

Abstract
Zebrafish, first introduced as a developmental model, have gained popularity in many other fields. The ease of rearing large numbers of rapidly developing organisms, combined with the embryonic optical clarity, served as initial compelling attributes of this model. Over the past two decades, the success of this model has been further propelled by its amenability to large-scale mutagenesis screens and by the ease of transgenesis. More recently, gene-editing approaches have extended the power of the model.
For neurodevelopmental studies, the zebrafish embryo and larva provide a model to which multiple methods can be applied. Here, we focus on methods that allow the study of an essential property of neurons, electrical excitability. Our preparation for the electrophysiological study of zebrafish spinal neurons involves the use of veterinarian suture glue to secure the preparation to a recording chamber. Alternative methods for recording from zebrafish embryos and larvae involve the attachment of the preparation to the chamber using a fine tungsten pin1,2,3,4,5. A tungsten pin is most often used to mount the preparation in a lateral orientation, although it has been used to mount larvae dorsal-side up4. The suture glue has been used to mount embryos and larvae in both orientations. Using the glue, a minimal dissection can be performed, allowing access to spinal neurons without the use of an enzymatic treatment, thereby avoiding any resultant damage. However, for larvae, it is necessary to apply a brief enzyme treatment to remove the muscle tissue surrounding the spinal cord. The methods described here have been used to study the intrinsic electrical properties of motor neurons, interneurons, and sensory neurons at several developmental stages6,7,8,9.
Keywords: Neuroscience, Issue 122, zebrafish, spinal neurons, motor neurons, sensory neurons, Rohon-Beard cell, CaP, electrophysiology, whole cell, patch clamp
Introduction
George Streisinger pioneered the use of Danio rerio, commonly known as zebrafish, as a model system for the genetic analysis of vertebrate development10. The model offers several advantages including: (1) relatively simple and inexpensive animal husbandry; (2) external fertilization, allowing easy access to embryos from the earliest developmental stages; and (3) a transparent embryo, permitting direct and repeated observations of cells, tissues, and organs as they form.
Over the ensuing decades, several advances further increased the power of the zebrafish model. In particular, forward genetic screens and whole-genome sequencing efforts played key roles in the identification of mutations and genes critical to many developmental processes11,12,13,14,15,16. Gateway cloning methods have allowed the routine application of transgenic approaches17,18. Recent advances in genome editing, exemplified by transcription activator-like (TALENs) and clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 nucleases, allow for the targeted introduction of mutations, as well as knock-out and knock-in approaches19,20,21,22. Combined, these methods make zebrafish a powerful model for the study of the genetic mechanisms underlying specific behaviors and several human diseases23,24,25,26,27.
This work focuses on developmental regulation and the role of electrical activity in neuronal development. The focus is on the spinal cord, for which the zebrafish model provides several advantages. First, it is relatively easy to access zebrafish at embryonic and larval stages; therefore, one can study spinal cord function during developmental stages that have fewer neurons and simpler circuitry28,29. Moreover, the zebrafish spinal cord has a diverse set of neurons, similar to other vertebrates, as demonstrated by characteristic and distinguishing patterns of transcription factors30,31,32,33,34,35.
The majority of studies in zebrafish that aim to uncover the mechanisms that underlie the function of spinal cord circuits, especially ones that support locomotion, are understandably focused on larval stages36,37,38,39,40,41,42,43. However, many of the neurons that form the spinal locomotive networks initiate their differentiation at early embryonic stages, ~9-10 h post-fertilization (hpf)44,45,46,47,48,49,50,51. In view of this, understanding how the morphological and electrical properties of spinal neurons arise and change between the embryonic and larval stages is important for an overall understanding of locomotor circuit formation and function.
The dissection methods described here allow patch clamp recordings from spinal neurons and have been successfully applied at embryonic stages (~17-48 hpf) and larval stages (~3-7 days post fertilization [dpf]). This approach limits the amount of dissection required to provide access to the neurons of interest. The protocol differs from the majority of the other published methods for recording from zebrafish spinal neurons in that veterinarian suture glue is used, rather than a fine tungsten pin, to attach the embryo or larva to the recording chamber. The availability of two different approaches (i.e., suture glue versus the tungsten pin) for mounting the zebrafish embryos or larvae for electrophysiological analysis provides researchers with alternative options to achieve their specific experimental goals.
First, procedures for accessing and recording from a population of primary sensory neurons, Rohon-Beard cells, are described. The cell bodies of these neurons lie within the dorsal spinal cord. Rohon-Beard cells exist in numerous vertebrate species, differentiate early in development, and underlie the embryonic touch response6,44,47,48.
Second, procedures for accessing and recording from spinal motor neurons are detailed. Zebrafish spinal motor neurons arise during two waves of neurogenesis. The earlier-born primary motor neurons arise at the end of gastrulation (~9-16 hpf), with only 3-4 primary motor neurons present per hemisegment45,46,49. In contrast, the later-born population of secondary motor neurons is more numerous and arises during a prolonged period, starting at ~14 hpf45,50. Secondary motor neuron genesis in mid-trunk segments is mostly completed by 51 hpf50. Secondary motor neurons are considered to be the counterpart of motor neurons in amniotes46. Interestingly, supraspinal neurons, via dopamine, regulate locomotion in the larva and secondary motor neuron genesis in the embryo and young larva50,51. Primary and secondary motor neurons each comprise several different subtypes. Each primary motor neuron subtype projects a peripheral axon that innervates a characteristic muscle group, resulting in a stereotypical, identifying axonal trajectory. Generally, secondary motor neurons follow the axonal pathways previously established by primary motor neurons. Thus, with respect to axonal trajectories, primary and secondary motor neurons are similar, with the exception that axonal thickness and somata size are greater for primary motor neurons45.
Third, methods for recording from a few types of interneurons are discussed. However, in these cases, a limited amount of removal of other spinal cord cells is required, and thus the spinal cord is less intact than for recordings from Rohon-Beard cells or motor neurons.
Protocol
All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC; Office of Laboratory Animal Resources, University of Colorado Anschutz Medical Campus).
1. Zebrafish Husbandry
Raise and maintain adult zebrafish (Danio rerio) at 28.5 °C on a 10 h dark/14 h light cycle and with appropriate water treatment and exchange52.
Raise zebrafish embryos/larvae at 28.5 °C in embryo medium until they reach the desired stage (e.g., 2 dpf).
2. Preparation of Dissection Materials
- Glue dispenser for zebrafish dissection NOTE: This protocol involves the use of veterinarian suture glue to attach the preparation to the recording chamber. Successful use of the suture glue requires the application of small amounts of glue in a controlled manner. The glue begins to harden as soon as it encounters an aqueous environment. Therefore, draw the glue into a micropipette and deliver small amounts using a home-made "glue dispenser" that allows the application of negative or positive pressure by mouth. The central part of the glue dispenser is a glass bore, a piece that is included within the package containing the borosilicate thin-wall glass capillaries (Figure 1A). On one end, the bore is connected via a piece of flexible tubing to a mouthpiece, while the other end holds the glass micropipette (Figure 1).
- Remove the black bulb from one glass bore and replace it with the white rubber adapter from another glass bore (Figure 1B and 1C). NOTE: This double-adapter-capped glass bore allows connection to a glass micropipette on one end and, at the other end, to a piece of flexible tubing via a small, straight polypropylene fitting (Figure 1B, inset).
- Cut a piece of flexible tubing to a length of ~38 cm. Attach the mouthpiece (e.g., a yellow 200 µL micropipette tip) to the end of the flexible tubing not connected to the glass bore. NOTE: The piece of tubing should be long enough to allow manipulation of the glass micropipette under a dissecting scope while the micropipette yellow tip is in the mouth (Figure 1D).
 Figure 1: Glue dispenser. (A-C) A glass bore connects to flexible tubing at one end and the glass micropipette at the other. The rubber adapters allow attachment via a small polypropylene fitting (B, inset) to the tubing and, eventually, to a glass micropipette at the other end. (D) The final glue dispenser has a mouthpiece (e.g., made from a plastic pipette tip) at one end of the tubing and the glass bore with the attached micropipette at the other (arrowhead).
