

Abstract
Vaccinia virus (vv), a member of the poxvirus family, is unique among most DNA viruses in that its replication occurs in the cytoplasm of the infected host cell. Although this viral process is known to occur in distinct cytoplasmic sites, little is known about its organization and in particular its relation with cellular membranes. The present study shows by electron microscopy (EM) that soon after initial vv DNA synthesis at 2 h postinfection, the sites become entirely surrounded by membranes of the endoplasmic reticulum (ER). Complete wrapping requires ∼45 min and persists until virion assembly is initiated at 6 h postinfection, and the ER dissociates from the replication sites. [3H]Thymidine incorporation at different infection times shows that efficient vv DNA synthesis coincides with complete ER wrapping, suggesting that the ER facilitates viral replication. Proteins known to be associated with the nuclear envelope in interphase cells are not targeted to these DNA-surrounding ER membranes, ruling out a role for these molecules in the wrapping process. By random green fluorescent protein-tagging of vv early genes of unknown function with a putative transmembrane domain, a novel vv protein, the gene product of E8R, was identified that is targeted to the ER around the DNA sites. Antibodies raised against this vv early membrane protein showed, by immunofluorescence microscopy, a characteristic ring-like pattern around the replication site. By electron microscopy quantitation the protein concentrated in the ER surrounding the DNA site and was preferentially targeted to membrane facing the inside of this site. These combined data are discussed in relation to nuclear envelope assembly/disassembly as it occurs during the cell cycle.
INTRODUCTION
Vaccinia virus (vv), the prototype member of the Poxviridae, is a large DNA virus with a double-stranded DNA genome encoding for >200 proteins. Poxviruses are unique among most DNA viruses in that DNA replication occurs in the cytoplasm, independent of the nucleus of the infected host cell. Accordingly, its genome encodes for factors required for both cytoplasmic transcription as well as DNA replication (Moss, 1990a).
The vv cytoplasmic life cycle is initiated upon virus entry at the plasma membrane in a poorly understood process (Krijnse Locker et al., 2000), but which results in the delivery of the viral core, containing >100 proteins, into the cytoplasm. Within 20 min of infection this core, in which the enzymes required for the process of viral early transcription are packaged during virion assembly, produces a defined set of so-called early mRNAs in which about half of the genome is transcribed (Moss, 1990b). The early vv genes encode for proteins required for the process of vv-driven cytoplasmic DNA replication that begins at ∼2 h postinfection. DNA replication initiates the transcription of late genes, which code for late proteins that are necessary for the assembly of new virions, starting from ∼6 h postinfection. Virion assembly is complex and involves the acquisition of a double-membraned cisterna derived from the smooth endoplasmic reticulum (ER) around the core to form the first infectious form of the virus, the intracellular mature virus (IMV; Sodeik et al., 1993). This latter viral form is fully infectious and stays mostly intracellular. A small percentage of the IMVs become enwrapped by a double membrane cisterna of the trans-Golgi network to form the intracellular enveloped virus (Schmelz et al., 1994). This form, like some intracellular bacteria, is capable of polymerizing actin tails (Cudmore et al., 1995), which facilitate the release of the extracellular enveloped virus into the extracellular space.
vv DNA replication may occur in discrete cytoplasmic structures called virosomes or factories (Cairns, 1960; Kit et al., 1963; Beaud and Beaud, 1997). Early electron microscopy (EM) observations showed these sites to contain dense filamentous material, resembling nuclear chromosomes (Dales and Siminovitch, 1961; Dales, 1963). The latter studies also concluded that the viral factory region was not bounded by any membrane, but appeared to lie “free” in the cytoplasm. vv Factories are also the site of viral intermediate and late transcription (Dahl and Kates, 1970), as well as the site where early vv proteins accumulate (Kovacs and Moss, 1996; Beaud and Beaud, 1997; Domi and Beaud, 2000). The kinetics of vv DNA replication was studied in detail by Joklik and Becker (1964). Infection of HeLa cells resulted in a cessation of cellular DNA replication, which appeared to be independent of viral early protein or DNA synthesis, because it was also seen in cells infected with UV-inactivated virus (Joklik and Becker, 1964). At the multiplicity of infection used in that study, cytoplasmic vv DNA synthesis was found to start as early as 90 min postinfection and then peaked at 2 h 30 min, before decreasing, such that at the time when virion assembly could be expected to start, DNA synthesis had ceased completely.
Cellular transcription as well as DNA replication occurs in the cell nucleus. This cellular compartment contains a number of subcompartments with distinct structures and functions (Lamond and Earnshaw, 1998; Misteli, 2000). One of these subcompartments is the nuclear envelope (NE) that is physically connected to the rough ER. In interphase cells the inner nuclear membrane (INM) facing the intranuclear space is distinct from the rest of the ER. It harbors a set of INM-specific membrane proteins that interact with a specialized structure underlying the INM composed of lamins, DNA-binding proteins, and heterochromatin (reviewed in Gant and Wilson, 1997; Wilson, 2000). One of the most dramatic features of the NE in mammalian cells is that it breaks down at the onset of mitosis and reassembles at late anaphase/telophase. NE disassembly is known to be regulated by phosphorylation of lamins as well as certain INM membrane proteins, whereas dephosphorylation may accompany NE reassembly (Gerace and Blobel, 1980; Peter et al., 1990, 1991; Pfaller et al., 1991; Nigg, 1992; Foisner and Gerace, 1993). NE reassembly around DNA of different sources has been successfully reconstituted in a number of in vitro systems (Wilson and Wiese, 1996; Gant and Wilson, 1997). Such studies have clearly established that efficient DNA replication not only needs factors required for chromatin organization and DNA synthesis but also requires intact nuclear membranes, functional nuclear pore complexes (NPCs), as well as nuclear lamina (Gant and Wilson, 1997, and references therein).
In the present study we show that vv DNA replication shows several interesting analogies to cellular DNA replication; soon after initial vv DNA synthesis, the sites become entirely enwrapped by membranes of the ER. We provide evidence that this wrapping is required for efficient vv DNA replication. Strikingly, at the onset of virion assembly, DNA synthesis ceases, coinciding with a dispersal of the ER from the viral DNA sites. Finally, we identify a novel early vv membrane protein that is targeted to the vv replication sites.
MATERIALS AND METHODS
Cells, Virus, and Antibodies
HeLa cells C3 obtained from American Type Culture Collection (Rockville, MD) were grown essentially as described (Sodeik et al., 1993). The WR strain of vv was grown, semipurified, and plaque titrated as described in Pedersen et al. (2000). The following antibodies were used throughout this study. Antibodies to p35 raised against the first 15 amino acids of the H5R gene were made as described in Cudmore et al. (1996). Affinity-purified antibodies to green fluorescent protein (GFP) were kindly provided by Nathalie LeBot (European Molecular Biology Laboratory, Heidelberg, Germany), and the rat monoclonal antibodies to bromodeoxyuridine (BrdU) (MAS 250) were from Harlan Seralab (Belton, United Kingdom). Antibodies to lamin B1 and lamin B receptor were provided by Spyros Georgatos (University of Crete, Crete, Greece) monoclonal antibodies to Nup153 (SA1) and to nucleoporin (QE5) were kindly given by Brian Burke (University of Calgary, Calgary, Alberta) (Pante et al., 1994; Bodoor et al., 1999); and the monoclonal antibody RL2 to O-linked N-acetylglucosamine was a gift from Larry Gerace (Scripps Research Institute, La Jolla, CA) (Snow et al., 1987). A peptide corresponding to amino acid residue 7–21 (extreme N terminus) of the E8R sequence was synthesized and an antibody raised to this peptide according to the instructions of the manufacturer (Neolab, Strasbourg, France). Sera were affinity purified with the use of the corresponding peptide. Anti-rabbit coupled to fluorescein isothiocyanate was from Jackson Immunoresearch Laboratories (Dianova, Hamburg, Germany). GFP-tagged constructs of lamin B receptor, emerin, and nurim were kindly provided by Jan Ellenberg (European Molecular Biology Laboratory, Heidelberg, Germany), (Ellenberg et al., 1997; Ötlund et al., 1999; Rolls et al., 1999).
