

SUMMARY
The pharyngeal arch arteries (PAAs) are transient embryonic blood vessels that mature into critical segments of the aortic arch and its branches. Although defects in PAA development cause life-threating congenital cardiovascular defects, the molecular mechanisms that orchestrate PAA morphogenesis remain unclear. Through small-molecule screening in zebrafish, we identified TGF-β signaling as indispensable for PAA development. Specifically, chemical inhibition of the TGF-β type I receptor ALK5 impairs PAA development because nkx2.5+ PAA progenitor cells fail to differentiate into tie1+ angioblasts. Consistent with this observation, we documented a burst of ALK5-mediated Smad3 phosphorylation within PAA progenitors that foreshadows angioblast emergence. Remarkably, premature induction of TGF-β receptor activity stimulates precocious angioblast differentiation, thereby demonstrating the sufficiency of this pathway for initiating the PAA progenitor to angioblast transition. More broadly, these data uncover TGF-β as a rare signaling pathway that is necessary and sufficient for angioblast lineage commitment.
In Brief
Improper embryonic development of the pharyngeal arch arteries (PAA) causes congenital defects characterized by discontinuity of the aorta and its branches. Abrial et al. identify TGF-β signaling as necessary and sufficient for inducing angioblast differentiation of nkx2.5+ PAA progenitors in zebrafish. Remarkably, premature activation of TGF-β signaling results in precocious angioblast differentiation.
INTRODUCTION
In mammals, specific segments of several large arteries including the aorta, pulmonary arteries, carotid arteries, and right subclavian artery derive from transient embryonic blood vessels termed pharyngeal arch arteries (PAAs) (Moore and Persaud, 1993; Sadler, 2006; Vincent and Buckingham, 2010). Although initially bilateral, the PAAs undergo extensive remodeling, a process that creates the asymmetric pattern present at birth. Defects in the formation, maintenance, or remodeling of the PAAs result in congenital cardiovascular malformations, including interrupted aortic arch type B (IAA-B), aberrant origin of the right subclavian artery (ARSA), and right aortic arch (RAA). While sig-nificant progress has been made in understanding the molecular pathways required for proper PAA maintenance and remodeling (Kameda, 2009), relatively little is known regarding the genetic control of PAA establishment as the cellular events, including the molecular characteristics, timing, and location of progenitor cell specification and angioblast commitment, preceding the initiation of blood flow were only recently described (Anderson et al., 2008; Li et al., 2012; Moore and Persaud, 1993; Sadler, 2006; Vincent and Buckingham, 2010).
The zebrafish model organism allows for unparalleled real-time visualization and genetic dissection of PAA formation, a process that has been highly conserved throughout vertebrate evolution (Anderson et al., 2008; Kameda, 2009; Li et al., 2012; Paffett-Lugassy et al., 2013). Prior work in zebrafish demonstrated that PAAs 3–6 arise in a cranial to caudal manner by vas-culogenesis (Anderson et al., 2008), the de novo differentiation of new endothelial cells from an undifferentiated progenitor population. Specifically, small clusters of tie1+ angioblasts appear in pharyngeal arches 3–6 and subsequently undergo angiogenic sprouting to give rise to patent vessels that support blood flow. This vasculogenic mechanism for PAA development was later reported to be conserved in mice (Li et al., 2012). Unlike in higher vertebrates, the zebrafish PAAs are not remodeled but mature instead into the gill circulation (Isogai et al., 2001). Despite these later differences, the early anatomic configurations of the PAAs in zebrafish and mammals are virtually indistinguishable (Anderson et al., 2008; Hiruma et al., 2002; Isogai et al., 2001; Moore and Persaud, 1993; Paffett-Lugassy et al., 2013), suggesting that the biological determinants of PAA establishment are also likely to be conserved.
