is possible to address this limitation by tracing a neuron across multiple adjacent brain sections, this approach introduces a significant technical challenge in aligning the sections accurately for tracing17. A more practical approach would be to visualize entire neurons contained within brain sections that are cut at a greater thickness.
We report here a technique to stain neurons within 400 - 500 μm thick brain sections of mice using the Golgi-Cox method, and to visualize their morphology using a high-resolution silicon oil-immersion objective that has an 800 μm working distance. The Golgi-Cox impregnation and processing protocol that we describe is modified from one of the most-cited modern protocols in the literature6. Our approach with thick brain sections provides the advantage of increasing the probability of identifying neurons of any type that are fully contained within the section. In addition to the main protocol, we also present two variations that provide unique advantages: (1) Golgi-Cox staining with the cresyl violet counterstain on the surface of mounted sections, in order to define boundaries of brain regions and to identify layers of the cerebral cortex, and (2) Golgi-Cox staining with an intermediate freezing step for the long-term storage of impregnated whole brains prior to sectioning and final processing.
Protocol
Adult female CD1-strain mice were used in this study. Similar staining can be accomplished using both sexes at various ages. Experimental animals were cared for according to the principles and guidelines of the Canadian Council on Animal Care, and the experimental protocol was approved by the University of Guelph Animal Care Committee.
1. Golgi-Cox Staining
- Golgi-Cox Impregnation of Brains
- Make the Golgi-Cox solution of 1% (w/v) potassium dichromate, 0.8% (w/v) potassium chromate and 1% (w/v) mercuric chloride by dissolving the potassium dichromate and potassium chromate into high-quality water separately. Mix the solutions, add the mercuric chloride and filter the final solution using grade 1 filter paper. Store the solution in the dark for up to one month.
- Anesthetize the mouse using 5% isoflurane.
- Euthanize the mouse by decapitation and quickly remove the brain.
- Place the brain into a 20 mL glass scintillation vial containing 17 mL of Golgi-Cox solution and incubate in the dark at RT for 25 d.
- Cryoprotect the brain by placing it into a 50 mL conical tube containing 40 mL of sucrose cryoprotectant (30% (w/v) sucrose in 0.1 M phosphate buffer, pH 7.4) in the dark at 4 °C for 24 h. NOTE: The following optional three steps may be employed as an alternative to the main protocol, in order to freeze the brain at this stage for long-term storage.
- Freeze the whole brain by immersing it into 200 mL isopentane that has been precooled on dry ice.
- Place the frozen brain into a 50 mL conical tube and store in the dark at -80 °C.
- When ready to proceed, thaw the brain by placing it into a 50 mL conical tube containing 40 mL of sucrose cryoprotectant in the dark at 4 °C for 24 h.
- Brain Sectioning
- Remove the brain from the sucrose and block it for sectioning by cutting off the cerebellum using a razor blade and leaving a flat edge at the remaining caudal end of the brain.
- Heat agar (3% (w/v) in water) until it is melted and let cool until it is slightly above its melting point.
- Place the brain in a small disposable weigh dish with its caudal end face-down and add a sufficient amount of melted agar to cover the brain.
- Once the agar has solidified, trim excess agar leaving approximately 2 - 4 mm surrounding the brain and glue the brain to the stage of a vibratome with its caudal end face-down using a small amount of ethyl cyanoacrylate glue.
- Fill the stage area of the vibratome with a sufficient amount of sucrose cryoprotectant to cover the brain, and section the brain at a slice thickness of 400 - 500 µm (depending on the brain region to be examined) using a vibration frequency of 86 Hz and a blade advancement speed of 0.125 mm/s.
- Using a small paint brush, place brain sections into a well of a 6-well tissue culture plate containing commercially-available mesh-bottom inserts and pre-filled with 10 mL of 6% (w/v) sucrose in 0.1 M phosphate buffer, pH 7.4.
- Incubate sections in the dark at 4 °C O/N.
- Developing Brain Sections
- Using the mesh-bottom inserts, transfer brain sections into a new well containing 5 mL of 2% (w/v) paraformaldehyde (PBD) in 0.1 M phosphate buffer, pH 7.4. Incubate on a rocker moving at slow speed in the dark at RT for 15 min.
- Wash sections twice by transferring them to new wells containing 5 mL of water. Wash on a rocker moving at a moderate speed in the dark at RT for 5 min.
- Transfer sections into a new well containing 5 mL of 2.7% (v/v) ammonium hydroxide. Incubate on a rocker moving at a slow speed in the dark at RT for 15 min.
- Wash sections twice by transferring them to new wells containing 5 mL of water. Wash on a rocker moving at a moderate speed in the dark at RT for 5 min.
