

Abstract
In response to cellular stress or damage, proliferating cells can induce a specific program that initiates a state of long-term cell-cycle arrest, termed cellular senescence. Accumulation of senescent cells occurs with organismal aging and through continual culturing in vitro. Senescent cells influence many biological processes, including embryonic development, tissue repair and regeneration, tumor suppression, and aging. Hallmarks of senescent cells include, but are not limited to, increased senescence-associated β-galactosidase activity (SA-β-gal); p16INK4A, p53, and p21 levels; higher levels of DNA damage, including γ-H2AX; the formation of Senescence-associated Heterochromatin Foci (SAHF); and the acquisition of a Senescence-associated Secretory Phenotype (SASP), a phenomenon characterized by the secretion of a number of pro-inflammatory cytokines and signaling molecules. Here, we describe protocols for both replicative and DNA damage-induced senescence in cultured cells. In addition, we highlight techniques to monitor the senescent phenotype using several senescence-associated markers, including SA-β-gal, γ-H2AX and SAHF staining, and to quantify protein and mRNA levels of cell cycle regulators and SASP factors. These methods can be applied to the assessment of senescence in various models and tissues.
Keywords: Cellular Biology, Issue 123, Aging, SASP, SA-β-gal, γ-H2AX, p16, p21, p53, IL6, SAHF, DNA damage, Senescence
Introduction
Over half a century ago, Hayflick and colleagues described how untransformed cells proliferate in culture, but for only a finite period of time1. Long-term culturing of human fibroblasts caused the cells to stop proliferating; however, they were metabolically active, and this was called cellular senescence. Senescence can be beneficial for inhibiting tumorigenesis, but it also can be detrimental, as it is thought to contribute to the loss of regenerative capacity that occurs with aging2,3. Senescent cells have been shown to accumulate in tissues as humans age4 and have been implicated in a number of biological processes, including embryonic development, wound healing, tissue repair, and age-related inflammation2.
Continual passaging of cells in culture induces replicative senescence, which has been linked to telomere attrition and genomic instability. Various cell stresses, including DNA damage and oncogenes, can also cause senescence3. Senescence caused by factors other than telomere attrition is often called stress-induced or premature senescence and generally depends on the p16INK4A/Rb pathway5. Although proliferating, untransformed cells typically appear spindle in shape, senescent cells can be identified as having certain characteristics, including a flat, large morphology and increased senescence-associated β-galactosidase activity (SA-β-gal) (Figures 1 & 2). Senescent cells also accumulate DNA damage markers, including γ-H2AX (Figure 3)6, and, potentially, senescence-associated heterochromatin foci (SAHF) (Figure 4)7. Senescent cells have higher levels of cell cycle regulators, including p16 (p16INK4A) and/or p21 and p53 (Figure 5)8,9. Moreover, recent data have shown that senescent cells can have non-autonomous effects by secreting a number of pro-inflammatory cytokines and chemokines called the senescence-associated secretory phenotype (SASP)10. Although this SASP phenomenon may vary from cell type to cell type, in general, it is demonstrated by an increase in Interleukin-6 (IL-6), IL-8, granulocyte-macrophage colony stimulating factor (GM-CSF), growth-regulated oncogene α (GRO-α), and GRO-β, among others (Figure 6). The particular stress or damage that induces senescence may also influence the secretory phenotype11,12,13. SASP can be detected by measuring the levels of secreted proteins using ELISAs or cytokine/protein arrays10,14. Although post-transcriptional mechanisms can regulate SASP protein levels11,15,16,17, changes in mRNA levels can also be detected in many cases. These changes are generally more sensitive and easier to quantify than protein level measurements. Other senescent markers can also be assessed, including persistent DNA damage nuclear foci, called DNA segments with chromatin alterations reinforcing senescence (DNA-SCARS)18, and various other markers3,19,20.
Here, we describe common techniques for inducing senescence in cells in culture and also for measuring several markers of senescence, including SA-β-Gal, γ-H2AX, SAHF, and the protein and mRNA of senescence-associated molecules.
Protocol
1. Inducing Replicative Senescence
- Thaw low-passage human diploid fibroblasts (e.g., WI-38 and IMR-90) or other cell lines. NOTE: Here, human diploid fibroblasts were used, but these protocols can be used to assess senescence in other cell types, such as endothelial, epithelial, or mesenchymal stem cells. Culturing conditions may be optimized for the different cell types used due to different growth rates or growth conditions. NOTE: Cells should be proliferating and have a spindle morphology (see Figure 1 for a schematic and Figure 2 for examples). Ideally, the cells should have a Population Doubling Level (PDL) of <30 at the start of experiments to avoid having possible replicative senescent cells.
- Calculate the PDL of the cells using the following equation PDL = log(Nf/N0)/log2, where Nf is the final cell number and N0 is the initial number of seeded cells21.
Culture WI-38 cells in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS), 1% nonessential amino acids, and 1% penicillin/streptomycin.
Grow IMR-90 cells in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin.
Grow all cells at 37 °C and 5% CO2. Continually grow the cells and split every 2 - 4 d when the cells reach ~70 - 80% confluence. Monitor cell confluence using a light microscope. Avoid cells with higher confluence, as this can affect cell growth due to contact inhibition.
Monitor the cells under a light microscope for the morphological appearance of senescent cells (see Figures 1 & 2) and for when there is no change in PDL from one subculture to the next.
2. DNA Damage-induced Senescence
Plate cells (e.g., WI-38, IMR-90, or another cell type) at a sparse density (i.e. ~300,000 cells/6 cm dish) in an appropriate dish for the type of analysis (i.e. a 6 cm dish for protein/RNA markers or a 6-well dish for SA-β-gal staining). Use early-passage cells that are young and proliferating (<30 PDL) and plate them so that the cells are roughly 50 - 60% confluent on the next day.
Remove the growth medium using a glass pipette connected to a vacuum and wash the cells by adding 1x Phosphate-buffered Saline (PBS). Repeat this step for a total of two washes. Add a thin layer of PBS (~750 µL for a 6 cm dish) for both a "mock" treated and for an Ionizing Radiation (IR) treated condition.
Use a gamma irradiator to expose cells to a single dose of 10 Gray (Gy) of IR; this is a typical exposure for most cell lines. In a hood, remove the PBS and replace it with growth medium. Alternatively, treat the cells with bleomycin at 20 µg/mL for 2 h.
Change the medium on the cells every 48 -7 2 h and split the cells if necessary.
Monitor the cells under a light microscope for morphological changes (see Figure 1 for a schematic and Figure 2 for representative cell images) and assay for senescent markers 7 - 10 d after the IR treatment. NOTE: This protocol describes senescence induced by IR. Senescence can also be induced by other stresses, including bleomycin (see above for conditions), H2O222,23, chemotherapeutic drugs24, cigarette smoke25, transforming growth factor (TGF)-β126,27, and other methods21,28,29.
3. Staining Cells for Senescence-associated β-Galactosidase
Prior to staining, examine the cells under a light microscope to monitor the morphological changes. NOTE: Senescent cells are typically enlarged and flat, unlike proliferating cells, which have a spindle morphology (See Figure 1 for a schematic and Figure 2 for representative results). Proliferating cells can be used as a negative control, and cells treated with IR or another DNA-damaging agent (see section 2) can be used as positive controls.