In 2013, we reported that the embryonic source of tie1+ PAA angioblasts are nkx2.5+ progenitor cells that are specified in the zebrafish anterior lateral plate mesoderm (Paffett-Lugassy et al., 2013). Using in situ hybridization, Kaede-mediated fate mapping, and Cre/loxP lineage tracing, we learned that nkx2.5+ progenitor clusters initiate tie1 expression sequentially in an anterior to posterior direction. After a transient period of overlap, nkx2.5 expression is downregulated while tie1 is maintained throughout PAA morphogenesis. A recent report in zebrafish highlighted the importance of Stat4 in this process (Meng et al., 2017). However, the molecular signals that trigger this progenitor to angioblast transition remain unknown.
To identify molecular pathways required for PAA development, we performed a small-molecule screen for suppressors of tie1+ angioblast formation in zebrafish embryos. Among several hits, a predicted antagonist of TGF-β signaling was identified. Through follow-up studies, we identify TGF-β signaling as necessary and sufficient for stimulating PAA progenitor cell differentiation toward the angioblast lineage.
RESULTS
A Chemical Screen in Zebrafish Implicates TGF-β Signaling in PAA Morphogenesis
To identify factors and pathways required for PAA morphogenesis, we screened 1,235 biologically active small molecules for suppressors of tie1+ PAA angioblast development in zebrafish embryos (Figure S1). Immediately following gastrulation (bud stage, 10 hpf), groups of 15–20 wild-type embryos were exposed in 48-well plates to individual chemicals diluted to a final concentration of 10 μM. Embryos were fixed at 36–38 hpf, processed for in situ hybridization with a tie1 riboprobe, and evaluated for angioblast number. We used the average number of clusters observed unilaterally per embryo per well to score each compound for suppressor activity. The negative control, DMSO, routinely scored between 2.8 and 3.0. Compounds with scores less than or equal to 1.5 were categorized as hits. From this effort, we identified 49 suppressors (4.0% of total) that did not grossly compromise blood vessel development elsewhere in the embryo (Table 1; Figures 1A and S1). The remaining compounds scored greater than 1.5 (90.8%; n = 1,121) or caused early embryonic lethality (5.2%; n = 65; Figure 1A).
Table 1.
Small Molecules Identified by Chemical Screening in Zebrafish for Suppressors of PAA Angioblast Differentiation
| Category | Compounds |
|---|---|
| Kinase and phosphatase inhibitors | |
| TGF-β receptor inhibitor | CB 41227199a |
| Protein kinase A and B inhibitor | CB 97929926 |
| Protein kinase C inhibitor | calphostin C |
| Thymidine kinase inhibitor | CB 17846240 |
| Cyclin-dependent kinase inhibitors | CB 42005171, CB 20372475, CB 14126598 |
| IκB kinase inhibitors | CB 15143266, CB 28324237 |
| c-Jun N-terminal kinase 3 inhibitor | CB 34431373 |
| ERBB2 kinase inhibitor | tyrphostin AG-825 |
| RAF kinase inhibitor | CB 14171361 |
| SRC kinase inhibitor | CB 14280993 |
| CD45 phosphatase inhibitor | RWJ-60475-(AM)3 |
| Bioactive lipids, modulators of lipid metabolism or signaling | |
| Omega-3, 6, 9 fatty acids | eicosatrienoic acid (20:3 n-3) (11,14,17-ETE) docosahexenoic (22:6, n-3) (4,7,10,13,16,19-DHA) arachidonic acid (20:4, n-6) (5,8,11,14-AA) eicosatriynoic acid (20:3, n-9) (5,8,11-ETI) |
| Cyclooxygenase inhibitors | indomethacin, indoprofen |
| Lipoxygenase metabolite | 12(R) = HETE |
| Prostaglandins | PGD2; 13,14-Dihydro-PGE1; 17-phenyl-trinor-PGE2; 9B,11A-PGF2 |
| Leukotriene B4 antagonist | U-75302 |
| Ion channel modulators and ionophores | |
| Potassium channel openers | cloxyquin, minoxidil, rottlerin |
| Calcium ionophore | ionomycin |
| Nuclear hormone receptor inhibitors | |
| Liver × receptor inhibitor | CB 78238822 |
| Retinoic acid receptor inhibitors | CB 70700976, CB 82029493 |
| Other signaling modulators | |
| Nuclear factor κB (NF-κB) inhibitor | caffeic acid phenethyl ester |
| Adenosine A3 receptor agonist | IB-MECA |
| Phosphodiesterase inhibitors | zardaverine, dyphylline |
| Histamine H1 receptor antagonists | doxylamine succinate, carbinoxamine maleate |
| Antimicrobials | fusidic acid, chloroxylenol, nitrofurantoin |
| Other | N-Acetyl-S-geranyl-L-cysteine, guanethidine sulfate, roxarsone, glucosamine hydrochloride, guaifenesin, lactulose, alrestatin |
Four compound libraries were screened, and hits (right column) were binned into categories (left column) based on biological activities.