- Transfer sections into a new well containing 5 mL of Fixative A (see Materials Table) that has been diluted in water 10x from its original purchased concentration. Incubate on a rocker moving at a slow speed in the dark at RT for 25 min.
- Wash sections twice by transferring them to new wells containing 5 mL of water. Wash on a rocker moving at a moderate speed in the dark at RT for 5 min.
- Mounting Brain Sections
- Using a small paint brush, mount sections onto microscope slides. Remove excess water and agar using tweezers and a small tissue. Ensure that all agar is removed before proceeding with dehydration.
- Allow sections to air dry at RT for approximately 45 min (400 μm sections) or 90 min (500 μm sections). NOTE: The timing and proper level of dryness is critical, and may need to be determined in each laboratory depending on ambient temperature and humidity level. Too short a drying time leads to sections falling off of slides during subsequent dehydration steps, and too long a drying time leads to sections cracking. Sections will still appear to be shiny at the appropriate level of dryness.
- Stain sections with cresyl violet by placing slides into Coplin staining jars as indicated. NOTE: This optional step may be employed for sections that had never been frozen, in order to stain neuronal nuclei with cresyl violet. We have found that clearing and rehydrating sections before incubation in cresyl violet leads to even staining and low background across sections.
- Place in clearing agent for 5 min. Repeat once.
- Place in 100% ethanol for 5 min. Repeat once.
- Place in 95% ethanol in water, then 75% ethanol in water, and then 50% ethanol in water for 2 min each.
- Place in water for 5 min.
- Place in 0.5% (w/v) cresyl violet in water for 7 min.
- Place in water for 2 min. Repeat once.
- Dehydrate sections by placing slides into Coplin staining jars as indicated.
- Place in 50% ethanol in water, then 75% ethanol in water, and then 95% ethanol in water for 2 min each.
- Place in 100% ethanol for 5 min. Repeat once.
- Place in clearing agent for 5 min. Repeat once. NOTE: These dehydration and clearing times are sufficient to process mouse brains as described in this manuscript. However, we have observed for other species including rat and cowbird, that the final clearing step may need to be extended up to 15 min total.
- Coverslip sections using an anhydrous mounting medium.
- Allow slides to dry horizontally in the dark at RT for at least 5 d.
2. Imaging Stained Neurons within Thick Brain Sections
- Capturing Image Stacks
- Turn on the microscope light bulb, camera, and stage controller.
- Open the microscope software (e.g., Neurolucida).
- Place a slide on the microscope stage.
- Capture a 2D wide-view image of the brain section using a low magnification objective such as 1.25X 0.4 N.A. PlanAPO or 4X 0.16 N.A.
- Focus image and adjust camera settings including the exposure time and white balance.
- Create a reference point by left-clicking anywhere on the section
- Capture the image by selecting "acquire single image" within the image acquisition window.
- Capture mid-resolution image stacks of the area containing the neurons(s) of interest, using a 10X 0.3 N.A. UPlan FL N objective.
- Focus the image and adjust camera settings including the exposure and white balance.
- Set the upper and lower boundaries for the image stack by focusing to the top of the section and selecting "set" next to "top of stack" within the image acquisition window, and then focusing to the bottom of the section and selecting "set" next to "bottom of stack" within the image acquisition window.
- Set the step distance to 5 μm by entering "5 μm" next to "distance between images" within the image acquisition window.
- Capture the image stack by selecting "acquire image stack" within the image acquisition window.
- Repeat the above steps to capture the entire area of interest, making sure that all image stacks overlap by at least 10% in the X and Y axes.
- Capture high-resolution image stacks of the area containing the neuron of interest, using a 30X 1.05 N.A. silicone oil-immersion objective.
- Apply 3 - 4 drops of silicone immersion oil to the slide and place the objective over the slide, ensuring to make contact between the objective and oil.
- Focus the image and adjust camera settings including the exposure and white balance.
- Set the upper and lower boundaries for the image stack by focusing to the top of the section and selecting "set" next to "top of stack" within the image acquisition window, and then focusing to the bottom of the section and selecting "set" next to "bottom of stack" within the image acquisition window.
- Set the step distance to 1 μm by entering "1 μm" next to "distance between images" within the image acquisition window.
- Capture the image stack by selecting "acquire image stack" within the image acquisition window.
- Repeat the above steps to capture the entire area of interest, making sure that all image stacks overlap by at least 10% in the X and Y axes.
- Save the data file and save all image files in TIFF format for external processing.
- Creating Z-projection Images and Image Montages
- Create Z-projection images in ImageJ
- Open ImageJ software that has the Bio-Formats plugin installed.