EM and Immunofluorescence
HeLa cells were grown overnight in 6-cm2 dishes to form a 70% confluent monolayer. Cells were infected at a multiplicity of infection of 50 for 30 min at 37°C. Cells were fixed and prepared for cryosectioning or Epon embedding as described (Ericsson et al., 1995; van der Meer et al., 1999). Indirect immunofluorescence was essentially as described (Den Boon et al., 1991). Transfection of GFP-tagged NE proteins was according to Krijnse Locker et al. (2000). Transfected HeLa cells were infected 24 h posttransfection, fixed at 3 h postinfection, and the viral DNA sites visualized with the use of Hoechst #33342 at 0.5 mg/ml (Sigma, St. Louis, MO). For the expression of GFP-tagged vv proteins, cells were infected and subsequently lipofected with the use of lipofectin (Life Technologies, Gaithersburg, MD) as described (Krijnse Locker et al., 1996).
Estimation of Fraction vv Factories Surrounded by ER and Quantitation of E8R Labeling
The percentage of ER wrapping was estimated by a standard stereological approach with the use of a lattice of orthogonal test lines (with a distance between the lines of 2 cm, which corresponds to 0.64 μm at the final magnification of 8000×; Griffiths, 1993). The fraction of the surface of the factory surrounded by ER was estimated by counting the number of intersections between the test lines and 1) the whole factory (as determined by p35 labeling on cryosections) and 2) the ER-enclosing parts. The percentage of the factory surface surrounded by ER is given by the ratio of the number of intersections over ER, divided by the total intersections times 100. This analysis was carried out on 15 micrographs from two different randomly sectioned blocks. To estimate the mean profile area of the factories, a series of test points was placed randomly on thin sections with the use of the same lattice grid as described above (a point is defined as a position where two orthogonal lines intersect). The average area of the profile is given by the number of points times the distance (d) between the points (given in μm2). For the quantitation of the E8R labeling in the ER, 18 random cell profiles of cryosections prepared from cells infected for 3 h and labeled with anti-E8R were photographed at a magnification of 8000×. Each micrograph was overlayed with an overhead sheet and the ER (either associated or not associated with a viral replication site), including 50 nm on both sides of the ER membrane, was marked. The distance of 50 nm from both sides of the membrane was included in the quantitation because the N-terminal epitope to which the antibody is raised was clearly cytosolically exposed and because of the predicted sequence of the E8R protein, this epitope can be expected to be located some distance away from the membrane. The overhead sheet was then overlayed with the lattice grid and the number of gold as well as the number of points (see above) in the marked area counted. Quantitation of gold per surface area (the surface of the marked area that included the ER membranes and the 50-nm distance on both sides of the membrane) was estimated according to Griffiths (1993).
Metabolic Labeling, Drug Treatment, TX-114 Extraction, and Western Blots
Monolayers of HeLa cells grown in 3.5-cm dishes were infected or mock-infected as described above. At the indicated times the culture medium was replaced with medium containing 4 μCi of [3H]thymidine (Amersham Pharmacia Biotech, Indianapolis, IN) per milliliter. Cells were metabolically labeled for 30 min at 37°C, washed three times with ice-cold phosphate-buffered saline, scraped from the dish in phosphate-buffered saline, and collected by centrifugation. The cell pellet was resuspended in 0.1 ml of HKM-buffer (50 mM HEPES-KOH, pH 7.4, 10 mM KCl, 2.5 mM MgCl2) containing protease inhibitors (aprotinin, leupeptin, pepstatin, and tosyl-phenylalanyl-chloromethyl ketone; all from Sigma) and left for 7 min on ice. The cells were broken by 23 strokes of a 25-gauge needle. The protein content was measured with the use of a Bio-Rad protein assay, equal amounts of OD595 units contained in each sample were trichloroacetic acid (TCA) precipitated and the amount of radioactivity determined by liquid scintillation counting. When [3H]thymidine incorporation was determined in ionomycin and/or N,N′-[1,2-ethanediylbis(oxy-2,1-phenylene)] bis[N-[2-[(acetyloxy)methoxy]-2-oxoethyl]] bis[(acetyloxy)methyl]ester- (BAPTA-AM) (both from Calbiochem, San Diego, CA) treated cells, the drug was added at a concentration of 5 μM, 10 min before metabolic labeling and kept on the cells throughout the labeling period. TX-114 extraction of cell lysates was essentially as described (Jensen et al., 1996). The proteins were detected by Western blots and enhanced chemiluminescence (Amersham Pharmacia Biotech) with the use of the indicated antibodies.
Cloning of vv E8R and A18R Genes and GFP Tagging
The vv E8R and A18R genes were amplified by polymerase chain reaction (PCR) with the use of appropriate primers, containing not I and HindIII restriction sites at the 5′ and 3′ ends, respectively. Purified PCR fragments were cloned in the pELGFP vector, generously provided by Michael Way (European Molecular Biology Laboratory, Heidelberg, Germany) (Frischknecht et al., 1999), cut with the same enzymes. Both constructs were verified by sequencing.
RESULTS
Gene Product of H5R, p35, Colocalizes with Vaccinia Virus DNA Replication Sites
The vv gene product H5R encodes for an early/late 22-kDa protein that by SDS-PAGE migrates at 35 kDa and will therefore be referred to as p35. Its precise function in the vv life cycle is not completely understood, although it has been proposed to bind single-stranded DNA in vitro (Beaud et al., 1994, 1995) and to be a vv intermediate/late transcription factor (Kovacs and Moss, 1996; Black et al., 1998). The protein is modified by phosphorylation, most likely mediated by a viral early kinase, the gene product of B1R (Beaud et al., 1995). Moreover, several studies have suggested that the protein associates with the sites of newly synthesized vv DNA (Kovacs and Moss, 1996; Beaud and Beaud, 1997; Domi and Beaud, 2000).
When infected cells were labeled with an antibody to p35, the protein appeared to localize to several distinct cytoplasmic spots and to colocalize with the Hoechst-positive DNA replication sites by immunofluorescence microscopy (IF; Figure 1, A and B). No such labeling was observed in uninfected cells (Figure 1C) or in cells infected in the presence of the DNA replication inhibitor hydroxyurea (our unpublished results). When infected cells were fixed at different times after infection the first p35-positive DNA sites were seen at 2 h postinfection at the multiplicity of infection used (our unpublished results), consistent with other studies showing that vv DNA replication may start around this time in HeLa cells (Joklik and Becker, 1964). With the use of p35 as a marker for the vv DNA replication sites we next examined them by EM.
Figure 1.

p35 colocalizes with the viral replication sites in the cytoplasm of infected cells. Monolayers of HeLa cells were infected (A and B) or mock infected (C) and fixed at 3 h postinfection. A is labeled with Hoechst at 0.5 mg/ml, whereas B and C are labeled with anti-p35 and donkey anti-rabbit-fluorescein isothiocyanate. Infected cells show prominent p35- (B) and Hoechst (A)-positive spots in the cytoplasm that entirely colocalize. Such spots are not present in uninfected cells in C. Bar, 5 μm.
Soon after Synthesis vv DNA Factories Become Enwrapped by Membranes of ER
When cells were fixed at 2 h postinfection and observed by EM, the p35-positive sites appeared as small electron-dense aggregates, apparently free in the cytoplasm. However, even at this early time of infection, ER membranes were observed close by (Figure 2A). Strikingly, when cells were fixed and observed 1 h later (3 h postinfection) the p35-positive sites were now almost completely surrounded by membranes typical of the ER (Figure 2B). Indeed, when sections were double-labeled with p35 and anti-protein disulfide isomerase, the surrounding membranes strongly labeled for the latter antigen, confirming their ER origin (our unpublished results; see also below). This ER wrapping was not seen by IF; because at this level the ER is distributed throughout the cytoplasm, the resolution of IF was not sufficient to observe an accumulation of ER markers around the p35-positive spots. That the p35 labeling was a bona fide marker of the vv replication sites was also confirmed by EM, by double-labeling with anti-p35 and either anti-DNA or anti-BrdU of cells incubated for short periods with BrdU, showing complete colocalization of the protein with the sites of cytoplasmic DNA accumulation (our unpublished results).