Figure 1: Glue dispenser. (A-C) A glass bore connects to flexible tubing at one end and the glass micropipette at the other. The rubber adapters allow attachment via a small polypropylene fitting (B, inset) to the tubing and, eventually, to a glass micropipette at the other end. (D) The final glue dispenser has a mouthpiece (e.g., made from a plastic pipette tip) at one end of the tubing and the glass bore with the attached micropipette at the other (arrowhead).
- Dissection/recording chamber NOTE: The dissection chamber also serves as the electrophysiology recording chamber. The chamber is formed on a glass slide using pre-cut pieces of cured silicone elastomer (Figure 2A).
- To prepare the silicone elastomer, add the base and the curing agent to a plastic conical tube at a 4:1 ratio, respectively.
- Mix the elastomer base and the curing agent thoroughly and pour the mix into two 100 mm Petri dishes. Pour the elastomer to a thickness of <1 mm in one Petri dish and ~2.5 mm in the other.
- Allow the silicone elastomer to cure by exposure to air for ~4-5 days. If cured elastomer is needed sooner, incubate it at 60 °C.
- Cut pieces of the cured silicone elastomer to the following sizes:
- From the ~1 mm-thick cured elastomer, cut out a ~3.8 x 6.3 cm rectangle; this piece serves as the bottom of the chamber (Figure 2A). From the ~2.5 mm-thick cured silicone, cut a rectangle ~3.8 x 6.3 cm. From the latter rectangle, cut out an internal rectangle ~2.5 x 5 cm; the resulting frame serves as the top of the chamber (Figure 2A).
- To make the dissection/recording chamber, place the thin silicone rectangle directly on top of a glass slide (5 x 7.6 cm), making sure to remove any air bubbles between the silicone and the glass (Figure 2A). Place the silicone rectangle frame, cut from the thicker elastomer, on top of the thin bottom layer of silicone.
- Verify that the silicone layers attach well to each other and that there are no air bubbles between the two layers (Figure 2A).
- After use, dismantle the chamber by removing the thick rectangle silicone frame from the bottom silicone layer that is attached to the glass slide. Rinse the silicone surfaces with ddH2O prior to use and after each recording session and dry with low-lint wipes.
- Store the chamber dry to prevent fungal growth between the two pieces of silicone. NOTE: If toxins or pharmacological agents that may not rinse readily from the silicone are used, dedicate specific chambers for those purposes.
 Figure 2: Electrophysiology chamber and dissection tools. (A)The chamber used for the dissections and electrophysiological recordings consists of a glass slide upon which are placed two pieces of cured silicone elastomer, layered on top of each other to provide a frame and a bottom for a well. The size of the well, ~2.5 x 5 cm, allows the use of small volumes (2-2.5 mL) of extracellular recording solution. The bottom silicone layer allows for secure positioning of the zebrafish embryo using tissue adhesive that does not adhere to glass. (B) A glass micropipette (top) is used for glue delivery during the dissection. The thin-wall glass is pulled to create a long, tapered end that is later cut, creating a tip with a diameter of ~75 µm. The tapered glass micropipette is attached to the free end of the glue dispenser (Figure 1D, arrowhead) and front-filled with glue through the application of suction. The other micropipette (bottom), pulled as for one used for a patch-clamp recording, is used for the transection of the hindbrain and for skin removal. (C) Under an upright microscope, a micromanipulator is used to maneuver the micropipette for the final dissection steps. A glass micropipette, as in B, bottom, is attached to the electrode holder (arrow). Muscle removal is achieved by applying suction through the tubing connected to the air outlet (arrowhead). At its other end, the tubing connects to a stopcock (black arrowhead) that, on its other side (asterisk), has tubing attached to a mouthpiece.
Figure 2: Electrophysiology chamber and dissection tools. (A)The chamber used for the dissections and electrophysiological recordings consists of a glass slide upon which are placed two pieces of cured silicone elastomer, layered on top of each other to provide a frame and a bottom for a well. The size of the well, ~2.5 x 5 cm, allows the use of small volumes (2-2.5 mL) of extracellular recording solution. The bottom silicone layer allows for secure positioning of the zebrafish embryo using tissue adhesive that does not adhere to glass. (B) A glass micropipette (top) is used for glue delivery during the dissection. The thin-wall glass is pulled to create a long, tapered end that is later cut, creating a tip with a diameter of ~75 µm. The tapered glass micropipette is attached to the free end of the glue dispenser (Figure 1D, arrowhead) and front-filled with glue through the application of suction. The other micropipette (bottom), pulled as for one used for a patch-clamp recording, is used for the transection of the hindbrain and for skin removal. (C) Under an upright microscope, a micromanipulator is used to maneuver the micropipette for the final dissection steps. A glass micropipette, as in B, bottom, is attached to the electrode holder (arrow). Muscle removal is achieved by applying suction through the tubing connected to the air outlet (arrowhead). At its other end, the tubing connects to a stopcock (black arrowhead) that, on its other side (asterisk), has tubing attached to a mouthpiece.
3. Dissection of Embryos and Larvae for Patch-clamp Recordings from Spinal Neurons
 Figure 3: Dorsal dissection of a zebrafish spinal cord. (A-A') After hindbrain transection (a) of a 2-dpf embryo, the skin is cut on the left and right sides of the embryo (b). A second cut, perpendicular to the first, is then performed (c). Next, the skin is lifted using a micropipette, allowing tweezers to grab and pull away the skin. (B) Removal of the skin exposes the dorsal spinal cord. Rostral to the black line, the skin has been removed and the surface of the spinal cord (asterisk), contained within the meninges, is exposed. The skin remains intact caudal to the black line (arrow). (C-C') The tip of the glass micropipette is pressed on the meninges, and swift, lateral, short movements are performed to pierce the meninges. (D-D' and E-E') Once the meninges are pierced (D-D'), the micropipette is advanced and (E-E') moved rostrally to tear the meninges into two segments. The somata of Rohon-Beard neurons typically emerge upon removal of the meninges (arrow). (F-G') In a 7-dpf larva, layers of muscle cover the dorsal aspect of the spinal cord, hindering access to Rohon-Beard neurons. Following the removal of the skin, the larva is treated with 0.05% collagenase. (F) A 5 min incubation with 0.05% collagenase is too stringent, resulting in excessive muscle damage, as evidenced by the frayed muscle (arrow and inset). (F') Excessive collagenase treatment may also damage Rohon-Beard neurons (arrow), revealed here by their expression of gfp in the Tg(islet2b:gfp) line.In theTg(islet2b:gfp) line, dorsal root ganglion neurons also express gfp (arrowhead). A briefer 1 min incubation with 0.05% collagenase sufficiently loosens the muscle (G) while conserving the myotome morphology (arrow and inset). (G') Pigment cells are present on top of the most dorsal muscle layer (arrowhead). (H and I) In the Tg(islet2b:gfp) line, Rohon-Beard neurons (arrows) and the dorsal root ganglion (arrowhead) continue to express gfp at 7 dpf. Dorsal views of Tg(islet2b:gfp) zebrafish embryos at 2 dpf (H) and 7 dpf (I). In Panel A Scale bars = 500 µm; B-E (shown in Panel B) Scale bars = 80 µm; F' and G' (shown in Panel F) Scale bars = 200 µm; H and I (shown in Panel H) Scale bars = 100 µm. Please click here to view a larger version of this figure.
