

Abstract
Myeloid cell leukemia-1 (Mcl-1) is an anti-apoptotic member of the Bcl-2 family of proteins that is overexpressed and amplified in many cancers. Overexpression of Mcl-1 allows cancer cells to evade apoptosis and contributes to the resistance of cancer cells to be effectively treated with various chemotherapies. From an NMR-based screen of a large fragment library, several distinct chemical scaffolds that bind to Mcl-1 were discovered. Here, we describe the discovery of potent tricyclic 2-indole carboxylic acid inhibitors that exhibit single digit nanomolar binding affinity to Mcl-1 and greater than 1700-fold selectivity over Bcl-xL and greater than 100 fold selectivity over Bcl-2. X-ray structures of these compounds when complexed to Mcl-1 provide detailed information on how these small-molecules bind to the target, which was used to guide compound optimization.
Keywords: Fragment-based screening, apoptosis, cancer, Mcl-1, inhibitor, drug discovery
INTRODUCTION
The intrinsic apoptosis pathway is tightly regulated by a balance of pro-apoptotic and anti-apoptotic proteins that respond to numerous extracellular and intracellular stresses, including growth factor and oxygen deprivation, DNA damage, oncogene induction, metabolic changes, and the effects of cytotoxic drugs.1 In normal cells, these stresses induce oligomerization of the pro-apoptotic proteins Bax and Bak at the mitochondrial outer membrane, which triggers cytochrome c release and caspase-dependent apoptosis.2 Anti-apoptotic Bcl-2 family proteins, including Bcl-2, Bcl-xL, Bcl-w, Bcl-A1, and Mcl-1, bind to and sequester pro-apoptotic members of the same family, leading to the inhibition of apoptosis.3 In cancers, anti-apoptotic Bcl-2-family proteins are often upregulated causing the enhancement of cancer cell survival in otherwise stressful conditions and contributes to increased resistance to anti-cancer therapeutics.4
Amplification of the gene encoding the anti-apoptotic protein Myeloid cell leukemia-1 (Mcl-1) is one of the most common genetic aberrations in human cancers.5,6 Mcl-1 overexpression is associated with high tumor grade and poor survival in lung7, breast8, prostate9, pancreatic10, ovarian11, and cervical cancers12, as well as melanoma13 and leukemia14. Mcl-1 activity has also been implicated in the resistance to multiple therapies, including microtubule-targeting agents like paclitaxel and vincristine, which are widely prescribed for cancer patients.15 Thus, neutralizing the function of Mcl-1 holds promise as an effective strategy to restore apoptotic signaling in cancer cells and enhance the response to currently approved chemotherapeutics. Despite this compelling rationale, no clinical therapeutic agent selectively targeting Mcl-1 is currently available.
Mcl-1 mediates its effects primarily through protein-protein interactions and is therefore considered difficult to target with small molecules.16 However, small molecule inhibitors that potently bind to the related family members Bcl-2 and Bcl-xL have been discovered.17–25 Among them, are the Bcl-2/Bcl-xL dual inhibitor ABT-263 and the selective Bcl-2 inhibitor ABT-199, both of which are in clinical trials.24,26,27 These inhibitors were reported to bind to their target proteins with 2–3 orders of magnitude higher affinities than the best Mcl-1 inhibitors reported to date in the literature.28–32
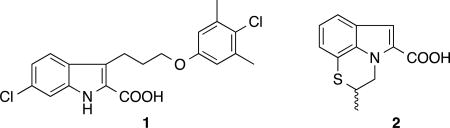
To discover Mcl-1 inhibitors, we have previously conducted a fragment-based screen of approximately 15,000 compounds using NMR and reported on the discovery of high-affinity (Ki <100 nM) inhibitors that bind to Mcl-1 starting from two classes of hits identified in the screen.28 The best inhibitor in this series was an optimized 2-indole carboxylic acid derivative 1 that binds to Mcl-1 with a Ki of 55 nM.28
Several additional structurally distinct small molecules were identified in the fragment screen with similar or higher binding affinities and/or ligand efficiency (LE). One of these series contains a tricyclic indole 2-carboxylic acid core, as found in the fragment hit 2. Here we describe the discovery of very potent Mcl-1 inhibitors that were derived from this fragment hit. These compounds have single digit nanomolar binding affinity and high selectivity for Mcl-1 over Bcl-xL and Bcl-2. Crystal structures of this series of inhibitors when bound to Mcl-1 provide detailed information on the molecular interactions that stabilize complex formation.
Structure of Compound 1 & 2
RESULTS AND DISCUSSION
Synthesis
The general synthetic approach used for synthesizing tricyclic 2-indole carboxylic acids derivatives is illustrated in Scheme 1 – 3. The preparation of the tricyclic analogs based on the fragment hit 2 that are listed in Table 1 is depicted in Scheme 1. The appropriate bicyclic hydrazine 10 was reacted with a selected α-ketoester 11 to give the tricyclic indole-2-carboxylic acid 3–8 via Fisher indole reaction33,34 followed by saponification.
Scheme 1.

Synthesis of tricyclic indole carboxylic acid cores
(a) EtOH, H2SO4; (b) KOH (aq.), THF, 56–90% (two steps).
Scheme 3.

Synthesis of tricyclic 6-Cl-2-indole carboxylic acid derivatives
(a) (1) NaNO2, HCl, sodium acetate, ethyl 2-oxocyclopentane carboxylate, H2O, 60% (two steps); (2) EtOH, H2SO4; (b) BH3-THF, THF, 83%; (c) Dt-BuAD, PPh3, THF, 82%; (d) Cs2CO3, DMF, 95%; (e) AIBN, Bu3SnH, toluene; (f) LiOH (aq.), THF, 50% (two steps); (g) bis(pinacolato)diboron, Pd(dppf)2Cl2:CH2Cl2, DMF, 35%; (h) H2O2, NaOH (aq.), THF, 28%; (i) Cs2CO3, DMF, 74%.
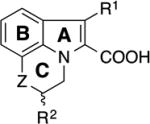
Table 1.
SAR of tricyclic indole 2-carboxylic acids for binding to Mcl-1.
|
|
||||||
|---|---|---|---|---|---|---|
| compd | Z | R1 | R2 | Ki (µM) | LE | |
|
|
||||||
|
2 | S | H | Me | 35 | 0.38 |
| 3 | S | H | H | 51 | 0.39 | |
| 4 | CH2 | H | H | 131 | 0.35 | |
| 5 | S | Me | H | 18 | 0.41 | |
| 6 | CH2 | Me | H | 90 | 0.35 | |
| 7 | 0 | Me | H | 18 | 0.41 | |
|
|
||||||
| 8 | 2-Indole-COOH | >1000 | ||||
|
|
||||||
The synthetic route illustrated in Scheme 2 was developed to prepare the compounds depicted in Table 2. Using the Fischer indole condition depicted in Scheme 1, the selected hydrazine 10 was cyclized with a selected α-ketoacid 12 to give a mixture of corresponding tricyclic indole diester 13a and indole acid 13b. Formation of the tricyclic indole diester 13a was improved by extending the reaction time, however, the reaction suffered from a poor yield. Therefore, the indole acid 13b in the mixture was converted to the methyl ester 13c using TMSCHN2 then both indole esters 13a and 13c were carried to the subsequent reaction without isolation. The alcohol 14 was prepared by selective reduction of the extended ethyl ester at the 3-position of the indole using BH3-THF. Mitsunobu condensation (as used in our previous report28) was used to couple a selected phenol to yield the phenyl ether 15. Subsequent saponification of the ester gave the desired carboxylic acid moiety in compounds 16–21, 23, 26–33. The sulfide groups in the tricyclic indole acids 19 and 23 were oxidized to the corresponding sulfoxide 24 or sulfone 22 and 25 using m-CPBA.
Scheme 2.

Synthesis of tricyclic indole carboxylic acid derivatives
(a) EtOH, H2SO4; (b) TMSCHN2 (2M in hexane), MeOH, benzene; (c) BH3-THF, THF; 20% (d) Dt-BuAD, PPh3, THF; (e) KOH (aq.), THF, 12–45% (six steps); (f) m-CPBA, CH2Cl2, 56–88%.
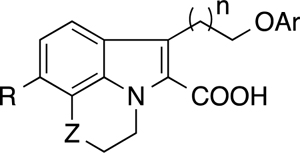
Table 2.
SAR of the merged tricyclic indole derivatives for binding to Mcl-1 and Bcl-xL
| ||||||||
|---|---|---|---|---|---|---|---|---|
| compd | Z | n | R | Ar | Mcl-1 |
Ki (µM) Bcl-xL |
Bcl-2 | |
| 16 | S | 1 | H | 1-naphthyl | 3.0 | 9.0 | ||
| 17 | CH2 | 1 | H | 1-naphthyl | 4.0 | >20 | ||
| 18 | 0 | 1 | H | 1-naphthyl | 1.6 | 12.7 | ||
| 19 | S | 2 | H | 1-naphthyl | 0.12 | >20 | 3.1 | |
| 20 | CH2 | 2 | H | 1-naphthyl | 0.31 | >20 | 7.3 | |
| 21 | 0 | 2 | H | 1-naphthyl | 0.11 | 1.9 | 5.7 | |
| 22 | S02 | 2 | H | 1-naphthyl | 0.088 | >20 | ||
| 23 | SCH2 | 2 | H | 1-naphthyl | 0.17 | 7.4 | 9.9 | |
| 24 | SOCH2 | 2 | H | 1-naphthyl | 0.074 | >20 | 16.0 | |
| 25 | S02CH2 | 2 | H | 1-naphthyl | 0.061 | >20 | 11.7 | |
| 26 | S | 2 | H | 1 -(5,6,7,8-tetrahy dronaphthyl) | 0.150 | 3.0 | 3.7 | |
| 27 | S | 2 | H | 1-(4-Cl-naphthyl) | 0.200 | 4.7 | 1.1 | |
| 28 | S | 2 | H | 2-(5,6,7,8-tetrahydronaphthyl) | 0.410 | >20 | 4.4 | |
| 29 | S | 2 | H | 3-Me4-a-phenyl | 0.210 | >20 | 6.3 | |
| 30 | S | 2 | H | 3,5-di-Me4-Cl-phenyl-phenyl | 0.071 | >20 | 2.3 | |
| 31 | CH2 | 2 | H | 3,5-di-Me4-Cl-phenyl-phenyl | 0.110 | 7.8 | ||
| 32 | 0 | 2 | H | 3,5-di-Me4-Cl-phenyl-phenyl | 0.065 | |||
| 33 | SCH2 | 2 | H | 3,5-di-Me4-Cl-phenyl-phenyl | 0.040 | >20 | 3.7 | |
| 34 | CH2 | 2 | CI | 3,5-di-Me4-Cl-phenyl-phenyl | 0.003 | 5.2 | 0.77 | |
| 35 | 0 | 2 | CI | 3,5-di-Me4-Cl-phenyl-phenyl | 0.009 | >20 | 1.3 | |
Tricyclic 6-Cl-2-indole carboxylic acid 34 and 35 were prepared as outlined in Scheme 3. 2-Br-3-Cl-aniline 38 was converted to the indole 39 utilizing the modified Japp−Klingemann reaction condition.28 Selective BH3 reduction followed by a Mitsunobu reaction gave the indole ester 41. Allylation at the indole nitrogen gave compound 42, which underwent a radical cyclization to yield the tricyclic indole ester containing the piperidine C-ring. Subsequent saponification gave the desired acid 34. The intermediate 41 was converted to the pinacol borate 43, which was oxidized to give the phenol 44. The morpholine C-ring was constructed using di-bromoethane followed by saponification to produce the 6-Cl-indole acid 35.
