

Abstract
Fibrosis is a hallmark of many cardiovascular diseases and is associated with the exacerbated secretion and deposition of the extracellular matrix (ECM). Using proteomics, we have previously identified more than 150 ECM and ECM-associated proteins in cardiovascular tissues. Notably, many ECM proteins are glycosylated. This post-translational modification affects protein folding, solubility, binding, and degradation. We have developed a sequential extraction and enrichment method for ECM proteins that is compatible with the subsequent liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis of intact glycopeptides. The strategy is based on sequential incubations with NaCl, SDS for tissue decellularization, and guanidine hydrochloride for the solubilization of ECM proteins. Recent advances in LC-MS/MS include fragmentation methods, such as combinations of higher-energy collision dissociation (HCD) and electron transfer dissociation (ETD), which allow for the direct compositional analysis of glycopeptides of ECM proteins. In the present paper, we describe a method to prepare the ECM from tissue samples. The method not only allows for protein profiling but also the assessment and characterization of glycosylation by MS analysis.
Keywords: Biochemistry, Issue 122, Extracellular matrix, fibrosis, glycoprotein, systems biology, proteomics, cardiovascular
Introduction
Fibrosis is a hallmark of many diseases. Fibroblasts proliferate and differentiate towards highly synthetic phenotypes, which are associated with the exacerbated secretion and deposition of extracellular matrix (ECM)1. Excessive ECM deposition can continue, even after the initial injury has abated, leading to functional impairment. Using proteomics, we have previously identified more than 150 ECM and ECM-associated proteins in cardiac tissue2,3. They are not only structural proteins, but also matricellular proteins and proteases that contribute to the continuous remodeling and dynamic adaptation of the heart. Notably, many ECM proteins are glycosylated4. This post-translational modification (PTM) involves the addition of sugar residues to certain amino acid positions, and it affects protein folding, solubility, binding, and degradation5.
There are two main glycosylation types that occur in mammals. (1) N-glycosylation occurs at the carboxamido nitrogen of asparagine residues (Asn) within the consensus sequence Asn-Xaa-Thr/Ser, where Xaa is any amino acid except for proline. (2) In O-glycosylation, sugar residues attach to serine and threonine residues (Ser, Thr) or, to a much lesser extent, to hydroxyproline and hydroxylysine. While O-glycosylation can occur in a variety of protein groups, N-glycosylation is restricted to secreted proteins or extracellular domains of membrane proteins5. This makes N-glycosylation an attractive target when studying the ECM.
Proteomics sets a new standard for the analysis of protein changes in disease. Thus far, most proteomics studies have been focused on intracellular proteins6. This is mainly due to the following reasons. First, abundant intracellular proteins hamper the identification of scarce ECM components. This is particularly crucial in cardiac tissue, in which mitochondrial and myofilament proteins account for a large proportion of the protein content7. Second, integral ECM proteins are heavily cross-linked and difficult to solubilize. Lastly, the presence of abundant PTMs (i.e., glycosylation) alters the molecular mass, charge, and electrophoretic properties of peptides, affecting both the separation and the identification by liquid chromatography tandem mass spectrometry (LC-MS/MS). Over recent years, we have developed and improved a sequential extraction and enrichment method for ECM proteins that is compatible with subsequent mass spectrometry (MS) analysis. The strategy is based on sequential incubations.
The first step is performed with NaCl, an ionic buffer that facilitates the extraction of ECM-associated and loosely bound ECM proteins, as well as newly synthesized ECM proteins. It is detergent free, non-denaturing, non-disruptive of cell membranes, and amenable for further biochemical assays8. Then, decellularization is achieved with sodium dodecyl sulfate (SDS). At this step, a low SDS concentration ensures membrane destabilization and the release of intracellular proteins whilst preventing the disruption of the more soluble non-integral ECM components. Finally, ECM proteins are extracted with a guanidine hydrochloride buffer (GuHCl). GuHCl is effective in extracting heavily cross-linked proteins and proteoglycans from tissues such as tendons9, cartilage10, vessels11,12,13 and the heart2,3. We applied this biochemical fractionation, in combination with LC-MS/MS, to explore ECM remodeling in cardiovascular disease2,3,11,12,13,14. Recent advances in MS include novel fragmentation methods, such as combinations of higher-energy collision dissociation (HCD) and electron transfer dissociation (ETD), which allow for the direct analysis of intact glycopeptides3,15.
Here we describe a methodology to prepare ECM for MS analysis that allows for the analysis of protein composition, the identification of glycosylation sites, and the characterization of glycan forms. Compared to previous analyses of ECM glycosylation16, this methodology allows for the direct assessment of compositional changes in glycosylation profiles in a site-specific manner using MS. We have applied this methodology to cardiovascular tissues. However, it can also be applied, with minor modifications, to the study of the ECM in other tissue specimens and can provide unprecedented insights into ECM biology.
Protocol
The study was approved by the Wandsworth Local Research Ethics Committee (reference number: 06/Q0803/37) and received institutional approval from the research and development office. All patients gave written informed consent.
1. Extraction of Extracellular Matrix Proteins
NOTE: The human atrial tissues used for these experiments were obtained from the atrial appendages during cardiopulmonary bypass, just after the cardioplegic arrest of the heart. All samples were collected at St George's Hospital, London, UK. All tissue samples must be frozen at -80 °C. Do not use samples preserved with fixatives, such as paraformaldehyde, that cross-link proteins.
Prepare all extraction buffers in advance of experiments, as per Table 1, in order to minimize the time between the extraction steps. Perform all incubations at room temperature (RT) in a temperature-controlled environment (i.e., ~20 °C) to ensure consistency between extractions.