- Quantify SA-β-gal activity using reagents from a commercial kit (see the Materials Table). Thaw all reagents and bring them to room temperature by warming them up to 37 °C.
- Calculate the amount of staining solution and fixative solution necessary. NOTE: Typically, a 1 mL volume is sufficient for one well in a 6-well plate. Different plate sizes can be used, but a 6-well plate size is well-suited to perform an assay in triplicate for each condition. This density usually provides a sufficient number of cells per field to count without using an excess number of cells.
- Dilute the staining solution 1:10 with distilled water.
- Dilute the fixative solution 1:10 with distilled water.
- Make up a 20x stock solution of X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside) in N,N-dimethylformamide (DMF; e.g., 20 mg of X-gal in 1 mL of DMF). To maximize use of the kit, measure out X-gal to the amount needed for each assay and then add the calculated amount of DMF to dissolve. Alternatively, store the stock solution at -20 °C in a light-protected tube for one month. Use only polypropylene (opaque) tubes for diluting and storing X-gal solutions.
- Prepare β-gal staining solution: 930 µL of 1x staining solution, 10 µL of staining supplement A, 10 µL of staining supplement B, and 50 µL of 20 mg/mL X-gal in DMF. Make a master mix depending on how many wells need to be stained.
Carefully remove the medium from the cells using a transfer pipette or the equivalent.
Gently wash the cells once with room-temperature 1x PBS.
Add 1 mL of 1x fixative solution to each well and incubate for 10 - 15 min at RT.
Remove the fixative solution with a transfer pipette. NOTE: The fixative solution (1x) contains 2% formaldehyde and 0.2% glutaraldehyde and should be disposed of as a hazardous waste according to the recommendations of the laboratory safety office.
Gently wash the cells twice with 1x PBS.
- Remove the PBS completely and add 1x β-gal staining solution to the wells (1 mL/well of a 6-well plate). Cover the plate completely with aluminum foil and incubate for 24 - 48 h in a 37 °C incubator (preferably in the absence of CO2, as this can lower the pH of the buffer and affect staining).
- Check the cells at 24 h for staining, and if the stain is too light, incubate for another 24 h.
After the incubation time, remove the β-gal solution and replace it with 1x PBS. NOTE: This step removes some of the background color when imaging and also prevents hazardous spilling of the X-gal solution. NOTE: Dispose of the β-gal solution properly, according to the recommendations of the laboratory safety office.
Examine the cells on a light microscope with an attached camera using a 10x or higher objective; SA-β-gal-positive cells will be blue. Acquire the desired number of images for the experiment. If counting the percentage of SA-β-gal-positive cells, acquire the appropriate number of images (>5) for quantification per well. Ensure that the images are non-overlapping from various fields of view within each well.
Count the number of SA-β-gal-positive (blue) cells versus the number of total cells to calculate the percent of SA-β-gal-positive cells. Count >100 cells per condition. NOTE: Representative pictures can also be used for figures. Note that cell confluency is critical if counting cells, as too many cells make it difficult to differentiate each cell and too few may not give a sufficient number of cells for quantification and statistics. Pictures for figures for publication are at the best resolution if saved as .tiff files, although .jpeg files are also suitable. Color images should be 300 dpi.
4. Staining Cells for γ-H2AX
Plate the cells from Sections 1 or 2 on glass coverslips in a 24-well plate.
The next day, wash the cells with 1x PBS. Rinse carefully, as senescent cells do not adhere well to coverslips and can easily be removed during washes. Alternatively, directly fix the cells without washing.
Remove the PBS and fix the cells with freshly prepared 3.7% formaldehyde in PBS for 10 min at RT.
Remove the solution and permeabilize the cells with 0.2% Triton X-100 in PBS for 10 min.
Remove the solution and block in 5% FBS in PBS for 2 h at RT.
Incubate with anti-phospho γ-H2AX (Ser139) and FITC conjugate in 5% FBS/PBS for 2 h at 37 °C or O/N at 4 °C. Protect from light.
Wash 3 - 6x with PBS at 37 °C for total of 30 - 60 min.
Stain with DAPI (diluted 1:15,000 in PBS) for 10 min at RT.
Wash 3x with PBS.
Quickly wash with water to remove the salts and mount the coverslip on a glass slide using 3 - 5 µL of antifade reagent. Alternatively, use a mounting solution containing DAPI.
Let it dry O/N and visualize the stained cells on a fluorescent or confocal microscope using a 63X or higher objective.
- Quantify the γ-H2AX foci by using either of two methods described below. Score for either protocol by eye using a fluorescent microscope. Ideally, use a confocal microscope. Take Z-sections and condense into one single plane in order to visualize all detectable foci (representative images for γ-H2AX staining in proliferating and senescent cells are shown in Figure 3). Count the foci by eye using imaging software; in general, ~100 - 200 nuclei are sufficient for counting.
- To quantify the percentage of cells that have γ-H2AX-positive foci, count each cell that has at least one focus visible and divide by the total number of DAPI-stained nuclei.
- To quantify the number of γ-H2AX foci, count the γ-H2AX foci per cell. NOTE: The levels of γ-H2AX may vary depending on the specific stressor or damage that induced senescence.
5. Staining Cells for SAHF Markers
NOTE: Human senescent cells will often have nuclear regions of condensed DNA/chromatin. In senescent cells, these heterochromatic regions are thought to inhibit the expression of proliferation-promoting genes, such as E2F family members. SAHF can be visualized by the reorganization of DAPI; by the presence of heterochromatin-associated histone marks, including the di- and tri-methylation of Lys9 on Histone H3 (H3K9Me2 and H3K9Me3); and by chromatin-reorganizing proteins, including HP-1 heterochromatin protein 1 (HP1), HIRA (histone repressor A), and ASF1a (anti-silencing function-1a) to chromatin30. We have chosen to use an oncogene-induced senescence model (OIS), as SAHF is more prominent in OIS models30. IDH4 cells proliferate in the presence of dexamethasone due to the presence of SV40 large T antigen but become senescent after the removal of dexamethasone from the medium31. SAHF can be detected by DAPI staining and by staining with antibodies against the specific markers listed above. Here, we describe DAPI and H3K9Me2 staining for the visualization of SAHF in cells28.
Grow IDH4 cells in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, and 1 µg/mL dexamethasone. To induce senescence, remove the dexamethasone and change the medium so that it contains charcoal-stripped FBS; after growing for ~10 days in this new medium, IDH4 cells become senescent31.
Plate IDH4 cells or cells from Sections 1 or 2, described above, on glass coverslips in a 24-well plate.
The next day, wash the cells with 1x PBS. Rinse carefully, as senescent cells do not adhere well to coverslips and can be easily removed during washes. Alternatively, directly fix cells without washing.
Remove the PBS and fix the cells with freshly prepared 3.7% formaldehyde in PBS for 10 min at RT.
Wash the cells 3 times with 1x PBS.
Remove the solution and permeabilize the cells with 0.2% Triton X-100 in PBS for 5 min.
Wash the cells with 1x PBS. Remove the PBS and block the coverslips by adding blocking buffer containing 3% BSA (w/v) in PBS for 5 min at RT. Always filter solutions containing BSA prior to use to remove particulate matter.