- Select "Plugins" -> "Bio-Formats" -> "Bio-Formats Importer".
- Select the image stack file to be opened.
- Once the file is opened, change the format to RGB by selecting "Image" -> "Type" -> "RGB Color".
- Create the Z-projection by selecting "Image" -> "Stacks" -> "Z Project…".
- Save two-dimensional Z-projection image as a TIFF file.
- Create a two-dimensional image montage of entire area of interest.
- Open the software (e.g., Adobe Photoshop).
- Select "File" -> "Automate" -> "Photomerge".
- Select "Browse" and then add all images files to be merged.
- Ensure that "Blend Images Together" is selected and then select "OK".
- Save the resulting montage image of the entire area of interest as a TIFF file.
Representative Results
This Golgi-Cox staining protocol and its two described optional variants may be employed to visualize individual neurons within 400 - 500 μm thick brain sections. Representative image montages of two-dimensional Z-projections captured using a 10X objective and 5 μm steps in the Z axis are shown in Figure 1: A1 - C1 for a large area of coronal brain sections that includes the anterior cingulate cortex area 1 and the secondary motor cortex18. Sections were cut at 500 μm. Note that the protocol variant that includes cresyl violet staining allows for the identification of cortical cell layers, for example as seen for cortical Layer 1 along the pial surface of the sections in Figure 1B1. Note also the similar appearance of stained sections when using the main protocol using all-fresh tissue (Figure 1A1) and the protocol variant in which brains are frozen part way through for long-term storage (Figure 1C1).
The use of a high-resolution 30X 1.05 N.A. silicone oil-immersion objective allows for the capture of image stacks that are larger in the X and Y axes than those captured using a higher-magnification objective, thus requiring fewer total stacks to image a given area of interest. The particular objective employed has an 800 μm working distance, which is more than sufficient to image thick brain sections. Image montages of two-dimensional Z-projections captured using this objective and 1 μm steps in the Z axis are shown in Figure 1 (A2-C2) within the anterior cingulate cortex area 1, and two-dimensional Z-projections of tracings for the indicated neurons are shown in Figure 1: A3 - C3. Note the high resolution images that allow for the visualization of neurons and their dendrites through the depth of each image stack. The protocol variant using cresyl violet adds purple Nissl staining across the slice, however with the parameters employed this cellular staining is only observed near the top of the slice. The protocol variant that includes the optional freezing step produces neuron staining that is similar to the main protocol, however it also introduces a diffuse background staining that creates the appearance of haze by diffracting light below the surface of the slice. This haze is most apparent when imaging deeper into the slice and is apparent in the Z-projection image in Figure 1C2. Images captured using the 30X objective may also be used to visualize and quantify fine neuronal structures such as dendritic spines. Single-plane images are shown in Figure 2 for dendrites stained using the main protocol and its two variants, with one image taken near the top of the section and one image taken near the bottom of the section. Note the purple cresyl violet staining of Nissl substance within individual neuronal nuclei near the top of the section in Figure 2B1, which is seen as diffuse purple background / scattered light near the bottom of the section in Figure 2B2. Note also that although dendrites and their spines are well-stained using the protocol with the freezing step, the previously-mentioned background haze that is apparent near the bottom of the slice in Figure 2C2 makes it more difficult to visualize spines by limiting the contrast between them and the adjacent background. Higher-magnification objectives can also be used to characterize dendritic spine morphology, as show in Figure 2: A3 - C3 where spines of differing types are clearly visible in sections stained using each of the three protocols. Here, a 60X 1.42 N.A. oil-immersion objective was used. We also employed this staining protocol and its variants to stain neurons within the hippocampus of sections cut at 400 μm. As shown in Figure 3, neuron dendrites and spines can be visualized both toward the more-medial areas of the section (e.g., the hippocampus dentate gyrus region in Figure 3: A2 - C2) and the more-lateral areas of the section (e.g., the hippocampus CA3 region in Figure 3: A3 - C3). It should also be noted that each staining condition produced similar results, advantages and disadvantages to those shown in Figures 1 & 2 for the cerebral cortex.
The final thickness of processed brain sections depends on the specific protocol variant employed. As shown in Figure 4A for sections containing the cerebral cortex and cut at 500 μm, there was a significant effect of protocol variant on the measured thickness of processed and mounted/coverslipped sections (one-way ANOVA, p < 0.0001). Here, the thickness of sections made using the main protocol (251.0 ± 12.5 (SEM) μm (n = 6)) and the protocol variant involving cresyl violet (219.3 ± 8.5 μm (n = 6)) was smaller than the thickness of sections made using the protocol variant involving the freezing step (340.6 ± 17.1 μm (n = 6)) (Bonferroni post-hoc test, each comparison p ≤ 0.0006). Very similar effects of protocol variant were observed for sections containing the hippocampus and cut at 400 μm (Figure 4B, one-way ANOVA, p < 0.0001). Here, the thickness of sections made using the main protocol (217.6 ± 19.2 μm (n = 6)) and the protocol variant involving cresyl violet (198.0 ± 14.8 μm (n = 6)) was smaller than the thickness of sections made using the protocol variant involving the freezing step (313.5 ± 8.4 μm (n = 6)) (Bonferroni post-hoc test, each comparison p ≤ 0.001).