Figure 3: Dorsal dissection of a zebrafish spinal cord. (A-A') After hindbrain transection (a) of a 2-dpf embryo, the skin is cut on the left and right sides of the embryo (b). A second cut, perpendicular to the first, is then performed (c). Next, the skin is lifted using a micropipette, allowing tweezers to grab and pull away the skin. (B) Removal of the skin exposes the dorsal spinal cord. Rostral to the black line, the skin has been removed and the surface of the spinal cord (asterisk), contained within the meninges, is exposed. The skin remains intact caudal to the black line (arrow). (C-C') The tip of the glass micropipette is pressed on the meninges, and swift, lateral, short movements are performed to pierce the meninges. (D-D' and E-E') Once the meninges are pierced (D-D'), the micropipette is advanced and (E-E') moved rostrally to tear the meninges into two segments. The somata of Rohon-Beard neurons typically emerge upon removal of the meninges (arrow). (F-G') In a 7-dpf larva, layers of muscle cover the dorsal aspect of the spinal cord, hindering access to Rohon-Beard neurons. Following the removal of the skin, the larva is treated with 0.05% collagenase. (F) A 5 min incubation with 0.05% collagenase is too stringent, resulting in excessive muscle damage, as evidenced by the frayed muscle (arrow and inset). (F') Excessive collagenase treatment may also damage Rohon-Beard neurons (arrow), revealed here by their expression of gfp in the Tg(islet2b:gfp) line.In theTg(islet2b:gfp) line, dorsal root ganglion neurons also express gfp (arrowhead). A briefer 1 min incubation with 0.05% collagenase sufficiently loosens the muscle (G) while conserving the myotome morphology (arrow and inset). (G') Pigment cells are present on top of the most dorsal muscle layer (arrowhead). (H and I) In the Tg(islet2b:gfp) line, Rohon-Beard neurons (arrows) and the dorsal root ganglion (arrowhead) continue to express gfp at 7 dpf. Dorsal views of Tg(islet2b:gfp) zebrafish embryos at 2 dpf (H) and 7 dpf (I). In Panel A Scale bars = 500 µm; B-E (shown in Panel B) Scale bars = 80 µm; F' and G' (shown in Panel F) Scale bars = 200 µm; H and I (shown in Panel H) Scale bars = 100 µm. Please click here to view a larger version of this figure.
- Dissection of embryos for recordings from Rohon-Beard cells
- Place an embryo in a dissection chamber containing ~2-3 mL of Ringer's solution (Figure 2A). Immobilize the embryo by adding ~100 µL of 0.4% tricaine solution to the chamber.
- Pull thin-wall glass micropipettes on a micropipette puller using a box filament to obtain a long, thin tip, similar to an injection micropipette (Figure 2B, top)53.
- Attach the pulled glass micropipette to the glass bore of the glue dispenser. Under the dissecting microscope, use tweezers to break the tip of the glass micropipette so that the tip is ~75 µm (Figure 2B, top). NOTE: The tip size should be large enough to allow efficient loading of the glue into the tip through the application of negative pressure (mouth suction) to front-fill the micropipette, but small enough to allow the precise application of the glue via positive pressure.
- Load the glass micropipette with ~3-5 µL of glue by applying suction through the mouthpiece. Bring the glue-filled tip to the dissection chamber. NOTE: Filling the micropipette with glue requires strong suction. If the micropipette fills quickly with weak suction, the tip is too large.
- Place the embryo in the recording chamber (Figure 3A and 3A'); for recordings from embryos and young larvae (≤72 hpf), there is no need to remove muscle tissue.
- Bring the tip of the glue-loaded micropipette near the head of the embryo/larva while maintaining slight positive pressure to the micropipette via the mouth tube (to prevent the entry of aqueous solution). Once the tip is near the head of the embryo, apply sufficient positive pressure to expel a small drop of glue onto the bottom of the chamber. NOTE: The glue hardens once in contact with the aqueous solution, so it is important to apply and maintain mild positive pressure as it is placed in the dissection solution.
- Use a dissecting pin tool to move the embryo/larva towards the drop of glue so that the head makes contact with the glue. Orient the embryo dorsal-side up and press on the head to ensure good contact with the glue.
- As the glue slowly hardens, use the dissecting pin to re-position the embryo/larva so that it lies ventral-side down, dorsal-side up. Dip the dissecting pin tool into the drop of glue and draw threads of the glue over and across the head of the embryo to further secure its position. NOTE: Tricaine speeds up the rate at which the glue hardens. To allow more time to work with the glue, use the minimal amount of tricaine required to immobilize the embryo/larva. Additionally, the hardening rate may vary with different lots of glue. Accordingly, when using a new batch of glue, ascertain its rate of hardening before using it for dissection.
- Once the head is securely attached to the silicone and the glue has solidified, sacrifice the embryo/larva by transection at the hindbrain level with another glass micropipette (Figure 3A-aand 4A-a). NOTE: Hindbrain transection is the method that is used here for humane animal sacrifice. However, depending on the experimental goals (e.g., the study of fictive swimming), another method may be needed.
- Before attaching the tail to the chamber, remove the skin from the trunk.
- Use a fresh glass micropipette (Figure 2B, bottom) to superficially cut the skin several times at a position caudal to the hindbrain (Figure 3A-band 4A-b). Pierce the skin superficially by moving the pipette perpendicular to the rostro-caudal axis on each side of the trunk for dorsally mounted (Figure 3A-b) specimens or on the exposed side of the trunk for laterally mounted specimens (Figure 4A-b).
- To create a flap of skin for tweezers to grab, scrape the skin several times with the micropipette at the level of and perpendicularly to the initial cut in step 3.1.10.1 (Figure 3A-cand 4A-c).
- Using tweezers, gradually lift the skin flap and pull the skin caudally. NOTE: This often results in the removal of the entirety of the skin from the trunk. However, it is sometimes necessary to remove the skin in several sections by performing reiterated scraping and pulling of the skin. For some applications, partial removal of the skin may allow sufficient access to the spinal cord segments of interest (Figures 3B and 4B).
- Deliver a small drop of glue near the tail of the embryo/larva. Use this glue to attach the tail to the bottom of the dissection chamber. During this step, as the glue hardens, adjust the position of the trunk with the dissecting pin tool to ensure that the trunk remains dorsally oriented and that it is firmly attached to the chamber.
- Following this initial dissection, rinse the preparation extensively with Ringer's solution to remove tricaine and debris. Allow the preparation to rest for ~5 min.
- Replace the dissection solution with extracellular recording solution. If needed, add an immobilizing agent to the preparation (e.g., α-bungarotoxin [final concentration of 1 µM]). NOTE: 1 µM α-bungarotoxin immobilizes 1 to 2 dpf embryos within ~30 min. For older larvae, a higher concentration of α-bungarotoxin may be needed. α-bungarotoxin is maintained in the bath solution during recordings, which are typically performed within a period of 1 h. Recording solutions that contain divalent cations, such as cobalt, do not require the addition of an immobilizing agent. For experiments requiring prolonged recording periods (over 1 h), the preparation is perfused with bath solution at a rate of 0.5-1 mL/min.
- Move the dissection chamber with the mounted embryo to the stage of an upright compound microscope equipped with a 40X water immersion long-working-distance objective. NOTE: This microscope is part of the rig where recordings will be performed. The rig should also be equipped with a headstage, a patch-clamp amplifier, a micromanipulator, and a data acquisition/computer system (Figure 2C).
- Mount an empty borosilicate thick-wall glass micropipette onto the electrode holder of the headstage (Figures 2B, bottom and 2C, arrow). Attach tubing (inner diameter: 0.16 cm, outer diameter: 0.32 cm, and length: ~90 cm) at one end to the air outlet of the electrode holder (arrowhead) and at the other end to a three-way stopcock (Figure 2C, black arrowhead).
- Place a mouthpiece at one end of another piece of tubing (~60-70 cm in length) and attach it to the three-way stopcock at the other end (Figure 2C, black asterisk). NOTE: This tubing system allows for the application of positive and negative pressure to the inside of the recording pipette during seal formation.
- Bring the micropipette tip to the most dorsal portion of the spinal cord and gently pierce the meninges. Follow with swift, short, sideways movements to loosen the meninges (Figure 3C and 3C').
- After the micropipette tip has traversed the meninges, advance and raise the micropipette to pull the meninges away from the spinal cord (Figure 3D and 3D').
- Move the micropipette rostrally, advancing over 1 to 2 hemisegments to expose Rohon-Beard cells (Figure 3E and 3E'). NOTE: Dissect the minimal amount of meninges required to expose only a few Rohon-Beard cells. After each recording, additional dissection is performed to reveal more Rohon-Beard cells. In both embryos and larvae, Rohon-Beard neurons may occasionally burst upon contact with the patch micropipette. Other spinal cord neurons do not behave this way, suggesting that this might reflect unique properties of the Rohon-Beard cells, such as their mechanosensitivity. In support of this, several solution compositions (e.g., ionic components and osmolarity) were tested, and none have prevented this behavior of Rohon-Beard cells.