Tricyclic Fragment Optimization
Among the initial hits obtained in the screen, we identified fragments containing a tricyclic indole 2-carboxylic acid that bind to Mcl-1 (Table 1). The thiazine-containing tricyclic fragment 2 displaces a FITC-labeled BAK peptide from Mcl-1 with a Ki of 35 µM in an FPA assay. SAR of the fragment core was investigated by using small tricyclic indoles with substitutions on rings A-C (Table 1) that were either purchased or synthesized as described in Scheme 1.
In general, all of the tricyclic indole acids exhibited similar binding affinity for Mcl-1 compared to the initial hit 2 with ligand efficiencies (LE) ≥ 0.35. Both compounds 3 and 4, containing C-ring moieties showed significantly enhanced binding affinity when compared to the unsubstituted indole 2-carboxylic acid 828. Improved affinity was also observed by substituting a methyl group at the R1 (compound 5, 6) or R2 (compound 2) positions compared to the unsubstituted fragments 3 and 4, suggesting that additional hydrophobic protein binding pockets may be accessible from these positions. In addition, C-ring analogs containing a thiazine moiety (3 and 5) or morpholine (7) were potent and preferred over the piperidine analogs 4 and 6. These results suggest that modifications in the C-ring are not only tolerated but can make additional interactions with Mcl-1 that result in improvements in binding affinity.
To determine how these tricyclic indole fragments bind to Mcl-1, we performed NMR-based structural studies using compound 2 bound to double-labeled (15N, 13C) Mcl-1. Several key NOEs were observed between the indole phenyl ring B and A227, M231, and F270 of Mcl-1. In addition, the thiazine methyl group at R2 exhibited weak NOEs involving T266 (Figure 1A). This pattern of intermolecular protein/ligand NOEs strongly suggests that compound 2 and similar analogs bind in the upper part of the second hydrophobic pocket17, P2, utilized by peptides that bind to Mcl-1. In peptides, this pocket is occupied by a highly conserved Leu residue.
Figure 1.

(A) Observed NOE interactions between Mcl-1 and compound 2. (B) Model structures of Mcl-1 complexed to fragment hit 2 using NMR-derived distance restraints. Residues (labeled) with NOEs to fragment 2 (green) are rendered as sticks (gray). R263 is also labeled.
The NMR-derived structural information was used to generate a model in which the carboxylic acid moiety of compound 2 on ring A points toward R263. In addition, the indole phenyl on ring B points towards A227 and M231 with ring C sitting next to the adjacent hydrophobic pocket (P3) (Figure 1B). This orientation places the indole 3-position (R1) to point into the lower P2 pocket. Thus, the model structure predicted that the R1 position is in an ideal location for extending deep into the pocket.
Fragment Merging with the Tricyclic Scaffold
We previously discovered that a substantial boost in affinity could be obtained by merging fragments that bind in the upper part of the P2 pocket to fragments that predominantly bind deeper into the pocket than Leu of the BH3 peptide.28 On the basis of the fragment SAR (Table 1), NMR-derived structural information, and molecular modeling, we designed and synthesized a new series of Mcl-1 inhibitors by merging molecules that bind deep into the P2 pocket to the tricyclic indoles. The binding affinity and selectivity of these compounds for binding to Mcl-1 are shown in Table 2. In an earlier study28, we found that both a 1-naphthyl and a 3,5-dimethyl-4-chloro-phenyl fragment bound to the lower P2 pocket and gave similar affinities for compounds containing both upper and lower P2 pocket binders. To generate the initial SAR in this series, a 1-naphthyl group as the lower P2 pocket binding group was connected to the R1 position of tricyclic 2-indole carboxylic acid cores bearing thiazine, piperidine or a morpholine C-ring moiety, using either a three- or four-atom linker. As expected, the merged compounds (16–21) exhibited markedly enhanced binding affinities compared to the parent compounds (2–7) that lack a lower pocket binding group. In particular, compounds containing a four-atom linker (19, 20 and 21) showed more than 2 orders of magnitude increased affinity for Mcl-1 compared to the initial fragment 2. The three-atom linker is not preferred, as compounds 16, 17 and 18 exhibited >10-fold higher Ki values compared to the corresponding four-atom linker analogs. Compounds containing different C-ring moieties had improved affinity from the original fragment and showed a parallel SAR trend with respect to linker length.
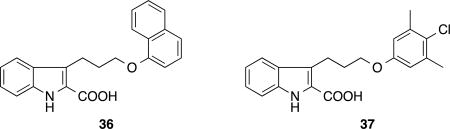
Structure of compounds 36 and 37
SAR of the C-ring Unit in the Merged Compounds
The influence of C-ring composition on binding to Mcl-1 was evaluated by preparing compounds 19–25 that contain heterocyclic units with varying ring sizes and functional groups. Similar to that observed for the fragments, the 6-membered C-ring moiety bearing S- (19) or O- (21) were slightly preferred to the compounds containing a carbon analog 20. The 7-membered thiazepine C-ring unit was well tolerated with compound 23 exhibiting near equal potency to the thiazine analog 19. We also explored the possibility of incorporating polar functional groups within the C-ring component. Interestingly, both sulfoxide and sulfone oxidation states (22, 24, 25) showed higher affinity to Mcl-1 than their parent sulfides (19 or 23). These observed trends in the SAR for the C-ring moiety were hypothesized to be advantageous for modulating the drug-like properties of ligands. For example, pharmaceutical agents containing a sulfone functional group generally exhibit more desirable properties, such as higher aqueous solubility and better metabolic stability, than the corresponding sulfide.35 Compound 25, the most potent Mcl-1 inhibitor (Ki = 61 nM) in this series, has more than a 5-fold improvement in potency when compared to the analog 36 (Ki = 330 nM)28, which contains the unsubstituted indole core lacking the C-ring moiety. These observations strongly suggest that introduction of a proper C-ring group in the series can enhance not only the binding affinity for Mcl-1 but also provides a pharmacophore unit which can be utilized to optimize the pharmaceutical properties of the molecules without sacrificing the potency of the parent ligand.
Optimization of the P2 Pocket Anchor Group
To further explore the SAR of the tricyclic indoles, compounds 26–30 were prepared by tethering lower pocket binding units that were identified in our earlier work28 along with new chemical moieties to the thiazine containing tricyclic indole fragment 3 using optimized four-atom linkers. Bicyclic aromatic P2 binding groups, such as 1′-(5,6,7,8-tetrahydronaphthyl) and 1′-(4′-Cl-naphthyl) contained in compounds 26 and 27, exhibited comparable potency to the 1-naphthyl analog 19. Conversely, compound 28 containing 2′-(5,6,7,8-tetrahydronaphthyl) showed a 3-fold reduction in affinity to Mcl-1 compared to its regioisomer 26 and the 1-naphthyl analog 19, indicating the importance of the attachment position for optimal binding. Compound 29 which contains a 3′-Me-4′-Cl-phenyl group displays a 240-fold improved potency over fragment 3, and compound 30 bearing a 3′,5′-di-Me-4′-Cl-phenyl moiety exhibited the most potent inhibitory activity against Mcl-1 with a Ki of 71 nM, representing a 720-fold improvement in affinity compared to the core unit 3.
To further investigate the effect of C-ring variations for compounds containing the 3,5′-di-Me-4′-Cl-phenyl group, compounds 30–33 were prepared. These inhibitors exhibited a parallel SAR trend compared to the corresponding 1-naphthyl analogs 19–21 and 23 with a 1.5–4.3 fold improvement in binding affinity. The thiazepine analog 33 displayed a Ki value (40 nM) which corresponds to a 7.5-fold increase in binding affinity compared to the indole analog 37 (Ki = 300 nM), which lacks a C-ring moiety.28
X-Ray structures of the Tricyclic Compounds
To guide further compound optimization, X-ray co-crystal structures of 17 and 24 complexed with Mcl-1 were obtained (Figure 2A–C). Both compounds 17 and 24 adopt similar binding poses as our earlier benzthiophene analog (PDB: 4HW3). In both structures, the naphthyl group penetrated into the deepest part of the hydrophobic pocket and induced a pronounced bend in helix 4 (α4) that likely helps to anchor the unit at this binding sub-site. In addition, the naphthyl group was positioned near F270, which causes favorable edge-to-face CH/π aromatic interactions with the phenyl group of this residue. The top of α4 sits close to the ligand causing the loop connecting helix 4–5 to be pulled towards the ligand allowing the carboxylic acid group of the ligand to engage in ion pairing interactions with the guanidinium moiety of R263.
Figure 2.

X-ray structures of tricyclic indole acids bound to Mcl-1. (A) Compounds 17 and 24 interact with Mcl-1 in the BH3-peptide binding cleft between helices 3, 4, and 5. The indole cores and naphthyl groups adopt almost the same binding poses while C-ring units adopt different binding conformations (inset). Shown is the surface depiction of Mcl-1 when complexed to (B) Compound 17 and (C) Compound 24 that illustrates how these ligands occupy the P2 pocket. Arrows indicate potential positions for substitutions to fill unoccupied sub-sites.
Interestingly, when the co-crystal structures of 17 and 24 are superimposed, the indole cores and naphthyl groups sit in almost the same position. However, the C-ring units adopt different binding conformations, and the larger thiazepane-oxide ring of 24 causes a pucker that localizes parts of the ring to be closer to A227 and T266 of Mcl-1 (Figure 2A). This may contribute to hydrophobic burial and could explain the slight improvement in binding affinity observed for this inhibitor. Finally, the X-ray structures predict that substitutions at the 6-position of the indole with small hydrophobic groups could fill the space between the ligands and helix 3 (α3) to gain additional binding affinity (Figure 2B and C).
Tricyclic Indole Core optimization by Substitutions of the B-Ring
From our X-ray structures and previously reported SAR28, we hypothesized that a substantial increase in binding affinity to Mcl-1 could be obtained by a substitution at the 6-position of the indole of the 6,5-fused heterocyclic carboxylic acid core. In a previously described series of compounds, a 6-Cl-indole addition (as exemplified in compound 1) exhibited greater than a 5-fold enhanced potency compared to the parent (37).28 To determine if this trend was the same for the tricyclic series, two 6-Cl-tricyclic indoles 34 and 35, containing piperidine and morpholine C-ring moieties respectively, were evaluated. Indeed, these modifications resulted in a >10-fold increase in binding affinities for both compounds, when compared to the des chloro analogs 31, 32. The affinity gain is likely the result of filling the pocket indicated by the arrow in Figure 2 (B,C). Our best inhibitor, 34, displayed a remarkable 36-fold higher potency over its des-Cl parent (31) and had a Ki of 3 nM and LE = 0.40. These results strongly suggest that binding contributions from the C-ring moiety and the 6-Cl group are additive and can even be synergistic for some combinations.