Weigh 20-50 mg of tissue. If several samples are to be extracted, cut and weigh them one by one to avoid the complete thawing of the tissues. Using a scalpel, dice the tissue into 3-4 smaller pieces (i.e., 2-3 mm) and place them together into 1.5 mL tubes.
Add 500 µL of ice-cold phosphate-buffered saline (PBS; see Table 1 and the Table of Materials) and perform five washes to minimize blood contamination.
- Extraction Step 1: Incubation with NaCl Buffer
- After washing with PBS, place the samples into 1.5 mL tubes with screw caps.Add NaCl buffer(see Table 1) at 10 times (v:w) the tissue weight. Vortex the tubes at RT for 1 h at minimum speed (i.e., 600 rpm). NOTE: A low vortex speed is critical to avoid the mechanical disruption of the tissue during this step. Use a foam adaptor to place all tubes during the extraction.
- Transfer the extracts to new tubes and centrifuge at 16,000 x g for 10 min at 4 °C. Store the extracts at −20 °C until use. Briefly wash the remaining tissue pellets with fresh NaCl buffer. Use the same type of buffer (i.e., 100 µL of NaCl buffer) for washing to prevent the extraction of other proteins with different solubilities (i.e., proteins not extractable with NaCl).
- After washing, ensure the complete removal of the buffer to minimize overlap in protein content with subsequent extraction steps. Discard the NaCl buffer used for washing. NOTE: The ratio between the buffer volume and tissue weight is important for a reproducible extraction. A 10:1 ratio (v:w) for the NaCl and SDS extractions and 5:1 for the GuHCl step provide sufficient amounts of protein without saturating the buffer. Protein concentrations are approximately 1-2 µg/µL after extraction.
- Extraction Step 2: Decellularization with SDS Buffer
- Add SDS buffer (Table 1) at ten times (v:w) the tissue weight; the use of low SDS concentrations (i.e., 0.1%) is critical to avoid the loss of ECM proteins during decellularization. Vortex the tubes at RT for 16 h at minimum speed (i.e., 600 rpm). NOTE: A low vortex speed minimizes mechanical disruption of the ECM.
- Transfer the extracts to new tubes. Centrifuge at 16,000 x g for 10 min at 4 °C; store at -20 °C until use. Briefly wash the remaining tissue pellets with ddH2O to remove the SDS. Ensure the complete removal of the liquid after washing.
- Extraction Step 3: Incubation with GuHCl Buffer
- Add GuHCl buffer (Table 1) at five times (v:w) the tissue weight. Vortex the tubes at RT for 72 h at maximum speed (i.e., 3,200 rpm); vigorous vortexing facilitates the mechanical disruption of the ECM.
- Transfer the extracts to new tubes. Centrifuge at 16,000 × g for 10 min at 4 °C and store at -20 °C until use.
2. Protein Quantification and Precipitation
NOTE: Due to the presence of detergents, the SDS buffer is incompatible with direct protein quantification based on measurements of absorbance at 280 nm. To ensure reproducible quantification, use colorimetric assays for all protein extracts17.
- Quantification.
- Prepare standards for a calibration curve using bovine serum albumin (BSA) serially diluted in the appropriate extraction buffer (i.e., NaCl, SDS, or GuHCl)17. During this time, thaw the sample extracts.
- Dilute the samples in extraction buffer to obtain concentrations within the linear range of absorbance; a 1:10 dilution (v:v) yields satisfactory results. Dilute the GuHCl samples with an equal amount of ddH2O for quantification, as the colorimetric assay is not compatible with concentrations of >4 M GuHCl.
- Use a bicinchoninic acid (BCA)-based colorimetric assay17 (see the Table of Materials), following the manufacturer's instructions for assays in 96-well plates; it is recommended to perform at least duplicate measurements.
- After incubating for 30 min, take absorbance readings at a wavelength of 570 nm in order to calculate the protein concentration using the BSA standard calibration curve17.
- Protein Precipitation
- Thaw GuHCl extracts at RT. Aliquot 10 µg of protein for each sample into new tubes. For a direct glycopeptide analysis, aliquot 50 µg. Add 10 times the volume of ethanol and incubate overnight at -20 °C. NOTE: GuHCl is not compatible with further enzymatic reactions and most electrophoretic applications. The removal of GuHCl is required before deglycosylation and trypsin digestion. The solubility of GuHCl in ethanol and, conversely, the low solubility of proteins allow for the effective removal of GuHCl while yielding approximately a 98% recovery of proteins18.
- Centrifuge the samples at 16,000 × g for 30 min at 4 °C and aspirate the supernatant. Take care not to disturb the precipitated pellet. Dry the pellets for 15 min using a vacuum concentrator (see the Table of Materials) at RT. NOTE: The protocol can be stopped here and the dried pellets stored at -20 °C until use.
- Optionally, run a gel electrophoresis as quality control (QC, see the Supplemental Methods).
3. Sequential Deglycosylation for the Assessment of N-glycosylation Site Occupancy
During sample drying (see step 2.2.2), prepare the deglycosylation buffer containing debranching deglycosylation enzymes, as per Table 1. See Table of Materials for product details.
Add 10 µL of deglycosylation buffer containing enzymes to each sample. Ensure the appropriate pellet resuspension by performing a quick vortex and spin-down of samples. NOTE: The removal of sugar monomers using debranching enzymes is essential for the subsequent and complete removal of O-linked complex saccharides and facilitates the later cleavage of N-linked sugars by PNGase-F.