- Dissection of larvae for recordings from Rohon-Beard cells NOTE: In 7 dpf larvae, muscle surrounding the dorsal spinal cord must be removed. Follow steps 3.1.1 to 3.1.15 (with a larva substituted for an embryo) before treatment with the enzyme.
- To remove the muscle, incubate the larva with 0.05% collagenase for 1 min.
- Remove the collagenase by rinsing the preparation ~5 times with Ringer's solution. Follow with ~5 rinses of extracellular solution to completely remove the collagenase.
- Carry out the remainder of the dissection after mounting the recording chamber on the stage of the microscope of the recording rig. Attach a borosilicate thick-wall patch micropipette (Figure 2B, bottom) to the electrode holder and break the tip of the micropipette by gently brushing it against the bottom of the silicone chamber.
- Use the slightly broken micropipette to tease the muscle away and expose the dorsal spinal cord. To remove muscle fibers from the preparation, apply suction through the tubing attached to the electrode holder outlet. Use the micromanipulator to move the micropipette along the length of a muscle fiber while applying suction. NOTE: The goal is to first use the micropipette to mechanically loosen the muscle and then to suck away and remove the individual muscle fibers. Occasionally, during the removal of muscle, the micropipette becomes clogged with the suctioned tissue.
- To unclog the micropipette, brush the micropipette against the bottom of the chamber, slightly breaking the tip, while blowing air through the tubing to expel the contents. NOTE: If the size of the micropipette tip becomes excessively large, a new micropipette may be needed. The size of the micropipette tip is more critical when removing the muscle layers closest to the membranes surrounding the spinal cord or meninges (small micropipette tips allow for more controlled work).
- Remove the meninges as described in steps 3.1.18-3.1.20 using a new micropipette (Figure 2B, bottom).
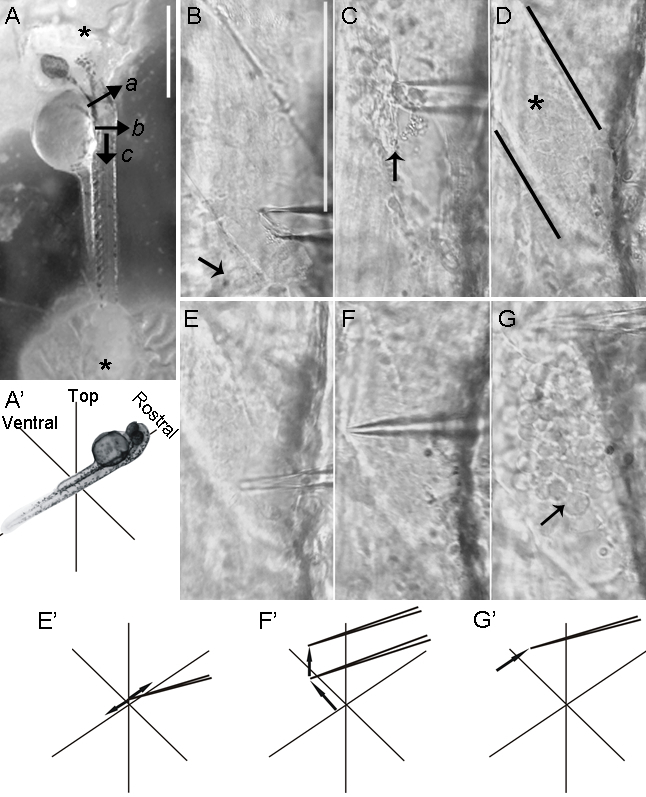 Figure 4: Lateral dissection of the zebrafish spinal cord. Mounting zebrafish embryos in a lateral orientation facilitates the access to motor neurons. The removal of the muscle and dissection of the meninges to expose the motor neurons is performed under an upright microscope adapted with a 40X water immersion objective (see Figure 2). (A) Motor neuron cell bodies are located ventrally and laterally within the spinal cord. Embryos are attached to the chamber so that their dorsal side faces the electrode holder. Note that the suture glue appears white once it hardens (asterisks). Once the hindbrain is transected (a), the skin is cut superficially several times at a site (b) caudal to the hindbrain using a glass micropipette. Additional superficial cuts (c), perpendicular to the first set (b), form a skin tab that tweezers can grab for the removal of the skin. (B-G) An empty glass micropipette, pulled to a short, tapered tip (Figure 2B, bottom), is attached to the electrode holder. The micropipette is maneuvered using the micromanipulator for the subsequent fine dissection and removal of muscle tissue. (B) The tip of the glass micropipette is first broken slightly by gently brushing it against the bottom of the chamber, creating a jagged end and a larger tip diameter. The micropipette is moved along the length of the muscle fibers while suction is applied. Muscle fibers are removed one layer at a time to prevent the disruption of the underlying meninges. In embryos, the most dorsal muscle layers are removed first, as these tend to be thinner. The skin is not removed from more caudal hemisegments (arrow). (C) The dorsal half of the muscle in one hemisegment has been removed (arrow). (D) Black lines demarcate a hemisegment devoid of muscle fibers, with intact meninges covering the spinal cord (asterisk). (E-E') Using a micropipette, pressure is applied to the meninges at a position slightly dorsal to motor neuron somata. Quick, short, lateral movements of the micropipette lead to the piercing of the meninges. (F-F') The micropipette is advanced ventrally, towards the ventral aspect of the hemisegment, and lifted to separate the meninges from the neuronal tissue. (G-G') Meninges are transected by moving the micropipette rostrally along the length of the hemisegment. Neurons immediately emerge from the exposed spinal cord and are now accessible to patch electrodes (arrow). Scale bars = 500 µm in (A); Scale bars = 100 µm in B-G (shown in Panel B).
Figure 4: Lateral dissection of the zebrafish spinal cord. Mounting zebrafish embryos in a lateral orientation facilitates the access to motor neurons. The removal of the muscle and dissection of the meninges to expose the motor neurons is performed under an upright microscope adapted with a 40X water immersion objective (see Figure 2). (A) Motor neuron cell bodies are located ventrally and laterally within the spinal cord. Embryos are attached to the chamber so that their dorsal side faces the electrode holder. Note that the suture glue appears white once it hardens (asterisks). Once the hindbrain is transected (a), the skin is cut superficially several times at a site (b) caudal to the hindbrain using a glass micropipette. Additional superficial cuts (c), perpendicular to the first set (b), form a skin tab that tweezers can grab for the removal of the skin. (B-G) An empty glass micropipette, pulled to a short, tapered tip (Figure 2B, bottom), is attached to the electrode holder. The micropipette is maneuvered using the micromanipulator for the subsequent fine dissection and removal of muscle tissue. (B) The tip of the glass micropipette is first broken slightly by gently brushing it against the bottom of the chamber, creating a jagged end and a larger tip diameter. The micropipette is moved along the length of the muscle fibers while suction is applied. Muscle fibers are removed one layer at a time to prevent the disruption of the underlying meninges. In embryos, the most dorsal muscle layers are removed first, as these tend to be thinner. The skin is not removed from more caudal hemisegments (arrow). (C) The dorsal half of the muscle in one hemisegment has been removed (arrow). (D) Black lines demarcate a hemisegment devoid of muscle fibers, with intact meninges covering the spinal cord (asterisk). (E-E') Using a micropipette, pressure is applied to the meninges at a position slightly dorsal to motor neuron somata. Quick, short, lateral movements of the micropipette lead to the piercing of the meninges. (F-F') The micropipette is advanced ventrally, towards the ventral aspect of the hemisegment, and lifted to separate the meninges from the neuronal tissue. (G-G') Meninges are transected by moving the micropipette rostrally along the length of the hemisegment. Neurons immediately emerge from the exposed spinal cord and are now accessible to patch electrodes (arrow). Scale bars = 500 µm in (A); Scale bars = 100 µm in B-G (shown in Panel B).
- Dissection of embryos for motor neuron and interneuron recordings
- Place the embryo in a dissection chamber containing Ringer's solution and immobilize the embryo with tricaine, as in step 3.1.1.
- Mount the embryo laterally, with its dorsal side facing the side of the chamber that is optimal for the user (typically depends upon handedness). Follow steps 3.1.2-3.1.17 and ensure that the embryo remains flat against the silicone (Figure 4A and 4A').