Mcl-1 Selectivity
Excessive inhibition of Bcl-xL in platelets causes mechanism-based, dose-dependent thrombocytopenia in preclinical studies24,36,37 and in clinical trials of the dual Bcl-2/Bcl-xL inhibitor ABT-263.24 Therefore, we sought to obtain selective Mcl-1 inhibitors that do not inhibit Bcl-xL to avoid these undesired adverse effects. To evaluate the selectivety of our series, binding affinities to the anti-apoptotic proteins Bcl-xL and Bcl-2 were measured in an FPA assay (Table 2). In all cases, the tricyclic 2-indole carboxylic acids (19–35) containing the 4-atom linker exhibited only weak affinity for both Bcl-xL (Ki ≥ 1.9 µM) and Bcl-2 (Ki ≥ 0.77 µM). Our most potent 6-Cl containing Mcl-1 inhibitors 34 and 35 exhibit >1700-fold binding selectivity over Bcl-xL and greater that 100-fold selectivity over Bcl-2. All structural modifications on the series that enhance the affinity for Mcl-1 had relatively little effect on improving binding to Bcl-xL or Bcl-2. As a result, the compounds became more selective against Bcl-xL as they became more potent for Mcl-1. Mcl-1 inhibitors likely achieve selectivity by filling the lower P2 pocket (Figure 2B) which is not present in Bcl-xL (PDB ID : 2YXJ) or BCL-2 (PDB ID : 4LVT) ligand structures.
Pulldown Experiment
To determine whether compound 34 could bind to cellular Mcl-1 and inhibit the binding of a peptide derived from a pro-apoptotic protein38, cell lysates from Human chronic myelogenous leukemia K562 cells were incubated with biotin labeled MS-1, a peptide that binds specifically to Mcl-1.39 This peptide selectively pulls down cellular Mcl-1. As shown in Figure 3, the addition of compound 34 blocks the ability of MS-1 to pulldown Mcl-1, demonstrating that the compound binds to cellular Mcl-1 and blocks the interaction with biotin MS-1 in a dose-dependent manner.
Figure 3.

Compound 34 inhibits binding of biotin-MS-1 to Mcl-1 in cell lysates. K562 cell lysates were incubated with compound or a Bim-BH3 peptide positive control at indicated concentrations. Streptavidin-conjugated beads bound with biotin-MS-1 were incubated with the lysates to pulldown cellular Mcl-1, which was visualized by Western blot.
Conclusion
We have described the optimization of a novel series of small molecule Mcl-1 inhibitors based on a hit obtained from an NMR-based fragment screen. NMR-based structural studies and SAR exploration resulted in a binding affinity increase of 100-fold over the initial micromolar fragment hit and further optimization, guided by structural data, resulted in an additional 30-fold gain in affinity. The resulting compound binds to Mcl-1 with a Ki = 3 nM, is selective over Bcl-2 and Bcl-xL, and binds to cellular Mcl-1 to prevent binding of a biotin labeled peptide that binds specifically to Mcl-1. An important result of this study is that the SAR knowledge learned from one fragment core could be rapidly translated to other related fragment hits. We were able to rapidly replace the core of our initial indole series28 with another fragment hit to obtain novel compounds with high Mcl-1 potency and selectivity.
While this manuscript was in review, Leverson et al published the characterization of a potent (Ki = 0.454 nM) small molecule inhibitor, A-1210477.40 This study emphasized the exquisite potency (“subnanomolar and often low picomolar affinities”) needed to demonstrate on-target cellular effects. Therefore, the low nanomolar compounds (e.g. 34, Ki = 3.0 nM) described here are not expected to display unambiguous on-target cellular activity. However, this tricyclic 2-indole carboxylic acid represents an excellent starting point for additional optimization to achieve these goals.
EXPERIMENTAL SECTION
Chemistry
General
All NMR spectra were recorded at room temperature on a 400 MHz AMX Bruker spectrometer. 1H chemical shifts are reported in δ values in ppm downfield with the deuterated solvent as the internal standard. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), integration, coupling constant (Hz). Low resolution mass spectra were obtained on an Agilent 1200 series 6140 mass spectrometer with electrospray ionization. All samples were of ≥95% purity as analyzed by LC−UV/vis-MS. Analytical HPLC was performed on an Agilent 1200 series with UV detection at 214 and 254 nm along with ELSD detection. LC/MS parameters were as follows: Phenomenex-C18 Kinetex column, 50 × 2.1 mm, 2 min gradient, 5% (0.1% TFA/MeCN) / 95% (0.1% TFA/ H2O) to 100% (0.1% TFA/MeCN). Preparative purification was performed on a Gilson HPLC (Phenomenex-C18, 100 × 30 mm, 10 min gradient, 5→95% MeCN/H2O with 0.1% TFA) or by automated flash column chromatography (Isco, Inc. 100sg Combiflash). Solvents for extraction, washing, and chromatography were HPLC grade. All reagents were purchased from chemical suppliers and used without purification.
6-Methyl-2,3-dihydro-[1,4]thiazino[2,3,4-hi]indole-5-carboxylic acid (5). General Procedure for the Fischer Indole Reaction
To a stirred solution of 2,3-dihydro-4H-benzo[b][1,4]thiazin-4-amine (83 mg, 0.50 mmol) in absolute EtOH (2 mL) was added methyl 2-oxobutanoate (58 mg, 0.50 mmol) at 20 °C. The reaction mixture was stirred for 30 min at 50 °C then cooled to 0 °C. Concentrated H2SO4 (0.2 mL) was added dropwise then the reaction mixture was warmed to 50 °C and stirred for an additional 4 h. The reaction mixture was cooled to 20 °C then concentrated in vacuo. The residue was dissolved in CH2Cl2 (5 mL), and the organic solution was washed with saturated aqueous NaHCO3 solution followed by brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was dissolved in 1:1 mixture of MeOH/THF (2 mL) then KOH (280 mg, 5.0 mmol) was added. The reaction mixture was stirred for 2 h at 50 °C then concentrated in vacuo. The residue was dissolved in H2O then acidified to pH 1 with concentrated HCl. The mixture was extracted with EtOAc, dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by reverse phase preparative HPLC (H2O/CH3CN gradient to 95% CH3CN/0.1% TFA) to yield the title compound (103 mg, 0.44 mmol) as a white solid. 1HNMR(400 MHz, DMSO-d6): δ (ppm) 7.45 (d, J = 8.0 Hz, 1 H), 7.09 (d, J = 7.7 Hz, 1H), 7.02 (dd, J = 8.0, 7.7 Hz, 1 H), 4.73−4.71 (m, 2 H), 3.32−3.30 (m, 2 H), 2.52 (s, 3 H): >98% at 215 nm, MS (ESI) m/z = 234.1 [M + H]+.
1-Methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinoline-2-carboxylic acid (6)
The general procedure for Fischer indole reaction was followed using 3,4-dihydroquinolin-1(2H)-amine (74 mg, 0.50 mmol) and methyl 2-oxobutanoate (58 mg, 0.50 mmol) to yield the title compound (97 mg, 0.45mmol). 1HNMR(400 MHz, DMSO-d6): δ (ppm) 7.45 (dd, J = 6.6, 2.6 Hz, 1 H), 7.01–6.96 (m, 2 H), 4.43 (t, J = 5.7 Hz, 2 H), 2.91 (t, J = 6.1 Hz, 2 H), 2.52 (s, 3 H), 2.15−2.09 (m, 2 H); >98% at 215 nm, MS (ESI) m/z = 216.1 [M + H]+.
6-Methyl-2,3-dihydro-[1,4]oxazino[2,3,4-hi]indole-5-carboxylic acid (7)
The general procedure for Fischer indole reaction was followed using 2,3-dihydro-4H-benzo[b][1,4]oxazin-4-amine (100 mg, 0.66 mmol) and methyl 2-oxobutanoate (85 mg, 0.73 mmol) to yield the title compound (81 mg, 0.37mmol). 1HNMR(400 MHz, DMSO-d6): δ (ppm) 7.20 (d, J = 8.0 Hz, 1 H), 6.95 (dd, J = 8.0, 7.6 Hz, 1H), 6.67 (d, J = 7.6 Hz, 1 H), 4.52–4.47 (m, 4 H), 2.52 (s, 3 H).; >98% at 215 nm, MS (ESI) m/z = 218.1 [M + H]+.
6-(2-(Naphthalen-1-yloxy)ethyl)-2,3-dihydro-[1,4]thiazino[2,3,4-hi]indole-5-carboxylic acid (16). General Procedure
A solution of 2,3-dihydro-4H-benzo[b][1,4]thiazin-4-amine (336 mg, 2.0 mmol) and 2-oxopentanedioic acid (325 mg, 2.2 mmol) in EtOH (5 mL) was stirred at 50 °C for 30 min then cooled to 0 °C. To the reaction mixture was added conc. H2SO4 (0.5 mL) dropwise at 0 °C. The reaction mixture was stirred for 2h at 60 °C then quenched by pouring into ice then extracted with CH2Cl2. The combined organic layer was washed with sat. NaHCO3, water, brine, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by filtering through a silica gel column using Et2O as an eluent. The filtrate was concentrated in vacuo to give 1:1 mixture of 6-(2-ethoxy-2-oxoethyl)-2,3-dihydro-[1,4]thiazino[2,3,4-hi]indole-5-carboxylic acid and ethyl 6-(2-ethoxy-2-oxoethyl)-2,3-dihydro-[1,4]thiazino[2,3,4-hi]indole-5-carboxylate as an off-white solid in 145 mg. This mixture of products was directly used for the subsequent step without further purification.
To a solution of a mixture of products (145 mg) from the previous step in MeOH and benzene mixture (1:10, 2.0 mL) was added TMSCHN2 (2M in hexane) until bubbling stopped and a yellow color of the reaction mixture persisted. The reaction mixture was stirred for additional 10 min at 20 °C then concentrated in vacuo to give a mixture of methyl and ethyl 6-(2-ethoxy-2-oxoethyl)-2,3-dihydro-[1,4]thiazino[2,3,4-hi]indole-5-carboxylate. They were directly used for the subsequent step without further purification.
To a solution of a mixture of methyl and ethyl 6-(2-ethoxy-2-oxoethyl)-2,3-dihydro-[1,4]thiazino[2,3,4-hi]indole-5-carboxylate from the previous step in THF (2.0 mL) was added BH3 in THF (2 mL, 2 mmol) at 20 °C. The reaction mixture was stirred for 5h at 20 °C and quenched by addition of MeOH then concentrated in vacuo. The residue was purified by flash chromatography (Combi-flash Rf Hexane/EtOAc gradient 0–100%) to give a mixture of methyl and ethyl 6-(2-hydroxyethyl)-2,3-dihydro-[1,4]thiazino[2,3,4-hi]indole-5-carboxylate as a colorless oil in 110 mg (0.40 mmol).