Figure 2.

Soon after initial DNA synthesis the sites become enwrapped by membranes typical of the ER. (A) Cryosections were fixed at 2 h postinfection and labeled with anti-p35. The labeling appears associated with a fine electron-dense structure apparently lying “free” in the cytoplasm. The arrowheads indicate putative ER membranes that appear in proximity to the p35-positive site. (B) Cryosections fixed at 3 h postinfection showing that the entire p35-positive area is now surrounded by a membrane typical of the ER (large arrowheads). The small arrow indicates a gap in the surrounding membrane. Bars, 200 nm.
The ultrastructure of the replication sites was subsequently analyzed in more detail with the use of sections of plastic-embedded infected and fixed cells. Although Epon embedding can usually not be combined with antibody labeling, the characteristic morphology of the vv factories made it possible to recognize them without any antibody marker. Figure 3, A and B, show low-magnification views of the vv replication sites at 2 h 45 min and their relation to other organelles. Although the ER often appeared to surround the DNA site completely, we also observed apparent gaps in the surrounding membrane (Figure 3B). These gaps presumably allowed exchange of molecules between the inner part of the DNA replication sites and the rest of the cytoplasm. Another striking feature of the replication sites was their close association with mitochondria; in some images these organelles virtually appeared to surround the viral DNA sites (Figure 3, A and C).
Figure 3.

Ultrastructure of the viral replication sites in Epon-embedded cells. (A and B) Cells infected and fixed at 2 h 45 min, postinfection. The stars indicate the factory region. Note in A the close association of a mitochondrium (M) with the ER membrane surrounding the factory site; B shows two DNA replication sites, one of which shows gaps in the surrounding membrane (large arrowheads); and C shows the factory region (large star in the center) at 6 h postinfection. The ER (small arrowheads) does not completely surround the factory region anymore. At this time of infection the DNA replication site also contains IVs (indicated), the precursors of the first infectious form of vv, the IMV. Note the close association of mitochondria with the factory region. G, Golgi complex; Nu, nucleus. Bars, 200 nm.
Figure 4 shows high-magnification views of the replication sites, in which we especially wanted to compare their ultrastructure to the nucleus and the nuclear envelope, known to enclose cellular DNA. The central part of the replication site showed a different electron density than the surrounding cytoplasm and resembled instead the inside of the nucleus (Figure 4, A–C). The ER around the factories looked mostly normal and not grossly modified by viral proteins, very much unlike the membranes that assemble from the smooth ER around the IMV during viral assembly late in infection (Sodeik et al., 1993). Although the INM was clearly decorated by an electron-dense meshwork adjacent to the membrane (presumably composed of heterochromatin and/or lamins), such electron-dense material appeared absent from the inner ER membrane around the vv replication sites (Figure 4, B and C). No evidence for pores typical of the NE in the ER membrane around the DNA site was seen (see below). In analogy to the NE, in most factories ribosomes were found predominantly on the side of the ER facing the cytoplasm. On closer inspection, however, we found that the ribosome distribution was different when the ER membrane completely or incompletely surrounded the replication site. On completely ER-enclosed factory profiles the ribosomes were predominantly located on the cytosolic side, whereas on incompletely enclosed sites, in which the ER membranes were apparently in the process of assembly, this segregation was much less evident (compare Figure 4, B and C, to 4A).
Figure 4.

High-magnification views of the DNA replication sites at 3 h postinfection in Epon-embedded infected HeLa cells. In these examples the fixed cells were treated with 1% tannic acid before Epon embedding to better reveal the ribosomes. The stars indicate the factory region. In B note the resemblance of the electron-dense structure of the viral factory to the interior of the nucleus. In A an incompletely closed ER membrane surrounds the factory region. The small arrows indicate ribosomes that appear present on both the inner and outer side of the ER membrane. In B and C the ER membrane completely encloses the factory region and the ribosomes (small arrows) are mostly on the side of the ER membrane facing the cytoplasm. Large arrowheads in B indicate rough ER studded with ribosomes on both sides of the membrane in the vicinity of the DNA replication site. Small arrowheads in B and C point to the electron-dense meshwork adjacent to the INM. Nu, nucleus. Bars, 200 nm.
To document this point further, ribosomes were counted on the inner and outer ER membrane of factories either completely or incompletely surrounded by the ER (Table 1). Such counts revealed that when the DNA site was completely ER enclosed 92% of the ribosomes were on the outer membrane, whereas when the ER membranes were still partly open, this number was only 64%.
Table 1.
Percentage of ribosomes on rER surrounding DNA-replication sites at 2h30 post-infection facing the cytoplasm
Closed ER: DNA-replication sites entirely surrounded by membranes of the rough ER.
Open ER: DNA-replication sites in which the surrounding ER is not (yet) surrounding the DNA-site completely.
For each DNA-replication site the amount of ribosomes on the ER facing the cytoplasm and facing the interior of the site were counted and the percentage of ribosomes on the outside (facing the cytoplasm) determined. The values represent the average percentage and the standard deviation. N=16.
These results suggest, as for the INM, that the inner ER membrane surrounding the vv DNA sites may be different from the outer membrane. Moreover, this putative segregation may occur gradually as the ER encloses the DNA replication site.
Complete ER Wrapping Takes ∼45 min, but at Onset of Virion Assembly the ER Disassembles Again
Next, a detailed time course of ER wrapping around the vv replication sites was conducted. Infected cells were fixed for EM from 2 to 3 h postinfection with 15-min intervals, as well as at 4 and 6 h postinfection. Sections were labeled with anti-p35 to unequivocally identify the replication sites. The extent of ER wrapping was subsequently quantified stereologically (in MATERIALS AND METHODS). The data are shown in Figure 5A; they demonstrate that at 2 h only 30% of the factory area was surrounded by ER membrane but this number amounted to 85% at 2 h 45 min and 3 h, postinfection. At these infection times the ER completely surrounded the DNA site in most profiles but in some images this was no so, resulting in an average ER wrapping of 85%. For simplicity, this percentage (85%) will subsequently be referred to as complete wrapping.
Figure 5.

Extent of ER wrapping coincides with the extent of viral DNA replication and factory growth. In A ER wrapping around the DNA replication sites was determined as described in MATERIALS AND METHODS for 15 randomly selected replication sites as detected by anti-p35 labeling on cryosections. The percentage of wrapping was determined for each site. The values represent the average and SDs of percentage of ER wrapped around 15 DNA sites at each time point tested. In B the extent of DNA replication during the course of infection. Infected (▪) or uninfected (░⃞) HeLa cells were labeled for 30 min with [3H]thymidine at different times of infection (indicated). At the end of the labeling period the cells were lysed and the protein content of each sample was determined with the use of a Bio-Rad protein assay. The amount of TCA-precipitable counts contained in 10 OD595 units was determined by liquid scintillation counting. In C the average size of the factories during the course of infection is represented. From the values obtained in A the average size of the factory region was calculated for 15 factories per time point. The values represent the average size (surface) and SDs of 15 factories for each time point tested. Note that complete (85%) ER wrapping coincides with a peak in viral DNA synthesis as well as an increase in the average size of the factory region. The decrease in factory size at 6 h postinfection is probably the result of the fact that at that time of infection the factory region is more difficult to define. This is because the p35-positive site is more dispersed and not bounded by ER membrane anymore.
These quantitative data revealed two interesting aspects. First, complete wrapping was obtained at 2 h 45 min, postinfection, implying that this process required ∼45 min from the time DNA synthesis was first detected by IF and EM. Second, as long as the wrapping process was not completed the factories did not seem to grow in size. However, from 2 h 45 min, postinfection, onward, when wrapping had reached its maximum, the DNA replication sites grew significantly in size (Figure 5C). These results may suggest that ER wrapping is required for DNA replication or at least for factory expansion (see below).