- Use a borosilicate thick-wall micropipette with a tip that has been broken to ~25 µm to scrape and suction the muscle away and expose the meninges, as described in steps 3.2.4-3.2.4.1 (Figure 4B-4D).
- Once the muscle layers are removed from the hemisegment(s) of interest (Figure 4D), replace the glass micropipette with a new one that has an intact tip (Figure 2B, bottom). Puncture the meninges at a position that is dorsal to the target neurons. Using the micromanipulator, push the micropipette down onto the meninges and then move it swiftly sideways to tear and traverse the meninges (Figure 4E and 4E').
- Upon rupturing the meninges, advance and raise the micropipette to lift the meninges away from the spinal cord (Figure 4F and 4F'). Move the micropipette rostrally to tear the membrane along the length of the hemisegment (Figure 4G and 4G'). NOTE: For recordings from Rohon-Beard cells and primary motor neurons, dissection is limited to clearing the meninges away from the immediate target region. In contrast, for recordings from interneurons and secondary motor neurons, it is necessary to remove neurons within the spinal cord that hinder access to the cell of interest. In the latter case, due to the extensive disruption of spinal cord circuitry, studies are limited to the analysis of intrinsic electrical membrane properties.
4. Electrophysiological Recordings from Spinal Neurons
- For recordings from Rohon-Beard cells and motor neurons, use borosilicate thick-wall glass capillaries pulled to a resistance of ~3 MΩ when filled with the pipette solution (Figure 2B, bottom).
- Apply positive pressure by blowing gently through the tubing attached to the electrode holder prior to immersing the micropipette in the bath. NOTE: The positive pressure prevents debris from clogging the micropipette tip and is maintained by turning the three-way stopcock to the off position. Once near the target neuron, the positive pressure will result in a distinctive indentation of the cell membrane, a helpful indicator that the micropipette is close enough to initiate seal formation.
- Release the positive pressure by turning the stopcock valve to the open position while applying additional light suction through the mouthpiece.
- After the formation of a GΩ-seal between the cell membrane and the micropipette tip, apply brief pulses of suction to rupture the membrane and achieve a whole-cell configuration.
- After establishing a stable whole-cell configuration, with an input resistance 500 MΩ and an access resistance 10 MΩ, obtain recordings in either voltage- or current-clamp mode.
Representative Results
We have successfully recorded from Rohon-Beard neurons in 17 hpf embryos through 7 dpf larvae (Figure 5A and 5B). When Rohon-Beard cells were recorded, the preparation was mounted dorsal-side up. Such mounting allows for the unambiguous identification of Rohon-Beard cells based on their superficial dorsal positions and large soma sizes. The identification is additionally confirmed by the stereotypical hyperpolarized resting membrane potential of these neurons (Figure 5, inset table)6,54. Moreover, as primary sensory neurons, Rohon-Beard neurons lack synaptic input. Therefore, in the absence of electrical stimulation, no changes in membrane potential should occur while recording in current-clamp mode (Figure 5B'). Since the initial recordings from Rohon-Beard cells in zebrafish were performed6, various transgenic lines (e.g., Tg(islet2b:gfp), Tg(ngn:gfp), and Tg(isletss:gfp)) have been generated that express fluorescent reporters in these neurons, further facilitating their identification55,56,57.
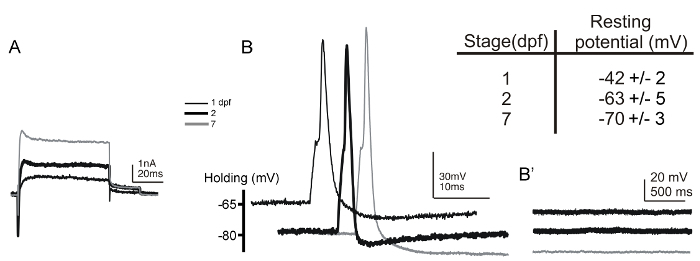 Figure 5: Whole-cell voltage- and current-clamp recordings from Rohon-Beard neurons in 1 and 2 dpf embryos and 7 dpf larvae. (A) Voltage-clamp recordings of outward and inward currents were obtained from Rohon-Beard neurons in 1- (thin black line), 2- (thick black line), and 7-dpf (gray line) embryos/larvae. The holding potential was -80 mV and currents were elicited by a depolarizing step to +20 mV. (B) Single action potentials are elicited by brief (1 ms) current injections (~0.35 nA) to Rohon-Beard neurons of 1- (thin black line), 2- (thick black line), and 7-dpf (gray line) embryos/larvae. (B') In the absence of electrical stimulation, no changes in membrane potential, such as spontaneous postsynaptic depolarizations, occur in Rohon-Beard neurons. The inset table summarizes the values of resting membrane potentials recorded from Rohon-Beard neurons of 1- (n = 21) and 2- (n = 9) dpf embryos and 7- (n = 7) dpf larvae. Please click here to view a larger version of this figure.
Figure 5: Whole-cell voltage- and current-clamp recordings from Rohon-Beard neurons in 1 and 2 dpf embryos and 7 dpf larvae. (A) Voltage-clamp recordings of outward and inward currents were obtained from Rohon-Beard neurons in 1- (thin black line), 2- (thick black line), and 7-dpf (gray line) embryos/larvae. The holding potential was -80 mV and currents were elicited by a depolarizing step to +20 mV. (B) Single action potentials are elicited by brief (1 ms) current injections (~0.35 nA) to Rohon-Beard neurons of 1- (thin black line), 2- (thick black line), and 7-dpf (gray line) embryos/larvae. (B') In the absence of electrical stimulation, no changes in membrane potential, such as spontaneous postsynaptic depolarizations, occur in Rohon-Beard neurons. The inset table summarizes the values of resting membrane potentials recorded from Rohon-Beard neurons of 1- (n = 21) and 2- (n = 9) dpf embryos and 7- (n = 7) dpf larvae. Please click here to view a larger version of this figure.
Transgenic lines that allow unequivocal identification of other spinal neuron subtypes are also available. Among these, the mnx1 transgenic line Tg(mnx1:gfp) expresses green fluorescent protein (gfp) in a subset of spinal motor neurons soon after their specification (~14-16 hpf)58,59. Due to the stereotypical positioning of the primary motor neurons within each hemisegment (Figure 6A), together with the expression of gfp in the mnx1 transgenic, it is possible to identify the various primary motor neuron subtypes (Figure 6B and 6C). Including a fluorescent dye in the recording electrode solution allows for the visualization of axonal trajectories, providing additional confirmation of motor neuron identity, as some interneurons also express gfp in the Tg(mnx1:gfp) line. Alternatively, another transgenic line that allows the identification of motor neurons is the ET2 line60.
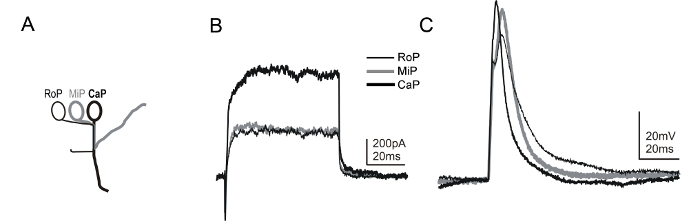 Figure 6: Whole-cell voltage and current-clamp recordings from motor neurons of 1 dpf zebrafish embryos. (A) A cartoon depicts the specific morphological features of the primary motor neuron subtypes present in the zebrafish spinal cord. Primary motor neurons are identified by the position of their soma within a segment (i.e., rostral [RoP], medial [MiP], or caudal [CaP])45. In addition, each subtype extends an axon to the periphery via a distinct path. The combined use of the Tg(mnx1:gfp) line and dye labeling reveals the stereotypical axonal arbor and the identity of the motor neuron subtype during a recording. Using the methods presented here, it is possible to sequentially record from three different primary motor neuron subtypes within the same hemisegment. (B) Voltage-clamp recordings are shown that were obtained from RoP, MiP, and CaP, all in a single hemisegment. A voltage step to +20 mV was used to elicit currents from a holding potential of -80 mV. (C) During current-clamp recordings from RoP, MiP, and CaP, brief (1 ms, ~0.4 nA) current injections were applied to trigger an action potential. The membrane potential was held at ~-65 mV. Please click here to view a larger version of this figure.