Remove the blocking buffer and incubate the cells with primary antibodies against SAHF markers such as anti-H3KMe2 antibodies (Figure 4; see the Materials Table for details). Dilute the antibodies in blocking buffer and incubate for 1 - 2 h at RT.
Remove the primary antibody solution and wash the coverslips 3 times with 1% (v/v) Triton X-100 in PBS.
Dilute the secondary antibodies in blocking buffer (Materials Table) and incubate for 1 h at RT. Protect from light.
Wash the cells twice with 1x PBS. Stain with DAPI (diluted 1:15,000 in PBS; see the Materials Table) for 10 min at RT.
- Wash the coverslips 3x with 1x PBS.
- Quickly wash with water to remove the salts and mount each coverslip on a glass slide using 3 - 5 µL of antifade reagent. Alternatively, use a mounting solution containing DAPI.
- Let dry O/N and visualize the stained cells on a fluorescent or confocal microscope using a 63X or higher objective (see the Representative Results in Figure 4). Quantify the SAHF as described in Section 4. NOTE: Isotype control primary antibodies or no primary antibody (i.e. secondary alone) should be used as a negative control for immunofluorescence staining.
6. Analyzing Senescence-associated Proteins by Immunoblotting
NOTE: The senescent phenotype is characterized by the upregulation of cell cycle regulators, including p16INK4A, p21, and p53. This protocol will describe the quantification of the levels of these proteins in cell lysates. These techniques can be used to assess senescence induced using a variety of methods21,28,29.
Induce cellular senescence as described in Sections 1 and 2 or by using another method21,28,29. To analyze cellular senescence markers, plate the cells in 6 cm dishes and analyze both the protein and the RNA simultaneously (see Section 7 for RNA isolation instructions).
- Remove the cell culture medium. Put the cells on a tray of ice.
- Wash the cells 2 times with cold 1x PBS. Tilt the dish to ensure that all the PBS is aspirated so as to be as precise in terms of the volumes used for the procedure.
- After aspirating the last wash of PBS, add 200 µL of cold 1x PBS (for a 6 cm dish) to the dish. Scrape the cells using a cell scraper.
- Pipette the cell/PBS mixture into an RNase-free tube for RNA isolation. Take approximately 150 µL of this mixture and pipette it into a new tube. This aliquot will be used for protein analysis, so be sure to pipette the same amount from each tube.
- Lyse the cells by adding 75 - 100 µL of modified 2x Laemmli's sample buffer (60 mM Tris-HCl pH 6.8, 12.5% glycerol, 2% SDS, 5% β-mercaptoethanol (BME), and bromophenol blue; can be made up as a big batch, aliquoted, and stored at -20 °C until use) or an equivalent sample buffer.
- Alternatively, lyse the cells in 75 - 100 µL of lysis buffer and centrifuge at 15,000 x g for 10 min at 4 °C to remove the cell debris. Remove the supernatant and pipette into a clean tube. A Bradford protein assay or an equivalent protein assay can be used on lysates to ensure the equal loading of protein. NOTE: Protein assays cannot be performed on samples lysed in sample buffer.
- Take out ~25 µL of lysate and add it to 15 µL of 2x sample buffer. Pipette up and down in the sample buffer to lyse the cells. NOTE: Volumes of lysis buffer or sample buffer may be adjusted depending on cell confluence. If the sample is "gooey" due to nuclear lysing, add more sample buffer or PBS to dilute. Samples can be stored at -20 °C until use.
- Boil the samples for 5 min at 95 °C on a heat block (or equivalent equipment) and load ~40 µL of lysed sample in sample buffer on an 18% Tris-Glycine gel. NOTE: A gradient gel (4-15%) or a 14% Tris-Glycine gel are also sufficient for detecting low molecular weight proteins. Different types of acrylamide gels can also be used, but ensure that the gels and system are adequate for detecting low molecular weight proteins.
- Run using 1x running buffer at a constant 100 V for 20 min (through the stacking gel) and ~60 min at 180 V. Monitor the gel so the dye front just runs off or is at the bottom of the gel so that the low molecular weight proteins do not run off of the gel.
Transfer the acrylamide gel to a PVDF membrane (pre-soaked with 100% methanol to activate). If doing a wet transfer, only 1 h is needed for transfer (versus the normal ~1.75 h). Adjust accordingly for the appropriate transfer system; using pre-stained molecular weight markers can aid in assessing transfer efficiency.
- Wash the membrane in water. Use a Ponceau stain to monitor for the equal loading of samples. Wash the Ponceau off with water.
- Block the membrane in 5% non-fat dry milk in TBST (50 mM Tris-HCl pH 7.5, 150 mM NaCl, and 0.1% Tween-20) for 1 h at RT or O/N at 4 °C with agitation.
Wash blot once with TBST and incubate with primary antibody diluted in 5% milk TBST (see the Materials Table for dilutions and antibody recommendations) for 1 h at RT.
Perform two quick washes with TBST and then wash twice on a shaker for total of ~30 min (~15 min each wash).
Incubate with a secondary antibody for 40 min at RT. Repeat step 6.10.
Incubate with freshly made Western blot substrate and develop the film or read it on a chemiluminescence reader.
Probe immunoblot with p16INK4A antibodies first (Figure 5). Strip the membrane by incubating for 15 min with water, 15 min with 0.2 N NaOH, and 15 min with water. Continue with Step 6.9 using anti-p21 antibodies.
Strip the membrane using 62.5 mM Tris-HCL pH 6.8 and 2% SDS with 300 µL of freshly added BME per 50 mL of stripping solution.
Incubate at 50 °C for 30 min and wash extensively with TBS and then TBST. Probe with anti-actin or anti-GAPDH antibodies for protein-loading controls (Figure 5 shows representative results). NOTE: p53 levels can also be assessed on the same membranes. However, since p53 is a higher molecular weight, it is better resolved on a lower-percentage gel (e.g., 10% Tris-Glycine or a gradient gel (4-15%)). Protein levels of other senescent markers can also be assessed by immunoblotting lysates, including γ-H2AX and SAHF markers.
7. Analyzing the mRNA Levels of Senescence Markers
NOTE: When isolating RNA, ensure that the work surfaces are cleaned with RNase removal solutions or an equivalent and that the materials are all RNase-free.
Using the cell/PBS mixture from Step 4.2, add 1 mL of RNA extraction solution and vortex for 30 s (there should be no visible clumps or cellular debris).
Add 200 µL of chloroform and shake vigorously for 15 s.
Centrifuge at max speed (>15,000 x g) for 15 min at 4 °C.
Remove ~400 µL of the top RNA aqueous layer and pipette it into a new microfuge tube.
Add 400 µL of isopropanol (1:1 with the total volume of aqueous layer pipetted in the previous step) and 4 µL of precipitant to the RNA layer.
Centrifuge for 15-30 min at maximum speed (>15,000 x g) and 4 °C to pellet the RNA.
Remove the supernatant and add 1 mL of 75% ice-cold ethanol.
Centrifuge for 1 min at maximum speed (>15,000 x g) and 4 °C.
Remove the supernatant and centrifuge for 1 min at maximum speed (>15,000 x g) and 4 °C.