To a solution of a mixture of methyl and ethyl 6-(2-hydroxyethyl)-2,3-dihydro-[1,4]thiazino[2,3,4-hi]indole-5-carboxylate (50 mg, 0.18 mmol), PPh3 (71 mg, 0.27 mmol) and naphthalen-1-ol (44 mg, 0.28mmol) in THF (1.0 mL) was added Dt-BuAD (62 mg, 0.27 mmol) at 20 °C. The reaction mixture was stirred for 15h at 20 °C. To the reaction mixture was added KOH (101 mg, 10 mmol) and MeOH (1.0 mL). The reaction mixture was stirred for 2h at 65 °C then concentrated in vacuo. The crude product was purified by reverse phase preparative HPLC (Phenomenex Gemini C18, H2O/CH3CN gradient to 95% CH3CN 0.1% TFA) to yield the title compound (43 mg, 0.11 mmol) as a light yellow solid. 1HNMR(400 MHz, DMSO-d6): δ 8.07 (d, J = 8.4 Hz, 1 H), 7.82(d, J = 8.0, Hz, 1H), 7.68 (dd, J = 7.4, 1.6 Hz, 1H), 7.49 (dd, J = 6.9, 1.6 Hz, 1H), 7.47−7.34 (comp, 3H), 7.14−7.08 (comp, 2H), 6.96 (d, J = 7.3 Hz, 1H), 4.77−4.74 (m, 2H), 4.36 (t, J = 6.6 Hz, 2H), 3.68 (t, J = 6.7 Hz, 2H), 3.35−3.10 (m, 2H); >98% at 215 nm, MS (ESI) m/z = 390.1 [M + H]+.
1-(2-(Naphthalen-1-yloxy)ethyl)-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinoline-2-carboxylic acid (17)
The title compound was prepared as a white solid according to procedures described for preparing compound 16 by substituting 2,3-dihydro-4H-benzo[b][1,4]thiazin-4-amine with 3,4-dihydroquinolin-1(2H)-amine in 23% overall yield. 1H NMR (400 MHz, DMSO-d6) δ 8.10 (d, J = 8.3 Hz, 1H), 7.83 (d, J = 8.1 Hz, 1H), 7.67 (dd, J = 7.7, 1.5 Hz, 1H), 7.49 (ddd, J = 8.2, 6.8, 1.3 Hz, 1H), 7.45 – 7.31 (m, 3H), 7.10 – 7.00 (m, 2H), 6.96 (d, J = 7.5 Hz, 1H), 4.50 – 4.42 (m, 2H), 4.35 (t, J = 6.7 Hz, 2H), 3.67 (t, J = 6.7 Hz, 2H), 2.92 (t, J = 6.1 Hz, 2H), 2.18 – 2.04 (m, 2H); >98% at 215 nm, MS (ESI) m/z = 372.2 [M + H]+.
6-(2-(Naphthalen-1-yloxy)ethyl)-2,3-dihydro-[1,4]oxazino[2,3,4-hi]indole-5-carboxylic acid (18)
The title compound was prepared as a white solid according to procedures described for preparing compound 16 by substituting 2,3-dihydro-4H-benzo[b][1,4]thiazin-4-amine with 2,3-dihydro-4H-benzo[b][1,4]oxazin-4-amin in 18% overall yield. 1H NMR (400 MHz, DMSO-d6) δ 8.09 (d, J = 8.1 Hz, 1H), 7.83 (d, J = 8.1 Hz, 1H), 7.51 − 7.46 (m, 1H), 7.45 − 7.32 (m, 4H), 7.02 (t, J = 7.9 Hz, 1H), 6.96 (d, J = 7.5 Hz, 1H), 6.71 (d, J = 7.3 Hz, 1H), 4.57 (t, J = 4.7 Hz, 2H), 4.48 (t, J = 4.6 Hz, 2H), 4.37 (t, J = 6.8 Hz, 2H), 3.67 (t, J = 6.7 Hz, 2H); >98% at 215 nm, MS (ESI) m/z = 374.1 [M + H]+.
6-(3-(Naphthalen-1-yloxy)propyl)-2,3-dihydro-[1,4]thiazino[2,3,4-hi]indole-5-carboxylic acid (19)
A mixture of methyl and ethyl 6-(3-hydroxypropyl)-2,3-dihydro-[1,4]thiazino[2,3,4-hi]indole-5-carboxylate (465 mg, 1.6 mmol) was prepared as a yellow oil according to procedures described for preparing compound 16 using 2,3-dihydro-4H-benzo[b][1,4]thiazin-4-amine (500 mg, 3.0 mmol) and 2-oxohexanedioic acid (722 mg, 4.5 mmol) in 53% overall yield.
The title compound was prepared (25 mg, 0.062 mmol) as an off-white solid following the Mitsunobu reaction procedure described for preparing compound 16 using a mixture of methyl and ethyl 6-(3-hydroxypropyl)-2,3-dihydro-[1,4]thiazino[2,3,4-hi]indole-5-carboxylate (56 mg, 0.2 mmol). 1H NMR (400 MHz, DMSO-d6) δ 8.21 – 8.15 (m, 1H), 7.90 – 7.82 (m, 1H), 7.57 – 7.34 (m, 5H), 7.06 (d, J = 7.2 Hz, 1H), 6.97 – 6.84 (m, 2H), 4.77 – 4.67 (m, 2H), 4.17 (t, J = 6.0 Hz, 2H), 3.32 – 3.28 (m, 4H), 2.25 – 2.14 (m, 2H); >98% at 215 nm, MS (ESI) m/z = 404.1 [M + H]+.
1-(3-(Naphthalen-1-yloxy)propyl)-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinoline-2-carboxylic acid (20)
A mixture of methyl and ethyl 1-(3-hydroxypropyl)-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinoline-2-carboxylate (72 mg, 0.25 mmol) was prepared as a pale yellow oil according to procedures described for preparing compound 16 using 3,4-dihydroquinolin-1(2H)-amine (74 mg, 0.50 mmol) and 2-oxohexanedioic acid (160 mg, 1.0 mmol) in 50% overall yield.
The title compound was prepared (20 mg, 0.052 mmol) as an off-white solid following the Mitsunobu reaction procedure described for preparing compound 16 using a mixture of methyl and ethyl 1-(3-hydroxypropyl)-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinoline-2-carboxylate (55 mg, 0.2 mmol). 1H NMR (400 MHz, DMSO-d6) δ 8.24 – 8.17 (m, 1H), 7.89 – 7.85 (m, 1H), 7.56 – 7.41 (m, 4H), 7.38 (t, J = 7.9 Hz, 1H), 6.97 (d, J = 6.9 Hz, 1H), 6.91 – 6.84 (m, 2H), 4.48 – 4.40 (m, 2H), 4.16 (t, J = 6.1 Hz, 2H), 3.32 – 3.30 (m, 2H), 2.91 (t, J = 6.1 Hz, 2H), 2.25 – 2.06 (m, 4H); >98% at 215 nm, MS (ESI) m/z = 386.2 [M + H]+.
6-(3-(Naphthalen-1-yloxy)propyl)-2,3-dihydro-[1,4]oxazino[2,3,4-hi]indole-5-carboxylic acid (21)
A mixture of methyl and ethyl 6-(3-hydroxypropyl)-2,3-dihydro-[1,4]oxazino[2,3,4-hi]indole-5-carboxylate (375 mg, 1.3 mmol) was prepared as a pale yellow oil according to procedures described for preparing compound 16 using 2,3-dihydro-4H-benzo[b][1,4]oxazin-4-amin (430 mg, 2.90 mmol) and 2-oxohexanedioic acid (595 mg, 3.7 mmol) in 45% overall yield.
The title compound was prepared (55 mg, 0.15 mmol) as an off-white solid following the Mitsunobu reaction procedure described for preparing compound 16 using a mixture of methyl and ethyl 6-(3-hydroxypropyl)-2,3-dihydro-[1,4]oxazino[2,3,4-hi]indole-5-carboxylate (75 mg, 0.27 mmol). 1H NMR (400 MHz, DMSO-d6) δ 8.18 (dd, J = 7.7, 1.8 Hz, 1H), 7.86 (dd, J = 7.4, 1.9 Hz, 1H), 7.56 − 7.42 (m, 3H), 7.38 (dd, J = 8.0, 7.6 Hz, 1H), 7.22 (d, J = 8.0 Hz, 1H), 6.91 − 6.82 (m, 2H), 6.65 (d, J = 7.4 Hz, 1H), 4.54 (t, J = 4.6 Hz, 2H), 4.47 (t, J = 4.8 Hz, 2H), 4.17 (t, J = 6.0 Hz, 2H), 3.36 − 3.31 (m, 2H), 2.26 − 2.17 (m, 2H); >98% at 215 nm, MS (ESI) m/z = 388.1 [M + H]+.
6-(3-(naphthalen-1-yloxy)propyl)-2,3-dihydro-[1,4]thiazino[2,3,4-hi]indole-5-carboxylic acid 1,1-dioxide (22)
To a solution of 6-(3-(naphthalen-1-yloxy)propyl)-2,3-dihydro-[1,4]thiazino[2,3,4-hi]indole-5-carboxylic acid (20 mg, 0.050 mmol) in CH2Cl2 (1.0 mL) and THF (1.0 mL) was added m-CPBA (17 mg (77% pure), 1.0 mmol) at 20 °C. The reaction mixture was stirred for 1 h at 20 °C then quenched by addition of H2O, extracted with CH2Cl2 and concentrated in vacuo. The residue was purified by reverse-phase preparative HPLC (Phenomenex Gemini C18, H2O/CH3CN gradient to 25–95% CH3CN 0.1% TFA) to give the title compound (12 mg, 0.028 mmol) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.13 (dd, J = 8.6, 1.8 Hz, 1H), 8.03 (d, J = 8.1 Hz, 1H), 7.89 − 7.83 (m, 1H), 7.72 (d, J = 7.4 Hz, 1H), 7.58 − 7.42 (m, 3H), 7.41 − 7.33 (m, 1H), 7.21 (t, J = 7.8 Hz, 1H), 6.88 (d, J = 7.3 Hz, 1H), 5.09 − 4.93 (m, 2H), 4.17 (t, J = 6.0 Hz, 2H), 3.91 − 3.80 (m, 2H), 3.41 − 3.35 (m, 2H), 2.29 − 2.14 (m, 2H); >98% at 215 nm, MS (ESI) m/z = 436.1 [M + H]+.
7-(3-(Naphthalen-1-yloxy)propyl)-3,4-dihydro-2H-[1,4]thiazepino[2,3,4-hi]indole-6-carboxylic acid (23)
A mixture of methyl and ethyl 7-(3-hydroxypropyl)-3,4-dihydro-2H-[1,4]thiazepino[2,3,4-hi]indole-6-carboxylate (211 mg, 0.72 mmol) was prepared as a pale yellow oil according to procedures described for preparing compound 16 using 2,3-dihydro-4H-benzo[b][1,4]oxazin-4-amin (261 mg, 1.45 mmol) and 2-oxohexanedioic acid (301 mg, 1.88 mmol) in 50% overall yield.
The title compound was prepared (76 mg, 0.18 mmol) as a white solid following the Mitsunobu reaction procedure described for preparing compound 16 using a mixture of methyl and ethyl 7-(3-hydroxypropyl)-3,4-dihydro-2H-[1,4]thiazepino[2,3,4-hi]indole-6-carboxylate (63 mg, 0.20 mmol). 1H NMR (400 MHz, DMSO-d6) δ 8.18 (dd, J = 7.6, 1.9 Hz, 1H), 7.86 (dd, J = 7.3, 2.0 Hz, 1H), 7.61 − 7.42 (m, 4H), 7.38 (dd, J = 8.0, 7.8 Hz, 1H), 7.10 (d, J = 7.3 Hz, 1H), 6.86 (dd, J = 8.0, 7.7 Hz, 2H), 4.86 (t, J = 5.8 Hz, 2H), 4.16 (t, J = 6.0 Hz, 2H), 3.34 (t, J = 6.6 Hz, 2H), 3.24 (t, J = 6.6 Hz, 2H), 2.34 − 2.23 (m, 2H), 2.22 − 2.13 (m, 2H); >98% at 215 nm, MS (ESI) m/z = 418.1 [M + H]+.