Figure 6: Whole-cell voltage and current-clamp recordings from motor neurons of 1 dpf zebrafish embryos. (A) A cartoon depicts the specific morphological features of the primary motor neuron subtypes present in the zebrafish spinal cord. Primary motor neurons are identified by the position of their soma within a segment (i.e., rostral [RoP], medial [MiP], or caudal [CaP])45. In addition, each subtype extends an axon to the periphery via a distinct path. The combined use of the Tg(mnx1:gfp) line and dye labeling reveals the stereotypical axonal arbor and the identity of the motor neuron subtype during a recording. Using the methods presented here, it is possible to sequentially record from three different primary motor neuron subtypes within the same hemisegment. (B) Voltage-clamp recordings are shown that were obtained from RoP, MiP, and CaP, all in a single hemisegment. A voltage step to +20 mV was used to elicit currents from a holding potential of -80 mV. (C) During current-clamp recordings from RoP, MiP, and CaP, brief (1 ms, ~0.4 nA) current injections were applied to trigger an action potential. The membrane potential was held at ~-65 mV. Please click here to view a larger version of this figure.
A principal difference between primary and secondary motor neurons is the larger somata of the earlier-born neurons. However, secondary motor neuron subtypes are not identifiable by soma size or position. For recordings from specific secondary motor neurons, two transgenic lines, Tg(gata2:gfp) and Tg(islet1:gfp), have been used for the identification of secondary motor neurons with ventrally and dorsally projecting axons, respectively55,61. However, a third secondary motor neuron subtype is present in the zebrafish spinal cord, with axons that project dorsally and ventrally62. Accordingly, dye can be used to fill secondary motor neurons during recordings to identify subtypes on the basis of morphology (Figure 7A)8,9. Often, during voltage-clamp (Figure 7B, asterisks) or current-clamp recordings (Figure 7C and 7C', arrowheads) from secondary motor neurons, spontaneous or synaptic events are recorded.
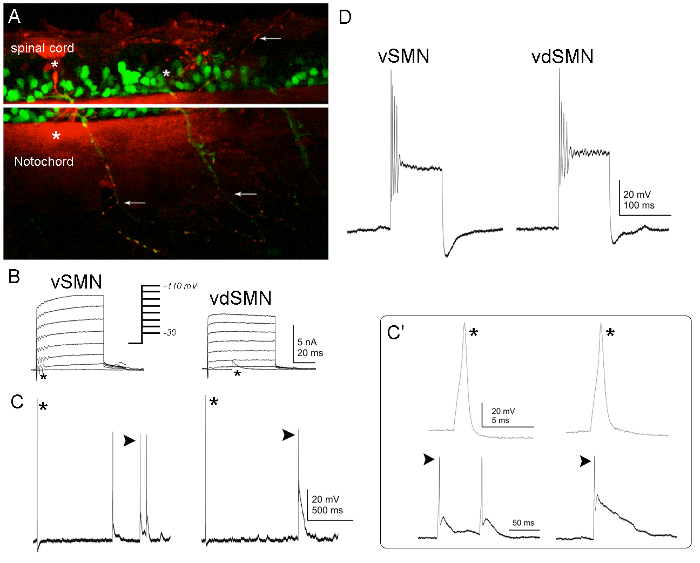 Figure 7: Whole-cell voltage and current-clamp recordings from secondary motor neurons of 2 dpf embryos. (A) In the Tg(gata2:gfp) line, two different secondary motor neuron subtypes express gfp62. In the left hemisegment, a ventral secondary motor neuron (asterisk and arrow indicate soma and axon, respectively). In the neighboring hemisegment, on the right (caudal), there is a ventral/dorsal secondary motor neuron (asterisk indicates soma; arrows indicate the two axons, one projecting ventrally [bottom arrow] and the other dorsally [top arrow]). These neurons were labeled with a red fluorescent dye during the recordings. To identify ventral/dorsal secondary motor neurons, it is critical to ensure that the dissection does not remove muscle in the adjacent caudal hemisegment, thereby damaging or removing the dorsal axon. After the recording, the neuron soma remains attached to the micropipette as it is pulled away from the preparation (right top asterisk). When using dyes to fill neurons during recordings, the dye often leaks while the electrode is in the bath, resulting in a red fluorescent background visible in the spinal cord and notochord (bottom asterisk in rostral [left] hemisegment). (B) Voltage-clamp recordings were obtained from ventral and ventral/dorsal secondary motor neurons. Voltage steps (to -30, -10, +10, +30, +50, +70, +90, and +110 mV) elicited outward and inward currents. Unclamped action potentials/depolarizations may be present in the recordings (asterisks). (C) During current-clamp recordings from secondary motor neurons, brief (1 ms) current injections of increasing amplitude were applied to the neurons to trigger an action potential (asterisks). (C') Examples of single action potentials triggered in secondary motor neurons by ~0.4-nA current injections are shown. At this stage, spontaneous action potentials are also observed (C and C', arrowheads). (D) Prolonged (100 ms) current injections (~0.35 nA) trigger the repetitive firing of action potentials. The membrane potential was held at ~-65 mV. Please click here to view a larger version of this figure.
Figure 7: Whole-cell voltage and current-clamp recordings from secondary motor neurons of 2 dpf embryos. (A) In the Tg(gata2:gfp) line, two different secondary motor neuron subtypes express gfp62. In the left hemisegment, a ventral secondary motor neuron (asterisk and arrow indicate soma and axon, respectively). In the neighboring hemisegment, on the right (caudal), there is a ventral/dorsal secondary motor neuron (asterisk indicates soma; arrows indicate the two axons, one projecting ventrally [bottom arrow] and the other dorsally [top arrow]). These neurons were labeled with a red fluorescent dye during the recordings. To identify ventral/dorsal secondary motor neurons, it is critical to ensure that the dissection does not remove muscle in the adjacent caudal hemisegment, thereby damaging or removing the dorsal axon. After the recording, the neuron soma remains attached to the micropipette as it is pulled away from the preparation (right top asterisk). When using dyes to fill neurons during recordings, the dye often leaks while the electrode is in the bath, resulting in a red fluorescent background visible in the spinal cord and notochord (bottom asterisk in rostral [left] hemisegment). (B) Voltage-clamp recordings were obtained from ventral and ventral/dorsal secondary motor neurons. Voltage steps (to -30, -10, +10, +30, +50, +70, +90, and +110 mV) elicited outward and inward currents. Unclamped action potentials/depolarizations may be present in the recordings (asterisks). (C) During current-clamp recordings from secondary motor neurons, brief (1 ms) current injections of increasing amplitude were applied to the neurons to trigger an action potential (asterisks). (C') Examples of single action potentials triggered in secondary motor neurons by ~0.4-nA current injections are shown. At this stage, spontaneous action potentials are also observed (C and C', arrowheads). (D) Prolonged (100 ms) current injections (~0.35 nA) trigger the repetitive firing of action potentials. The membrane potential was held at ~-65 mV. Please click here to view a larger version of this figure.
Discussion
The methods described here allow for the electrical and morphological characterization of sensory and motor neurons of zebrafish embryos after minimal dissection of the spinal cord. Neurons remain healthy for at least 1 h, the time limit imposed on these recordings. Neurons have been recorded using the standard whole-cell configuration, as well as from nucleated patches; the latter method minimizes space-clamp issues that can preclude a detailed biophysical study of ion currents9.
An important challenge is to achieve the firm attachment of the embryo or larva to the chamber in order to remove the skin and to perform the limited dissection required to provide access to the neurons of interest. The preparation also needs to be properly secured to the recording chamber for the whole-cell patch-clamp methods. A method that meets this challenge via the use of veterinarian suture glue to attach the embryo or larva to the dissection/recording chamber is described here, an approach that has been used for the dissection of other model organisms (e.g., Drosophila)63. From our experience training others, we find that the most critical step to master is the controlled and precise delivery of small amounts of glue. Here, a glue dispenser device that allows a user to apply negative and positive pressure to load glue into or to expel it from the tip of a micropipette is discussed. Using the suture glue, embryos and larvae can be firmly attached to the chamber and oriented either dorsal-side up or laterally. In this way, different access options for a variety of neurons are available. Also, the layer of silicone elastomer on the chamber bottom can be even thinner than the 1 mm specified here, providing potential optical advantages. Another method, more commonly used to attach the preparation to a recording chamber, involves use of fine tungsten pins1,2,3,4,5. While that methods differ, both allow electrophysiological access to zebrafish spinal neurons, affording researchers with options that can be selected based on the goals and challenges of the experiment.