 Figure 1. Exemplary Photomicrographs of Stained Neurons in the Anterior Cingulate Area 1. A1 - C1, Merged Z-projections of image stacks acquired using a 10X objective are shown for one hemisphere of the cerebral cortex that includes the anterior cingulate cortex area 1 and secondary motor cortex at approximately Bregma +1.18 mm18. These sections were cut at 500 μm. Photomicrographs are shown using the main all-fresh protocol (A1), for the fresh protocol variant with cresyl violet staining (B1) and for the protocol variant in which brains are frozen following Golgi-Cox impregnation (C1). Scale bars are 500 μm. Higher-resolution merged Z-projection image stacks acquired using a 30X silicon oil-immersion objective are shown in A2 - C2 for each area indicated by the red square in the photomicrographs in A1 - C1. Scale bars are 100 μm. 2D Z-projections of neuron tracings are shown in A3 - C3 for the neurons indicated by the red arrow in the photomicrographs in the A2 - C2. The diameter of all dendrites were set to 2 μm for ease of illustration. Scale bars are 100 μm. Please click here to view a larger version of this figure.
Figure 1. Exemplary Photomicrographs of Stained Neurons in the Anterior Cingulate Area 1. A1 - C1, Merged Z-projections of image stacks acquired using a 10X objective are shown for one hemisphere of the cerebral cortex that includes the anterior cingulate cortex area 1 and secondary motor cortex at approximately Bregma +1.18 mm18. These sections were cut at 500 μm. Photomicrographs are shown using the main all-fresh protocol (A1), for the fresh protocol variant with cresyl violet staining (B1) and for the protocol variant in which brains are frozen following Golgi-Cox impregnation (C1). Scale bars are 500 μm. Higher-resolution merged Z-projection image stacks acquired using a 30X silicon oil-immersion objective are shown in A2 - C2 for each area indicated by the red square in the photomicrographs in A1 - C1. Scale bars are 100 μm. 2D Z-projections of neuron tracings are shown in A3 - C3 for the neurons indicated by the red arrow in the photomicrographs in the A2 - C2. The diameter of all dendrites were set to 2 μm for ease of illustration. Scale bars are 100 μm. Please click here to view a larger version of this figure.
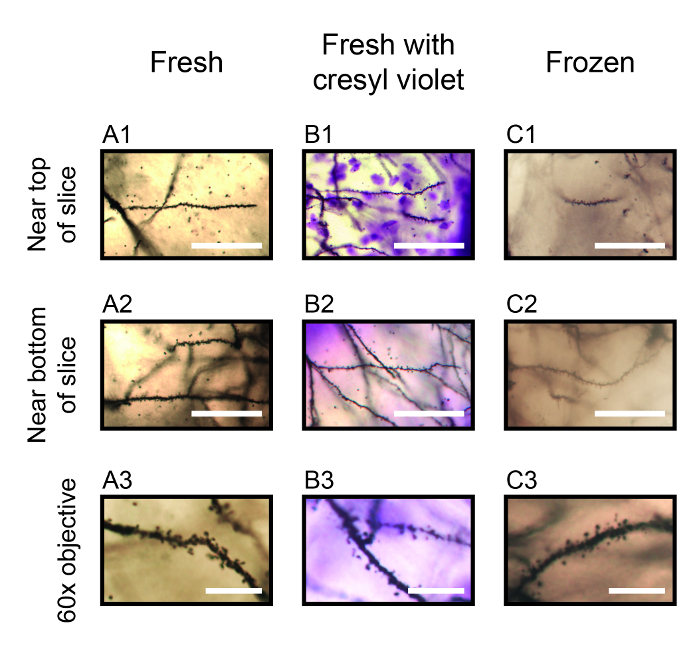 Figure 2. Exemplary Photomicrographs of Dendritic Spines for Stained Neurons. Photomicrographs captured using a 30X silicon oil-immersion objective are shown in one focal plane for neuron dendrites located within layer 1 of the anterior cingulate cortex area 1. These sections were cut at 500 μm. Neurons were stained using the fresh protocol (A1, A2), the protocol variant with cresyl violet staining (B1, B2) and for the protocol variant in which brains are frozen following Golgi-Cox impregnation (C1, C2). Photomicrographs were taken near the top of the slice (A1 - C1) and near the bottom of the slice next to the microscope slide (A2 - C2). Scale bars for A1-2, B1-2 and C1-2 are 50 μm. Photomicrographs are shown in A3, B3 and C3 for one focal plane using a higher-magnification 60X oil-immersion objective, in order to identify different spine types. Scale bars for A3, B3 and C3 are 10 μm. Please click here to view a larger version of this figure.