Incubate for 2 h at 25 °C to allow for the removal of heparan sulfate by heparinase II.Increase the temperature to 37 °C and incubate for 36 h with gentle agitation. NOTE: Given the low reaction volumes and prolonged incubation times, use incubator shakers and tightly pack multiple 1.5 mL tubes inside a 50-mL conical tube leaning inside the incubator at approximately 45°.
After 36 h, centrifuge the samples for 1 min at 16,000 x gand evaporate theH2O from the samples by using a vacuum concentrator at RT for approximately 45 min.
Resuspend the dried samples with 10 µL of H218O containing 50 U/mL PNGase-F, which cleaves all asparagine-linked glycans in a deamidation reaction. NOTE: The resulting aspartic acid carries an excess mass of 2.98 Da, indicative of the presence of N-glycosylation during MS analysis.
Incubate for 36 h at 37 °C under constant agitation in the incubator shaker.
4. In-solution Trypsin Digestion
NOTE: This step should be carried out for both non-deglycosylated (i.e., used for direct glycopeptide analysis) and deglycosylated samples (i.e., used for the assessment of glycan occupancy).
Use 10 µg of total protein for the assessment of N-glycosylation site occupancy (as indicated in the previous step). For samples intended for direct glycopeptide analysis, use 50 µg of protein as the starting amount. NOTE: The following steps are described for 10 µg of protein. Scale up th evolumes (i.e., 5 times for 50 µg) as required.
Denature the proteins on each sample aliquot using 9 M urea and 3 M thiourea, with final concentrations of 6 M urea and 2 M thiourea, respectively (e.g., for a 10 µL sample, 20 µL of urea/thiourea).
Reduce the proteins by adding 100 mM dithiotreitol (DTT, 3.33 µL, final concentration: 10 mM). Incubate at 37 °C for 1 h with agitation at 240 rpm.
Cool down the samples to RT before performing the alkylation by adding 0.5 M iodoacetamide (3.7 µL, final concentration: 50 mM). Incubate in the dark for 1 h.
Use pre-chilled (-20 °C) acetone (6 times the volume of the sample) to incubate the samples overnight at -20 °C. Precipitate by centrifugation at 14,000 x g for 25 min at 4 °C.
Aspirate the supernatant. Take care not to disturb the precipitated pellet. Dry the protein pellets using a vacuum concentrator for 30 min at RT.
Resuspend in 20 µL of 0.1 M triethylammonium bicarbonate (TEAB) buffer, pH 8.2, containing trypsin (0.01 µg/µL) and digest overnight at 37 °C and 240 rpm.
Stop the digestion by acidifying the samples with 10% trifluoroacetic acid (TFA, 2 µL for a final concentration of 1% TFA).
5. Peptide Cleanup Using C18 Columns
NOTE: The removal of interfering contaminants from the peptide mixture after digestion reduces ion suppression and improves signal-to-noise ratios and sequence coverage. This step should be carried out for both non-deglycosylated and deglycosylated samples.
Activate the resin on the C18 spin plate (see the Table of Materials) using 200 µL of methanol per well and centrifuge at 1,000 x g for 1 min.
Wash by adding 200 µL per well of 80% acetonitrile (ACN) and 0.1% TFA in H2O. Centrifuge at 1,000 x g for 1 min.
Equilibrate by adding 200 µL per well of 1% ACN and 0.1% TFA in H2O. Centrifuge at 1,000 x g for 1 min. Repeat this step two more times.
Load the samples (the entire volume) from step 4 into the wells containing the resin and centrifuge at 1,500 x g for 1 min. Reload the flow-through a second time and repeat the centrifugation.
Wash by adding 200 µL per well of 1% ACN and 0.1% TFA in H2O. Centrifuge at 1,500 x g for 1 min. Repeat this step two more times.
Elute the samples with 170 µL per well of 50% ACN, 0.1% TFA in H2O. Centrifuge at 1,500 x g for 1 min. Repeat the previous step and combine the collected eluate.
Dry the eluate using a vacuum concentrator for 2 h at RT. If it is not being used immediately, keep the dried samples at -80 °C until use. NOTE: Deglycosylated samples intended for protein identification only are ready to use for LC-MS/MS after this step. Steps 6 and 7 are not required for these samples.
Thaw and resuspend the deglycosylated samples in 2% ACN and 0.05% TFA in H2O to a final protein concentration of 0.5 µg/µL. Proceed with step 6 for the direct glycopeptide analysis of non-deglycosylated samples.
Optionally, filter the peptides prior to labeling with tandem mass tags (TMT); see the Supplemental Methods for peptide fitration.
6. Labeling with TMT (for Direct Glycopeptide Analysis Only)
Thaw and resuspend the dried pellets in 50 µL of 50 mM TEAB to obtain a concentration of 1 µg/µL.
Resusped the 0.8-mg vial of TMT Zero (TMT0, see the Table of Materials) reagent in 41 µL of ACN. Follow the manufacturer's recommendantions for resuspension.
Label the peptide samples at a ratio of 50 µg peptides to 0.4 mg TMT0 ( i.e., 50 µL of peptides to 20.5 µL of TMT0). Incubate at RT for 1 h.
Quench the labeling reaction by adding 5% hydroxylamine at a ratio 6:100 (i.e., 4.23 µL of 5% hydroxylamine). Incubate at RT for 15 min.
Dry the TMT0-labelled peptide samples for 1 h at RT using a vacuum concentrator. Resuspend in 10 µL of ddH2O. NOTE: Due to the glycan residues, glycopeptides display a higher molecular mass than non-glycosylated peptides. TMT0 increases the charge state of glycopeptides. This reduces their relative mass to charge (m/z) ratios and facilitates ETD fragmentation.
7. Glycopeptide Enrichment
Use reaction buffers provided in the kit (see the Table of Materials).