One hit, CB 41227199, highlighted the importance of TGF-β signaling in PAA angioblast differentiation.
Figure 1. A Small-Molecule Screen in Zebrafish Implicates TGF-β Signaling in PAA Morphogenesis.

(A) 1,235 small molecules were screened for activities that suppress the formation of tie1+ PAA angioblasts in zebrafish. Shown are the percentages of compounds that fell into each of three categories.
(B and C) Left lateral views of tie1+ PAA angioblast clusters in 36-hr post-fertilization (hpf) embryos treated from bud stage with DMSO (B) or 10 μM CB 41227199 (C), a predicted inhibitor of TGF-β signaling. The angioblast clusters are numbered as shown (B) according to the pharyngeal arches within which they form. Seven of 14 chemically treated embryos were devoid of tie1+ clusters.
(D–L) Left lateral views of (D–G) tie1+ or (I–L) kdrl+ PAA angioblasts in 36-hpf embryos treated from 20 hpf with DMSO or the indicated, increasing concentrations of LY-364947. The angioblast clusters that did form in LY-364947-treated embryos were smaller and/or failed to sprout (asterisks in F, G, K, and L). Scale bars, 200 μm.
(H and M) Bar graphs showing the average number of tie1+ or kdrl+ angioblast clusters ±SD that formed in each experimental group (*p < 0.05; **p < 0.01; ***p < 0.001). n = 40–50 embryos per group with a total of three biological replicates.
(N–Q) Left lateral views of 60-hpf Tg(kdrl:GFP),Tg(gata1:DsRed) embryos treated from 20 hpf with DMSO (N) or LY-364947 (O and P) imaged in the green and red channels to visualize endothelium (pseudocolored magenta) and red blood cells (RBC) (pseudocolored yellow), respectively. In DMSO-treated embryos, four patent PAAs supported robust blood flow as evidenced by strong luminal RBC fluorescence (N). LY-364947-treated embryos displayed a reduced number (O; class I) or complete absence (P; class II) of patent PAAs. Asterisks in (P) highlight compensatory sprouting of endothelial cells from the LDA. (Q) Bar graph showing the percentages of DMSO (n = 30) and LY-36497-treated (n = 40) embryos with four patent PAAs (wild-type), one to two patent PAAs (class I), or zero PAAs (class II). ***p < 0.001. No animals contained three patent PAAs. Scale bars, 25 μm.
We binned the small molecules identified by our primary screen into broad categories based on biological activity (Table 1). Compounds targeting receptors or downstream effectors for multiple pathways were identified as were bioactive lipids and modulators of lipid metabolism or signaling. We identified two antagonists predicted to inhibit retinoic acid signaling, a genetic pathway previously implicated in PAA morphogenesis in mice (Li et al., 2012). Although secondary screening will be required to validate hits from the primary screen, our efforts uncover several candidate pathways deserving of further attention.