Figure 2. Exemplary Photomicrographs of Dendritic Spines for Stained Neurons. Photomicrographs captured using a 30X silicon oil-immersion objective are shown in one focal plane for neuron dendrites located within layer 1 of the anterior cingulate cortex area 1. These sections were cut at 500 μm. Neurons were stained using the fresh protocol (A1, A2), the protocol variant with cresyl violet staining (B1, B2) and for the protocol variant in which brains are frozen following Golgi-Cox impregnation (C1, C2). Photomicrographs were taken near the top of the slice (A1 - C1) and near the bottom of the slice next to the microscope slide (A2 - C2). Scale bars for A1-2, B1-2 and C1-2 are 50 μm. Photomicrographs are shown in A3, B3 and C3 for one focal plane using a higher-magnification 60X oil-immersion objective, in order to identify different spine types. Scale bars for A3, B3 and C3 are 10 μm. Please click here to view a larger version of this figure.
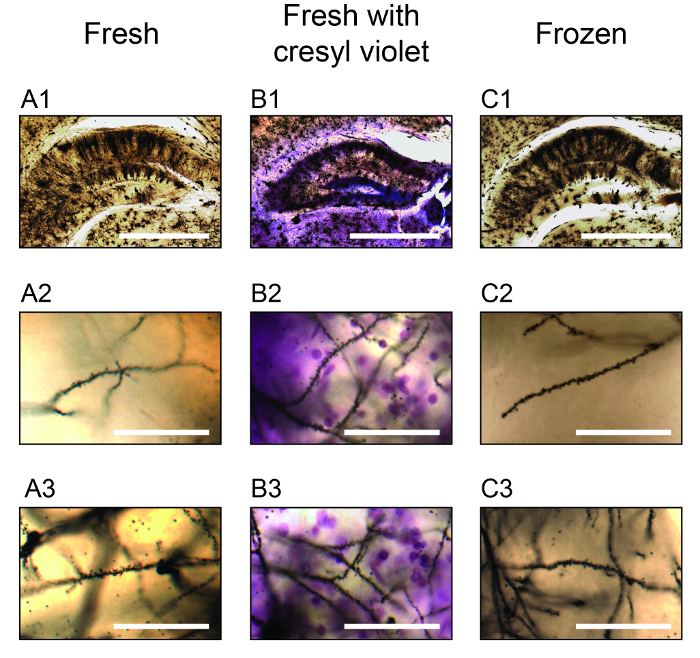 Figure 3. Exemplary Photomicrographs of Stained Neurons in the Hippocampus. Photomicrographs captured using a 4X objective are shown in one focal plane for neurons stained using the fresh protocol (A1), the protocol variant with cresyl violet staining (B1) and for the protocol variant in which brains are frozen following Golgi-Cox impregnation (C1). Sections were cut at 400 μm and are shown at approximately Bregma -2.18 mm18. Scale bars are 500 μm for A1 - C1. Higher magnification photomicrographs were captured using a 30X silicon oil-immersion objective and are shown in one focal plane for neuron dendrites located within the dentate gyrus region of the hippocampus (A2 - C2) and the CA3 region of the hippocampus (A3 - C3). Scale bars are 50 μm for A2 - C2 and A3 - C3. Please click here to view a larger version of this figure.
Figure 3. Exemplary Photomicrographs of Stained Neurons in the Hippocampus. Photomicrographs captured using a 4X objective are shown in one focal plane for neurons stained using the fresh protocol (A1), the protocol variant with cresyl violet staining (B1) and for the protocol variant in which brains are frozen following Golgi-Cox impregnation (C1). Sections were cut at 400 μm and are shown at approximately Bregma -2.18 mm18. Scale bars are 500 μm for A1 - C1. Higher magnification photomicrographs were captured using a 30X silicon oil-immersion objective and are shown in one focal plane for neuron dendrites located within the dentate gyrus region of the hippocampus (A2 - C2) and the CA3 region of the hippocampus (A3 - C3). Scale bars are 50 μm for A2 - C2 and A3 - C3. Please click here to view a larger version of this figure.