7-(3-(Naphthalen-1-yloxy)propyl)-3,4-dihydro-2H-[1,4]thiazepino[2,3,4-hi]indole-6-carboxylic acid 1-oxide (24)
To a cooled (−78 °C) solution of 7-(3-(naphthalen-1-yloxy)propyl)-3,4-dihydro-2H-[1,4]thiazepino[2,3,4-hi]indole-6-carboxylic acid (35 mg, 0.084 mmol) in THF/CH2Cl2 (1 mL, 1:1) was added m-CPBA (19 mg, 0.084 mmol), and the mixture was stirred for 1 h at −78 °C. The reaction was quenched by addition of saturated aqueous NaHCO3 solution (0.5 mL) followed by H2O (12 mL). The quenched reaction mixture was extracted with CH2Cl2 (2×10 mL), dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by reverse-phase preparative HPLC (Phenomenex Gemini C18, H2O/CH3CN gradient to 30–80% CH3CN 0.1% TFA) to give the title compound (33 mg, 0.076 mmol) as a white amorphous solid. 1H NMR (400 MHz, DMSO-d6) δ 8.15 (dd, J = 7.6, 1.7 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.86 (dd, J = 7.5, 1.7 Hz, 1H), 7.61 (d, J = 7.3 Hz, 1H), 7.57 − 7.42 (m, 3H), 7.38 (dd, J = 8.0, 7.6 Hz, 1H), 7.16 (dd, J = 7.8, 7.6 Hz, 1H), 6.88 (d, J = 7.4, 1H), 4.73 (dt, J = 9.1, 4.2 Hz, 1H), 4.64 (dt, J = 9.1, 4.2 Hz, 1H), 4.18 (t, J = 5.9 Hz, 2H), 3.55 − 3.48 (m, 2H), 3.39 − 3.24 (m, 2H), 3.05 (dt, J = 7.3, 6.8 Hz, 1H), 2.73 − 2.57 (m, 1H), 2.29 − 2.13 (m, 2H); >98% at 215 nm, MS (ESI) m/z = 434.1 [M + H]+.
7-(3-(Naphthalen-1-yloxy)propyl)-3,4-dihydro-2H-[1,4]thiazepino[2,3,4-hi]indole-6-carboxylic acid 1,1-dioxide (25)
To a cooled solution (0 °C) of 7-(3-(naphthalen-1-yloxy)propyl)-3,4-dihydro-2H-[1,4]thiazepino[2,3,4-hi]indole-6-carboxylic acid (15 mg, 0.036 mmol) in CH2Cl2 (1 mL) was added m-CPBA (18 mg, 0.072 mmol), and the mixture was stirred for 2 h at 0 °C. The reaction was quenched by addition of saturated aqueous NaHCO3 solution (2 mL). The quenched reaction mixture was extracted with CH2Cl2 (3×10 mL), dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by reverse-phase preparative HPLC (Phenomenex Gemini C18, H2O/CH3CN gradient to 30–85% CH3CN 0.1% TFA) to give the title compound (14 mg, 0.031 mmol) as an off-white amorphous solid. 1H NMR (400 MHz, DMSO-d6) δ 8.14 (dd, J = 7.8, 1.5 Hz, 1H), 8.06 (d, J = 8.0 Hz, 1H), 7.86 (dd, J = 7.4, 1.6 Hz, 1H), 7.82 (dd, J = 7.5, 1.0 Hz, 1H), 7.59 − 7.42 (m, 3H), 7.38 (dd, J = 8.1, 7.7 Hz, 1H), 7.22 (dd, J = 7.8, 7.6 Hz, 1H), 6.88 (d, J = 7.2 Hz, 1H), 4.89 − 4.68 (m, 2H), 4.18 (t, J = 5.9 Hz, 2H), 3.77 (t, J = 6.8 Hz, 2H), 3.33 (t, J = 7.1 Hz, 2H), 2.42 − 2.28 (m, 2H), 2.27 − 2.12 (m, 2H); >98% at 215 nm, MS (ESI) m/z = 450.1 [M + H]+.
Compounds 26−33 were prepared following the general procedure outlined above in library format. Purity of all final compounds was determined by HPLC analysis and is >95%. All compounds were isolated as solids.
6-(3-((5,6,7,8-Tetrahydronaphthalen-1-yl)oxy)propyl)-2,3-dihydro-[1,4]thiazino[2,3,4-hi]indole-5-carboxylic acid (26)
Coupling of a mixture of methyl and ethyl 6-(3-hydroxypropyl)-2,3-dihydro-[1,4]thiazino[2,3,4-hi]indole-5-carboxylate and 5,6,7,8-tetrahydronaphthalen-1-ol followed by saponification yielded 26. >98% at 215 nm, MS (ESI) m/z = 408.2 [M + H]+.
6-(3-((4-Chloronaphthalen-1-yl)oxy)propyl)-2,3-dihydro-[1,4]thiazino[2,3,4-hi]indole-5-carboxylic acid (27)
Coupling of a mixture of methyl and ethyl 6-(3-hydroxypropyl)-2,3-dihydro-[1,4]thiazino[2,3,4-hi]indole-5-carboxylate and 4-chloronaphthalen-1-ol followed by saponification yielded 27. >98% at 215 nm, MS (ESI) m/z = 438.1 [M + H]+.
6-(3-((5,6,7,8-Tetrahydronaphthalen-2-yl)oxy)propyl)-2,3-dihydro-[1,4]thiazino[2,3,4-hi]indole-5-carboxylic acid (28)
Coupling of a mixture of methyl and ethyl 6-(3-hydroxypropyl)-2,3-dihydro-[1,4]thiazino[2,3,4-hi]indole-5-carboxylate and 5,6,7,8-tetrahydronaphthalen-2-ol followed by saponification yielded 28. >98% at 215 nm, MS (ESI) m/z = 408.1 [M + H]+.
6-(3-(4-Chloro-3-methylphenoxy)propyl)-2,3-dihydro-[1,4]thiazino[2,3,4-hi]indole-5-carboxylic acid (29)
Coupling of a mixture of methyl and ethyl 6-(3-hydroxypropyl)-2,3-dihydro-[1,4]thiazino[2,3,4-hi]indole-5-carboxylate and 4-chloro-3-methylphenol followed by saponification yielded 29. >98% at 215 nm, MS (ESI) m/z = 402.1 [M + H]+.
6-(3-(4-chloro-3,5-dimethylphenoxy)propyl)-2,3-dihydro-[1,4]thiazino[2,3,4-hi]indole-5-carboxylic acid (30)
Coupling of a mixture of methyl and ethyl 6-(3-hydroxypropyl)-2,3-dihydro-[1,4]thiazino[2,3,4-hi]indole-5-carboxylate and 4-chloro-3,5-dimethylphenol followed by saponification yielded 30. >98% at 215 nm, MS (ESI) m/z = 416.1 [M + H]+.
1-(3-(4-Chloro-3,5-dimethylphenoxy)propyl)-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinoline-2-carboxylic acid (31)
Coupling of a mixture of methyl and ethyl 1-(3-hydroxypropyl)-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinoline-2-carboxylate and 4-chloro-3,5-dimethylphenol followed by saponification yielded 31. 1H NMR (400 MHz, DMSO-d6) δ 7.43 (d, J = 7.6 Hz, 1H), 7.00 − 6.90 (m, 2H), 6.74 (s, 2H), 4.43 (t, J = 5.6 Hz, 2H), 3.92 (t, J = 6.4 Hz, 2H), 3.17 (t, J = 7.2 Hz, 2H), 2.91 (t, J = 5.9 Hz, 2H), 2.27 (s, 6H), 2.17 − 2.08 (m, 2H), 2.07 − 1.96 (m, 2H); >98% at 215 nm, MS (ESI) m/z = 398.2 [M + H]+.
6-(3-(4-Chloro-3,5-dimethylphenoxy)propyl)-2,3-dihydro-[1,4]oxazino[2,3,4-hi]indole-5-carboxylic acid (32)
Coupling of a mixture of methyl and ethyl 6-(3-hydroxypropyl)-2,3-dihydro-[1,4]oxazino[2,3,4-hi]indole-5-carboxylate and 4-chloro-3,5-dimethylphenol followed by saponification yielded 32. >98% at 215 nm, MS (ESI) m/z = 400.1 [M + H]+.
7-(3-(4-Chloro-3,5-dimethylphenoxy)propyl)-3,4-dihydro-2H-[1,4]thiazepino[2,3,4-hi]indole-6-carboxylic acid (33)
Coupling of a mixture of methyl and ethyl 7-(3-hydroxypropyl)-3,4-dihydro-2H-[1,4]thiazepino[2,3,4-hi]indole-6-carboxylate and 4-chloro-3,5-dimethylphenol followed by saponification yielded 33. 1H NMR (400 MHz, DMSO-d6) δ 7.46 (d, J = 8.0 Hz, 1H), 7.13 (d, J = 7.2 Hz, 1H), 6.91 (t, J = 7.6 Hz, 1H), 6.75 (s, 2H), 4.90 – 4.81 (m, 2H), 3.94 (t, J = 6.3 Hz, 2H), 3.39 – 3.34 (m, 2H), 3.13 – 3.07 (m, 2H), 2.34 – 2.22 (m, 8H), 2.04 – 1.95 (m, 2H); >98% at 215 nm, MS (ESI) m/z = 430.1 [M + H]+.
7-chloro-1-(3-(4-chloro-3,5-dimethylphenoxy)propyl)-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinoline-2-carboxylic acid (34)
To a stirring mixture of 2-bromo-3-chloroaniline (4.1 g, 20 mmol) in 1M HCl (25 mL) and water (5 mL) at 0 °C was added NaNO2 (1.38 g, 20 mmol) in water (20 mL), NaCH3COOH (9.23 g, 112 mmol) in water (25 mL) and ethyl 2-oxocyclopentane carboxylate (3.0 mL, 20 mmol) in sequence. The reaction mixture was stirred for 15 min at 0 °C then warmed to 20 °C over 2h and extracted with CH2Cl2, dried over MgSO4, filtered and concentrated in vacuo to give 5-(2-(2-bromo-3-chlorophenyl)hydrazono)-6-ethoxy-6-oxohexanoic acid as a red oil in 7.0 g (90% crude).
To a solution of 5-(2-(2-bromo-3-chlorophenyl)hydrazono)-6-ethoxy-6-oxohexanoic acid (7.0 g, 18 mmol) in EtOH (30 mL) was added conc. H2SO4 (7.5 mL), slowly. The reaction mixture was refluxed for 1.5 h. The reaction was quenched by pouring into ice then extracted with CH2Cl2. The combined organic layer was washed with saturated aqueous NaHCO3 solution, H2O, brine, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash chromatography (Combi-flash Rf Hex/EtOAc 0–25% gradient) to give ethyl 7-bromo-6-chloro-3-(3-ethoxy-3-oxopropyl)-1H-indole-2-carboxylate (4.8 g, 12 mmol) as an off-white solid. MS (ESI) m/z = 402.0 [M + H]+.