One suppressor identified by our screen, CB 41227199 (Table 1), is predicted to antagonize the TGF-β type I receptor ALK5 (Figures 1B and 1C). To corroborate that blocking ALK5 inhibits PAA angioblast formation, we treated embryos between 20 and 36–38 hpf with increasing doses of two selective ALK5 antagonists, LY-364947 and SB-505124 (Li et al., 2006; Vogt et al., 2011; Zhou et al., 2011). Both chemicals significantly reduced the number of tie1+ angioblast clusters in a dose-dependent fashion (Figures 1D–1H and S1). LY-364947 also decreased the expression of kdrl (Figures 1I–1M), a second marker of PAA angioblasts. When clusters did form, they were often smaller and/or failed to sprout. These data uncover TGF-β signaling as a pathway required for PAA angioblast cluster formation.
To determine whether phenotypic recovery and/or compensatory angiogenesis from the lateral dorsal aorta (LDA) (Nagelberg et al., 2015) might permit chemically treated embryos to form patent PAAs despite the angioblast phenotype, we analyzed LY-364947-treated embryos for PAA defects at 60 hpf using fluores-cent reporters of the endothelial (kdrl:GFP) (Jin et al., 2005) and red blood cell (gata1:dsRed) (Traver et al., 2003) lineages. Whereas control embryos exhibited robust blood flow through PAAs 3–6 (Figures 1N and 1Q), significant reductions in patent vessel number were observed in LY-364947-treated animals (Figures 1O–1Q). Approximately one-quarter lacked PAAs altogether with many showing evidence of incomplete compensatory angiogenesis. Consistent with our screening criteria, the vascular defects in LY-364947-treated embryos were localized specifically to the pharyngeal arches. The head and body vasculature appeared largely unaffected by LY-364947 treatment and blood flow through the cranial vessels remained intact (Figure S2). Importantly, we ruled out a pharyngeal segmentation defect (Figure S3) as an indirect cause of the tie1+;kdrl+ PAA cluster phenotype (Piotrowski et al., 2003; Schilling et al., 1996). Taken together, these data demonstrate that TGF-β signaling is required for PAA angioblast formation and PAA morphogenesis.
TGF-β Promotes the PAA Progenitor to Angioblast Transition
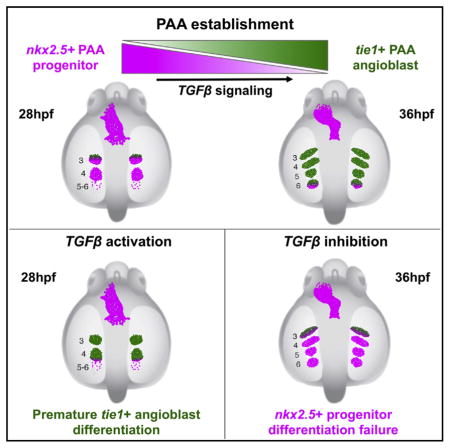
We considered two potential explanations for the LY-364947-mediated reduction in tie1+;kdrl+ angioblast clusters (Figures 1D–1M) including failed specification of nkx2.5+ PAA progenitors in the anterior lateral plate mesoderm (ALPM) (Paffett-Lugassy et al., 2013) or a reduced capacity of progenitors to differentiate into angioblasts in the arches proper. To distinguish these possibilities, we inhibited TGF-β signaling during shorter intervals (Figure 2A), timed to overlap with progenitor specification or differentiation, and evaluated animals for angioblast formation. TGF-β suppression during progenitor cell specification (10–16 hpf) did not impair tie1+ angioblast formation (Figures 2A, 2B, 2G, and 2L). By contrast, TGF-β inhibition during progenitor cell differentiation (16 hpf+) effectively suppressed angioblast emergence (Figures 2A and 2H–2L) compare to DMSO vehicle control (Figures 2A, 2C–2F, and 2L). In fact, significant reductions in tie1+ clusters were observed in embryos treated as late as 24 and 28 hpf, which coincides temporally with the anterior to posterior wave of PAA cluster differentiation (Paffett-Lugassy et al., 2013). Together, these data suggest that TGF-β pathway inhibition might impair the progenitor to angioblast transition.
