

Abstract
The ability to probe the structure and physiology of individual nerve cells in culture is crucial to the study of neurobiology, and allows for flexibility in genetic and chemical manipulation of individual cells or defined networks. Such ease of manipulation is simpler in the reduced culture system when compared to the intact brain tissue. While many methods for the isolation and growth of these primary neurons exist, each has its own limitations. This protocol describes a method for culturing low-density and high-purity rodent embryonic hippocampal neurons on glass coverslips, which are then suspended over a monolayer of glial cells. This 'sandwich culture' allows for exclusive long-term growth of a population of neurons while allowing for trophic support from the underlying glial monolayer. When neurons are of sufficient age or maturity level, the neuron coverslips can be flipped-out of the glial dish and used in imaging or functional assays. Neurons grown by this method typically survive for several weeks and develop extensive arbors, synaptic connections, and network properties.
Keywords: Neuroscience, Issue 122, primary neurons, astroglial monolayer, Sandwich culture, Banker method, hippocampus, low-density culture, cellular neurobiology
Introduction
The brain is organized into intricate networks of neurons. The contribution of individual neurons to network activity and brain function can be studied by selective alteration of their molecular composition and perturbance of their physiological properties. Genetic and chemical manipulation of individual neurons is arguably easier in cultured neurons than in intact brain tissue, unencumbered by the latter's cellular heterogeneity and complexity. Neurons in culture develop well-defined axonal and dendritic arbors and form extensive synaptic connections with each other.
While neuron culture from adult animals or from other regions of the nervous system is possible, embryonic hippocampal cultures are frequently preferred due to their defined pyramidal cell population and relatively low glial density 1,2. Hippocampal neurons grown at low density in culture are particularly amenable to the study of subcellular localization, protein trafficking, neuronal polarity and synapse development. Neurons in culture have also been extensively employed in studying molecular processes in synaptic plasticity 3,4,5,6. Neuron culture preparations from mice with global genetic deletions that do not survive postnatally have been especially useful in studying cellular and synaptic roles of certain genes 7.
As in the brain, cultured hippocampal neurons are dependent on trophic support from glial cells. This complicates their culture, and has led to the development of several different methods by which this support is supplied. One commonly used method involves plating neurons directly onto a monolayer of glial cells 8, or allowing contaminant glial cells from the acquired hippocampal tissue to proliferate and form a monolayer beneath the neurons 9. While this method has found some success, the impurity of the resulting neuronal culture is disadvantageous for imaging experiments. Another commonly used method of neuron culture is to leave-out the glial feeder layer altogether, and instead provide trophic support in the form of a defined growth medium 10.
Here, we describe the "sandwich" or "Banker" method of neuron culture 2,11. This method involves plating the hippocampal neurons on glass coverslips, which are then suspended over a monolayer of glial cells separated by paraffin wax beads. This facilitates long-term culture of a homogenous population of neurons without contaminating glia while allowing for trophic support from the underlying glial monolayer. When neurons are of sufficient age or maturity level, the neuron coverslips can be flipped-out of the glial dish and used in imaging or functional assays.
Protocol
All experiments and protocols using laboratory animals were approved by the University of Manitoba animal ethics committee and were compliant with the guidelines of the Canadian Council on Animal Care.
1. Preparation of Instruments, Buffers and Solutions
Sterilize by autoclaving all dissection equipment, glass Pasteur pipettes, pipette tips, filter apparatus, and deionized water.
Prepare a 20% (w/v; 1.1 M) stock glucose solution in deionized water and filter-sterilize. Store at 4 °C.
Prepare a 100 mM sodium pyruvate solution in deionized water and filter-sterilize. Store at 4 °C.
Prepare calcium- and magnesium-free Hank's Balanced Salt Solution (CMF-HBSS) by making a solution of 10% HBSS (10x), 10 mM HEPES, and 100 U/mL penicillin-streptomycin in deionized water. Store at 4 °C for no more than 1 to 2 months.