To a solution of ethyl 7-bromo-6-chloro-3-(3-ethoxy-3-oxopropyl)-1H-indole-2-carboxylate (2.0 g, 4.8 mmol) in THF (20 mmol) was added BH3 in THF (20 mL, 20 mmol) at 20 °C. The reaction mixture was stirred for 15h at 20 °C and quenched by addition of MeOH then concentrated in vacuo. The residue was purified by flash chromatography (Combi-flash Rf Hexane/EtOAc gradient 0–50%) to give ethyl 7-bromo-6-chloro-3-(3-hydroxypropyl)-1H-indole-2-carboxylate (1.44 g, 4.0 mmol) as a white solid. MS (ESI) m/z = 360.0 [M + H]+.
To a solution of ethyl 7-bromo-6-chloro-3-(3-hydroxypropyl)-1H-indole-2-carboxylate (1.0 g, 0.28 mmol), PPh3 (1.1 g, 5.1 mmol) and 3,5-diMe-4-Cl-phenol (810 g, 5.2 mmol) in THF (35 mL) was added Dt-BuAD (990 mg, 5.1 mmol) at 20 °C. The reaction mixture was stirred for 15h at 20 °C then concentrated in vacuo. The residue was purified by flash chromatography (Combi-flash Rf Hexane/EtOAc gradient 0–10%) to give ethyl 7-bromo-6-chloro-3-(3-(4-chloro-3,5-dimethylphenoxy)propyl)-1H-indole-2-carboxylate (1.15 g, 2.3 mmol) as a white solid. MS (ESI) m/z = 498.0 [M + H]+.
To A solution of ethyl 7-bromo-6-chloro-3-(3-(4-chloro-3,5-dimethylphenoxy)propyl)-1H-indole-2-carboxylate (100 mg, 0.20 mmol) and 3-bromoprop-1-ene (0.026 mL, 0.30 mmol) in DMF (1.5 mL) was added Cs2CO3 (196 mg, 0.60 mmol), and the mixture was stirred for 3 h at 80 °C. The reaction mixture was cooled to 20 °C then concentrated in vacuo. The residue was purified by flash chromatography (Combi-flash Rf Hexane/EtOAc gradient 0–70%) to give ethyl 1-allyl-7-bromo-6-chloro-3-(3-(4-chloro-3,5-dimethylphenoxy)propyl)-1H-indole-2-carboxylate (105 mg 0.19 mmol) as a yellow oil. MS (ESI) m/z = 538.1 [M + H]+.
To a solution of ethyl 1-allyl-7-bromo-6-chloro-3-(3-(4-chloro-3,5-dimethylphenoxy)propyl)-1H-indole-2-carboxylate (105 mg, 0.19 mmol) and Bu3SnH (0.105 ml, 0.39 mmol) in toluene (1.0 mL) was added AIBN (1.6 mg, 9.7 µmol), and the mixture was stirred for 90 min at 100 °C. The reaction mixture was cooled to 20 °C then concentrated in vacuo. The residue was dissolved in THF (1 mL) and LiOH (0.5 mL, 2N) was added. The reaction mixture was stirred for 24 h at 40 °C then 20 °C then concentrated in vacuo. The residue was purified by reverse-phase preparative HPLC (Phenomenex Gemini C18, H2O/CH3CN gradient to 60–95% CH3CN 0.1% TFA) to give the title compound (41 mg, 0.095 mmol) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 7.48 (d, J = 8.0 Hz, 1 H), 7.00 (d, J = 8.0 Hz, 1 H), 6.73 (s, 2 H), 4.42 (t, J = 6.0 Hz, 2 H), 3.91 (t, J = 6.0 Hz, 2 H), 3.15 (t, J = 6.0 Hz, 2 H), 2.90 (t, J = 6.0 Hz, 2 H), 2.27 (s, 6 H), 2.15 (m, 2 H), 2.00 (m, 2 H); >98% at 215 nm, MS (ESI) m/z = 432.1 [M + H]+.
9-Chloro-6-(3-(4-chloro-3,5-dimethylphenoxy)propyl)-2,3-dihydro-[1,4]oxazino[2,3,4-hi]indole-5-carboxylic acid (35)
A solution of ethyl 7-bromo-6-chloro-3-(3-(4-chloro-3,5-dimethylphenoxy)propyl)-1H-indole-2-carboxylate (250 mg, 0.50 mmol) and bis(pinacolato)diboron (153 mg, 0.60 mmol) in DMF (2.5 mL) was degased at 20 °C then potassium acetate (226 mg, 2.3 mmol) and 1,1'-bis(diphenylphosphino)ferrocenedichloro palladium(ii) dichloromethane complex (18.32 mg, 0.025 mmol) was added. The reaction mixture was stirred for 20 h at 90 °C under Ar then cooled to 20 °C and diluted with Et2O (100 mL). The combined organic solution was washed with H2O (4 × 10 mL), brine (10 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash chromatography (Combi-flash Rf Hexane/EtOAc gradient 0–15%) to give ethyl 6-chloro-3-(3-(4-chloro-3,5-dimethylphenoxy)propyl)-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole-2-carboxylate (115 mg, 0.21 mmol) as a white solid. MS (ESI) m/z = 546.0 [M + H]+.
To a solution of ethyl 6-chloro-3-(3-(4-chloro-3,5-dimethylphenoxy)propyl)-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole-2-carboxylate (63 mg, 0.12 mmol) in THF (1.2 mL) was added 0.5 M NaOH aqueous solution (1.2 mL, 0.60 mmol) followed by 30% H2O2 (118 µL, 1.2 mmol) at 20 °C. The reaction mixture was stirred for 15 h at 20 °C then quenched with sat NH4Cl/NH4OH buffer solution. The mixture was extracted with EtOAc (2 × 10 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by reverse-phase preparative HPLC (Phenomenex Gemini C18, H2O/CH3CN gradient to 5–65% CH3CN 0.1% TFA) to give ethyl 6-chloro-3-(3-(4-chloro-3,5-dimethylphenoxy)propyl)-7-hydroxy-1H-indole-2-carboxylate (15 mg, 0.034 mmol). MS (ESI) m/z = 436.0 [M + H]+.
A mixture of ethyl 6-chloro-3-(3-(4-chloro-3,5-dimethylphenoxy)propyl)-7-hydroxy-1H-indole-2-carboxylate (10 mg, 0.023 mmol), cesium carbonate (37 mg, 0.12 mmol) and dibromoethane (4.0 µL, 0.046 mmol) in DMF (460 µL) was stirred at 100 °C for 15 h. The mixture was cooled to 20 °C, filtered through a hydrophobic frit and concentrated in vacuo. The residue was dissolved in THF (230 µL) and 2N aqueous LiOH solution (115 µL, 0.23 mmol) was added. The reaction mixture was stirred for 15 h 20 °C, acidified with TFA and concentrated in vacuo. The residue was purified by reverse-phase preparative HPLC (Phenomenex Gemini C18, H2O/CH3CN gradient to 50–95% CH3CN 0.1% TFA) to give the title compound (7.5 mg, 0.017 mmol) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 7.23 (d, J = 8.6 Hz, 1H), 6.98 (d, J = 8.6 Hz, 1H), 6.71 (s, 2H), 4.57 (m, 4H), 3.93 (t, J = 6.2 Hz, 2H), 3.31 (s, 3H), 3.15 (t, J = 7.6 Hz, 2H), 2.25 (s, 6H), 2.00 (t, J = 7.1 Hz, 2H); >98% at 215 nm, MS (ESI) m/z = 434.1 [M + H]+.
Protein expression and purification
A codon-optimized gene sequence encoding residues 172–327 of human Mcl-1 (Uniprot: Q07820) was purchased (Genscript) and cloned into a Gateway entry vector (pDONR-221, Invitrogen) using the protocols provided. This construct was further sub-cloned into an expression vector (pDEST-HisMBP) containing a maltose binding protein (MBP) tag for increased solubility, a tobacco etch virus (TEV) protease recognition site for tag-removal and an N-terminal His-tag to facilitate purification. The integrity of all plasmids was checked by sequencing. Soluble Mcl-1 protein was expressed in Escherichia coli BL21 CodonPlus (DE3) RIL (Stratagene) using ampicillin and chloramphenicol for selection.
In brief, a colony from a fresh transformation plate was picked to inoculate 100 mL of LB medium (37°C). The overnight culture was used to start a 10 L fermentation (BioFlo 415, New Brunswick Scientific) grown at 37°C. For NMR studies, uniformly 15N and 15N/13C isotopically labeled protein samples were produced in minimal M9 media, where 15NH4Cl and [U-13C]-D-glucose were used as sole nitrogen and carbon sources (Cambridge Isotope Laboratories). When the cell density corresponded to OD600=2, the temperature was lowered to 20°C. After one hour, protein expression was induced with 0.5 mM IPTG. Cells were harvested after 16 h by centrifugation. Pellets were frozen and re-dissolved in lysis buffer (20 mM TRIS pH 7.5, 300 mM NaCl, 20 mM imidazole, 5 mM BME), approximately 100 mL / 10 g pellet, before the cells were broken by homogenization (APV-2000, APV). Prior to application to an affinity column (140 mL, ProBond, Invitrogen), lysate was cleared by centrifugation (18,000 rpm) and filtration (0.44 µm). Bound protein was washed on the column and then eluted by a gradient (20 mM TRIS pH 7.5, 300 mM NaCl, 500 mM imidazole, 5 mM BME). To enhance TEV protease cleavage, samples were buffer exchanged (50 mM TRIS pH 7.5, 100 mM NaCl, 5 mM BME) on three serially connected columns (HiPrep 26/10 Desalting, GE Healthcare). TEV protease was added to a molar ratio of 1:10 (TEV:Mcl-1) and incubated at room temperature until cleavage was complete. After adding 20 mM imidazole to the samples, they were passed over a subtractive second nickel-column (120 mL, Ni-NTA Superflow, Qiagen) to remove the MBP-tag, non-cleaved protein, and TEV protease. Mcl-1 protein for NMR screening was buffer exchanged into an optimized NMR buffer (25 mM sodium phosphate pH 6.3, 25 mM NaCl, 1 mM DTT, 0.01% NaN3). To achieve highly pure samples (e.g. for crystal screening), a supplementary step of size-exclusion chromatography (HiLoad 26/60, Superdex 75, GE Healthcare) was implemented. The running buffer also acted as the Mcl-1 storage and crystallography buffer (20 mM HEPES pH 6.8, 50 mM NaCl, 3 mM DTT, 0.01% NaN3). Purifications were done at 4°C, and concentration steps were performed in stirred ultrafiltration cells (Amicon, Millipore).
To improve protein sample quality and to increase crystal diffraction, protein mutants were created by site-directed mutagenesis (QuikChange, Agilent Technologies). Primers for a C-terminal deletion (Δ5) were designed using their online tool and ordered from Eurofins MWG Operon. Mutations were made on the entry vector above, analyzed by in-house sequencing, and subsequently transferred into the pDEST-HisMBP expression vector. Mutant proteins were purified in the same way as wild-type (WT) proteins.