Figure 2. TGF-β Signaling Is Required during PAA Progenitor Differentiation.

(A–L) Stage-specific LY-364947 exposure reveals a requirement for TGF-β signaling after PAA progenitor specification. (A) Schematic diagram showing drug treatment regimens designed to inhibit TGF-β signaling during (10–16 hr post-fertilization; hpf) and/or after (+16 hpf) progenitor specification. (B–K) Left lateral views of tie1+ PAA angioblast clusters in 36-hpf embryos treated during the indicated developmental windows with DMSO or 50 μM LY-364947. The angioblast clusters that did form in LY-364947-treated embryos appeared smaller and/or failed to sprout (asterisks in H, J, and K). Scale bars, 200 μm. (L) Bar graph showing the average number of tie1+ angioblast clusters ±SD within each experimental group (*p < 0.05; **p < 0.01; ***p < 0.001). n = 20–50 embryos per treatment group with a total of three biological replicates.
To test this hypothesis directly, we evaluated TGF-β-suppressed animals for expression of an earlier marker of the angio-blast lineage, etsrp, that precedes tie1 and kdrl expression (Paffett-Lugassy et al., 2013). Dose-dependent reductions in etsrp+ cluster numbers were observed following treatment with LY-364947 (Figures 3A–3E), indicating that initiation of the angio-blast program requires TGF-β signaling. Next, we analyzed expression of the PAA progenitor marker nkx2.5 that disappears in a cranial to caudal sequence as tie1 expression initiates in a reciprocal fashion (Paffett-Lugassy et al., 2013). In control embryos at 36 hpf, we observed nkx2.5 expression exclusively in progenitors residing in pharyngeal arch 6 because expression in arches 3–5 has downregulated by this stage (Figure 3F) (Paffett-Lugassy et al., 2013). By contrast, TGF-β-inhibited embryos expressed nkx2.5 in pharyngeal arches 3–6 indicating that nkx2.5 transcripts persisted abnormally in the anterior three arches (Figure 3G). We quantified this phenotype using double transgenic embryos, [Tg(nkx2.5:nZsYellow) (Paffett-Lu-gassy et al., 2013); Tg(fli1a:nEGFP) (Yaniv et al., 2006)], that express fluorescent proteins in the nuclei of PAA progenitor or endothelial cells (Figures 3H–3J). This analysis revealed a 3-fold increase in the number of nkx2.5+ PAA progenitor cells resulting from TGF-β suppression (Figure 3J). These data demonstrate that TGF-β signaling is required for nkx2.5+ PAA progenitors to differentiate into angioblasts.
Figure 3. TGF-β Signaling Is Required for the PAA Progenitor to Angioblast Transition.

(A–D) Left lateral views of etsrp+ PAA angioblasts in 36-hpf embryos treated from 20-hpf stage with DMSO or the indicated, increasing concentrations of LY-364947. The angioblast clusters that did form in LY-364947-treated embryos were smaller and/or failed to sprout (asterisks in C and D). Scale bars, 200 μm.
(E) Bar graph showing the average number of etsrp+ angioblast clusters ±SD within each experimental group (**p < 0.01; ***p < 0.001). n = 40–50 embryos per treatment group with a total of four biological replicates.
(F and G) Left lateral views of nkx2.5+ PAA progenitors in 36-hpf embryos treated from 20 hpf with DMSO (F, n = 43) or LY-364947 (G, n = 38/54 with the phenotype shown, 14/54 showed a milder phenotype, 2/54 appeared normal). The line in (G) highlights persistent expression of nkx2.5 transcripts in PAA progenitors of arches 3–6 in LY-364947-treated embryos.