Pipette out as much liquid as possible and air dry for 2 min.
Re-suspend in 10 µL of 10x DNase buffer, 85 µL of H2O, and 5 µL of DNase 1.
Incubate at 37 °C for 30 min.
Add 200 µL NT2 buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM MgCl2, and 0.05% Nonidet P-40) and add 300 µL of the lower layer of acid phenol-CHCl2.
Vortex for 1 min and centrifuge at room temperature for 5 min at maximum speed (>15,000 x g).
Collect 250 µL of the upper RNA layer (2 x 125 µL), add 25 µL of 3 M sodium acetate pH 5.2, 625 µL of 100% ice-cold ethanol, and 5 µL of precipitant. Mix well and precipitate O/N at -20 °C.
The next day, mix and spin at maximum speed (>15,000 x g) and 4 °C for 30 min and discard the supernatant.
Repeat steps 6.8-6.11.
Re-suspend the pellet in 12 µL of nuclease-free water and store at -80 °C until use. NOTE: Other RNA isolation procedures and kits can be used as well.
Perform reverse transcription with random hexamers and SSII reverse transcriptase according to the manufacturer's protocol. NOTE: Given the low amount of RNA isolated from senescent cells, it is best to use all of the RNA (12 µL) for the reverse transcription synthesis. Alternatively, RNA can be quantified using a spectrophotometer, and equal amounts of RNA can be used for reverse transcription.
- Analyze the mRNA levels using real-time quantitative qPCR (RT-qPCR) amplification and SYBR-green PCR master mix with gene-specific primers.
- As in a typical RT-qPCR reaction, dilute the cDNA 1:30 (in RNase/DNase-free water) and add 3 µL to the real-time 96-well plate for better pipetting accuracy. Prepare a reaction master mix of gene-specific forward and reverse primers (2.5 µM concentration, add 5 µL/reaction), SYBR-green PCR mix (6.25 µL/reaction; use less than the manufacturer's recommendation), and 5.75 µL of RNase/DNase-free water for a final reaction volume of 20 µL (17 µL of primers/SYBR-green/water and 3 µL of cDNA).
- Perform RT-qPCR amplification using a real-time PCR machine; follow the manufacturer's protocol. NOTE: A list of validated human forward and reverse real-time primers can be found in Table 1. These genes include SASP proteins and other senescence-associated markers and/or genes. Representative results from IR-induced senescence are shown in Figure 6.
8. Quantifying SASP Protein Levels
Induce senescence using Sections 1 or 2 or by using another method. Plate the cells on either 6 or 10 cm plates, as described above (~250,000 cells/6 cm plate or ~650,000/10 cm plate).
Either with cells 10 d post-IR treatment or with replicative senescent cells (and appropriate control cells), wash the cells three times with 1x PBS and add serum-free medium with antibiotics in a small volume (2.5 mL for 6 cm or 5 mL for 10 cm).
Incubate overnight at 37 °C and 5% CO2 in a tissue culture incubator. The next day, collect the medium and put it in a conical tube on ice.
Wash the cells 2 times with PBS and then add 1 mL of trypsin (for a 6 cm dish). Count the cells using a hemocytometer to later adjust the concentrations according to cell number.
Centrifuge the medium for 5 min at 300 x g.
Filter the medium using a 0.45-µm syringe filter.
Aliquot the medium and freeze at -80 °C until use or assay directly using ELISA or equivalent assays.
Calculate the number of cells/mL concentration by taking the total cell number/volume of medium collected.
Perform an appropriate enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions. See the Materials Table for recommended ELISA kits for assaying for IL-6, IL-8, GROα, and other cytokines/chemokines (Figure 6B).
To ensure that the factors that are measured are within the linear range of the ELISA kit, make dilutions of the senescent cell medium compared to the proliferating cell medium. Account for these dilutions and the volume when calculating the amount of IL-6. To acquire the amount of IL-6 secreted per cell per day, calculate the IL-6 value obtained from the kit (in pg) per diluted cell concentration per mL (from step 7.8). NOTE: Other methods, such as cytokine arrays, can be used to detect SASP protein levels and may be suitable for screening a large number of SASP factors. The protein levels of SASP factors may vary based on the time after senescence induction, the cell type, or the type of senescence induced.
Representative Results
Figures 2 - 6 show representative results from SA-β-gal staining; staining for γ-H2AX and SAHF; assessment of protein levels of p16INK4A, p21, and p53; and mRNA and protein levels of senescent-associated molecules. Increased SA-β-gal staining occurs with replicative and DNA damage-induced senescence. Also, observe the morphological changes that occur with senescence. Cells become enlarged and flat compared to the spindle appearance of proliferating fibroblasts. For this experimental paradigm, cells were stained and RNA/protein were isolated 7 d after IR exposure. More robust results may occur by waiting longer after IR exposure (i.e. 10 d). Conditions need to be determined for each condition and experimental setup. These results are representative and these markers can also be detected when senescence is induced using other methods.
For Figure 6, gene names are listed and correspond to CXCL1 (GROα), CXCL2 (GROβ), and CSF2 (GM-CSF). The upregulation of mRNA levels of some, but not all, of the SASP factors were observed after IR-induced senescence in WI-38 cells. Other senescence-associated mRNAs, including CDKN1A (encodes p21) and CDKN2A (encodes p16INK4A), were upregulated. Other mRNAs (EDN1 and ANKRD1) have also been included and have previously been shown to be higher in senescent cells15. Primers are listed in Table 1.