Add 50 µL of binding buffer to each 10 µL sample from step 6.5. Vortex the glycocapture resin solution until it becomes homogeneous. Use a zwitterionic hydrophilic interaction liquid chromatography (ZIC-HILIC)-based capture15.
Aliquot 50 µL of the resin suspension to new 1.5 mL tubes. Spin for 1 min at 2,500 x g and remove the supernatant. Add 60 µL of sample (i.e., the sample with binding buffer) to the tubes containing the resin pellets. Mix using a pipette and incubate at RT for 20 min in agitation at 1,200 rpm.
Centrifuge for 2 min at 2,000 x g and transfer the supernatant to new tubes. Keep the tubes. Add 150 µL of wash buffer to the resin tubes. Mix using a pipette and incubate at RT for 10 min in agitation at 1,200 rpm.
Spin for 2 min at 2,500 x g. Transfer the supernatant to the same tubes (from step 7.4). Repeat the washing steps two times.
Add 75 µL of elution buffer and mix using a pipette. Agitate at 1,200 rpm for 5 min at RT and then centrifuge the tubes for 2 min at 2,500 x g. Transfer the supernatants to new 1.5 mL tubes. Repeat the washing steps and then transfer the eluate supernatant to the same tube.
Centrifuge the tubes containing the eluate (i.e., glycopeptides) for 2 min at 2,500 x g. Transfer the supernatant to new tubes to ensure the removal of any remaining resin from the previous steps.
Dry the total 150 µL of eluate using a vacuum concentrator for approximately 2 h at RT. Resuspend the dried-down glycopeptides in 15 µL of 2% ACN and 0.05% TFA in ddH2O.
Proceed to perform LC-MS/MS using HCD fragmentation to analyse the ECM protein composition, and LC-MS/MS using HCD and ETD fragmentation for glycopeptide characterization. See section 8.
8. Mass Spectrometry Analysis
Perform LC-MS/MS using HCD fragmentation to analyse the ECM protein composition; see the Supplemental Methods for details.
Perform LC-MS/MS using HCD and ETD fragmentation for glycopeptide characterization (see the Supplemental Methods for details); the enriched sample should be compared to the non-enriched input material15. NOTE: Detailed descriptions of LC-MS/MS methods for indirect glycopeptide analysis, direct glycopeptide analysis, and database search are provided in the Supplemental Methods. Researchers interested in characterizing ECM protein and glycan composition using mass spectrometry are encouraged to refer to previous publications3,11,15.
Representative Results
A schematic workflow of the protocol is provided in Figure 1.
ECM extraction protocol
The efficiency of the extraction can be monitored by running aliquots form each extract on Bis-Tris acrylamide gels and using silver staining for visualization. Figure 2A shows the complementarity of the NaCl, SDS and GuHCl extracts after sequential extraction. This QC allows for identification of potential issues with sample quality such as excessive protein degradation. After extraction, ECM glycoproteins are abundant in the GuHCl extracts (Figure 2B).
Deglycosylation
To assess the efficiency of deglycosylation, a non-deglycosylated control should be run in parallel. Deglycosylation times have to be suitable to achieve a complete and homogeneous removal of sugar residues, as exemplified in Figure 3A. Figure 3B shows a representative example of samples efficiently deglycosylated by the addition of enzymes for GAG removal and deglycosylation enzymes that target smaller N- and O-linked oligosaccharides.
Glycoproteomics
The protocol for assessment of the occupancy of NxT/S sequons improves protein sequence coverage for ECM glycoproteins after MS (Figure 3C) and allows for an initial screening of the presence of glycoproteins. This helps to reduce the search time for glycopeptides, as databases can be customized to contain previously identified glycoproteins. HCD-ETD fragmentation is used for identification and compositional characterization of oligosaccharides attached to ECM glycoproteins. Figure 4A displays a representative spectrum obtained for a peptide labeled with 18O after deglycosylation (indirect glycopeptide analysis). Figure 4B is a representative example of a spectrum obtained after analysis of intact glycopeptides from ECM extracts (direct glycopeptide analysis).
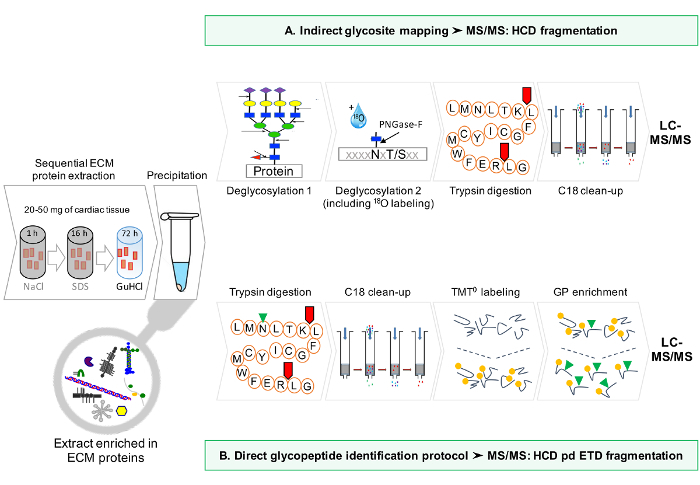 Figure 1: Method Overview. (A) After sequential enrichment for ECM proteins, LC-MS/MS analyses are performed on the deglycosylated extracts. (B) Alternatively, non deglycosylated ECM extracts are further enriched for glycopeptides. Please click here to view a larger version of this figure.
Figure 1: Method Overview. (A) After sequential enrichment for ECM proteins, LC-MS/MS analyses are performed on the deglycosylated extracts. (B) Alternatively, non deglycosylated ECM extracts are further enriched for glycopeptides. Please click here to view a larger version of this figure.