The fate of the ER wrapped around the vv DNA sites was also analyzed at later times of infection. At 4 h postinfection the factories had grown to huge areas in infected cells that, like at 3 h of infection, were almost completely surrounded by ER membranes (80% wrapping; Figure 5A). At 6 h., however, the factory region had clearly changed its aspects. Immature virions (IVs), the precursors of the first infectious form of vv, the IMV, could now be observed in the factory region. Although ER cisternae were still found close by, this organelle had mostly dispersed away from the DNA replication sites (for an example, see Figure 3C). Indeed, quantification showed that only 39% of the factory region was surrounded by the ER at this time of infection (Figure 5A).
These combined data show that the viral DNA replication sites become entirely surrounded by membranes of the ER early in infection, resembling small cytoplasmic nuclei. Remarkably, however, this ER wrapping is reversed late in infection, when virion assembly starts (in DISCUSSION).
ER Wrapping Correlates with Efficient DNA Replication
Because the EM data strongly suggested that ER wrapping was required for factory growth and perhaps for efficient viral DNA replication, next a detailed time course of [3H]thymidine incorporation was performed to determine the extent of viral DNA synthesis over the course of infection. Infected (as well as uninfected control cells) cells were labeled with this radioactive compound for 30 min at different times of infection, starting from 1 h postinfection onward, before the onset of vv DNA replication (Figure 5B). When infected cells were labeled at 1 h postinfection (a time at which no vv DNA synthesis can be detected by IF or EM), the incorporation had decreased more than fivefold compared with uninfected cells, consistent with the notion that vv efficiently switches off host DNA replication (Joklik and Becker, 1964; Figure 5B). At a time point when DNA factories can first be detected by IF and EM, but before ER wrapping was completed (2 h 15 min-2 h 45 min, postinfection), [3H]thymidine incorporation had increased to levels similar to those in uninfected cells. Viral DNA synthesis peaked between 3 h 45 and 5 h 30 min, postinfection, times that coincide with complete enclosure of the DNA sites, to reach a level that was 5 times higher than that in uninfected cells. At 6 h postinfection, however, at the onset of virion assembly and ER disassembly, DNA synthesis had decreased dramatically (Figure 5, A and B).
Although these data provide no direct proof for the requirement of ER in vv DNA replication, they do show a strong correlation between complete ER wrapping and the extent of vv DNA synthesis as well as factory growth (Figure 5, A–C).
Disruption of ER Structure Results in a Significant Decrease in vv DNA Replication
To provide further evidence that ER wrapping was required for efficient vv replication we used drugs known to disrupt the ER structure and tested whether they could interfere with the ER wrapping around the viral replication sites. The calcium ionophore ionomycin has been shown to induce ER vesiculation while leaving the nuclear envelope intact (Subramanian and Meyer, 1997). Indeed, when infected cells were treated for 10 min with 5 μM ionomycin at 3 h postinfection, the NE remained intact while the ER appeared as large vacuoles and no wrapping around the replication sites was observed anymore, as assessed by EM (our unpublished results). Ionomycin treatment strongly inhibited [3H]thymidine incorporation in both infected and uninfected cells (Figure 6). Because we considered that this inhibition was caused by increased levels of cytoplasmic calcium released from the ER, the experiment was also conducted in the presence of both ionomycin and the membrane-permeable chelator BAPTA-AM. EM analysis showed that simultaneous addition of the ionophore and BAPTA-AM resulted in the same ER vesiculation (but leaving the NE intact) as seen in the presence of ionomycin alone (our unpublished results). Under these conditions cellular replication was now inhibited only twofold, whereas vv replication was inhibited by >90% compared with untreated infected cells (Figure 6). Although it cannot be excluded that ionomycin has some indirect effect on cellular as well as viral DNA synthesis, these data suggest that disrupting the ER wrapping around the viral factories severely inhibits vv DNA replication.
Figure 6.

Ionomycin treatment severely affects DNA replication. Infected or uninfected HeLa cells (indicated) were treated for 10 min with 5 μm ionomycin (grey shaded bar), ionomycin and BAPTA-AM (dotted bar), or not treated (solid black bar) at 3 h postinfection. Cells were then metabolically labeled with [3H]thymidine for 30 min in the presence or absence of the drugs. The cells were lysed and equal amounts of OD595 units determined by a Bio-Rad protein assay were TCA precipitated and the extent of [3H]thymidine incorporation determined by liquid scintillation counting. The values obtained from untreated control samples were expressed as 100%. The values of the treated samples are expressed as the percentage of [3H]thymidine incorporation relative to the untreated control. All values show the averages and SDs of duplicate samples.
Membrane Proteins or Membrane-associated Proteins of Inner NE May Be Absent from Viral Factories
A possibility that could account for the observed wrapping of the ER around the vv replication sites was that vv infection leads to (aberrant) targeting of INM proteins to these ER membranes, where these proteins might interact with the viral DNA or DNA-associated proteins. To test this possibility proteins of the NE were either visualized by transient transfection of GFP-tagged proteins or by antibody labeling. Their IF pattern in uninfected control cells was compared with cells infected for 3 h with vv in which the replication sites were visualized with Hoechst. Table 2 lists the GFP-tagged proteins and antibodies that were used. All five GFP-tagged proteins tested behaved identical; upon vv infection the proteins remained entirely associated with the NE with no hint of labeling around the viral factory region. These results were confirmed with antibodies to endogenous LBR and lamin B1. Proteins known to be associated with the NPC also appeared to be absent from the replication sites; none of the antibodies tested decorated the viral factory region. In addition, a monoclonal antibody recognizing cytoplasmic O-linked N-acetylglucosamine, a modification typical of NPC proteins (Snow et al., 1987), failed to label the DNA sites although it gave a typical nuclear pore pattern (Table 2). These data suggest that NE proteins are not involved in the ER wrapping process around the viral DNA.
Table 2.
Overview of the NE associated proteins tested in this study
| Protein | Detectiona | Component ofb | Reference |
|---|---|---|---|
| Lamin B receptor | T/A | NE/MP | (Ellenberg et al., 1997) |
| Nurim | T | NE/MP | (Rolls et al., 1999) |
| Emerin | T | NE/MP | (Östlund et al., 1999) |
| Lamin B1 | T/A | NE/MA | |
| LAP 2β | T | NE/MP | |
| O-GlcNac/RL2c | A | NPC | (Snow et al., 1987) |
| Nup 153/SA1 | A | NPC | (Bodoor et al., 1999) |
| Nucleoporin/QE5d | A | NPC | (Pante et al., 1994) |
The proteins were detected by transient transfection (T) of GFP-tagged proteins and/or by antibody (A) labeling.
The proteins were classified as member of the nuclear envelope (NE) or nuclear pore complex (NPC) and as membrane protein (MP) or membrane-associated protein (MA).
O-GlcNac/RL2: monoclonal antibody recognizing O-linked N-acetylglucosamine.
Nucleoporin: monoclonal antibody QE5 nucleoporin, recognizing p62, Nup 153, Nup 214 and Nup 358.
vv E8R Gene Is a Candidate Membrane Protein Involved in ER Wrapping
Because the above-mentioned experiments suggested that the process of ER wrapping was not mediated by typical INM-associated proteins, we sought to find candidate vv membrane or membrane-associated proteins involved in this process. For this, the entire vv genome was inspected for genes of unknown function fulfilling two criteria: the presence of a putative transmembrane domain with the use of the “dense alignment surface” method (Cserzo et al., 1994) and the PHDhtm program (our unpublished results; Rost et al., 1995) and preceeded by a sequence typical for an early vv promotor (Moss, 1990a). Several genes fulfilled both criteria and the corresponding DNAs were subsequently amplified by PCR from vv DNA. They were cloned into a pELGFP vector, which resulted in N-terminal GFP-tagging of the gene, whereas their expression was driven by a vv early/late (7.5 kDa) promotor (Frischknecht et al., 1999). The proteins could thus be expressed in the background of vv infection and because expression was driven by a viral early/late promotor, the GFP-tagged proteins could be detected by IF as early as 3 h postinfection.