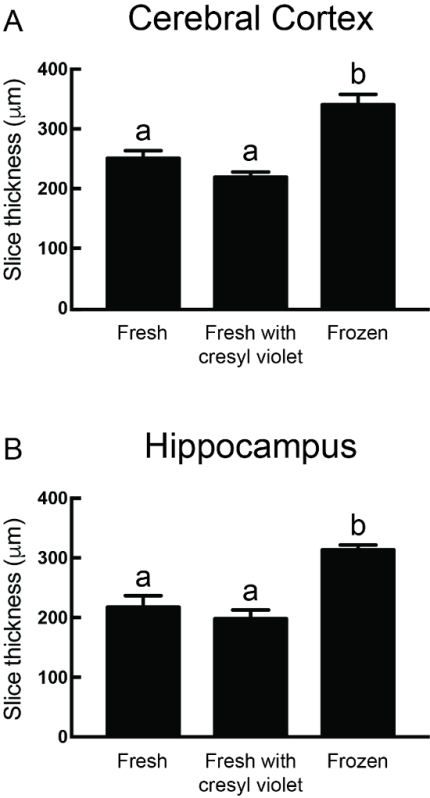 Figure 4. Thickness of Mounted Sections. The measured thickness of mounted sections is shown in A for sections containing the cerebral cortex at approximately Bregma +1.18 mm18 and cut at 500 μm thickness, and in B for sections containing the hippocampus at approximately Bregma -2.18 mm18 and cut at 400 μm thickness. Measured sections were stained using either the fresh protocol, the protocol variant with cresyl violet staining, or the protocol variant in which brains are frozen following Golgi-Cox impregnation. For both brain regions, there was a significant effect of processing protocol (one-way ANOVA, p < 0.0001), where sections from brains that were frozen part way through the protocol were significantly thicker than those that were not frozen (Bonferroni post-hoc test, all p < 0.001). Data are shown as mean ±SEM for six sections in each group, and data sets with different letters indicate significant differences. Please click here to view a larger version of this figure.
Figure 4. Thickness of Mounted Sections. The measured thickness of mounted sections is shown in A for sections containing the cerebral cortex at approximately Bregma +1.18 mm18 and cut at 500 μm thickness, and in B for sections containing the hippocampus at approximately Bregma -2.18 mm18 and cut at 400 μm thickness. Measured sections were stained using either the fresh protocol, the protocol variant with cresyl violet staining, or the protocol variant in which brains are frozen following Golgi-Cox impregnation. For both brain regions, there was a significant effect of processing protocol (one-way ANOVA, p < 0.0001), where sections from brains that were frozen part way through the protocol were significantly thicker than those that were not frozen (Bonferroni post-hoc test, all p < 0.001). Data are shown as mean ±SEM for six sections in each group, and data sets with different letters indicate significant differences. Please click here to view a larger version of this figure.
Discussion
We describe here a Golgi-Cox staining protocol along with two useful variants for visualizing neurons within thick brain sections. As shown in the Representative Results, the use of a high-resolution objective that has a long 800 μm working distance allows for the reliable visualization of entire neurons throughout the depth of brain sections cut at 500 μm. This study of relatively thick brain sections increases the probability that stained neurons of any type are fully contained within the slice, which is especially important for pyramidal neurons with long and complicated apical dendrite trees. For example, in coronal sections of rodent brain, this allows for the inclusion of neurons that extend farther in the rostral or caudal direction, and also increases the room for error when blocking the brain squarely at the sectioning step. Although we describe brain sectioning in the coronal plane only, this protocol may be adapted for sectioning in other planes as required to fully contain labeled neurons within single sections. The plane of sectioning employed will depend on the morphological properties of the neuron population under investigation. Dendritic spines can be visualized and quantified using this protocol, although higher-magnification objectives are required to assess spine type/morphology such as the 60X oil-immersion objective used to produce the photomicrographs in Figure 2: A3 - C3. This can be employed in thick sections but is limited to the top of the slice within the working distance of the objective. This protocol can also be adapted for use in different species of similar brain size, as we have found that a standard 25-day impregnation period will fully stain neurons throughout the mouse, rat and cowbird brain without leading to excess non-specific staining or background that may occur with impregnation periods beyond one month. Shorter impregnation periods may be sufficient for species with smaller brains or in dissected samples of brains from the above-mentioned species, which could be optimized by the individual investigator.