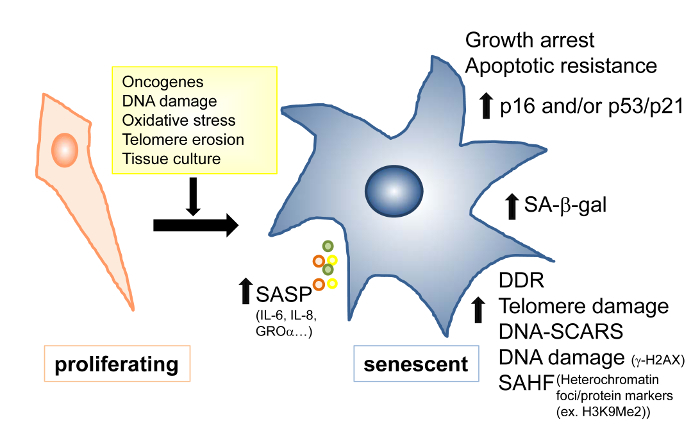 Figure 1: Proliferating and Senescent Fibroblasts. A schematic representation of proliferating (left) and senescent (right) fibroblasts. Examples of senescence inducers are shown in the yellow box. Senescence is associated with higher levels of the indicated markers. SA-β-gal, senescence-associated β-galactosidase activity; SASP, senescence-associated secretory phenotype; SAHF, senescence-associated heterochromatin foci; DDR, DNA-damage response. Please click here to view a larger version of this figure
Figure 1: Proliferating and Senescent Fibroblasts. A schematic representation of proliferating (left) and senescent (right) fibroblasts. Examples of senescence inducers are shown in the yellow box. Senescence is associated with higher levels of the indicated markers. SA-β-gal, senescence-associated β-galactosidase activity; SASP, senescence-associated secretory phenotype; SAHF, senescence-associated heterochromatin foci; DDR, DNA-damage response. Please click here to view a larger version of this figure
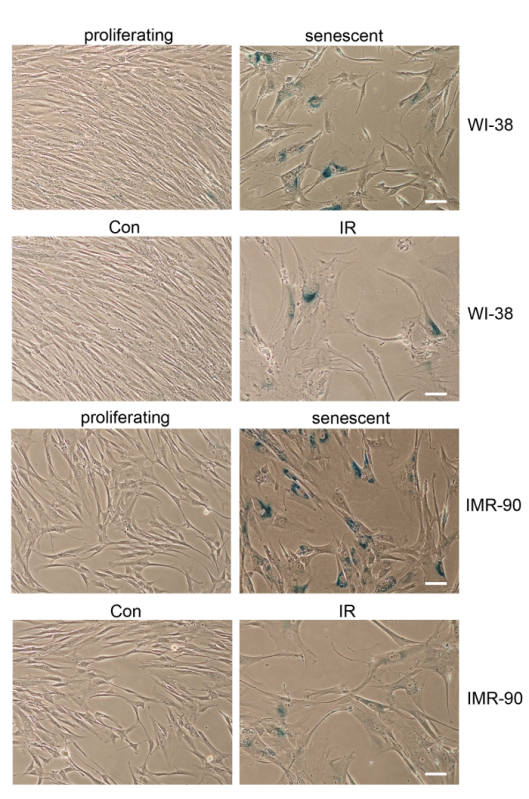 Figure 2. Morphological Characteristics and Increased SA-β-gal Activity of Senescent Cells. WI-38 cells, either proliferating "young" (PDL 27), replicative senescent (PDL 40), or those exposed to ionizing radiation (IR), were stained for SA-β-gal activity. IMR-90 cells, either proliferating "young" (PDL 30), replicative senescent (PDL 52), or those exposed to ionizing radiation (IR), were stained for SA-β-gal activity. Sections 1 - 2 describe inducing replicative and DNA damage-induced senescence, and Section 3 explains staining for SA-β-gal. Cells were stained 7 d after IR. Con, control. Scale bar = 50 µm. Please click here to view a larger version of this figure.
Figure 2. Morphological Characteristics and Increased SA-β-gal Activity of Senescent Cells. WI-38 cells, either proliferating "young" (PDL 27), replicative senescent (PDL 40), or those exposed to ionizing radiation (IR), were stained for SA-β-gal activity. IMR-90 cells, either proliferating "young" (PDL 30), replicative senescent (PDL 52), or those exposed to ionizing radiation (IR), were stained for SA-β-gal activity. Sections 1 - 2 describe inducing replicative and DNA damage-induced senescence, and Section 3 explains staining for SA-β-gal. Cells were stained 7 d after IR. Con, control. Scale bar = 50 µm. Please click here to view a larger version of this figure.
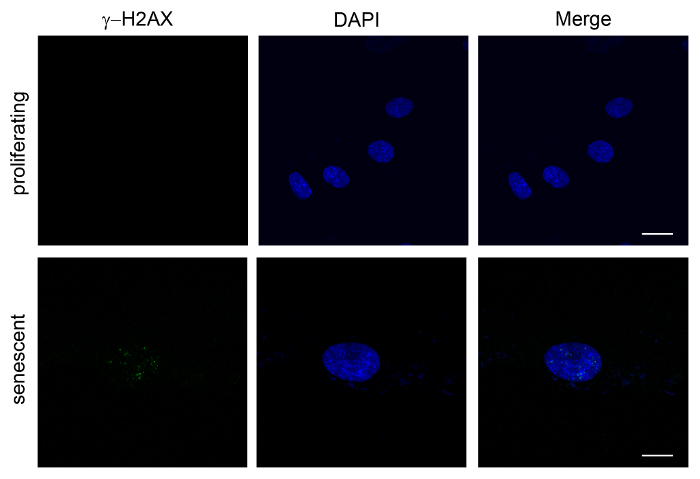 Figure 3. Increased γ-H2AX Foci in Senescent Cells. WI-38 cells, either proliferating "young" (PDL 30) or replicative senescent (PDL 53), were fixed and stained with anti-γ-H2AX antibodies and DAPI. Note the increase in the number of γ-H2AX foci in senescent cells. Images were obtained on a confocal microscope. Z-sections were taken, and the maximum intensity projection was used to obtain a single plane image to visualize all detectable foci. Scale bar = 20 µm. Please click here to view a larger version of this figure.
Figure 3. Increased γ-H2AX Foci in Senescent Cells. WI-38 cells, either proliferating "young" (PDL 30) or replicative senescent (PDL 53), were fixed and stained with anti-γ-H2AX antibodies and DAPI. Note the increase in the number of γ-H2AX foci in senescent cells. Images were obtained on a confocal microscope. Z-sections were taken, and the maximum intensity projection was used to obtain a single plane image to visualize all detectable foci. Scale bar = 20 µm. Please click here to view a larger version of this figure.
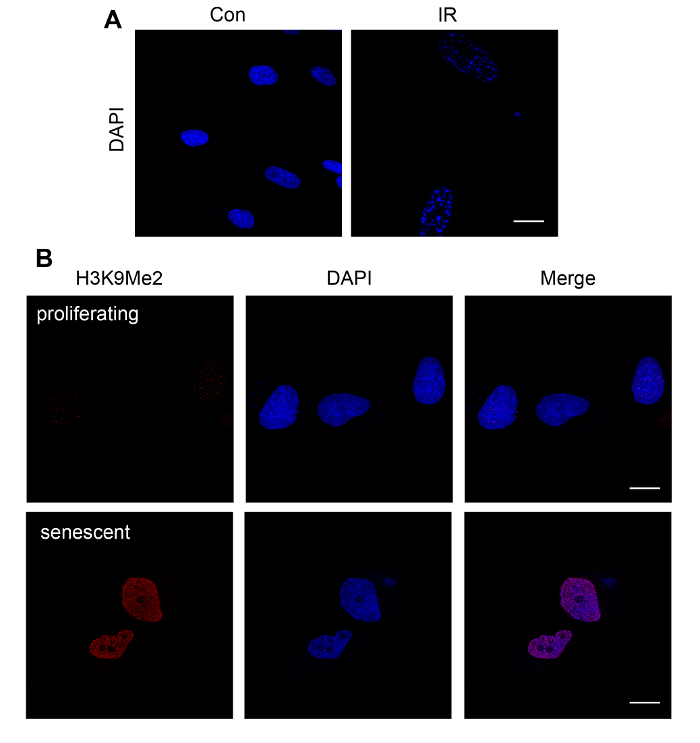 Figure 4. Fluorescent Staining of SAHF in Senescent Cells. (A) Proliferating WI-38 cells (PDL 27) were untreated or exposed to ionizing radiation (IR). After 10 days, the cells were stained with DAPI to visualize SAHF (note dense/punctate DAPI staining in IR-treated cells) (B) IDH4 were grown in the presence of dexamethasone (proliferating) or in charcoal-stripped FBS to induce senescence (senescent). After 10 d, the cells were stained with antibodies against H3K9Me2 and DAPI. Images were obtained on a confocal microscope. Z-sections were taken, and the maximum-intensity projection was used to obtain a single-plane image. Proliferating cells in (A) and (B) have diffuse DAPI staining, whereas senescent cells have punctate DAPI staining. In (B), control cells have diffuse, very light staining for H3K9Me2, whereas H3K9Me2 staining is more robust and colocalizes with DAPI-stained foci in senescent cells. Scale bar = 20 µm. Please click here to view a larger version of this figure.