Prepare 10 mg/mL deoxyribonuclease I (DNase) in CMF-HBSS, and then filter-sterilize and store at -20 °C.
Prepare glial medium by making a solution of 30 mM glucose (from the sterile solution), 95 U/mL penicillin-streptomycin, and 10-15% bovine growth serum (BGS) in Minimum Essential Medium (MEM). NOTE: As indicated in the protocol, a solution of 5% BGS can be used if required to slow cell proliferation. Horse Serum may be used instead of BGS, but note that each batch must be tested before use due to inter-batch variation. Store the prepared solution at 4 °C for no more than 1 to 2 months. Though many laboratories use FBS, we consistently observed that glial growth in BGS is as robust as in FBS. BGS is derived from fetal calf serum which is supplemented with chemically defined components. In addition, BGS is substantially more economical than FBS.
Prepare the neuron growth and maintenance medium (neurobasal-B27, NBG) by making a solution of 1x B27 supplement and 0.5 mM L-Glutamine in 500 mL Neurobasal Medium. Store at 4 °C for no more than 1 to 2 months. L-Glutamine can be substituted with GlutaMAX, which has been suggested to be more stable in the growth medium.
Prepare the neuron plating medium by making a solution of 30 mM glucose (from the sterile solution), 1 mM sodium pyruvate (from the sterile solution), and 10% BGS in MEM. Store at 4 °C for no more than 1 to 2 months.
Prepare a solution of 1 mM Cytarabine (Ara-C) and filter-sterilize. Aliquot into 1 mL portions and store at -20 °C.
Prepare a solution of 100 mM APV in 100 mN filter-sterilized NaOH. Store at -20 °C.
Dissolve 1 mg/mL poly-L-lysine in borate buffer, pH 8.5 (50 mM boric acid and 12 mM borax) and filter-sterilize. Aliquot and store at -20 °C. Instead of poly-L-lysine, one may use poly-L-ornithine which may be less toxic to cells.
2. Glia Culture Preparation
- Prepare cortical astroglia cell culture
- Obtain rat pups within 24 h of birth. Place the pups on ice to anaesthetize them.
- Spray the pups with 70% ethanol and decapitate them.
- Place the heads in a 100 mm dish on ice containing 15 mL CMF-HBSS.
- Dissect out each brain by cutting the scalp, opening the skull, severing the brainstem, and then transferring the brain to 60 mm Petri dishes on ice containing 3 mL CMF-HBSS.
- Separate the cerebral hemispheres from the hippocampi and then strip away the meninges surrounding them. Exercise adequate precaution to ensure minimal contamination from meningeal fibroblasts. Meningeal fibroblasts in the culture can compete with glia for growth and be detrimental for neuron health. Collect all the cerebral hemispheres in a 60 mm Petri dish on ice containing 5 mL CMF-HBSS.
- Remove the CMF-HBSS and then mince the collected cortical tissue as finely as possible using micro-dissecting spring-scissors.
- Add sufficient CMF-HBSS to the chopped tissue. Using a glass Pasteur pipette, transfer the tissue pieces to a 15 mL tube. Bring the total volume to 12 mL by adding CMF-HBSS.
- Add 1.5 mL each of 2.5% trypsin and 10 mg/mL DNase solution. Incubate the resulting mixture at 37 °C in a water bath for 12 min.
- Triturate the tissue 10-15 times using a glass Pasteur pipette, and further incubate it in a 37 °C water bath for 3 min.
- Filter the supernatant through lens paper or a filter mesh into a 50 mL tube containing 5 mL of BGS. This is done to remove chunks of undissociated tissue. Alternatively, use commercially available cell strainers.
- Add 13.5 mL of CMF-HBSS and 1.5 mL of 2.5% trypsin to the tube containing the remaining tissue pieces and further incubate it at 37 °C in a water bath for an additional 10 min.