The zebrafish preparation described here allows for the electrical and morphological study of spinal neurons in situ during their earliest stages of differentiation. By recording from spinal neurons using these methods, we have gained insight into the cellular effects of several mutations, even prior to the identification of the lesioned gene6,64,65.
Disclosures
The authors declare no competing financial interests.
Acknowledgments
This work was supported by grants from the NIH (F32 NS059120 to RLM and R01NS25217 and P30NS048154 to ABR).
References
- Drapeau P, Ali DW, Buss RR, Saint-Amant L. In vivo recording from identifiable neurons of the locomotor network in the developing zebrafish. J Neurosci Methods. 1999;88(1):1–13. doi: 10.1016/s0165-0270(99)00008-4. [DOI] [PubMed] [Google Scholar]
- Drapeau P, Saint-Amant L, Buss RR, Chong M, McDearmid JR, Brustein E. Development of the locomotor network in zebrafish. Prog Neurobiol. 2002;68(2):85–111. doi: 10.1016/s0301-0082(02)00075-8. [DOI] [PubMed] [Google Scholar]
- Saint-Amant L, Drapeau P. Whole cell patch-clamp recordings from identified spinal neurons in the zebrafish embryo. Methods Cell Sci. 2003;25:59–64. doi: 10.1023/B:MICS.0000006896.02938.49. [DOI] [PubMed] [Google Scholar]
- Masino MA, Fetcho JR. Fictive Swimming Motor Patterns in Wild Type and Mutant Larval Zebrafish. J Neurophysiol. 2005;93(6):3177–3188. doi: 10.1152/jn.01248.2004. [DOI] [PubMed] [Google Scholar]
- Wen H, Brehm P. Paired motor neuron-muscle recordings in zebrafish test the receptor blockade model for shaping synaptic current. J Neurosci. 2005;25(35):8104–8111. doi: 10.1523/JNEUROSCI.2611-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribera AB, Nüsslein-Volhard C. Zebrafish Touch-Insensitive Mutants Reveal an Essential Role for the Developmental Regulation of Sodium Current. J Neurosci. 1998;18:9181–9191. doi: 10.1523/JNEUROSCI.18-22-09181.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda RH, Heiser RA, Ribera AB. Developmental, molecular, and genetic dissection of INa in vivo in embryonic zebrafish sensory neurons. J Neurophysiol. 2005;93:3582–3593. doi: 10.1152/jn.01070.2004. [DOI] [PubMed] [Google Scholar]
- Moreno RL, Ribera AB. Zebrafish motor neuron subtypes differ electrically prior to axonal outgrowth. J Neurophysiol. 2009;102:2477–2484. doi: 10.1152/jn.00446.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno RL, Ribera AB. Spinal neurons require Islet1 for subtype-specific differentiation of electrical excitability. Neural Dev. 2014;9(1):19. doi: 10.1186/1749-8104-9-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streisinger G, Walker C, Dower N, Knauber D, Singer F. Production of clones of homozygous diploid zebra fish (Brachydanio rerio) Nature. 1981;291(5813):293–296. doi: 10.1038/291293a0. [DOI] [PubMed] [Google Scholar]
- Driever W, et al. A genetic screen for mutations affecting embryogenesis in zebrafish. Development. 1996;123:37–46. doi: 10.1242/dev.123.1.37. [DOI] [PubMed] [Google Scholar]
- Haffter P, et al. The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Development. 1996;123:1–36. doi: 10.1242/dev.123.1.1. [DOI] [PubMed] [Google Scholar]
- Vogel G. Genomics: Sanger will sequence zebrafish genome. Science. 2000;290(5497):1671. [PubMed] [Google Scholar]
- Patton EE, Zon LI. The art and design of genetic screens: zebrafish. Nature Reviews Genetics. 2001;2(12):956–966. doi: 10.1038/35103567. [DOI] [PubMed] [Google Scholar]
- Lawson ND, Wolfe SA. Forward and reverse genetic approaches for the analysis of vertebrate development in the zebrafish. Dev Cell. 2011;21(1):48–64. doi: 10.1016/j.devcel.2011.06.007. [DOI] [PubMed] [Google Scholar]
- Yates A, et al. Ensembl. Nucleic Acids Res. 2016;44(D1):D710–D716. doi: 10.1093/nar/gkv1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K. Transgenesis and gene trap methods in zebrafish by using the Tol2 transposable element. Methods Cell Biol. 2004;77:201–222. doi: 10.1016/s0091-679x(04)77011-9. [DOI] [PubMed] [Google Scholar]
- Kwan KM, et al. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn. 2007;236(11):3088–3099. doi: 10.1002/dvdy.21343. [DOI] [PubMed] [Google Scholar]
- Huang P, Xiao A, Zhou M, Zhu Z, Lin S, Zhang B. Heritable gene targeting in zebrafish using customized TALENs. Nat Biotechnol. 2011;29:699–700. doi: 10.1038/nbt.1939. [DOI] [PubMed] [Google Scholar]
- Sander JD, et al. Targeted gene disruption in somatic zebrafish cells using engineered TALENs. Nat Biotechnol. 2011;29(8):697–698. doi: 10.1038/nbt.1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang N, et al. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res. 2013;23(4):465–472. doi: 10.1038/cr.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WY, et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol. 2013;31(3):227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieschke GJ, Currie PD. Animal models of human disease: zebrafish swim into view. Nature Rev Genet. 2007;8(5):353–367. doi: 10.1038/nrg2091. [DOI] [PubMed] [Google Scholar]
- Levin ED, Cerutti DT. Behavioral neuroscience of zebrafish. In: Buccafusco JJ, editor. Methods of behavior analysis in neuroscience. CRC Press; 2009. [PubMed] [Google Scholar]
- Stewart A, Gaikwad S, Kyzar E, Green J, Roth A, Kalueff A. Modeling anxiety using adult zebrafish: A conceptual review. Neuropharmacology. 2012;62:135–143. doi: 10.1016/j.neuropharm.2011.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mushtaq MY, Verpoorte R, Kim HK. Zebrafish as a model for systems biology. Biotechnol Genet Eng Rev. 2013;29(2):187–205. doi: 10.1080/02648725.2013.801238. [DOI] [PubMed] [Google Scholar]
- Phillips JB, Westerfield M. Zebrafish models in translational research: tipping the scales toward advancements in human health. Dis Model Mech. 2014;7(7):739–743. doi: 10.1242/dmm.015545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis JS. Analysis of the development of nervous system of the zebrafish, Brachydanio rerio. I. The normal morphology and development of the spinal cord and ganglia of the zebrafish. J Embryol Exp Morphol. 1968;19(2):109–119. [PubMed] [Google Scholar]
- Bernhardt RR, Chitnis AB, Lindamer L, Kuwada JY. Identification of spinal neurons in the embryonic and larval zebrafish. J Comp Neurol. 1990;302(3):603–616. doi: 10.1002/cne.903020315. [DOI] [PubMed] [Google Scholar]
- Tsuchida T, et al. Topographic organization of embryonic motor neurons defined by expression of LIM homeobox genes. Cell. 1994;79(6):957–970. doi: 10.1016/0092-8674(94)90027-2. [DOI] [PubMed] [Google Scholar]
- Guillemot F. Spatial and temporal specification of neural fates by transcription factor codes. Development. 2007;134:3771–3780. doi: 10.1242/dev.006379. [DOI] [PubMed] [Google Scholar]
- Goulding M. Circuits controlling vertebrate locomotion: Moving in a new direction. Nat Rev Neurosci. 2009;10:507–518. doi: 10.1038/nrn2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetcho JR, McLean DL. Some principles of organization of spinal neurons underlying locomotion in zebrafish and their implications. Ann N Y Acad Sci. 2010;1198:94–104. doi: 10.1111/j.1749-6632.2010.05539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Barrio MG, et al. A transcription factor code defines nine sensory interneuron subtypes in the mechanosensory area of the spinal cord. PLoS One. 2013;8(11) doi: 10.1371/journal.pone.0077928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satou C, Kimura Y, Hirata H, Suster ML, Kawakami K, Higashijima S. Transgenic tools to characterize neuronal properties of discrete populations of zebrafish neurons. Development. 2013;140(18):3927–3931. doi: 10.1242/dev.099531. [DOI] [PubMed] [Google Scholar]