Figure 4. Fluorescent Staining of SAHF in Senescent Cells. (A) Proliferating WI-38 cells (PDL 27) were untreated or exposed to ionizing radiation (IR). After 10 days, the cells were stained with DAPI to visualize SAHF (note dense/punctate DAPI staining in IR-treated cells) (B) IDH4 were grown in the presence of dexamethasone (proliferating) or in charcoal-stripped FBS to induce senescence (senescent). After 10 d, the cells were stained with antibodies against H3K9Me2 and DAPI. Images were obtained on a confocal microscope. Z-sections were taken, and the maximum-intensity projection was used to obtain a single-plane image. Proliferating cells in (A) and (B) have diffuse DAPI staining, whereas senescent cells have punctate DAPI staining. In (B), control cells have diffuse, very light staining for H3K9Me2, whereas H3K9Me2 staining is more robust and colocalizes with DAPI-stained foci in senescent cells. Scale bar = 20 µm. Please click here to view a larger version of this figure.
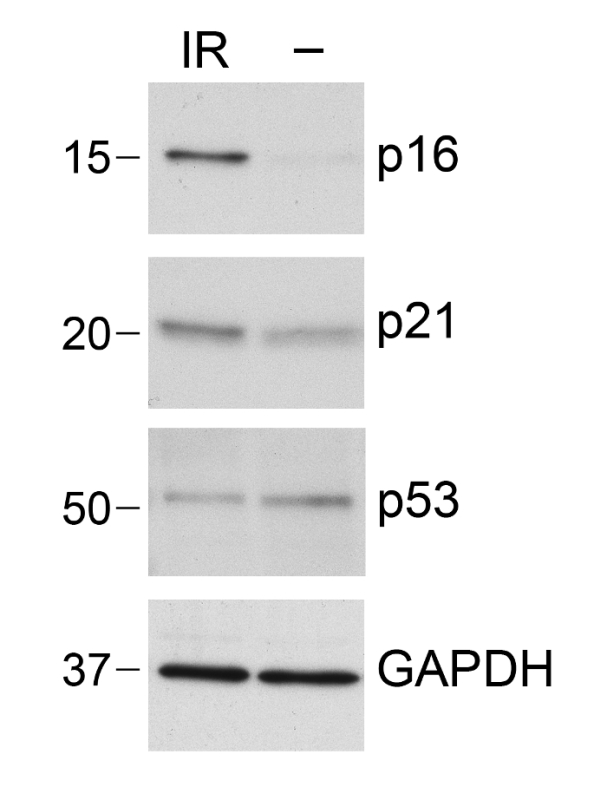 Figure 5. Quantification of Senescence-associated Proteins. Protein was isolated from either proliferating "young" (PDL 30) IMR-90 cells (-) or IMR-90 cells that were exposed to 10 Gy of ionizing radiation (IR). The medium was changed every 48 h and the cells were lysed 7 d after IR. Samples were analyzed by SDS-PAGE, immunoblotted with anti-p16INK4A antibodies, and re-probed with anti-p21 and then anti-p53 antibodies. GAPDH was used as a protein loading control. Please click here to view a larger version of this figure.
Figure 5. Quantification of Senescence-associated Proteins. Protein was isolated from either proliferating "young" (PDL 30) IMR-90 cells (-) or IMR-90 cells that were exposed to 10 Gy of ionizing radiation (IR). The medium was changed every 48 h and the cells were lysed 7 d after IR. Samples were analyzed by SDS-PAGE, immunoblotted with anti-p16INK4A antibodies, and re-probed with anti-p21 and then anti-p53 antibodies. GAPDH was used as a protein loading control. Please click here to view a larger version of this figure.
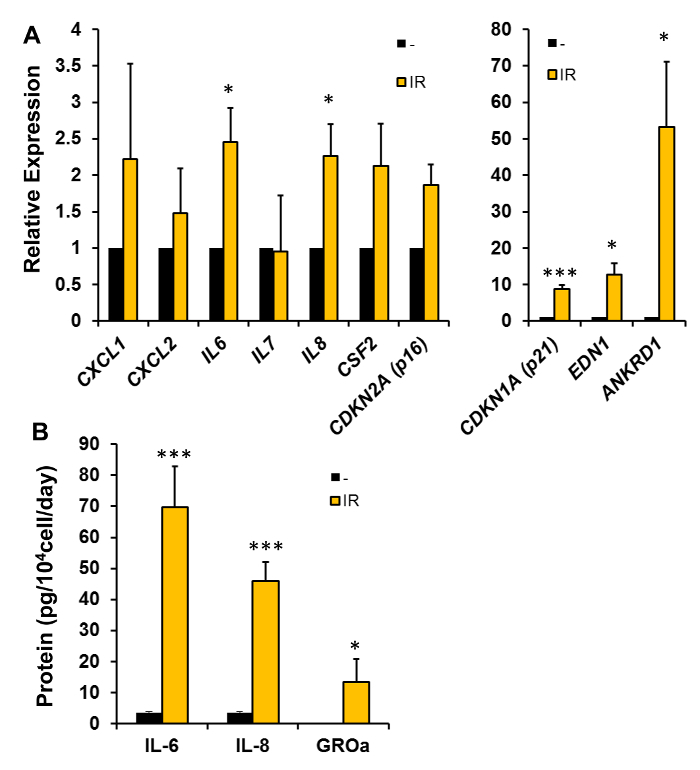 Figure 6. Quantification of Senescence-associated mRNAs and SASP Proteins. (A) RNA was isolated from either proliferating "young" (PDL 27 or 34) WI-38 cells (-) or WI-38 cells that were exposed to 10 Gy of ionizing radiation (IR). The medium was changed every 48 h and RNA isolation was performed 7 d after IR. RT-qPCR was used to quantify the levels of the indicated senescence-associated mRNAs using the gene-specific primers listed in Table 1. 18S levels were used for normalization. The histograms represent the mean normalized to untreated ±SEM from 5 independent experiments. (B) WI-38 cells (PDL 26) were exposed to 10 Gy of IR; 10 d later, the medium was changed to serum-free medium (see Section 7). After 24 h, conditioned medium was collected and IL-6, IL-8, and GROα levels were quantified using ELISA assays. The histogram represents the mean ±SEM from 3 experiments. * P < 0.05 and *** P < 0.001 by Student's t-test. Please click here to view a larger version of this figure.