- Triturate the remaining tissue once more, and then filter the suspension again into the 50 mL tube containing the initial filtrate. Next, rinse the lens paper with 3 mL of BGS. The final volume should be approximately 38 mL.
- Centrifuge the suspension containing the dissociated glial cells for 6 min at 130 x g. Carefully discard the supernatant using a glass Pasteur pipette. Ensure that the pelleted cells at the bottom are not disturbed. The bottom of the pellet appears slightly reddish containing blood components.
- Transfer the glial cells in 1 mL of glial medium into a fresh tube, taking care not to disturb the reddish portion of the pellet.
- Add up to 2 mL of glial medium per pup to resuspend the combined dissociated glial cells. For example, if 10 pups were used, and then the total resuspension volume would be 20 mL.
- Prepare one 75 cm2 cell culture flask per pup. Add 18 mL pre-warmed glial medium to each flask.
- Transfer 2 mL of cell suspension to each flask and incubate in a 37 °C, 5% CO2 incubator.
- After one day, replace with 20 mL glial medium. At this stage, the culture is a mix of glial and other unwanted cell-types.
- Four days after seeding the cells, vigorously shake the flasks to dislodge non-astrocyte cells. To do this, rinse the flasks once with CMF-HBSS, and then add 10 mL fresh CMF-HBSS to each flask. Shake the flasks vigorously 5-10 times, and then aspirate off the CMF-HBSS. Rinse twice with CMF-HBSS, and then add 20 mL glial medium. NOTE: This step is crucial to ensure the removal of non-target cell types, and should not result in the loss of the desired astrocytes.
- Replace with fresh glial medium twice a week until the cells are confluent (Figure 1).
- Freezing glia
- Once the cultures are confluent, aspirate off the medium and wash the cell monolayer with 10 mL CMF-HBSS.
- Add 10 mL of 0.25% trypsin/EDTA to each flask and incubate at 37 °C for 3 min.
- Tap flasks gently to dislodge the cells. Add 1 mL BGS to each flask and transfer the cell suspensions into 15 mL tubes.
- Centrifuge the tubes at 130 x g for 6 min. Discard the resulting supernatant.
- Resuspend the cell pellet in 2 mL glial medium.
- Prepare the glial freezing medium (1-part dimethyl sulfoxide and 4-parts BGS). Add 0.5 mL of this solution to each of 4 cryogenic vials per flask.
- Transfer 0.5 mL of cell suspension to each vial, and then freeze the cells slowly by placing the vials in an insulated container in a -80 °C freezer overnight. After one day, transfer the vials to a liquid nitrogen freezer for long-term storage.
- Plating revived glia
- Plate glia 1-2 weeks before the neuron culture day.
- Prepare a 50 mL tube with 10 mL warmed glial medium.
- Thaw 1 frozen vial of glia in a 37 °C water bath for 2-3 min.
- Transfer the contents of the vial into the 50 mL tube containing 10 mL glial medium.
- Centrifuge the tube for 5 min at 130 x g and aspirate off the supernatant.
- Resuspend the pellet in 20 mL glial medium.
- Add 3 mL fresh glial medium to each of the culture dishes (60 mm), and then transfer 1 mL of the cell suspension to each dish. Incubate the cultures in a 37 °C, 5% CO2 incubator.
- Feed the cells twice a week. If the cell density reaches 50% confluence well before the neuron culture day, switch to glial medium containing 5% BGS to slow the glial growth rate. The desired confluence of the glial monolayer for neuron culture is 50% - 70%.
- Replace the glial medium with NBG medium (6 mL/dish) 1-2 days before the neuron culture.
3. Coverslip Preparation
- Cleaning and Preparation of Paraffin Beads
- Submerge coverslips in concentrated nitric acid (70% wt/wt; CAUTION: highly corrosive) and sonicate them for 30 min at room temperature. Other laboratories have found it beneficial to incubate the coverslips in 70% nitric acid in ceramic racks for 12-24 h at room temperature instead.