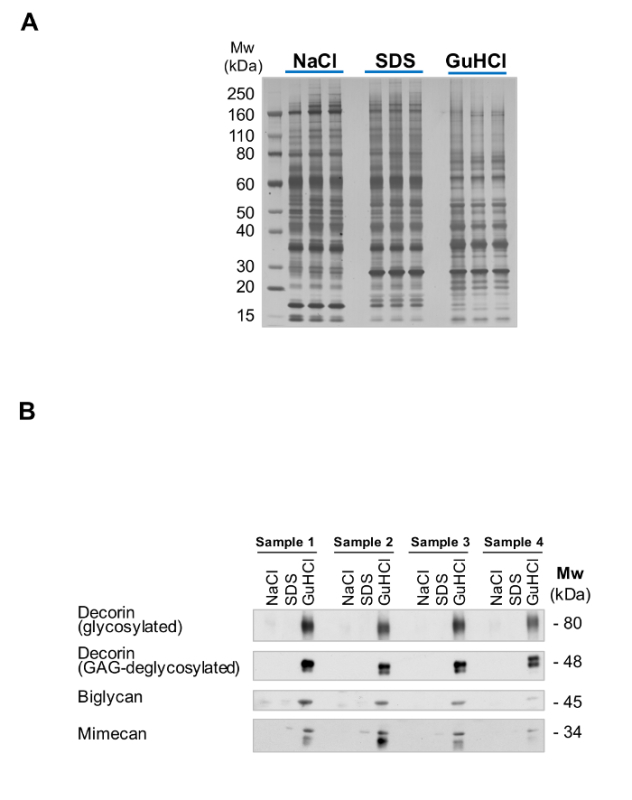 Figure 2: Extraction of ECM Proteins. (A) The 3 different extracts from the sequential extraction procedure ("English Quickstep") are complementary in their protein content. While SDS extracts are enriched in intracellular proteins, GuHCl extracts contain the majority of ECM proteins. Successful fractionation is visualized by the different silver staining pattern. (B) ECM proteins such as the small leucine-rich proteoglycans decorin, biglycan and mimecan are predominantly detected in the GuHCl extracts, with little presence in the SDS and NaCl extracts. Please click here to view a larger version of this figure.
Figure 2: Extraction of ECM Proteins. (A) The 3 different extracts from the sequential extraction procedure ("English Quickstep") are complementary in their protein content. While SDS extracts are enriched in intracellular proteins, GuHCl extracts contain the majority of ECM proteins. Successful fractionation is visualized by the different silver staining pattern. (B) ECM proteins such as the small leucine-rich proteoglycans decorin, biglycan and mimecan are predominantly detected in the GuHCl extracts, with little presence in the SDS and NaCl extracts. Please click here to view a larger version of this figure.
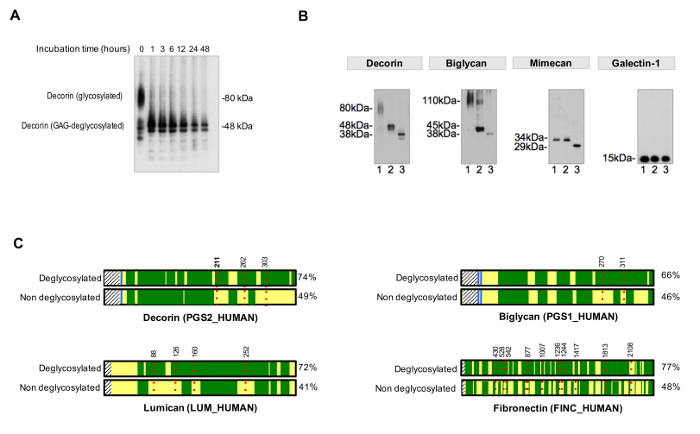 Figure 3. Analysis of Glycosylation. (A) Appropriate incubation times are required for complete deglycosylation. The example shows the effect of incubation time during removal of glycosaminoglycan chains from the glycoprotein decorin. (B) ECM glycoproteins are decorated with large and repetitive glycosaminoglycan chains and short and diverse N- and O-linked oligosaccharides. Lane 1 on each of the immunoblots represents untreated cardiac extracts. Lane 2 contains extracts treated with enzymes that digest glycosaminoglycans. Samples in lane 3 contain, in addition, enzymes for the removal of N- and O-linked oligossacharides. Galectin-1 is not glycosylated, hence there is no shift in protein size. Adapted from Lynch M, et al.4 (C) In LC-MS/MS analysis, samples treated with PNGase-F in the presence of H218O achieve better sequence coverage (%, on the right side) compared to non-deglycosylated samples. Dark green areas represent sequence coverage by LC-MS/MS. The red, dotted lines represent glycosites, with numbers indicating their amino acid position. Detection of glycosylation of decorin at position Asn211 (N, highlighted in bold) is shown in detail as an example in Figure 4. Please click here to view a larger version of this figure.
Figure 3. Analysis of Glycosylation. (A) Appropriate incubation times are required for complete deglycosylation. The example shows the effect of incubation time during removal of glycosaminoglycan chains from the glycoprotein decorin. (B) ECM glycoproteins are decorated with large and repetitive glycosaminoglycan chains and short and diverse N- and O-linked oligosaccharides. Lane 1 on each of the immunoblots represents untreated cardiac extracts. Lane 2 contains extracts treated with enzymes that digest glycosaminoglycans. Samples in lane 3 contain, in addition, enzymes for the removal of N- and O-linked oligossacharides. Galectin-1 is not glycosylated, hence there is no shift in protein size. Adapted from Lynch M, et al.4 (C) In LC-MS/MS analysis, samples treated with PNGase-F in the presence of H218O achieve better sequence coverage (%, on the right side) compared to non-deglycosylated samples. Dark green areas represent sequence coverage by LC-MS/MS. The red, dotted lines represent glycosites, with numbers indicating their amino acid position. Detection of glycosylation of decorin at position Asn211 (N, highlighted in bold) is shown in detail as an example in Figure 4. Please click here to view a larger version of this figure.