Figure 6. Quantification of Senescence-associated mRNAs and SASP Proteins. (A) RNA was isolated from either proliferating "young" (PDL 27 or 34) WI-38 cells (-) or WI-38 cells that were exposed to 10 Gy of ionizing radiation (IR). The medium was changed every 48 h and RNA isolation was performed 7 d after IR. RT-qPCR was used to quantify the levels of the indicated senescence-associated mRNAs using the gene-specific primers listed in Table 1. 18S levels were used for normalization. The histograms represent the mean normalized to untreated ±SEM from 5 independent experiments. (B) WI-38 cells (PDL 26) were exposed to 10 Gy of IR; 10 d later, the medium was changed to serum-free medium (see Section 7). After 24 h, conditioned medium was collected and IL-6, IL-8, and GROα levels were quantified using ELISA assays. The histogram represents the mean ±SEM from 3 experiments. * P < 0.05 and *** P < 0.001 by Student's t-test. Please click here to view a larger version of this figure.
| Human Primers | Forward | Reverse |
| 18S | CCCTATCAACTTTCGATGGTAGTCG | CCAATGGATCCTCGTTAAAGGATTT |
| ANKRD1 | AGT AGA GGA ACT GGT CACTGG | TGG GCT AGA AGT GTCTTC AGA T |
| CDKN1A (p21) | GAC ACCACT GGA GGG TGA CT | CAGGTC CAC ATG GTC TTC CT |
| CDKN2A (p16) | CCAACGCACCGAATAGTTACG | GCGCTGCCCATCATCATG |
| CSF2 (GM-CSF) | GGCCCCTTGACCATGATG | TCTGGGTTGCACAGGAAGTTT |
| CXCL1 | GAAAGCTTGCCTCAATCCTG | CACCAGTGAGCTTCCTCCTC |
| CXCL2 | AACTGCGCTGCCAGTGCT | CCCATTCTTGAGTGTGGCTA |
| EDN1 | CAG CAGTCT TAG GCG CTG AG | ACTCTT TAT CCA TCA GGG ACG AG |
| IL6 | CCGGGAACGAAAGAGAAGCT | GCGCTTGTGGAGAAGGAGTT |
| IL7 | CTCCAGTTGCGGTCATCATG | GAGGAAGTCCAAAGATATACCTAAAAGAA |
| IL8 | CTTTCCACCCCAAATTTATCAAAG | CAGACAGAGCTCTCTTCCATCAGA |
Table 1: RT-qPCR Primers for Senescence-associated mRNAs. Forward and reverse primers for RT-qPCR using SYBR-green technology are indicated.
Discussion
Here, we have described methods for replicative and DNA-damage induced senescence using human diploid fibroblasts. In addition, techniques for quantifying protein and mRNA levels of various senescence-associated proteins are included, as well as staining for SA-β-gal and for the DNA-damage marker γ-H2AX. These protocols can be widely used to assess senescent phenotypes both in vitro and in vivo, although many caveats exist for characterizing senescence in vivo20. Other cells may express senescent markers, even if they are not senescent cells. For example, cells such as osteoclasts and macrophages have higher levels of β-gal activity32,33. In addition, many cancer cells have upregulated p16INK4A 34. Cells can also accumulate DNA damage (e.g., γ-H2AX foci) and not be senescent. SASP factors can also be upregulated by various normal physiological or pathological conditions that may or may not involve senescent cells. Therefore, multiple senescence markers should be utilized, especially when assessing senescence in vivo.
Although senescent cells were found to be increased in normal aging tissues more than 20 years ago4, it is only now that we are fully appreciating the plethora of roles that senescent cells play in both normal tissue homeostasis and in pathological conditions. The recent employment of mouse models to eliminate p16INK4A-positive senescent cells has allowed researchers to more clearly and precisely appreciate the role of senescent cells in tissues and organs35,36,37. These models have uncovered that senescent cells contribute to a multitude of age-related pathologies, tumorigenesis, and mouse lifespan35,36,37. Given these recent discoveries and the validation of the importance of senescent cells in physiology, the time is ripe for more research on these specific cells. For example, studies employing strategies to therapeutically remove senescent cells or to delay senescence could be beneficial for both healthspan and lifespan studies.
Recently, our laboratory found that metformin, a drug commonly used to treat type 2 diabetes mellitus, decreases cellular senescence38. As metformin is currently being proposed as an anti-aging therapy and has anti-cancer properties39,40, it will be interesting in the future to explore whether this drug, or other aging interventions, affect health outcomes by modulating cellular senescence.
The protocols we discuss here are common and widely accepted techniques to assess the senescent phenotype. For cells grown in vitro, it is critical to monitor cell changes from a spindle-like to a flattened morphology in order to ensure that the cells are senescent (See Figures 1 & 2). If cells are not positive for SA-β-gal or other markers described here, assess senescence at higher cell PDLs (for replicative senescence) or at an increased time after DNA damage-induced senescence. In addition, these protocols may need to be optimized for different cell types. For assessing SASP protein levels, it is critical to remove the serum from the cell medium, as this will cause high background levels in the ELISA or protein arrays. Therefore, keep this in mind when designing experiments, as it may not be ideal for simultaneously assessing multiple markers.
Staining for SA-β-gal, as described here, is limited by the fact that it requires the fixation of the cells. In addition, cell confluence has been shown to influence SA-β-gal activity. Protocols have been developed to assess SA-β-Gal activity using a fluorogenic substrate for β-gal activity, which allows for the quantification of this marker in live cells using flow cytometry41,42,43. SA-β-gal activity can also be measured in cell lysates using a chemiluminescent method44. In addition, γ-H2AX can also be measured using various other methods, including flow cytometry or the immunoblotting of cell lysates45.
It should be noted that SAHF formation is cell-line dependent and is mostly associated with oncogene-induced senescence20,30,46. Therefore, SAHF may not be present in all senescence models. The utility of SAHF as a senescent marker in mouse models may also be limited by the fact that SAHF is generally not observed in mouse senescent cells30,46,47. Here, antibodies against H3K9Me2 were used, but other molecular markers of SAHF can be used, including macroH2A, high mobility group A (HMGA) proteins, and tri-methylated lysine 9 histone H3 (H3K9Me3) and bound HP1 proteins. In addition, chromatin immunoprecipitation assays can be used to assess histone marks or the association of chromatin binding proteins to E2F target genes.
One should be aware that there are various limitations to characterizing senescence, including heterogeneity for markers and the fact that these markers are not expressed exclusively in senescent cells. Multiple (in general, 2 - 3) markers should be employed to assess senescence, including markers not described here. For example, DNA-SCARS, telomere damage, and/or DNA damage signaling can also be quantified. Furthermore, as senescence and, in particular, the SASP phenotype, has recently been shown to be affected by the specific stressor or damage that induces senescence11,12,13, it is important to assess multiple markers in each experimental paradigm.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This study was supported by the Intramural Research Program of the National Institutes of Health, National Institute on Aging. The authors wish to thank Myriam Gorospe and Kotb Abdelmohsen for many helpful discussions about senescence and Kotb Abdelmohsen for also critically reading the manuscript. We also thank our laboratory members, especially Douglas Dluzen for critically reading the manuscript.