- Carefully discard the nitric acid in a designated chemical fume hood in accordance to institutional guidelines. Rinse the coverslips 3-5 times with deionized water.
- Submerge the coverslips in deionized water and sonicate them again for 30 min (skip if following the alternative method).
- Remove the deionized water and rinse the coverslips another three times.
- Submerge the coverslips in 50 mL deionized water and sterilize them by autoclaving. Alternatively, sterilize the coverslips at 225 °C in an oven for 6 h. If using this alternative approach, skip to step 3.1.9.
- Once autoclaved, transfer the coverslips to a sterile laminar flow hood for the following steps.
- Remove the deionized water and rinse with 20-30 mL of 100% ethanol.
- Add 20-30 mL of 100% ethanol and transfer the coverslips and ethanol to a 100 mm Petri dish.
- Use blunt tweezers to transfer coverslips to 60 mm Petri dishes, 4-5 coverslips per dish, and then allow them to air-dry by leaning the coverslips against the edge of the dish. Once the ethanol has evaporated, lay the coverslips flat on the surface.
- Transfer 10 g paraffin to a suitable heat-resistant glass bottle. Melt the paraffin and maintain it at about 150-200 °C on a hot plate for at least 1 h. The ideal temperature to which the paraffin should be heated may take some tweaking. Non-ideal temperatures can lead to difficulty producing the correct size of the paraffin beads. The waiting period after melting helps to ensure consistent temperature throughout the liquid.
- Dip a Pasteur pipette into the paraffin, and then quickly touch it to three spots near the edge of each coverslip (each spot being approximately 120° apart). The drops will solidify into beads approximately 0.3-0.5 mm high and 1.0-2.0 mm in diameter and will function to provide a space between the neurons on the coverslip and the glial monolayer below (Figure 2).
- Sterilize the dishes under UV light for 30 min, and then cover them with aluminum foil and store at room temperature. Prepared coverslips can be stored and used for up to one month. Paraffin beads should be allowed to adhere to the coverslips for at least one week before use. This will prevent the paraffin from detaching during the remaining steps of the protocol.
- Coating Coverslips with Poly-L-Lysine
- Obtain Petri dishes containing previously prepared coverslips with the paraffin beads.
- Add 200 µL of poly-L-lysine in borate buffer to the surface of each coverslip and let the solution sit on the coverslips overnight, covered, at room temperature. Coat the same surface as the one on which the paraffin beads are situated. Note that if properly cleaned, the poly-L-lysine should spread easily to cover the surface of the coverslip.
- After one day, wash the coverslips twice by soaking them in sterile deionized water for 2 h each time. After the final wash, replace the water with 4 mL of neuronal plating medium per dish. Note that it is important that the surface of the coverslips not be allowed to dry after having been coated with poly-L-lysine.
- Place the coverslip-containing dishes in a 37 °C, 5% CO2 incubator. The coated coverslips should be incubated for at least 24 h before the neuron culture. This is done to prime the medium for neuron culture.
4. Dissection of Hippocampus and Plating Neurons
Obtain a pregnant rat (embryonic day 18-19, as measured from the day of vaginal plug discovery) and euthanize by isoflurane-anesthetized decapitation or other approved method.
Spray the underside of the rat with 70% ethanol, and then make an incision from the pubic region through to the end of the abdominal cavity. To avoid contamination, take care to cut only through the skin at this step.
Rinse the dissection instruments again with 70% ethanol, and then cut through the abdominal wall to expose the uterus. Remove the two uterine horns and place them in a sterile 100 mm Petri dish.
Perform the remaining steps in a laminar flow hood. Remove the uterine membrane surrounding the fetuses. Decapitate them and place the heads in a 100 mm Petri dish containing 20 mL CMF-HBSS.
Dissect out the brains and immediately place them in a 60 mm Petri dish containing 2 mL CMF-HBSS on ice. Collect only 2-3 brains per dish to allow sufficient space for the next step.