Of the three variants presented for this staining protocol, the main protocol using fresh brain samples that are not counterstained yields the best results. Photomicrographs in Figure 2A demonstrate that this main protocol produces well-labeled dendrites and spines that are easily visible near the top and bottom of mounted thick sections. Adding the cresyl violet counterstain has the advantage of facilitating the definition of brain regions and cerebral cortex layers, but also added a diffuse purple background when visualizing deep into thick sections. However, since this protocol variant only stains the surface of mounted thick sections with cresyl violet, dendrites and their spines were clearly visible near the bottom of sections (Figure 2B). This also appears to produce a clearer pattern of Nissl staining near the surface of thick sections than that shown in previously reports, even when using much thinner sections19,20,21. The protocol variant that includes the freezing step yields acceptable results for the analysis of dendritic tree morphology. However, the background staining is much darker than that in fresh tissue, which is most noticeable when imaging deeper into the slice thus making it more difficult to visualize and quantify dendrite spines (Figure 2C). We attempted the cresyl violet counterstain in sections from brains that had been frozen, and this produced an even-darker purple background haze that made it extremely difficult to visualize and quantify spines near the bottom of the section (data not shown). Another unsuccessful protocol variant that was assessed included freezing mouse brains prior to Golgi-Cox impregnation. This resulted in the staining of cellular objects that may have been glia, but did not stain any type of neuron (data not shown).
There are two critical steps in this protocol that must be followed to obtain successful results. (1) It is critical to verify the drying time of sections within each laboratory, because this is highly dependent on temperature and humidity. If the drying time is too short, sections will fall off the slides during the dehydration process; if the drying time is too long, sections will crack. It would be advantageous to verify drying time in a set of "test" brains just prior to preforming a research study, as this can vary by season even with the same laboratory. (2) All excess agar must be removed from the section and microscope slide before the dehydration steps. If this is not done, the agar will deform and may pull the section off of the slide.
Another important factor for the histological analysis of biological samples is tissue shrinkage. We found that after processing, mounting, dehydrating and coverslipping brain sections using the main Golgi-Cox protocol that the final section thickness was reduced by approximately half of the original cut value (Figure 4). Also note that the sections processed using the protocol variant that included the freezing step were significantly thicker than the fresh sections that were processed via the main protocol (Figure 4). This was both interesting and unexpected because both protocols involved the same cryoprotection and processing steps, and only differed by the actual freezing of brains. It therefore is critical to account for potential differences in protocols when comparing neuron morphology data generated using Golgi-Cox staining from different studies or from different laboratories. Although we were not able to find data for final section thickness in the literature, it would be beneficial for investigators to report this data and/or correct for tissue shrinkage when reporting measures of neuron morphology. It should also be noted that although the final thickness of >200 μm for sections cut at 500 μm is greater than the working distance of most high-magnification objectives, this is still well within the working distance of the 30X objective used in our laboratory. Since dendrites and spines are clearly visible near the bottom of these slices using the main Golgi-Cox protocol, it may be possible to visualize neurons contained within sections cut at a much greater thickness up to and including the 1 mm range.
Although this protocol offers a number of advantages including the ability to trace long dendritic arbors within single sections, the option to delay slicing and processing of impregnated brains, and the option of counterstaining with cresyl violet to better identify brain structures, there are some disadvantages that should be considered. The 25-day incubation period may be considered too long for some investigators. The ability to image deep into mounted sections depends on access to a high-resolution immersion objective with a long working distance, which is relatively expensive. In addition, the ability to visualize fine structures with high resolution decreases as one images near the bottom of the mounted slice. The implementation of this protocol therefore depends on the morphological properties of the neurons and brain region to be studied, and the equipment available to the investigator. We recommend the use of "test" samples to optimize protocol variables prior to running a research study, such as Golgi-Cox impregnation time for the size of the brain and region of interest, and the plane and thickness of sectioning for the dendritic arbor to be visualized.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
This work was supported by a Discovery Grant to CDCB from the Natural Sciences and Engineering Research Council of Canada (NSERC), a John R. Evans Leaders Fund research infrastructure grant to CDCB from Canada Foundation for Innovation (CFI project number 30381), and by a Discovery Grant to NJM from NSERC. ELL was supported by an Ontario Graduate Scholarship and by an OVC Scholarship from the Ontario Veterinary College at the University of Guelph. CDS was supported by an Undergraduate Student Research Assistantship from NSERC. ALM was supported by an Alexander Graham Bell Scholarship from NSERC and by an OVC Scholarship from the Ontario Veterinary College at the University of Guelph.