 Figure 4. Glycopeptide Analysis by MS. (A) Using a shotgun proteomics approach on ECM enriched extracts, glycopeptides can be identified by the presence of deamidated asparagines within NxT/S sequons and labeled with 18O. The example shows a HCD MS/MS spectrum for a peptide of decorin containing the previously glycosylated Asn211. The data obtained can be used to create a customized database of ECM glycoproteins. (B) HCD-ETD fragmentation is used to analyze the glycopeptide enriched ECM extract. The ETD MS/MS spectrum allows the characterization of glycan composition. Please click here to view a larger version of this figure.
Figure 4. Glycopeptide Analysis by MS. (A) Using a shotgun proteomics approach on ECM enriched extracts, glycopeptides can be identified by the presence of deamidated asparagines within NxT/S sequons and labeled with 18O. The example shows a HCD MS/MS spectrum for a peptide of decorin containing the previously glycosylated Asn211. The data obtained can be used to create a customized database of ECM glycoproteins. (B) HCD-ETD fragmentation is used to analyze the glycopeptide enriched ECM extract. The ETD MS/MS spectrum allows the characterization of glycan composition. Please click here to view a larger version of this figure.
| A. Stock solutions | |
| DTT (Dithiotreitol, C4H10O2S2) | 100 mM DTT in ddH2O.1 |
| EDTA (Ethylenediaminetetraacetic acid, C10H16N2O8) | 250 mM EDTA in ddH2O, pH 8.0. |
| GuHCl (Guanidine hydrochloride, CH6ClN3) | 8 M GuHCl in ddH2O. |
| IAA (Iodoacetamide, C2H4INO) | 500 mM IAA in ddH2O.1,2 |
| Na acetate (Sodium acetate, C2H3NaO2) | 1 M Na acetate in ddH2O, pH 5.8. |
| NaCl (Sodium chloride, NaCl) | 1 M NaCl in ddH2O. |
| Na phosphate dibasic (Disodium phosphate, Na2H2PO4) | 1 M Na phosphate dibasic in ddH2O, pH 6.8. |
| SDS (Sodium dodecyl sulfate, NaC12H25SO4) | 1% SDS (35 mM) in ddH2O.3 |
| TFA (Trifluoroacetic acid, C2HF3O2) | 10% TFA (1.2 M) in ddH2O. |
| TEAB (Triethylammonium bicarbonate, C7H17NO3) | 1M TEAB in in ddH2O, pH 8.5 |
| Thiourea (Thiourea, CH4N2S) | 3 M thiourea in ddH2O. |
| Tris-HCl (Tris-hydrochloride (NH11C4O3[HCl]) | 100 mM Tris–HCl in ddH2O, pH 7.5. |
| Urea (Urea, CH4N2O) | 9 M urea in ddH2O. |
| B. Reaction buffers | |
| C18 clean-up equilibration buffer | 1% ACN, 0.1% TFA in ddH2O |
| C18 clean-up column wash buffer | 80% ACN, 0.1% TFA in H2O |
| C18 clean-up elution buffer | 50% acetonitrile, 0.1% TFA in ddH2O |
| Deglycosylation Buffer (4x) | 600 mM NaCl and 200 mM Na phosphate in ddH2O, pH 6.8. |
| GuHCl buffer4 | 4 M guanidine hydrochloride, 50 mM Na acetate and 25 mM EDTA in ddH2O, pH 5.8. Add 1:100 (v:v) of cocktail of proteinase inhibitors before use. |
| NaCl buffer4 | 0.5 M NaCl, 10 mM Tris-HCl and 25 mM EDTA in ddH2O, pH 7.5. Add 1:100 (v:v) of cocktail of proteinase inhibitors before use. |
| PBS (1x) | 1.7 mM KH2PO4, 5 mM Na2HPO4, 150 mM NaCl, pH 7.4. Add 25 mM EDTA and 1:100 (v:v) of cocktail of proteinase inhibitors before use. |
| Sample buffer (4x) | 100 mM Tris, 2% SDS, 40% Glycerol, 0.02% bromophenol blue in ddH2O, pH 6.8. Add 10% ß-mercaptoethanol before use. |
| SDS buffer4 | 0.1 % SDS and 25 mM EDTA in ddH2O. Add 1:100 (v:v) of cocktail of proteinase inhibitors before use. |
| C. Enzymes | |
| Chondroitinase ABC5 | 0.5 U/mL in deglycosylation buffer (1x) |
| Keratanase5 | 0.1 U/mL in deglycosylation buffer (1x) |
| Heparinase II5 | 0.1 U/mL in deglycosylation buffer (1x) |
| α2-3,6,8,9-Neuraminidase (sialidase)5 | 0.025 U/mL in deglycosylation buffer (1x) |
| β1,4-Galactosidase5 | 0.015 U/mL in deglycosylation buffer (1x) |
| β-N-Acetylglucosaminidase5 | 0.25 U/mL in deglycosylation buffer (1x) |
| Endo-α-N-acetylgalactosaminidase (O-glycosidase)5 | 0.013 U/mL in deglycosylation buffer (1x) |
| PNGase-F(N-glycosidase-F)6 | 50 U/mL in H218O |
| Trypsin | 0.01 µg/µL in TEAB buffer |
| Table NOTES. | |
| 1 Keep stock solution frozen at -20 °C. | |
| 2 IAA should be kept protected from light. | |
| 3 SDS readily crystallizes at < 20 °C. In order to facilitate solubilization of 1% SDS (stock solution), warm the buffer under hot tap water. | |
| 4 Extraction buffers can be stored at RT. Add broad-spectrum cocktail of proteinase inhibitors as indicated before use. | |
| 5 These enzymes should be added during the first deglycosylation step. | |
| 6 PNGase-F should be only added during the second deglycosylation step. |
Table 1: Stock Solutions, Reaction Buffers and Enzymes. This table lists the composition of each stock solution and reaction buffer required for the extraction and subsequent processing (including enzymatic digestions) of cardiac ECM proteins prior to MS analysis.