Under a dissecting microscope strip away the meninges from the midline of the cerebral hemispheres, and then dissect out the hippocampi and place them in a 60 mm Petri dish containing 4.5 mL CMF-HBSS. Note that the hippocampus can be distinguished as a crescent-shaped structure in the medial surface of the cortical hemisphere, and is easy to remove as it is bordered laterally by a ventricle.
Transfer the collected hippocampal tissue and CMF-HBSS to a 15 mL tube. Add 0.5 mL of 2.5% trypsin, and then incubate at 37 °C for 10 min. Papain may be used instead of trypsin because it is gentler and may increase yield.
Gently remove the trypsin solution by Pasteur pipette, leaving the hippocampal tissue undisturbed at the bottom of the tube.
Gently wash the tissue twice with 5 mL CMF-HBSS while incubating it at 37 °C for 5 min. After the second wash, bring the total volume to at least 5 mL by adding a sufficient volume of CMF-HBSS to the tissue. If there are hippocampi from more than 10 pups, up to 8 mL CMF-HBSS can be added.
- Prepare two variants of fire-polished pipettes for tissue dissociation. The tip of one variant is of the normal diameter of the pipette but with fire-polished smoothened edges. The other variant is one with the tip diameter reduced by half. Briefly expose the tip of a glass Pasteur pipette diagonally to a flame source, and then examine the tips of the pipettes.
- Repeat this step until the tip edge had been smoothened or the diameter has been reduced. It is important to practice to find the ideal diameter to ensure that the tissue is dissociated to yield single cells without damaging these cells.
Triturate the hippocampal tissue first by about 10 passes through a smooth-edged normal glass Pasteur pipette, and then a further 5 to 10 passes through the pipette with the reduced diameter. The goal is to obtain a homogenous solution of cells with no or very few tissue clumps.
Determine the cell density using a hemocytometer or an automated cell counter. Typically, 0.8 to 1.0 million cells per pup are obtained when hippocampi from the two hemispheres are combined.
Transfer a sufficient volume of the cell suspension to dishes containing poly-L-lysine-coated coverslips in 4 mL plating medium to obtain a plating density of 300,000 cells per 60 mm dish. The density of the cells can vary from 100,000 to 500,000, depending upon the intended experiment.
After 3-4 h, transfer the coverslips with neurons attached to dishes containing glial cells in NBG medium. This should be done such that the side on which the neurons were plated is facing down towards the glial cell layer. Add a total of 4-5 coverslips per glia dish.
Two or three days after plating, add the Ara-C solution to a final concentration of 5 µM per dish to arrest glial growth.
Feed the cells once a week by replacing 2 mL of the old medium with 2.5 mL of fresh medium at each feeding. The extra medium added is to compensate for evaporation over time, and only a portion of the medium is changed to ensure consistent conditioning of the medium by the glial cells. These cultures are typically healthy for at least one month (Figure 3). Neuron survival can be improved by adding APV to the culture medium to a final concentration of 100 µM. APV is an NMDA receptor antagonist and promotes survival by reducing glutamate excitotoxicity.
Representative Results
In this "sandwich" method of primary nerve cell culture, hippocampal neurons (Figure 3) grow on a bed of glial cells (Figure 1) separated by paraffin beads (Figure 2). This ensures that neurons selectively grow on glass coverslips with minimal glial cell contamination but receive adequate trophic support from glia growing on the tissue culture dish. Typically, neurons can be maintained in culture for >3 weeks and develop extensive arborisation with well-developed axons and dendrites identified by their typical dephospho-Tau and MAP2 immunostaining, respectively (Figure 4). Mature neurons develop synaptic spines identified by selective localization of postsynaptic marker Drebrin1 to these compartments. These synaptic spines are closely apposed to presynaptic specializations, which are identified by synaptic vesicle marker Synapsin1 immunostaining. The proximity of pre- and postsynaptic sites indicates well-developed and correctly aligned synapses. These primary neurons are well-suited for functional and structural studies of neurons and synapses. These include, but are not limited to, studies of signal transduction, neuronal polarity, subcellular trafficking and synapse development and plasticity.