References
- Hayflick L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509(7501):439–446. doi: 10.1038/nature13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- Dimri G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24(22):2463–2479. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pospelova TV, et al. Pseudo-DNA damage response in senescent cells. Cell Cycle. 2009;8(24):4112–4118. doi: 10.4161/cc.8.24.10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113(6):703–716. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005;37(5):961–976. doi: 10.1016/j.biocel.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120(4):513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Coppe JP, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga-Yamanaka K, et al. NF90 coordinately represses the senescence-associated secretory phenotype. Aging (Albany NY) 2012;4(10):695–708. doi: 10.18632/aging.100497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley CD, et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016;23(2):303–314. doi: 10.1016/j.cmet.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoare M, et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat Cell Biol. 2016;18(9):979–992. doi: 10.1038/ncb3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F. Detection of the senescence-associated secretory phenotype (SASP) Methods Mol Biol. 2013;965:165–173. doi: 10.1007/978-1-62703-239-1_10. [DOI] [PubMed] [Google Scholar]
- Srikantan S, Marasa BS, Becker KG, Gorospe M, Abdelmohsen K. Paradoxical microRNAs: individual gene repressors, global translation enhancers. Cell Cycle. 2011;10(5):751–759. doi: 10.4161/cc.10.5.14825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelmohsen K, Gorospe M. Noncoding RNA control of cellular senescence. Wiley Interdisciplinary Reviews: RNA. 2015. [DOI] [PMC free article] [PubMed]
- Bhaumik D, et al. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging. 2009;1(4):402–411. doi: 10.18632/aging.100042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, et al. DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J Cell Sci. 2011;124(PT 1):68–81. doi: 10.1242/jcs.071340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardes de Jesus B, Blasco MA. Assessing cell and organ senescence biomarkers. Circ Res. 2012;111(1):97–109. doi: 10.1161/CIRCRESAHA.111.247866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015;15(7):397–408. doi: 10.1038/nrc3960. [DOI] [PubMed] [Google Scholar]
- Rubio MA, Kim S-H, Campisi J. Reversible Manipulation of Telomerase Expression and Telomere Length: implications for the ionizing radiation response and replicative senescence of human cells. J Biol Chem. 2002;277(32):28609–28617. doi: 10.1074/jbc.M203747200. [DOI] [PubMed] [Google Scholar]
- Chen QM, Prowse KR, Tu VC, Purdom S, Linskens MHK. Uncoupling the Senescent Phenotype from Telomere Shortening in Hydrogen Peroxide-Treated Fibroblasts. Experimental Cell Research. 2001;265(2):294–303. doi: 10.1006/excr.2001.5182. [DOI] [PubMed] [Google Scholar]
- Dumont P, et al. Induction of replicative senescence biomarkers by sublethal oxidative stresses in normal human fibroblast. Free Radic Biol Med. 2000;28(3):361–373. doi: 10.1016/s0891-5849(99)00249-x. [DOI] [PubMed] [Google Scholar]
- Roninson IB. Tumor Cell Senescence in Cancer Treatment. Cancer Research. 2003;63(11):2705–2715. [PubMed] [Google Scholar]
- Nyunoya T, et al. Cigarette Smoke Induces Cellular Senescence. J Respir Cell Mol Biol. 2006;35(6):681–688. doi: 10.1165/rcmb.2006-0169OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai H, Gao Y, Hoyle DL, Cheng T, Wang ZZ. Suppression of Transforming Growth Factor-β Signaling Delays Cellular Senescence and Preserves the Function of Endothelial Cells Derived From Human Pluripotent Stem Cells. Stem Cells Transl Med. 2016. [DOI] [PMC free article] [PubMed]
- Senturk S, et al. Transforming growth factor-beta induces senescence in hepatocellular carcinoma cells and inhibits tumor growth. Hepatology. 2010;52(3):966–974. doi: 10.1002/hep.23769. [DOI] [PubMed] [Google Scholar]
- Aird KM, Zhang R. Detection of senescence-associated heterochromatin foci (SAHF) Methods Mol Biol. 2013;965:185–196. doi: 10.1007/978-1-62703-239-1_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pospelova TV, Chitikova ZV, Pospelov VA. An integrated approach for monitoring cell senescence. Methods Mol Biol. 2013;965:383–408. doi: 10.1007/978-1-62703-239-1_26. [DOI] [PubMed] [Google Scholar]
- Di Micco R, et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat Cell Biol. 2011;13(3):292–302. doi: 10.1038/ncb2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright WE, Pereira-Smith OM, Shay JW. Reversible cellular senescence: implications for immortalization of normal human diploid fibroblasts. Mol Cell Biol. 1989;9(7):3088–3092. doi: 10.1128/mcb.9.7.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bursuker I, Rhodes JM, Goldman R. Beta-galactosidase--an indicator of the maturational stage of mouse and human mononuclear phagocytes. J Cell Physiol. 1982;112(3):385–390. doi: 10.1002/jcp.1041120312. [DOI] [PubMed] [Google Scholar]
- Kopp HG, Hooper AT, Shmelkov SV, Rafii S. Beta-galactosidase staining on bone marrow. The osteoclast pitfall. Histol Histopathol. 2007;22(9):971–976. doi: 10.14670/HH-22.971. [DOI] [PubMed] [Google Scholar]
- Witkiewicz AK, Knudsen KE, Dicker AP, Knudsen ES. The meaning of p16(ink4a) expression in tumors: functional significance, clinical associations and future developments. Cell Cycle. 2011;10(15):2497–2503. doi: 10.4161/cc.10.15.16776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530(7589):184–189. doi: 10.1038/nature16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479(7372):232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria M, et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev Cell. 2014;31(6):722–733. doi: 10.1016/j.devcel.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noren Hooten N, et al. Metformin-mediated increase in DICER1 regulates microRNA expression and cellular senescence. Aging Cell. 2016;15(3):572–581. doi: 10.1111/acel.12469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA. Metformin as a Tool to Target Aging. Cell Metab. 2016;23(6):1060–1065. doi: 10.1016/j.cmet.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell Metab. 2014;20(6):953–966. doi: 10.1016/j.cmet.2014.09.018. [DOI] [PubMed] [Google Scholar]
- Cahu J, Sola B. A sensitive method to quantify senescent cancer cells. J Vis Exp. 2013. [DOI] [PMC free article] [PubMed]
- Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc. 2009;4(12):1798–1806. doi: 10.1038/nprot.2009.191. [DOI] [PubMed] [Google Scholar]
- Noppe G, et al. Rapid flow cytometric method for measuring senescence associated beta-galactosidase activity in human fibroblasts. Cytometry A. 2009;75(11):910–916. doi: 10.1002/cyto.a.20796. [DOI] [PubMed] [Google Scholar]
- Bassaneze V, Miyakawa AA, Krieger JE. Chemiluminescent detection of senescence-associated beta galactosidase. Methods Mol Biol. 2013;965:157–163. doi: 10.1007/978-1-62703-239-1_9. [DOI] [PubMed] [Google Scholar]
- Redon CE, et al. gamma-H2AX detection in peripheral blood lymphocytes, splenocytes, bone marrow, xenografts, and skin. Methods Mol Biol. 2011;682:249–270. doi: 10.1007/978-1-60327-409-8_18. [DOI] [PubMed] [Google Scholar]
- Kosar M, et al. Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner and follow expression of p16(ink4a) Cell Cycle. 2011;10(3):457–468. doi: 10.4161/cc.10.3.14707. [DOI] [PubMed] [Google Scholar]
- Kennedy AL, et al. Senescent mouse cells fail to overtly regulate the HIRA histone chaperone and do not form robust Senescence Associated Heterochromatin Foci. Cell Div. 2010;5:16. doi: 10.1186/1747-1028-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]