References
- Golgi C. Sulla struttura della sostanza grigia del cervello. Gazzetta Medica Italiana. Lombardia. 1873;33:244–246. [Google Scholar]
- Cox WH. Imprägnation des centralen Nervensystems mit Quecksilbersalzen. Archiv f. mikrosk. Anat. 1891;37:16–21. [Google Scholar]
- Zaqout S, Kaindl AM. Golgi-Cox Staining Step by Step. Front Neuroanat. 2016;10:38. doi: 10.3389/fnana.2016.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ND, et al. Advances in thin tissue Golgi-Cox impregnation: fast, reliable methods for multi-assay analyses in rodent and non-human primate brain. J Neurosci Methods. 2013;213(2):214–227. doi: 10.1016/j.jneumeth.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjan A, Mallick BN. A modified method for consistent and reliable Golgi-Cox staining in significantly reduced time. Front Neurosci. 2010;1:157. doi: 10.3389/fneur.2010.00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb R, Kolb B. A method for vibratome sectioning of Golgi-Cox stained whole rat brain. J Neurosci Methods. 1998;79(1):1–4. doi: 10.1016/s0165-0270(97)00163-5. [DOI] [PubMed] [Google Scholar]
- Friedland DR, Los JG, Ryugo DK. A modified Golgi staining protocol for use in the human brain stem and cerebellum. J Neurosci Methods. 2006;150(1):90–95. doi: 10.1016/j.jneumeth.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Narayanan SN, et al. Appraisal of the effect of brain impregnation duration on neuronal staining and morphology in a modified Golgi-Cox method. J Neurosci Methods. 2014;235:193–207. doi: 10.1016/j.jneumeth.2014.07.007. [DOI] [PubMed] [Google Scholar]
- Das G, Reuhl K, Zhou R. The Golgi-Cox method. Methods Mol Biol. 2013;1018:313–321. doi: 10.1007/978-1-62703-444-9_29. [DOI] [PubMed] [Google Scholar]
- Sutherland R, Gibb R, Kolb B. The hippocampus makes a significant contribution to experience-dependent neocortical plasticity. Behav Brain Res. 2010;214(1):121–124. doi: 10.1016/j.bbr.2010.05.051. [DOI] [PubMed] [Google Scholar]
- Hamilton GF, Whitcher LT, Klintsova AY. Postnatal binge-like alcohol exposure decreases dendritic complexity while increasing the density of mature spines in mPFC Layer II/III pyramidal neurons. Synapse. 2010;64(2):127–135. doi: 10.1002/syn.20711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashed MG, Seidlitz EP, Frey BN, Singh G. Depressive-like behaviours and decreased dendritic branching in the medial prefrontal cortex of mice with tumors: A novel validated model of cancer-induced depression. Behav Brain Res. 2015;294:25–35. doi: 10.1016/j.bbr.2015.07.040. [DOI] [PubMed] [Google Scholar]
- Frauenknecht K, et al. Mice with experimental antiphospholipid syndrome display hippocampal dysfunction and a reduction of dendritic complexity in hippocampal CA1 neurones. Neuropathol Appl Neurobiol. 2015;41(5):657–671. doi: 10.1111/nan.12180. [DOI] [PubMed] [Google Scholar]
- Camacho-Abrego I, et al. Rearrangement of the dendritic morphology of the neurons from prefrontal cortex and hippocampus after subthalamic lesion in Sprague-Dawley rats. Synapse. 2014;68(3):114–126. doi: 10.1002/syn.21722. [DOI] [PubMed] [Google Scholar]
- Redila VA, Christie BR. Exercise-induced changes in dendritic structure and complexity in the adult hippocampal dentate gyrus. Neuroscience. 2006;137(4):1299–1307. doi: 10.1016/j.neuroscience.2005.10.050. [DOI] [PubMed] [Google Scholar]
- Bailey CD, Alves NC, Nashmi R, De Biasi M, Lambe EK. Nicotinic alpha5 subunits drive developmental changes in the activation and morphology of prefrontal cortex layer VI neurons. Biol Psychiatry. 2012;71(2):120–128. doi: 10.1016/j.biopsych.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytton WW. From computer to brain : foundations of computational neuroscience. Springer; 2002. [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. 2nd. Academic; 2001. [Google Scholar]
- Ha H. A modified Golgi-Cox method with counterstain for the study of synapses. Anat Rec. 1966;155(1):59–64. doi: 10.1002/ar.1091550107. [DOI] [PubMed] [Google Scholar]
- Swanson LW. The locus coeruleus: a cytoarchitectonic, Golgi and immunohistochemical study in the albino rat. Brain Res. 1976;110(1):39–56. doi: 10.1016/0006-8993(76)90207-9. [DOI] [PubMed] [Google Scholar]
- Pilati N, Barker M, Panteleimonitis S, Donga R, Hamann M. A rapid method combining Golgi and Nissl staining to study neuronal morphology and cytoarchitecture. J Histochem Cytochem. 2008;56(6):539–550. doi: 10.1369/jhc.2008.950246. [DOI] [PMC free article] [PubMed] [Google Scholar]