An IF screen of the expressed proteins proved to be unsatisfactory, however, because most of the tagged proteins localized uniformly to the ER, with no clear concentration around the DNA factory region (our unpublished results). Different results were obtained when the same proteins were localized by EM, because most of them now localized to specific subcellular locations. The entire screen will be published elsewhere (Krijnse Locker, Schleich, and Doglio, unpublished data). We believe that this discrepancy between the IF and EM results can be explained by the fact that the latter technique will detect the GFP-tagged protein at locations where it concentrates, whereas the IF signal is amplified to show the protein in all possible locations.
By EM two proteins were found to be localized to the DNA replication sites, the gene products of A18R and E8R (see below). The A18R gene encodes for a protein identified before as a putative transcription factor interacting with p35 (H5R; Black et al., 1998). The sequence predicts this protein to have a region of ∼40 residues at the C terminus that is moderately hydrophobic, but perhaps not hydrophobic enough to be a true membrane-spanning domain (Figure 7A). In contrast, the gene product of E8R, a protein of unknown function, predicted to have two transmembrane domains in the C-terminal half of the protein (Figure 7A). To test whether A18R and E8R were membrane proteins, lysates of infected and transfected cells were extracted with TX-114 to separate membrane from nonmembrane proteins (Bordier, 1981), and the proteins detected by Western blotting. As shown in Figure 7C, the GFP-tagged A18R did not partition into the detergent fraction, whereas E8R did, confirming the sequence prediction data.
Figure 7.

The gene product of E8R is a membrane protein. In A hydrophobicity plots of the gene products of E8R and A18R with the use of the dense alignment surface method. The dotted line on the graphs indicates the loose and the upper uninterrupted line the strict cut-off for the determination of a putative transmembrane region. The amino acid sequence of the E8R gene is depicted under its respective “dense alignment surface” plot. The putative transmembrane domains are underlined and the peptide sequence used to raise antibodies is indicated with a hatched line above the sequence. In B HeLa cells were infected (I) or mock infected (U) for 3 h. and postnuclear supernatants were prepared. Equal amounts of protein were loaded onto a 12.5% SDS-PAGE, blotted onto polyvinylidene difluoride membrane and the E8R protein detected by enhanced chemiluminescence with the use of the peptide antibody. On the right side the positions of the 34-, 38-, and 54-kDa marker proteins are indicated. In C infected and transfected cells were lysed at 6 h postinfection and the lysate subjected to extraction with TX-114. A is the aqueous phase and D the detergent phase. E8R- and A18R-GFP refers to detection of the transfected GFP-tagged proteins with the use of anti-GFP antibodies, whereas E8R represents the untagged vv protein detected by anti-E8R. The A14L and H5R gene products serve as a control for a membrane (Salmons et al., 1997) and a soluble protein, respectively, and were detected with antibodies raised against the respective proteins.
Gene Product of E8R Concentrates in ER Surrounding the Viral Replication Site
Subsequently, a peptide corresponding to the extreme N terminus of E8R was made and corresponding antibodies raised in rabbits. By Western blotting the antibody recognized a band in infected but not in uninfected cells that migrated around 30 kDa, consistent with the predicted molecular weight of E8R of 32 kDa (Figure 7B). By immunofluorescence microscopy the GFP-tagged protein showed a general ER pattern and some concentration around the DNA sites (our unpublished results). When cells infected for 3 h (coinciding with complete wrapping) were labeled with the antibody to E8R, however, we observed a striking ring-like pattern around the DNA site with little accumulation in any other part of the cell, including the ER or in the central part of the DNA site (Figure 8, A–C). Subsequently, the E8R protein was localized by EM on cryosections prepared from cells fixed at 3 h, or from infected/E8R-GFP-transfected cells fixed at 4 h postinfection. Sections were then labeled with either affinity-purified anti-E8R or with anti-GFP. E8R localized as expected of a membrane protein because it was detected in the NE, the ER, and in the ER around the DNA factories. No significant labeling was seen on other organelles, such as the Golgi, endosomes, or the plasma membrane (Figure 9C; our unpublished results), suggesting that the protein behaved like an ER resident. The labeling for both anti-GFP and anti-E8R was found predominantly on the cytosolic side of the membranes, suggesting that the N-terminal part of the protein was exposed in the cytoplasm (Figure 9, A–C; in DISCUSSION). Moreover, the labeling was always some distance away from the membranes, consistent with the possibility that the ∼120-amino-acid-long N terminus of the E8R gene, preceding the first putative transmembrane domain, may separate the extreme N terminus (to which either the antibody was increased or that was GFP-tagged) from the membrane. In the membranes surrounding the DNA sites the protein was clearly segregated, with more labeling in the ER membrane facing the inner side of the factory than the one facing the cytosol. Quantitation of this distribution on randomly photographed factories showed that 72% of the labeling for both anti-GFP and for anti-E8R (detecting the GFP-tagged and the wild-type vv protein) was associated with the inner ER membrane (our unpublished results). Although on the replication site most of the labeling was associated with the ER, labeling was also occasionally observed in the central part of the factory region (Figure 9B). The nature of this labeling was not further investigated, but may reflect the presence of membranes located in the inner part of the DNA site.
Figure 8.
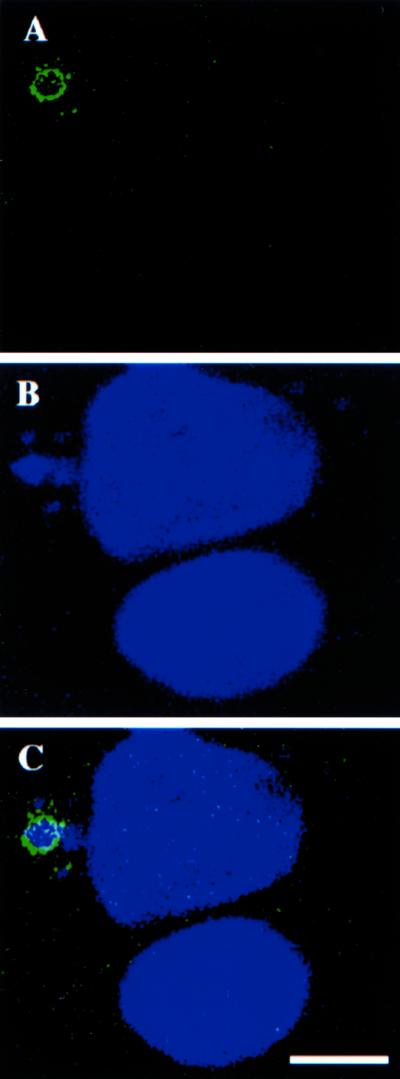
Localization of E8R by immunofluorescence microscopy. HeLa cells grown on coverslips were infected and fixed at 3 h postinfection. Cells were double-labeled with anti-E8R antibody (A) and Hoechst (B). C shows the merged image. The image shows two cells and only the upper cell is infected. In this cell the E8R antibody labels around the Hoechst-positive replication site. This same cell has also several small DNA-sites that are not surrounded by E8R labeling. Bar, 2.5 μm.
Figure 9.

EM localization of E8R. HeLa cells were infected with vv and fixed at 3 h postinfection (B), or infected and lipofected with E8R-GFP and fixed at 4 h postinfection (A and C). Sections were labeled with affinity purified anti-E8R (B) or anti-GFP (A and C; anti-GFP or anti-E8R labeling is indicated with small arrowheads). In all three images the factory region is indicated with a star. In A labeling is observed on the inner ER membrane and in ER membranes in continuity with the membranes enclosing the factory region. In B cells were infected for 3 h and labeled with anti-E8R. The image shows one entire replication site and part of a second site in the top right of the picture. Labeling is found on the ER membrane enclosing the DNA site as well as on the inside of the factory region (see text). C shows a part of a factory region of which the inner membrane is labeled. The image also shows an unlabeled Golgi stack (G) as well as the NE. The large arrowhead indicates the electron-dense structure underlying the INM. Nu, nucleus. Bars, 200 nm.