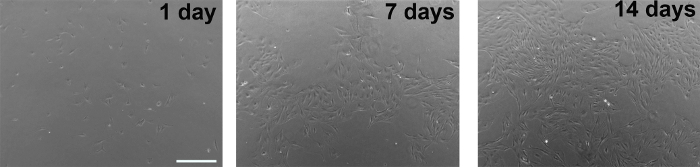 Figure 1. Phase contrast images of glial cultures taken 1 day, 7 days, or 14 days after plating from frozen glial stock. Scale bar, 500 µm. Please click here to view a larger version of this figure.
Figure 1. Phase contrast images of glial cultures taken 1 day, 7 days, or 14 days after plating from frozen glial stock. Scale bar, 500 µm. Please click here to view a larger version of this figure.
 Figure 2. High-resolution images of coverslips prepared with paraffin beads. (A, B) Examples of properly applied paraffin beads. (C, D) Examples of incorrectly applied paraffin beads. α: More than one bead was applied at the same location. β: The bead failed to be properly situated on the coverslip. χ: The bead was applied too close to the center of the coverslip. δ: The bead was too high. Please click here to view a larger version of this figure.
Figure 2. High-resolution images of coverslips prepared with paraffin beads. (A, B) Examples of properly applied paraffin beads. (C, D) Examples of incorrectly applied paraffin beads. α: More than one bead was applied at the same location. β: The bead failed to be properly situated on the coverslip. χ: The bead was applied too close to the center of the coverslip. δ: The bead was too high. Please click here to view a larger version of this figure.
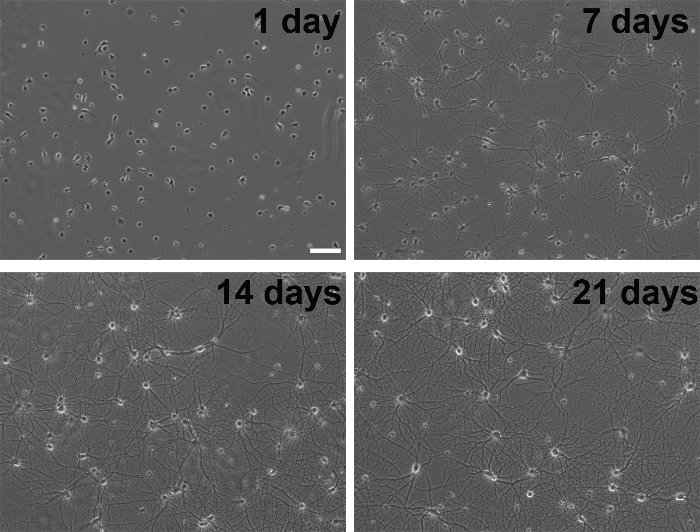 Figure 3. Phase contrast images of the developmental stages of primary neurons in culture. Scale bar, 100 µm. Please click here to view a larger version of this figure.
Figure 3. Phase contrast images of the developmental stages of primary neurons in culture. Scale bar, 100 µm. Please click here to view a larger version of this figure.