Discussion
This proteomics protocol has been optimized over the last few years in our laboratory. Here, we used cardiac tissue, but only minor adjustments may be required for its application to other tissues. For example, the extraction protocol needs to take the cellularity of the tissue into consideration. Cardiac tissue is highly cellular compared to vascular tissue. When using vascular tissue, the SDS concentration can be lower (i.e. 0.08%) and the decellularization time is shorter (i.e. 4 h)11,12,13. The use of deglycosylation enzymes is crucial for LC-MS/MS analysis of ECM composition. However, incubation times need to be adjusted for different tissue types. For example, heparinase II required extended incubation times at 25 °C when using samples such as skin, which are rich in basement membrane proteins (e.g. agrin, perlecan) (data not shown). Direct glycopeptide analysis can be performed on conditioned media from cells in culture15. Enrichment steps may not be required for the analysis of this simplified subproteome. Similar to GuHCl extracts, NaCl extracts are also amenable for glycoproteomics analysis with minor modifications. Other extraction protocols for enrichment of ECM proteins can be adapted to characterize ECM glycopeptides19,20.
Glycosylation is the most complex PTM5. Indirect identification of glycopeptides is achieved by the detection of deamidated Asn with incorporated 18O at a NxT/S sequon. Deamidated Asn at other positions may represent false positives. Likewise, N-glycosylation must be considered in the context of protein ontologies: intracellular proteins containing a NxT/S sequon will not be glycosylated but might give rise to false positives. As current search algorithms do not allow for the screening of PTMs at pre-determined sequences only (i.e. Asn at NxT/S), manual filtering of the data is required. Identification of presence/absence of glycosylation at these positions can be compared between disease and control samples. There is no enzyme equivalent to PNGase F for O-deglycosylation (i.e. introducing a mass shift on threonine or serine). Therefore, the identification of O-glycosylation is restricted to direct glycopeptide analysis. Direct glycopeptide analysis is used to obtain compositional information of sugars attached to proteins, but does not provide structural information of the glycan. Moreover, the glycan composition is the result of glycan synthesis and processing after secretion.
Our 3-step extraction method for ECM proteins ("English Quickstep")6 has allowed characterizing the ECM in a variety of cardiovascular tissues. Fractionation of the tissue into several extracts is required to obtain a simplified ECM proteome as discussed elsewhere6. Intracellular proteins would otherwise contribute to an excessive dynamic range of protein abundances within the extracts that would hinder identification of less abundant ECM proteins. Moreover, intracellular proteins carry O-glycosylations5 that would complicate ECM glycopeptide enrichment and subsequent MS analysis. Other authors applied similar extraction methodologies to characterize for example lung21 and cartilage tissues10, however they did not pursue the analysis of glycosylation. Previous analysis of glycosylation focused on identification of glycosites only, require removal of the glycan from the protein core, and cannot assess O-glycosylation22,23. Lectin arrays and chemical enrichment are available for assessment of glycan types on biological samples based on their binding specificity, but these techniques cannot assign glycan types to specific proteins24 nor can they assess glycosylation sites.
Initially, we used gel electrophoresis prior to LC-MS/MS of ECM proteins. Although gel separation is useful in obtaining simplified protein fractions amenable to LC-MS/MS analysis, the latest instruments offer faster scan speeds. Thus, the electrophoretic separation step can be omitted. This provides an additional advantage as large ECM proteins, which are retained on top of the gel, are analyzed more efficiently. However, information regarding the Mw of the intact proteins is lost. The evaporation step prior to PNGase F deglycosylation ensures complete removal of regular H2O to minimize false negatives. Sugar residues (i.e. variable glycan masses) interfere with the separation by LC and compromise subsequent peptide identification by MS/MS. A pan-deglycosylation protocol is also recommended for proteomics analysis of ECM proteins not focused on glycosylation.
Proteomics can provide unprecedented insights into the ECM. Beyond structural support, glycans attached to the ECM are essential for host-pathogen interaction, cell-cell communication and the immune response25, i.e. allograft rejection after organ transplantation. Glycoproteomics will be an essential tool in glycobiology.
Disclosures
None.
Acknowledgments
JBB is a Career Establishment Fellow in the King's British Heart Foundation Centre. MM is a Senior Fellow of the British Heart Foundation (FS/13/2/29892). The study was supported by an excellence initiative (Competence Centers for Excellent Technologies - COMET) of the Austrian Research Promotion Agency FFG: "Research Center of Excellence in Vascular Ageing - Tyrol, VASCage" (K-Project number 843536) and the NIHR Biomedical Research Center based at Guy's and St. Thomas' National Health Service Foundation Trust and King's College London in partnership with King's College Hospital.