The above-mentioned data suggested that the E8R gene product might play a role in the ER-wrapping process. If so, we expected the protein to concentrate in the ER around the replication site as the immunofluorescence data suggested. Therefore, E8R labeling associated with the ER that was either surrounding the DNA site or not was counted and compared (in MATERIALS AND METHODS). Table 3 shows that the gold labeling in the ER surrounding the viral replication sites compared with the rest of the ER was indeed more than twice as high. Collectively, these data show that the viral membrane protein, the gene product of E8R, is targeted to the ER around the factory region.
Table 3.
The gene product of E8R is concentrated in the ER surrounding the factory site
| Au/μm2 ER not associated with a DNA sitea | 2.29 ± 0.57 |
| Au/μm2 of ER around the DNA site | 4.78 ± 0.87 |
The values are the averages and the standard deviations of the means of 18 randomly photographed cell-profiles of cryosections prepared from cells infected for 3 h and labeled with the anti-E8R antibody.
Gold per μm2 (corresponding to the surface of the ER membrane, including a distance of 50 nm on both sides of the membrane; see MATERIALS AND METHODS) was estimated according to Griffiths, 1993.
DISCUSSION
ER Wrapping and vv DNA Replication
In the present study we show that the sites of vv cytoplasmic DNA synthesis, soon after initial replication, become entirely enwrapped by membranes of the ER. Several lines of evidence show that this wrapping may be required for efficient vv DNA replication. First, incompletely wrapped factories early in infection did not grow in size, whereas soon after completion of the wrapping process, the factories started to significantly expand. Second, we were able to show that complete wrapping coincided with a peak in viral DNA synthesis. Most interestingly, when assembly started DNA replication rapidly declined, a process that was accompanied by the disassembly of the ER around the factory region. Finally, when ER wrapping was disrupted with the use of ionomycin, vv DNA replication was significantly decreased. In summary, our data strongly suggest a role for the ER in vv DNA replication.
Analogies to NE Dynamics during Cell Cycle
Our data point to some intriguing analogies to at least two events occurring during M-phase of the cell cycle. Early in infection small DNA replication units became surrounded by membranes of the ER in analogy to NE assembly in late anaphase/telophase. Late in infection, however, when virion assembly started, this ER underwent disassembly, resembling the process of prophase. Our extensive EM observations showed that the ER disassembly always coincided with the formation of viral precursors of the virion assembly process, the immature viruses (IVs). In the absence of such precursors, the ER entirely surrounded the replication site, whereas when IVs were detected in and around the factory region the ER had disassembled. Thus, virion formation, known to be driven by vv late proteins, coincided with ER disassembly, strongly suggesting that the switch from “early” to “late” vv protein synthesis regulated ER dispersal. As discussed in INTRODUCTION, entry into and exit from mitosis is closely linked to phosphorylation/dephosphorylation of lamins as well as NE membrane proteins. Interestingly, the viral genome encodes for two early kinases, one late kinase, and one early/late phosphatase (Howard and Smith, 1989; Guan et al., 1991; Banham and Smith, 1992; Lin et al., 1992; Banham et al., 1993; Lin and Broyles, 1994). Whether a similar phosphorylation/dephosphorylation cycle regulates the ER assembly/disassembly or whether these enzymes, directly or indirectly, play a role in this process remains to be tested.
Which Proteins Drive ER Assembly? A Possible Role for E8R Gene
An intriguing question is which proteins drive the wrapping event. A possibility is that the ER (and/or its associated proteins) has an intrinsic property to wrap around DNA, if present in the cytoplasm. In support of this idea was the fact that the ER membrane did not look grossly modified by viral proteins and our unpublished results show that ER resident proteins such protein disulfide isomerase and calnexin are not excluded from this domain. This is very much in contrast to the formation of viral membrane precursors made during virion assembly, which are derived from the smooth ER and that wrap around the vv core. These typical crescent-shaped membranes are highly modified by viral membrane proteins and exclude cellular proteins (Sodeik et al., 1993).
The collective data on the assembly of the NE at the end mitosis suggest that this is a highly regulated process, driven by specific interactions between INM proteins, chromatin and lamins. As a result, NE membrane proteins that may initially be mixed with ER proteins (Ellenberg et al. 1997; Yang et al.,1997) gradually segregate from the latter, most likely as a result of binding of NE integral proteins (and subsequent immobilization) to chromatin/lamins (Foisner and Gerace, 1993; Ellenberg et al., 1997). These studies strongly suggest that although NE membrane or membrane-associated proteins do have affinity for chromatin and/or lamins at late anaphase/telophase, resident ER proteins do not. Because the present study showed that none of the NE membrane and membrane-associated proteins that were tested appeared to be targeted to the viral replication sites, the ER-wrapping process could in principle be driven by two possible mechanisms. On viral infection host resident ER proteins could become modified (perhaps by phosphorylation or dephosphorylation), such that they are now able to specifically bind to (viral) DNA and/or its associated proteins. Alternatively, this process is predominantly driven by viral proteins; viral ER resident membrane proteins could play a role similar to INM proteins by binding to viral DNA and/or DNA-binding proteins and in this process would segregate to the ER membrane facing the inside of the factory region. Our results on the E8R gene product would be consistent with such a possibility. By EM the E8R gene had all the hallmarks of an ER resident protein, which was moreover concentrated in the ER surrounding the replication site. In addition, the protein localized preferentially to the inner of the two membranes facing the DNA and its associated proteins. Extensive two-dimensional gel analysis of metabolically labeled infected cells early in infection suggest the existance of >10 early proteins with hallmarks of membrane proteins (Krijnse Locker, unpublished observations). These proteins, however, will have to await identification and characterization to determine whether they play any role in the observed wrapping process. Finally, a combination of both scenarios can be envisioned as well, in which one or several viral membrane proteins would initiate DNA-binding, whereas ER host proteins would assist in subsequent wrapping and fusion to form an almost completely sealed envelope around the viral DNA (see below).
Several characteristics of the E8R sequence are interesting to note. Having two transmembrane domains the protein could potentially span the membrane twice, exposing the N and C terminus either cytosolic or luminal. Our EM data strongly suggested that the N-terminal part containing the GFP tag, or to which the peptide antibody was increased, was cytoplasmic. Thus, if the protein traverses the membrane twice, it could expose an ∼120-residue-long N-terminal part as well as 25 C-terminal amino acids into the cytoplasm that could potentially interact with the vv DNA and/or its associated proteins. The N-terminal part of the protein contains multiple serine and threonine residues (17 in total) that could be the target of phosphorylation and dephosphorylation events. Finally, the predicted pI of the E8R gene is extremely basic (pI = 9.3) as expected for a putative DNA-binding protein. The precise role of this protein in vv life cycle will be elucidated in a separate study (Doglio, Schleich, and Krijnse Locker, unpublished data).
A Model on ER Assembly/Disassembly around vv DNA
In summary, we propose that the observed wrapping and disassembly of the ER around the viral DNA site may occur in analogy to NE dynamics during the cell cycle, although it may not use the same molecules. Our current working model is that one or several ER-resident (viral and/or cellular) membrane proteins bind to the newly synthesized viral DNA and/or its associated proteins. As more of such membrane proteins bind, the ER membrane might become segregated, in analogy to the formation of the inner and outer NM, with the inner side of the ER containing proteins that mediate DNA binding. This process of segregation may not only be aided by membrane protein–DNA interactions but also perhaps by lateral interactions among membrane proteins within the segregating ER membrane. Evidence for segregation of the ER membrane around the factories was that ribosomes were mainly present on the outer ER membrane and that the E8R protein localized to the inner membrane, showing strong analogies to the inner and outer NM. As more ER membrane associates with the DNA sites, we speculate that these membranes will (homotypically) fuse to make a completely sealed envelope around the factory region. This fusion is most likely mediated by the cellular machinery required for ER homotypic fusion that is perhaps specifically recruited to the ER membranes surrounding the DNA site. It is interesting to note in this respect that the GTPase RAN, as well as its effector RCC-1, thought to be required for NE fusion in Xenopus extracts (Hetzer et al., 2000), are not targeted to the replication sites (Krijnse Locker, unpublished observations), suggesting no role for these proteins in this event. Another intriguing aspect of the viral-induced ER wrapping is that the process is entirely reversed 3 h later in the viral life cycle, when assembly of new virions starts. Apparently, the synthesis of a completely different set of late proteins, now promotes a fission process leading to the disassembly of the previously assembled ER. We believe that unraveling the molecular details of this ER assembly and disassembly process might not only shed light on vv DNA replication but also on the fundamental cellular process of mitosis.