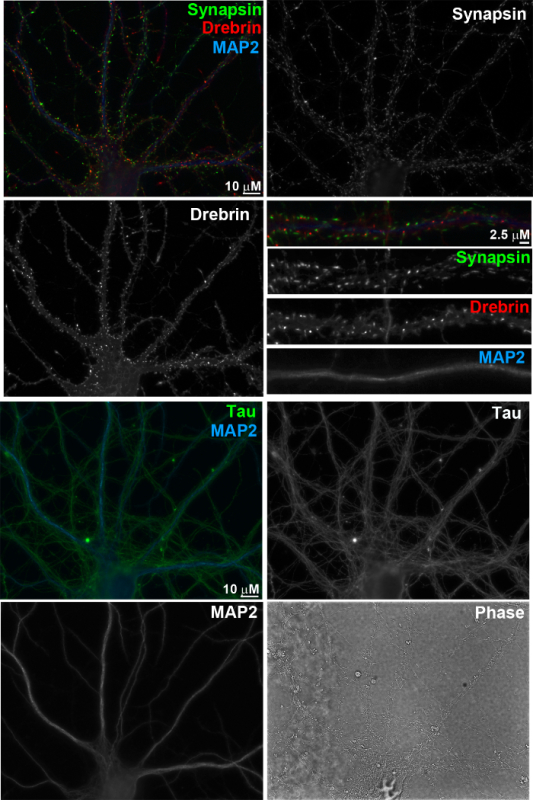 Figure 4. Immunofluorescence images of primary neurons at 17 days in vitro. The neurons were fixed with 4% formaldehyde and 4% sucrose in PBS for 12 min, and then permeabilized with 0.25% Triton X-100 for 5 min. After blocking with 10% BSA in PBS for 30 min, the neurons were incubated overnight at 4 °C with primary antibodies applied in 3% BSA in PBS. Primary antibodies were used as follows: anti-synapsin I (rabbit, 1:2,000), anti-drebrin (mouse IgG1, 1:8, clone M2F6, hybridoma supernatant), anti-MAP2 (chicken polyclonal IgY, 1:5,000), and anti-Tau-1 (mouse IgG2a, 1:2,000, clone PC1C6; recognizes dephosphorylated tau). The cells were then washed with PBS and incubated with appropriate species-or isotype-specific secondary antibodies in 3% BSA in PBS for 1 h at 37 °C. Secondary antibodies used were as follows: Alexa 488-conjugated anti-mouse IgG2a (1:500), Alexa 568-conjugated anti-rabbit (1:500), Alexa 647-conjugated anti-mouse IgG1 (1:500), and AMCA-conjugated anti-chicken IgY (donkey IgG, 1:200). Scale bar, 10 µm and 2.5 µm, as indicated. Please click here to view a larger version of this figure.
Figure 4. Immunofluorescence images of primary neurons at 17 days in vitro. The neurons were fixed with 4% formaldehyde and 4% sucrose in PBS for 12 min, and then permeabilized with 0.25% Triton X-100 for 5 min. After blocking with 10% BSA in PBS for 30 min, the neurons were incubated overnight at 4 °C with primary antibodies applied in 3% BSA in PBS. Primary antibodies were used as follows: anti-synapsin I (rabbit, 1:2,000), anti-drebrin (mouse IgG1, 1:8, clone M2F6, hybridoma supernatant), anti-MAP2 (chicken polyclonal IgY, 1:5,000), and anti-Tau-1 (mouse IgG2a, 1:2,000, clone PC1C6; recognizes dephosphorylated tau). The cells were then washed with PBS and incubated with appropriate species-or isotype-specific secondary antibodies in 3% BSA in PBS for 1 h at 37 °C. Secondary antibodies used were as follows: Alexa 488-conjugated anti-mouse IgG2a (1:500), Alexa 568-conjugated anti-rabbit (1:500), Alexa 647-conjugated anti-mouse IgG1 (1:500), and AMCA-conjugated anti-chicken IgY (donkey IgG, 1:200). Scale bar, 10 µm and 2.5 µm, as indicated. Please click here to view a larger version of this figure.
Discussion
While the "sandwich" method of culturing neurons has been well-described elsewhere 2,11, there are several steps throughout the protocol that are quite difficult to describe in text alone, which can lead to frustration for investigators who wish to adopt it.