NMR experiments NOE-guided fragment docking
NOE-derived distance restraints were acquired to enable NMR-based docking of fragments into a previously determined X-ray structure of a Mcl-1/ligand complex (PDB: 4HW2). Spectra were recorded on a Bruker 800-MHz spectrometer equipped with a cryo-probe and pulsed field gradients. 300 µM 15N/13C-labeled samples of Mcl-1 was prepared in a D2O-based NMR buffer and mixed with fragment 2 at a concentration of 1 mM. Side-chain 1H and 13C NMR signals were assigned from 13C-edited NOE and HCCH-TOCSY experiments.41,42 NOE distance restraints were obtained from three-dimensional 13C-edited NOESY spectra, as well as three-dimensional 15N- and 13C-filter/edited NOESY spectra acquired with a mixing time of 80 ms. Compounds were docked into a X-ray structure using the NMR-derived restraints and a simulated annealing protocol using the program Xplor-NIH.43 A square-well potential (FNOE = 50 kcal mol−1) was employed to constrain NOE-derived distances. Five low energy models from this process were energy minimized using MOE 2011.10 (Chemical Computing Group Inc., Montreal). The lowest energy structures obtained by this method were consistent with the observed NOEs.
Protein crystallization, data collection and structure refinement
Fresh batches of Mcl-1 proteins, WT and Δ5, were concentrated to 600 µM (10.7 mg/mL) and 1 mM (17.4 mg/mL), respectively, and screened for crystallization conditions with a 1.2× excess of ligand. Crystals were obtained by mixing 1 µL protein with 1 µL reservoir solution (25–30% PEG 3350, 0.1 M Bis-TRIS pH 6.5, 0.2 M MgCl2) as a hanging drop at 4°C or 18°C. Crystals appeared within the first week and were flash frozen in liquid nitrogen after cryoprotection using 10–20% glycol.
Data were collected on the Life Sciences Collaborative Access Team (LS-CAT) 21-ID-D beamline at the Advanced Photon Source (APS), Argonne National Laboratory. Indexing, integration and scaling was performed with HKL2000.44 Using a previously determined ligand-bound structure (PDB: 4HW2), phasing was done by molecular replacement with Phaser45 as implemented in CCP4.46 Refinement of the structural models were performed with Phenix47 and Refmac48, and included rounds of manual model building in COOT.49 Figures were prepared in PyMOL50 and MOE.
FPA Competition Assays
A fluorescein isothiocyanate (FITC)-labeled BH3 peptide derived from Bak (FITC-Bak-BH3; FITC-AHx-GQVGRQLAIIGDDINR-NH2) was purchased from GenScript and used without further purification. FPA measurements were carried out in 384-well, black, flat-bottom plates (Greiner Bio-One) using the EnVision plate reader (PerkinElmer). All assays were conducted in assay buffer containing 20 mM TRIS pH 7.5, 50 mM NaCl, 3 mM DTT, and 5% final DMSO concentration. To measure displacement of the FITC-Bak-BH3 peptide from Bcl-2 family members, 10 nM FITC-Bak-BH3 peptide was incubated with either 15 nM Mcl-1 or 4 nM Bcl-xL. For IC50 determination, compounds were diluted in DMSO in a 10-point, 3-fold serial dilution scheme, added to assay plates, and incubated for 1.5 h at room temperature. The change in anisotropy was measured and used to calculate an IC50 (inhibitor concentration at which 50% of bound peptide is displaced) by fitting the anisotropy data using XLFit (IDBS) to a four parameter dose-response (variable slope) equation. This was converted into a binding dissociation constant (Ki) according to the formula51:
where [I]50 is the concentration of the free inhibitor at 50% inhibition, [L]50 is the concentration of the free labeled ligand at 50% inhibition, [P]0 is the concentration of the free protein at 0% inhibition and Kd represents the dissociation constant of the FITC-labeled peptide probe. Compounds were evaluated using replicate measurements, in duplicate; Ki values shown are the average of duplicate values.
Biotin streptavidin pull-down experiment
Human chronic myelogenous leukemia K562 cells were grown in RPMI-8226 complete media, lysed in NP-40 buffer (150mM NaCl, 1% NP-40, 50mM Tris, pH 8.0), cleared by centrifugation, and incubated with indicated concentrations of compound or Bim-BH3 peptide (FITC-Ahx-EARIAQELRRIGDEFNETYTR, Genscript) for 60 minutes at room temperature. Separately, Dynabeads M-280 streptavidin conjugated beads (Life Technologies) were incubated with biotin-Ahx-MS-1, an Mcl-1 specific peptide39, for 30 minutes and washed four times with PBS plus 0.01% TWEEN-20. The loaded beads were then incubated with lysate at room temperature for ten minutes, washed twice, boiled in SDS-PAGE loading buffer, and analyzed by Western blot (Licor Odyssey) with an Mcl-1 specific antibody (Y37, Abcam).
Acknowledgments
The authors thank co-workers at the High-Throughput Screening Core facility of Vanderbilt University, TN, for compound management and providing the instrumentation to perform the binding assay. This research was supported by the U.S. National Institutes of Health, NIH Director’s Pioneer Award DP1OD006933/DP1CA174419 to S.W.F., The NCI Experimental Therapeutics (NExT) Program BOA29XS129TO22 under the Leidos Biomed Prime Contract No. HHSN261200800001E, and a career development award to S.W.F. from a NCI SPORE grant in breast cancer (Grant P50CA098131) to C. L. Arteaga. This work was also funded by the American Cancer Society (Postdoctoral Fellowship, Grant PF1110501CDD) to J.P.B. The Biomolecular NMR Facility at Vanderbilt University is supported in part by a NIH SIG Grant 1S-10RR025677-01 and Vanderbilt University matching funds. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
Funding Sources
Any funds used to support the research of the manuscript should be placed here (per journal style).
ABBREVIATIONS
- Mcl-1
myeloid cell leukemia 1
- Bcl-2
B-cell lymphoma 2
- Bcl-xL
B-cell lymphoma extra large
- BH3
Bcl-2 homology domain 3
- Bax
Bcl-2-associated X protein
- Bak
Bcl-2 homologous antagonist killer
- LE
ligand efficiency
- m-CPBA
meta-chloroperoxybenzoic acid
- NOE
Nuclear Overhauser Effect
- FITC
fluorescein isothiocyanate
- FPA
fluorescence polarization anisotropy
Footnotes
ASSOCIATED CONTENT
PDB ID codes: XXXX (in process)
The authors declare no competing financial interest.
References
- 1.Danial NN, Korsmeyer SJ. Cell Death: Critical Control Points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 2.Kroemer G, Galluzzi L, Brenner C. Mitochondrial Membrane Permeabilization in Cell Death. Physiol. Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 3.Cory S, Adams JM. The Bcl2 Family: Regulators of the Cellular Life-or-Death Switch. Nat. Rev. Cancer. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 4.Adams JM, Cory S. The Bcl-2 Apoptotic Switch in Cancer Development and Therapy. Oncogene. 2007;26:1324–1337. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wei G, Margolin AA, Haery L, Brown E, Cucolo L, Julian B, Shehata S, Kung AL, Beroukhim R, Golub TR. Chemical Genomics Identifies Small-Molecule MCL1 Repressors and BCL-xL as a Predictor of MCL1 Dependency. Cancer Cell. 2012;21:547–562. doi: 10.1016/j.ccr.2012.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, McHenry KT, Pinchback RM, Ligon AH, Cho Y-J, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, Tanaka KE, Chiang DY, Bass AJ, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q, Yecies D, Signoretti S, Maher E, Kaye FJ, Sasaki H, Tepper JE, Fletcher JA, Tabernero J, Baselga J, Tsao M-S, Demichelis F, Rubin MA, Janne PA, Daly MJ, Nucera C, Levine RL, Ebert BL, Gabriel S, Rustgi AK, Antonescu CR, Ladanyi M, Letai A, Garraway LA, Loda M, Beer DG, True LD, Okamoto A, Pomeroy SL, Singer S, Golub TR, Lander ES, Getz G, Sellers WR, Meyerson M. The Landscape of Somatic Copy-Number Alteration Across Human Cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song L, Coppola D, Livingston S, Cress D, Haura EB. Mcl-1 Regulates Survival and Sensitivity to Diverse Apoptotic Stimuli in Human Non-Small Cell Lung Cancer Cells. Cancer Biol. Ther. 2005;4:267–276. doi: 10.4161/cbt.4.3.1496. [DOI] [PubMed] [Google Scholar]
- 8.Ding Q, He X, Xia W, Hsu J-M, Chen C-T, Li L-Y, Lee D-F, Yang J-Y, Xie X, Liu J-C, Hung M-C. Myeloid Cell Leukemia-1 Inversely Correlates with Glycogen Synthase Kinase-3β Activity and Associates with Poor Prognosis in Human Breast Cancer. Cancer Res. 2007;67:4564–4571. doi: 10.1158/0008-5472.CAN-06-1788. [DOI] [PubMed] [Google Scholar]
- 9.Krajewska M, Krajewski S, Epstein JI, Shabaik A, Sauvageot J, Song K, Kitada S, Reed JC. Immunohistochemical Analysis of Bcl-2, Bax, Bcl-X, and Mcl-1 Expression in Prostate Cancers. Am. J. Pathol. 1996;148:1567–1576. [PMC free article] [PubMed] [Google Scholar]
- 10.Miyamoto Y, Hosotani R, Wada M, Lee JU, Koshiba T, Fujimoto K, Tsuji S, Nakajima S, Doi R, Kato M, Shimada Y, Imamura M. Immunohistochemical Analysis of Bcl-2, Bax, Bcl-X, and Mcl-1 Expression in Pancreatic Cancers. Oncology. 1999;56:73–82. doi: 10.1159/000011933. [DOI] [PubMed] [Google Scholar]
- 11.Brotin E, Meryet-Figuie-re M, Simonin K, Duval RE, Villedieu M, Leroy-Dudal J, Saison-Behmoaras E, Gauduchon P, Denoyelle C, Poulain L. Bcl-XL and MCL-1 Constitute Pertinent Targets in Ovarian Carcinoma and Their Concomitant Inhibition Is Sufficient to Induce Apoptosis. Int. J. Cancer. 2010;126:885–895. doi: 10.1002/ijc.24787. [DOI] [PubMed] [Google Scholar]
- 12.Zhang T, Zhao C, Luo L, Zhao H, Cheng J, Xu F. The Expression of Mcl-1 in Human Cervical Cancer and Its Clinical Significance. Med. Oncol. 2012;29:1985–91. doi: 10.1007/s12032-011-0005-y. [DOI] [PubMed] [Google Scholar]
- 13.Derenne S, Monia B, Dean NM, Taylor JK, Rapp M-J, Harousseau J-L, Bataille R, Amiot M. Antisense Strategy Shows that Mcl-1 Rather than Bcl-2 or Bcl-xL Is an Essential Survival Protein of Human Myeloma Cells. Blood. 2002;100:194–199. doi: 10.1182/blood.v100.1.194. [DOI] [PubMed] [Google Scholar]