ACKNOWLEDGMENTS
We thank the following people for antibodies and plasmids: Jan Ellenberg, Nathalie LeBot, Spyros Georgatos, Brian Burke, Larry Gerace, and Michael Way. Kai Simons, Martin Hetzer, Jan Ellenberg, and Gareth Griffiths are kindly acknowledged for critical reading of the manuscript. We also thank Anja Haberman for advice on the stereology, and Gareth Griffiths for support. Part of this work was supported by a European Union Training and Mobility Research and Biotech Grant.
REFERENCES
- Banham AH, Leader DP, Smith GL. Phosphorylation of ribosomal proteins by the vaccinia virus B1R protein kinase. FEBS Lett. 1993;321:27–31. doi: 10.1016/0014-5793(93)80614-z. [DOI] [PubMed] [Google Scholar]
- Banham AH, Smith GL. Vaccinia virus gene B1R encodes a 34-kDa serine/threonine protein kinase that localizes in cytoplasmic factories and is packaged into virions. Virology. 1992;191:803–812. doi: 10.1016/0042-6822(92)90256-o. [DOI] [PubMed] [Google Scholar]
- Beaud G, Beaud R. Preferential virosomal location of underphosphorylated H5R protein synthesized in vaccinia virus-infected cells. J Gen Virol. 1997;78:3297–3302. doi: 10.1099/0022-1317-78-12-3297. [DOI] [PubMed] [Google Scholar]
- Beaud G, Beaud R, Leader DP. Vaccinia virus gene H5R encodes a protein that is phosphorylated by the multisubstrate vaccinia virus B1R protein kinase. J Virol. 1995;69:1819–1826. doi: 10.1128/jvi.69.3.1819-1826.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaud G, Sharif A, Topa-Masse A, Leader DP. Ribosomal protein S2/Sa kinase purified from HeLa cells infected with vaccinia virus corresponds to the B1R kinase and phosphorylates in vitro the viral ssDNA-binding protein. J Gen Virol. 1994;75:283–293. doi: 10.1099/0022-1317-75-2-283. [DOI] [PubMed] [Google Scholar]
- Black EP, Moussatche N, Condit RC. Characterization of the interaction among vaccinia virus transcription factors G2R, A18R and H5R. Virology. 1998;245:313–322. doi: 10.1006/viro.1998.9166. [DOI] [PubMed] [Google Scholar]
- Bodoor K, Shaikh S, Salina D, Raharjo WH, Bastos R, Lohka M, Burke B. Sequential recruitment of NPC proteins to the nuclear periphery at the end of mitosis. J Cell Sci. 1999;112:2253–2264. doi: 10.1242/jcs.112.13.2253. [DOI] [PubMed] [Google Scholar]
- Bordier C. Phase separation of integral membrane proteins in Triton X-114 solution. J Biol Chem. 1981;256:1604–1607. [PubMed] [Google Scholar]
- Cairns HJF. The initiation of vaccinia infection. Virology. 1960;11:603–623. doi: 10.1016/0042-6822(60)90103-3. [DOI] [PubMed] [Google Scholar]
- Cserzo M, Bernassau JM, Simon I, Maigret B. New alignment strategy for transmembrane proteins. J Mol Biol. 1994;243:388–396. doi: 10.1006/jmbi.1994.1666. [DOI] [PubMed] [Google Scholar]
- Cudmore S, Blasco R, Vincentelli R, Esteban M, Sodeik B, Griffiths G, Krijnse Locker J. A vaccinia virus core protein, p39, is membrane associated. J Virol. 1996;70:6909–6921. doi: 10.1128/jvi.70.10.6909-6921.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cudmore S, Cossart P, Griffiths G, Way M. Actin-based motility of vaccinia virus. Nature. 1995;378:636–638. doi: 10.1038/378636a0. [DOI] [PubMed] [Google Scholar]
- Dahl R, Kates JR. Intracellular structures containing vaccinia DNA: isolation and characterization. Virology. 1970;42:453–462. doi: 10.1016/0042-6822(70)90288-6. [DOI] [PubMed] [Google Scholar]
- Dales S. The uptake and development of vaccinia virus in strain L cells followed with labeled viral deoxyribonucleic acid. J Cell Biol. 1963;18:51–72. doi: 10.1083/jcb.18.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dales S, Siminovitch L. The development of vaccinia virus in Earl's L strain cells as examined by electron microscopy. J Biophys Biochem Cytol. 1961;10:475–503. doi: 10.1083/jcb.10.4.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Den Boon JA, Snijder EJ, Krijnse Locker J, Horzinek MC, Rottier PJM. Another triple-spanning envelope protein among intracellular budding RNA viruses: the torovirus E protein. Virology. 1991;182:655–663. doi: 10.1016/0042-6822(91)90606-C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domi A, Beaud G. The punctate sites of accumulation of vaccinia virus early proteins are precursors of sites of viral DNA synthesis. J Gen Virol. 2000;81:1231–1235. doi: 10.1099/0022-1317-81-5-1231. [DOI] [PubMed] [Google Scholar]
- Ellenberg J, Siggia ED, Moreira JE, Smith CL, Presley JF, Worman HJ, Lippincott-Schwartz J. Nuclear membrane dynamics and reassembly in living cells: targeting of an inner nuclear membrane proteins in interphase and mitosis. J Cell Biol. 1997;138:1193–1206. doi: 10.1083/jcb.138.6.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericsson M, Cudmore S, Shuman S, Condit RC, Griffiths G, Krijnse Locker J. Characterization of ts 16, a temperature sensitive mutant of vaccinia virus. J Virol. 1995;69:7072–7086. doi: 10.1128/jvi.69.11.7072-7086.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foisner R, Gerace L. Integral membrane proteins of the nuclear envelope interact with lamins and chromosomes, and binding is modulated by mitotic phosphorylation. Cell. 1993;73:1267–1279. doi: 10.1016/0092-8674(93)90355-t. [DOI] [PubMed] [Google Scholar]
- Frischknecht F, Moreau V, Rottger S, Gonfloni S, Reckman I, Superti-Furga G, Way M. Actin-based motility of vaccinia virus mimics receptor tyrosine kinase signaling. Nature. 1999;401:926–929. doi: 10.1038/44860. [DOI] [PubMed] [Google Scholar]
- Gant TM, Wilson KL. Nuclear assembly. Annu Rev Cell Dev Biol. 1997;13:669–695. doi: 10.1146/annurev.cellbio.13.1.669. [DOI] [PubMed] [Google Scholar]
- Gerace L, Blobel G. The nuclear envelope lamina is reversibly depolymerized during mitosis. Cell. 1980;19:277–287. doi: 10.1016/0092-8674(80)90409-2. [DOI] [PubMed] [Google Scholar]
- Griffiths G. Fine Structure Immunocytochemistry. Heidelberg, Germany: Springer-Verlag; 1993. [Google Scholar]
- Guan KL, Broyles SS, Dixon JE. A Tyr/Ser protein phosphatase encoded by vaccinia virus. Nature. 1991;350:359–362. doi: 10.1038/350359a0. [DOI] [PubMed] [Google Scholar]
- Hetzer M, Bilbao-Cortes D, Walther TC, Gruss OJ, Mattaj I. GTP hydrolysis by Ran is required for nuclear envelope assembly. Mol Cell. 2000;5:1013–1024. doi: 10.1016/s1097-2765(00)80266-x. [DOI] [PubMed] [Google Scholar]
- Howard ST, Smith GL. Two early vaccinia virus genes encode polypeptides related to protein kinases. J Gen Virol. 1989;70:3187–3201. doi: 10.1099/0022-1317-70-12-3187. [DOI] [PubMed] [Google Scholar]
- Jensen ON, Houthaeve T, Shevchenko A, Cudmore S, Mann M, Griffiths G, Krijnse Locker J. Identification of the major membrane and core proteins of vaccinia virus by two-dimensional electrophoresis. J Virol. 1996;70:7485–7497. doi: 10.1128/jvi.70.11.7485-7497.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joklik WK, Becker Y. The replication and coating of vaccinia DNA. J Mol Biol. 1964;10:452–474. doi: 10.1016/s0022-2836(64)80066-8. [DOI] [PubMed] [Google Scholar]