The method can be divided into three broad workflows: glial culture, coverslip preparation and neuron culture and maintenance. Each of the three preparations are critical for high-quality neuron cultures and several important considerations must be kept in mind. Before plating neurons, the glial feeder layer should ideally be a homogenous population of astrocytes and be between 50-70% confluence. This would ensure sufficient trophic support from the glial feeder layer. It is important to ensure that the astroglial culture has minimal contamination of other cell types, particularly microglia, which are loosely attached and rounded cells. Microglia release cytokines, which are detrimental to neuron health and survival 12. The serum, either horse serum or bovine growth serum, can be variable from lot to lot. Careful screening of several lots of serum should be performed before deciding to use a specific lot.
Coverslip preparation is another important step. The quality of the glass and the protocol used in its cleaning are critical for healthy well-developed neurons. If the method is being newly adopted in a lab, it would be worth testing glass coverslips from several suppliers. For cleaning of the coverslips, some labs soak coverslips in 70% nitric acid for 18-36 h while others do so for only 1 h in a sonicator. An important criterion of clean coverslips is that the poly-L-lysine solution should spread evenly on the surface. Another important consideration is that the paraffin wax beads applied to the coverslips should be of appropriate height and diameter and be heated to the right temperature, as outlined in the protocol.
For obtaining neurons, hippocampi can be dissected from embryonic day 17-19 rats. Though dissection at this stage leads to very few glial cells, glial proliferation can be arrested by adding the anti-mitotic agent Ara-C after 2-3 days of culture. Trituration of trypsinized hippocampal tissue by fire-polished pipette tips is critical to dissociate the tissue into individual cells without damaging the cells themselves. The neuronal growth and maintenance medium used in this protocol is Neurobasal + L-Glutamine + B27 supplement. L-Glutamine may be substituted by GlutaMAX, which is more stable in the culture medium. The quality of the B27 supplement can vary from lot to lot, and so it should be tested before using a specific lot for longer term culture. A poor lot of B27 may cause neurons to clump. Alternate products such as GS21 and SM1 have been found to be good substitutes for B27. Neurons grown for longer than a week should be fed with fresh medium every week, wherein a third of the medium is replaced every week.
This method of culturing nerve cells can prove invaluable to experiments that rely on pure neuronal populations with little or no glial contamination. Low-density neurons grown using this method typically survive for several weeks with well-developed arbors, synaptic connections and network properties.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by CIHR MOP-142209 to TJS.
References
- Banker GA, Cowan WM. Rat hippocampal neurons in dispersed cell culture. Brain Res. 1977;126:397–425. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Adamantidis A, et al. Optogenetics: 10 years after ChR2 in neurons--views from the community. Nat Neurosci. 2015;18:1202–1212. doi: 10.1038/nn.4106. [DOI] [PubMed] [Google Scholar]
- Ho VM, Lee JA, Martin KC. The cell biology of synaptic plasticity. Science. 2011;334:623–628. doi: 10.1126/science.1209236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huganir RL, Nicoll RA. AMPARs and synaptic plasticity: the last 25 years. Neuron. 2013;80:704–717. doi: 10.1016/j.neuron.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C, et al. Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron. 1999;24:649–658. doi: 10.1016/s0896-6273(00)81119-8. [DOI] [PubMed] [Google Scholar]
- Varoqueaux F, et al. Neuroligins determine synapse maturation and function. Neuron. 2006;51:741–754. doi: 10.1016/j.neuron.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Huettner JE, Baughman RW. Primary culture of identified neurons from the visual cortex of postnatal rats. J Neurosci. 1986;6:3044–3060. doi: 10.1523/JNEUROSCI.06-10-03044.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester RA, Quarum ML, Parker JD, Weber E, Jahr CE. Interaction of 6-cyano-7-nitroquinoxaline-2,3-dione with the N-methyl-D-aspartate receptor-associated glycine binding site. Mol Pharmacol. 1989;35:565–570. [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Goslin K, Asmussen H, Banker G. Banker G, Goslin K. Rat hippocampal neurons in low-density culture. Culturing nerve cells. 1998. pp. 339–370.
- Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]