- 14.Andersen MH, Becker JC, Thor Straten P. The Antiapoptotic Member of the Bcl-2 family Mcl-1 Is a CTL Target in Cancer Patients. Leukemia. 2005;19:484–485. doi: 10.1038/sj.leu.2403621. [DOI] [PubMed] [Google Scholar]
- 15.Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, Belmont LD, Kaminker JS, O’Rourke KM, Pujara K, Kohli PB, Johnson AR, Chiu ML, Lill JR, Jackson PK, Fairbrother WJ, Seshagiri S, Ludlam MJC, Leong KG, Dueber EC, Maecker H, Huang DCS, Dixit VM. Sensitivity to Antitubulin Chemotherapeutics Is Regulated by MCL1 and FBW7. Nature. 2011;471:110–114. doi: 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- 16.Arkin MR, Wells JA. Small-Molecule Inhibitors of Protein-Protein Interactions: Progressing Towards the Dream. Nat. Rev. Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 17.Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nat. Rev. Drug Discovery. 2008;7:989–1000. doi: 10.1038/nrd2658. [DOI] [PubMed] [Google Scholar]
- 18.Lessene G, Czabotar PE, Sleebs BE, Zobel K, Lowes KN, Adams JM, Baell JB, Colman PM, Deshayes K, Fairbrother WJ, Flygare JA, Gibbons P, Kersten WJ, Kulasegaram S, Moss RM, Parisot JP, Smith BJ, Street IP, Yang H, Huang DC, Watson KG. Structure-guided design of a selective BCL-X(L) inhibitor. Nat. Chem. Biol. 2013;9:390–397. doi: 10.1038/nchembio.1246. [DOI] [PubMed] [Google Scholar]
- 19.Wendt MD, Wang Shen W, Kunzer A, McClellan WJ, Bruncko M, Oost TK, Ding H, Joseph MK, Zhang H, Nimmer PM, Ng S-H, Shoemaker AR, Petros AM, Oleksijew A, Marsh K, Bauch J, Oltersdorf T, Belli BA, Martineau D, Fesik SW, Rosenberg SH, Elmore SW. Discovery and Structure-Activity Relationship of Antagonists of B-Cell Lymphoma 2 Family Proteins with Chemopotentiation Activity in Vitro and in Vivo. J. Med. Chem. 2006;49:1165–1181. doi: 10.1021/jm050754u. [DOI] [PubMed] [Google Scholar]
- 20.Bruncko M, Oost TK, Belli BA, Ding H, Joseph MK, Kunzer A, Martineau D, McClellan WJ, Mitten M, Ng S-C, Nimmer PM, Oltersdorf T, Park C-M, Petros AM, Shoemaker AR, Song X, Wang X, Wendt MD, Zhang H, Fesik SW, Rosenberg SH, Elmore SW. Studies Leading to Potent, Dual Inhibitors of Bcl-2 and Bcl-xL. J. Med. Chem. 2007;50:641–662. doi: 10.1021/jm061152t. [DOI] [PubMed] [Google Scholar]
- 21.Park C-M, Bruncko M, Adickes J, Bauch J, Ding H, Kunzer A, Marsh KC, Nimmer P, Shoemaker AR, Song X, Tahir SK, Tse C, Wang X, Wendt MD, Yang X, Zhang H, Fesik SW, Rosenberg SH, Elmore SW. Discovery of an Orally Bioavailable Small Molecule Inhibitor of Prosurvival B-Cell Lymphoma 2 Protein. J. Med. Chem. 2008;51:6902–6915. doi: 10.1021/jm800669s. [DOI] [PubMed] [Google Scholar]
- 22.Petros AM, Huth JR, Oost T, Park C-M, Ding H, Wang X, Zhang H, Nimmer P, Mendoza R, Sun C, Mack J, Walter K, Dorwin S, Gramling E, Ladror U, Rosenberg SH, Elmore SW, Fesik SW, Hajduk PJ. Discovery of a Potent and Selective Bcl-2 Inhibitor Using SAR by NMR. Bioorg. Med. Chem. Lett. 2010;20:6587–6591. doi: 10.1016/j.bmcl.2010.09.033. [DOI] [PubMed] [Google Scholar]
- 23.Zhou H, Chen J, Meagher JL, Yang C-Y, Aguilar A, Liu L, Bai L, Cong X, Cai Q, Fang X, Stuckey JA, Wang S. Design of Bcl-2 and Bcl-xL Inhibitors with Subnanomolar Binding Affinities Based upon a New Scaffold. J. Med. Chem. 2012;55:4664–4682. doi: 10.1021/jm300178u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang CCS, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park C-M, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse RA, Rosenberg SH, Elmore SW. ABT-199, a Potent and Selective BCL-2 Inhibitor, Achieves Antitumor Activity While Sparing Platelets. Nature Medicine. 2013;19:202–210. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 25.Tao Z-F, Hasvold L, Wang L, Wang X, Petros AM, Park CH, Boghaert ER, Catron ND, Chen J, Colman PM, Czabotar PE, Deshayes K, Fairbrother WJ, Flygare JA, Hymowitz SG, Jin S, Judge RA, Koehler MFT, Kovar PJ, Lessene G, Mitten MJ, Ndubaku CO, Nimmer P, Purkey HE, Oleksijew A, Phillips DC, Sleebs BE, Smith BJ, Smith ML, Tahir SK, Watson KG, Xiao Y, Xue J, Zhang H, Zobel K, Rosenberg SH, Tse C, Leverson JD, Elmore SW, Souers AJ. Discovery of a Potent and Selective BCL-XL Inhibitor with in Vivo Activity. ACS Med. Chem. Lett. 2014;5:1088–1093. doi: 10.1021/ml5001867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilson WH, O’Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP, Tulpule A, Dunleavy K, Xiong H, Chiu YL, Cui Y, Busman T, Elmore SW, Rosenberg SH, Krivoshik AP, Enschede SH, Humerickhouse R. Navitoclax, a Targeted High-Affinity Inhibitor of BCL-2, in Lymphoid Malignancies: a Phase 1 Dose-Escalation Study of Safety, Pharmacokinetics, Pharmaco-Dynamics, and Antitumour Activity. Lancet Oncol. 2010;11:1149–1159. doi: 10.1016/S1470-2045(10)70261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, Carney DA, He SZ, Huang DCS, Xiong H, Cui Y, Busman TA, McKeegan EM, Krivoshik AP, Enschede SH, Humerickhouse R. Substantial Susceptibility of Chronic Lymphocytic Leukemia to BCL2 Inhibition: Results of a Phase I Study of Navitoclax in Patients with Relapsed or Refractory Disease. J. Clin. Oncol. 2012;30:488–496. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Friberg A, Vigil D, Zhao B, Daniels RN, Burke JP, Garcia-Barrantes PM, Camper D, Chauder BA, Lee T, Olejniczak ET, Fesik SW. Discovery of Potent Myeloid Cell Leukemia 1 (Mcl-1) Inhibitors Using Fragment-Based Methods and Structure-Based Design. J. Med. Chem. 2013;56:15–30. doi: 10.1021/jm301448p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abulwerdi FA, Liao C, Mady A, Gavin J, Shen C, Cierpicki T, Stuckey JA, Showalter HDH, Nikolovska-Coleska Z. 3-Substituted-N-(4-Hydroxynaphthalen-1-yl)arylsulfonamides as a Novel Class of Selective Mcl-1 Inhibitors: Structure-Based Design, Synthesis, SAR and Biological Evaluation. J. Med. Chem. 2014;57:4111–4133. doi: 10.1021/jm500010b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Z, Li X, Song T, Zhao Y, Feng Y. An Anthraquinone Scaffold for Putative, Two-Face Bim BH3 α-Helix Mimic. J. Med. Chem. 2012;55:10735–10741. doi: 10.1021/jm301504b. [DOI] [PubMed] [Google Scholar]
- 31.Petros AM, Swann SL, Song D, Swinger K, Park C, Zhang H, Wendt MD, Kunzer AR, Souers AJ, Sun C. Fragment-Based Discovery of Potent Inhibitors of the Anti-Apoptotic MCL-1 Protein. Bioorg. Med. Chem. Lett. 2014;24:1484–1488. doi: 10.1016/j.bmcl.2014.02.010. [DOI] [PubMed] [Google Scholar]
- 32.Rega MF, Wu B, Wei J, Zhang Z, Cellitti JF, Pellecchia M. SAR by Interligand Nuclear Overhauser Effects (ILOEs) Based Discovery of Acylsulfonamide Compounds Active against Bcl-xL and Mcl-1. J. Med. Chem. 2011;54:6000–6013. doi: 10.1021/jm200826s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bian Z, Marvin C, Pettersson M, Martin SF. Enantioselective Total Syntheses of Citrinadins A and B. Stereochemical Revision of their Assigned Structures. J. Am. Chem. Soc. 2014;136:14184–14192. doi: 10.1021/ja5074646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bian Z, Marvin C, Martin SF. Enantioselective Total Synthesis of (−)-Citrinadin A and Revision of its Stereochemical Structure. J. Am. Chem. Soc. 2013;135:10886–10889. doi: 10.1021/ja405547f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caron G, Gaillard P, Carrupt P-A, Testa B. Lipophilicity Behavior of Model and Medicinal Compounds Containing a Sulfide, Sulfoxide, or Sulfone Moiety. Helv. Chim. Acta. 1997;80:449–462. [Google Scholar]
- 36.Zhang H, Nimmer PM, Tahir SK, Chen J, Fryer RM, Hahn KR, Iciek LA, Morgan SJ, Nasarre MC, Nelson R, Preusser LC, Reinhart GA, Smith ML, Rosenberg SH, Elmore SW, Tse C. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007;14:943–951. doi: 10.1038/sj.cdd.4402081. [DOI] [PubMed] [Google Scholar]
- 37.Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, Kelly PN, Ekert PG, Metcalf D, Roberts AW, Huang DCS, Kile BT. Programmed Anuclear Cell Death Delimits Platelet Life Span. Cell. 2007;128:1173–1186. doi: 10.1016/j.cell.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 38.Abulwerdi FA, Liao C, Mady AS, Gavin J, Shen C, Cierpicki T, Stuckey JA, Showalter HD, Nikolovska-Coleska Z. 3-Substituted-N-(4-hydroxynaphthalen-1-yl)arylsulfonamides as a novel class of selective Mcl-1 inhibitors: structure-based design, synthesis, SAR, and biological evaluation. J. Med. Chem. 2014;57:4111–4133. doi: 10.1021/jm500010b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Foight GW, Ryan JA, Gulla SV, Letai A, Keating AE. Designed BH3 Peptides with High Affinity and Specificity for Targeting Mcl-1 in Cells. ACS Chem. Bio. 2014;9:1962–1968. doi: 10.1021/cb500340w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, Nimmer S, Jin P, Smith M, Xiao Y, Kovar P, Tanaka A, Bruncko M, Sheppard GS, Wang L, Gierke S, Kategaya L, Anderson DJ, Wong C, Eastham-Anderson J, Ludlam MJC, Sampath D, Fairbrother WJ, Wertz I, Rosenberg SH, Tse C, Elmore SW, Souers AJ. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax) Cell Death & Disease. 2015;6:e1590. doi: 10.1038/cddis.2014.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clore GM, Gronenborn A. M. Methods Enzymol. 1994;239:349–363. doi: 10.1016/s0076-6879(94)39013-4. [DOI] [PubMed] [Google Scholar]
- 42.Sattler M, Schleucher J, Griesinger C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed. Prog. Nucl. Magn. Reson. Spectrosc. 1999;34:93–158. [Google Scholar]
- 43.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 44.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CW Jr, Sweet RM, editors. Macromolecular Crystallography. Part A. Vol. 276. Academic Press; San Diego, CA: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 45.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 48.Murshudov GN, Skubaák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 50.The PyMOL Molecular Graphics System, version 1.5. Schrödinger, LLC; New York: 2010. [Google Scholar]
- 51.Wang ZX, Jiang RF. A novel two-site binding equation presented in terms of the total ligand concentration. FEBS Lett. 1996;392:245–249. doi: 10.1016/0014-5793(96)00818-6. [DOI] [PubMed] [Google Scholar]