- Kit S, Dubbs DR, Hsu TC. Biochemistry of vaccinia-infected mouse fibroblasts (strain L-M). III. Radioautographic and biochemical studies of thymidine-3H uptake into DNA of L-M cells and rabbit cells in primary culture. Virology. 1963;19:13–22. doi: 10.1016/0042-6822(63)90019-9. [DOI] [PubMed] [Google Scholar]
- Kovacs GR, Moss B. The vaccinia virus H5R gene encodes late gene transcription factor 4: purification, cloning and overexpression. J Virol. 1996;70:6796–6802. doi: 10.1128/jvi.70.10.6796-6802.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijnse Locker J, Kuehn A, Rutter G, Hohenberg H, Wepf R, Griffiths G. Vaccinia virus entry at the plasma membrane is signaling-dependent for the IMV but not the EEV. Mol Biol Cell. 2000;11:2497–2511. doi: 10.1091/mbc.11.7.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijnse Locker J, Schleich S, Rodriguez D, Goud B, Snijder EJ, Griffiths G. The role of a 21-kDa viral membrane protein in the assembly of vaccinia virus from the intermediate compartment. J Biol Chem. 1996;271:14950–14958. doi: 10.1074/jbc.271.25.14950. [DOI] [PubMed] [Google Scholar]
- Lamond AI, Earnshaw WC. Structure and function in the nucleus. Science. 1998;280:547–553. doi: 10.1126/science.280.5363.547. [DOI] [PubMed] [Google Scholar]
- Lin S, Broyles SS. Vaccinia protein kinase 2: a second essential serine/threonine protein kinase encoded by vaccinia virus. Proc Natl Acad Sci USA. 1994;91:7653–7657. doi: 10.1073/pnas.91.16.7653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Chen W, Broyles SS. The vaccinia virus B1R gene product is a serine/threonine protein kinase. J Virol. 1992;66:2717–2723. doi: 10.1128/jvi.66.5.2717-2723.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T. Cell biology of transcription and pre-mRNA splicing: nuclear architecture meets nuclear function. J Cell Sci. 2000;113:1841–1849. doi: 10.1242/jcs.113.11.1841. [DOI] [PubMed] [Google Scholar]
- Moss B. Poxviridae and their replication. In: Fields BN, Knipe DM, Chanock RM, Hirsch MS, Melnick JL, Monath TP, Roizman B, editors. Fields Virology. New York: Raven Press; 1990a. [Google Scholar]
- Moss B. Regulation of vaccinia virus transcription. Annu Rev Biochem. 1990b;59:661–688. doi: 10.1146/annurev.bi.59.070190.003305. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Assembly-disassembly of the nuclear lamina. Curr Opin Cell Biol. 1992;4:105–109. doi: 10.1016/0955-0674(92)90066-l. [DOI] [PubMed] [Google Scholar]
- Östlund C, Ellenberg J, Hallberg E, Lippincott-Schwartz J, Worman HJ. Intracellular trafficking of emerin, the Emery-Dreifuss muscular dystrophy protein. J Cell Sci. 1999;112:1709–1719. doi: 10.1242/jcs.112.11.1709. [DOI] [PubMed] [Google Scholar]
- Pante N, Bastos R, McMorrow I, Burke B, Aebi U. Interactions and three-dimensional localization of a group of nuclear pore complex proteins. J Cell Biol. 1994;126:603–617. doi: 10.1083/jcb.126.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen K, Snijder EJ, Schleich S, Roos N, Griffiths G, Krijnse Locker J. Characterization of vaccinia virus intracellular cores: implications for viral uncoating and core structure. J Virol. 2000;74:3525–3536. doi: 10.1128/jvi.74.8.3525-3536.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter M, Heitlinger E, Häner M, Aebi U, Nigg EA. Disassembly of in vitro formed lamin head-to-tail polymers by CDC2 kinase. EMBO J. 1991;10:1535–1544. doi: 10.1002/j.1460-2075.1991.tb07673.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter M, Nakagawa J, Dorée M, Labbé JC, Nigg EA. In vitro disassembly of the nuclear lamina and M phase-specific phosphorylation of lamins by cdc2 kinase. Cell. 1990;61:591–602. doi: 10.1016/0092-8674(90)90471-p. [DOI] [PubMed] [Google Scholar]
- Pfaller R, Smythe C, Newport JW. Assembly/disassembly of nuclear envelope membrane: cell cycle-dependent binding of nuclear membrane vesicles to chromatin in vitro. Cell. 1991;65:209–217. doi: 10.1016/0092-8674(91)90155-r. [DOI] [PubMed] [Google Scholar]
- Rolls MM, Stein PA, Taylor SS, Ha E, McKeon F, Rapoport TA. A visual screen of a GFP-fusion library identifies a new type of nuclear envelope membrane protein. J Cell Biol. 1999;146:29–43. doi: 10.1083/jcb.146.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost B, Casadio R, Fariselli P, Sander C. Transmembrane helices predicted at 95% accuracy. Protein Sci. 1995;4:521–533. doi: 10.1002/pro.5560040318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmons T, Kuhn A, Wylie F, Schleich S, Rodriguez JR, Rodriguez D, Esteban M, Griffiths G, Krijnse Locker J. Vaccinia virus membrane proteins p8 and p16 are co-translationally inserted into the rough ER and retained in the intermediate compartment. J Virol. 1997;71:7404–7420. doi: 10.1128/jvi.71.10.7404-7420.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmelz M, Sodeik B, Ericsson M, Wolffe E, Shida H, Hiller G, Griffiths G. Assembly of vaccinia virus: the second wrapping cisterna is derived from the trans Golgi network. J Virol. 1994;68:130–147. doi: 10.1128/jvi.68.1.130-147.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow CM, Senior A, Gerace L. Monoclonal antibodies identify a group of nuclear pore complex glycoproteins. J Cell Biol. 1987;104:1143–1156. doi: 10.1083/jcb.104.5.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodeik B, Doms RW, Ericsson M, Hiller G, Machamer CE, van't Hof W, van Meer G, Moss B, Griffiths G. Assembly of vaccinia virus: role of the intermediate compartment between the endoplasmic reticulum and the Golgi stacks. J Cell Biol. 1993;121:521–541. doi: 10.1083/jcb.121.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian K, Meyer T. Calcium-induced restructuring of nuclear envelope and endoplasmic reticulum calcium stores. Cell. 1997;89:963–971. doi: 10.1016/s0092-8674(00)80281-0. [DOI] [PubMed] [Google Scholar]
- van der Meer Y, Snijder EJ, Dobbe JC, Schleich S, Denison MR, Spaan WJM, Krijnse Locker J. The localization of mouse hepatitis virus nonstructural proteins and RNA synthesis indicates a role for late endosomes in viral replication. J Virol. 1999;73:7641–7657. doi: 10.1128/jvi.73.9.7641-7657.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson KL. The nuclear envelope, muscular dystrophy and gene expression. Trends Cell Biol. 2000;10:125–129. doi: 10.1016/s0962-8924(99)01708-0. [DOI] [PubMed] [Google Scholar]
- Wilson KL, Wiese C. Reconstituting the nuclear envelope and the endoplasmic reticulum in vitro. Semin Cell Dev Biol. 1996;7:487–496. [Google Scholar]
- Yang L, Guan T, Gerace L. Integral membrane proteins of the nuclear envelope are dispersed throughout the endoplasmic reticulum during mitosis. J Cell Biol. 1997;137:119–1210. doi: 10.1083/jcb.137.6.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]