- McLean DL, Fan J, Higashijima S, Hale ME, Fetcho JR. A topographic map of recruitment in spinal cord. Nature. 2007;446:71–75. doi: 10.1038/nature05588. [DOI] [PubMed] [Google Scholar]
- McLean DL, Masino MA, Koh IY, Lindquist WB, Fetcho JR. Continuous shifts in the active set of spinal interneurons during changes in locomotor speed. Nat. Neurosci. 2008;11:1419–1429. doi: 10.1038/nn.2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean DL, Fetcho JR. Spinal interneurons differentiate sequentially from those driving the fastest swimming movements in larval zebrafish to those driving the slowest ones. J. Neurosci. 2009;29:13566–13577. doi: 10.1523/JNEUROSCI.3277-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ampatzis K, Song J, Ausborn J, El Manira A. Separate microcircuit modules of distinct V2a interneurons and motoneurons control the speed of locomotion. Neuron. 2014;83:934–943. doi: 10.1016/j.neuron.2014.07.018. [DOI] [PubMed] [Google Scholar]
- Ljunggren EE, Haupt S, Ausborn J, Ampatzis K, El Manira A. Optogenetic activation of excitatory premotor interneurons is sufficient to generate coordinated locomotor activity in larval zebrafish. J. Neurosci. 2014;34:134–139. doi: 10.1523/JNEUROSCI.4087-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menelaou E, VanDunk C, McLean DL. Differences in the morphology of spinal V2a neurons reflect their recruitment order during swimming in larval zebrafish. J Comp Neurol. 2014;522:1232–1248. doi: 10.1002/cne.23465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard JM, et al. Intraspinal Sensory Neurons Provide Powerful Inhibition to Motor Circuits Ensuring Postural Control during Locomotion. Curr Biol. 2016;26(21):2841–2853. doi: 10.1016/j.cub.2016.08.026. [DOI] [PubMed] [Google Scholar]
- Song J, Ampatzis K, Björnfors ER, El Manira A. Motor neurons control locomotor circuit function retrogradely via gap junctions. Nature. 2016;529(7586):399–402. doi: 10.1038/nature16497. [DOI] [PubMed] [Google Scholar]
- Lamborghini JE. Rohon-beard cells and other large neurons in Xenopus embryos originate during gastrulation. J. Comp. Neurol. 1980;189:323–333. doi: 10.1002/cne.901890208. [DOI] [PubMed] [Google Scholar]
- Myers PZ, Eisen JS, Westerfield M. Development and axonal outgrowth of identified motoneurons in the zebrafish. J Neurosci. 1986;6:2278–2289. doi: 10.1523/JNEUROSCI.06-08-02278.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel CB, Westerfield M. Primary neurons of the zebrafish. In: Edelman GM, Gall WE, Cowan WM, editors. Signals and Sense: Local and Global Order in Perceptual Maps. New York: Wiley-Liss; 1990. pp. 561–588. [Google Scholar]
- Metcalfe WK, Myers PZ, Trevarrow B, Bass MB, Kimmel CB. Primary neurons that express the L2/HNK-1 carbohydrate during early development in the zebrafish. Development. 1990;110(2):491–504. doi: 10.1242/dev.110.2.491. [DOI] [PubMed] [Google Scholar]
- Rossi CC, Kaji T, Artinger KB. Transcriptional control of Rohon-Beard sensory neuron development at the neural plate border. Dev Dyn. 2009;238:931–943. doi: 10.1002/dvdy.21915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis KE, Eisen JS. From cells to circuits: development of the zebrafish spinal cord. Progress in Neurobiology. 2003;69(6):419–449. doi: 10.1016/s0301-0082(03)00052-2. [DOI] [PubMed] [Google Scholar]
- Reimer MM, et al. Dopamine from the brain promotes spinal motor neuron generation during development and adult regeneration. Dev Cell. 2013;25(5):478–491. doi: 10.1016/j.devcel.2013.04.012. [DOI] [PubMed] [Google Scholar]
- Lambert AM, Bonkowsky JL, Masino MA. The conserved dopaminergic diencephalospinal tract mediates vertebrate locomotor development in zebrafish larvae. J Neurosci. 2012;32(39):13488–13500. doi: 10.1523/JNEUROSCI.1638-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M. The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio) University of Oregon Press; 1995. [Google Scholar]
- Oesterle A. Pipette Cookbook 2015 P-97 & P-1000 Micropipette pullers. Sutter Instrument; 2015. [Google Scholar]
- Spitzer NC. The ionic basis of the resting potential and a slow depolarizing response in Rohon-Beard neurons of Xenopus tadpoles. J Physiol. 1976;255(1):105–135. doi: 10.1113/jphysiol.1976.sp011272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashijima S, Hotta Y, Okamoto H. Visualization of cranial motor neurons in live transgenic zebrafish expressing green fluorescent protein under the control of the islet-1 promoter/enhancer. J Neurosci. 2000;20:206–218. doi: 10.1523/JNEUROSCI.20-01-00206.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blader P, Plessy C, Strahle U. Multiple regulatory elements with spatially and temporally distinct activities control neurogenin1 expression in primary neurons of the zebrafish embryo. Mech Dev. 2003;120:211–218. doi: 10.1016/s0925-4773(02)00413-6. [DOI] [PubMed] [Google Scholar]
- Palanca AM, et al. New transgenic reporters identify somatosensory neuron subtypes in larval zebrafish. Dev Neurobiol. 2013;73:152–167. doi: 10.1002/dneu.22049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan-Street H, Fox MA, Meyer D, Sanes JR. Neuromuscular synapses can form in vivo by incorporation of initially aneural postsynaptic specializations. Development. 2005;132:4471–4481. doi: 10.1242/dev.02044. [DOI] [PubMed] [Google Scholar]
- Arkhipova V, Wendik B, Devos N, Ek O, Peers B, Meyer D. Characterization and regulation of the hb9/mnx1 beta-cell progenitor specific enhancer in zebrafish. Dev Biol. 2012;365:290–302. doi: 10.1016/j.ydbio.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balciunas D, Davidson AE, Sivasubbu S, Hermanson SB, Welle Z, Ekker SC. Enhancer trapping in zebrafish using the Sleeping Beauty transposon. BMC Genomics. 2004;5(1):62. doi: 10.1186/1471-2164-5-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng A, Tang H, Ong BA, Farrell MJ, Lin S. Promoter analysis in living zebrafish embryos identifies a cis-acting motif required for neuronal expression of GATA-2. Proc. Natl. Acad. Sci. USA. 1997;94:6267–6272. doi: 10.1073/pnas.94.12.6267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menelaou E, McLean DL. A gradient in endogenous rhythmicity and oscillatory drive matches recruitment order in an axial motor pool. J Neurosci. 2012;32:10925–10939. doi: 10.1523/JNEUROSCI.1809-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrbough J, Pinto S, Mihalek RM, Tully T, Broadie K. latheo, a Drosophila gene involved in learning, regulates functional synaptic plasticity. Neuron. 1999;23(1):55–70. doi: 10.1016/s0896-6273(00)80753-9. [DOI] [PubMed] [Google Scholar]
- McKeown KA, Moreno R, Hall VL, Ribera AB, Downes GB. Disruption of Eaat2b, a glutamate transporter, results in abnormal motor behaviors in developing zebrafish. Dev Biol. 2012;362(2):162–171. doi: 10.1016/j.ydbio.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmean V, et al. pigk mutation underlies macho behavior and affects Rohon-Beard cell excitability. J Neurophysiol. 2015;114(2):1146–1157. doi: 10.1152/jn.00355.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]