References
- Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol. Ther. 2009;123(2):255–278. doi: 10.1016/j.pharmthera.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Barallobre-Barreiro J, et al. Proteomics analysis of cardiac extracellular matrix remodeling in a porcine model of ischemia/reperfusion injury. Circulation. 2012;125(6):789–802. doi: 10.1161/CIRCULATIONAHA.111.056952. [DOI] [PubMed] [Google Scholar]
- Barallobre-Barreiro J, et al. Glycoproteomics reveals decorin peptides with anti-myostatin activity in human atrial fibrillation. Circulation. 2016;134(11):817–832. doi: 10.1161/CIRCULATIONAHA.115.016423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Barallobre-Barreiro J, Jahangiri M, Mayr M. Vascular proteomics in metabolic and cardiovascular diseases. J. Intern. Med. 2016;280(4):325–338. doi: 10.1111/joim.12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A, Lowe JB, et al. Biological Roles of Glycans. In: Varki A, et al., editors. Essentials of glycobiology. 2nd ed. Cold Spring Harbor NY: Cold Spring Harbor Laboratory Press; 2009. [PubMed] [Google Scholar]
- Barallobre-Barreiro J, Lynch M, Yin X, Mayr M. Systems biology - opportunities and challenges: The application of proteomics to study the cardiovascular extracellular matrix. Cardiovasc. Res. 2016. Sep 15, [DOI] [PMC free article] [PubMed]
- Agnetti G, Husberg C, Van Eyk JE. Divide and conquer: the application of organelle proteomics to heart failure. Circ. Res. 2011;108(4):512–526. doi: 10.1161/CIRCRESAHA.110.226910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason RM, Mayes RW. Extraction of cartilage protein-polysaccharides with inorganic salt solutions. Biochem. J. 1973;131(13):535–540. doi: 10.1042/bj1310535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel KG, Peters JA. Isolation of proteoglycans from tendon. Methods. Mol. Biol. 2001;171:9–17. doi: 10.1385/1-59259-209-0:009. [DOI] [PubMed] [Google Scholar]
- Wilson R, et al. Comprehensive profiling of cartilage extracellular matrix formation and maturation using sequential extraction and label-free quantitative proteomics. Mol. Cell. Proteomics. 2010;9(6):1296–1313. doi: 10.1074/mcp.M000014-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barallobre-Barreiro J, et al. Extracellular matrix remodeling in response to venous hypertension: proteomics of human varicose veins. Cardiovasc. Res. 2016;110(3):419–430. doi: 10.1093/cvr/cvw075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didangelos A, Yin X, Mandal K, Baumert M, Jahangiri M, Mayr M. Proteomics characterization of extracellular space components in the human aorta. Mol. Cell. Proteomics. 2010;9(9):2048–2062. doi: 10.1074/mcp.M110.001693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didangelos A, et al. Extracellular matrix composition and remodeling in human abdominal aortic aneurysms: a proteomics approach. Mol. Cell. Proteomics. 2011;10(8) doi: 10.1074/mcp.M111.008128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandoch M, et al. Loss of biglycan enhances thrombin generation in apolipoprotein E-deficient mice: Implications for inflammation and atherosclerosis. Arterioscler Thromb. Vasc. Biol. 2016;36(5):e41–e50. doi: 10.1161/ATVBAHA.115.306973. [DOI] [PubMed] [Google Scholar]
- Yin X, Bern M, Xing Q, Ho J, Viner R, Mayr M. Glycoproteomic analysis of the secretome of human endothelial cells. Mol. Cell. Proteomics. 2013;12(4):956–978. doi: 10.1074/mcp.M112.024018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker BL, et al. Quantitative N-linked glycoproteomics of myocardial ischemia and reperfusion injury reveals early remodeling in the extracellular environment. Mol. Cell. Proteomics. 2011;10(8) doi: 10.1074/mcp.M110.006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PK, et al. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985;150(1):76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Pepinsky RB. Selective precipitation of proteins from guanidine hydrochloride-containing solutions with ethanol. Anal. Biochem. 1991;195(1):177–181. doi: 10.1016/0003-2697(91)90315-k. [DOI] [PubMed] [Google Scholar]
- de Castro-Brás LE, et al. Texas 3-step decellularization protocol: looking at the cardiac extracellular matrix. J. Proteomics. 2013;86:43–52. doi: 10.1016/j.jprot.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naba A, Clauser KR, Hynes RO. Enrichment of extracellular matrix proteins from tissues and digestion into peptides for mass spectrometry analysis. J Vis Exp. 2015. p. e53057. [DOI] [PMC free article] [PubMed]
- Decaris ML, et al. Proteomic analysis of altered extracellular matrix turnover in bleomycin-induced pulmonary fibrosis. Mol. Cell. Proteomics. 2014;13(7):1741–1752. doi: 10.1074/mcp.M113.037267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Li XJ, Martin DB, Aebersold R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol. 2003;21(6):660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- Parker BL, et al. Site-specific glycan-peptide analysis for determination of N-glycoproteome heterogeneity. J. Proteome Res. 2013;12(12):5791–5800. doi: 10.1021/pr400783j. [DOI] [PubMed] [Google Scholar]
- Li Y, et al. Simultaneous analysis of glycosylated and sialylated prostate-specific antigen revealing differential distribution of glycosylated prostate-specific antigen isoforms in prostate cancer tissues. Anal. Chem. 2011;83(1):240–245. doi: 10.1021/ac102319g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rienks M, Papageorgiou AP, Frangogiannis NG, Heymans S. Myocardial extracellular matrix: an ever-changing and diverse entity. Circ. Res. 2014;114(5):872–888. doi: 10.1161/CIRCRESAHA.114.302533. [DOI] [PubMed] [Google Scholar]