(H and I) Left lateral views of 38-hpf Tg(fli1a:nGFP),Tg(nkx2.5:ZsYellow) embryos treated from 20 hpf with DMSO or LY-364947, immunostained for GFP and ZsYellow, and imaged in the green and red channels to visualize PAA endothelial (pseudocolored magenta) and progenitor (pseudocolored yellow) cell nuclei, respectively.
(J) The bar graph shows the average numbers ±SD of nkx2.5:ZsYellow+ PAA progenitors in DMSO and LY-364947-treated embryos. n = 4 for both experimental groups. ***p < 0.0001. Scale bars, 25 μm.
TGF-β Signaling Components Are Expressed in the Pharynx during PAA Angioblast Differentiation
To visualize the pharyngeal expression patterns of TGF-β pathway components, we performed in situ hybridization for five TGF-β ligands, four TGF-β receptors, and one TGF-β chaperone at 28 hpf, a time point immediately preceding PAA angioblast differentiation (Figure 4) (Paffett-Lugassy et al., 2013). Transcripts encoding the type I (ALK5) and type II TGF-β receptor paralogs were broadly distributed at varying levels throughout the pharynx (Figures 4A–4D). Although expression of TGF-β1b did not appear robust (Figure 4F), mRNAs encoding TGF-β1a, 2a, 2b, and 3 were more pronounced (Figures 4E, 4G, 4I, and 4J). Specifically, TGF-β1a and 2b transcripts were broadly distributed (Figures 4E and 4I), while 2a and 3 co-localized with the endodermal pouch marker nkx2.3 (Figures 4H and 4K). Transcripts encoding the TGF-β chaperone, Latent TGF-β Protein 3 (LTBP3), were also observed in the pharyngeal pouches (Figure 4L). Taken together, these data suggest that multiple cell types in the pharynx are capable of producing and responding to TGF-β ligands during the PAA angioblast differentiation window. Moreover, the overlapping distributions of multiple receptors and ligands indicate that considerable genetic redundancy exists within the TGF-β signaling pathway to ensure proper PAA angioblast differentiation.
Figure 4. TGF-β Pathway Components Are Expressed in the Pharynx during PAA Angioblast Differentiation.

(A–L) Left lateral views of indicated TGF-β pathway component expression in 28 hr post-fertilization (hpf) embryos. (A–D) Transcripts encoding the canonical TGF-β receptor paralogs RIa/b and RIIa/b are broadly distributed in the pharyngeal region. (E–G, I, and J) Expression of the indicated TGF-β ligands in the pharynx. (H, K, and L) Double in situ hybridization highlighting overlap of indicated TGF-β pathway component transcripts with transcripts encoding the pharyngeal endodermal pouch marker, nkx2.3. (n = 30–40 embryos per in situ hybridization probe). Scale bars, 200 μm.
A Wave of TGF-β Signaling in the Pharynx Precedes the Differentiation of PAA Angioblasts from nkx2.5+ Precursors
To better characterize the timing and location of TGF-β receptor activity in pharyngeal tissues during PAA development, we stained embryos for phosphorylated-Smad3 epitopes (Carlson et al., 2008; Schmierer and Hill, 2007; Shi and Massagué, 2003), prior to and during the emergence of tie1+ PAA angioblast clusters. At these stages, pharyngeal arches 3–6 comprise mesodermal cores enveloped by sox10+ neural-crest-derived cells that are bounded medially by sox17+ endodermal pouches (Figures 5A–5C) (Piotrowski et al., 2003). Approximately 4 hr prior to the appearance of tie1 expression in arch 3 (28 hpf) (Paffett-Lugassy et al., 2013), we observed pSmad3 staining in a majority of nkx2.5+ PAA progenitor cells in the same location (Figures 5D and 5E). By contrast, progenitors in arch 4 were devoid of pSmad3 at this developmental stage (Figures 5D and 5E). Four hours later, pSmad3 staining was observed in arch 4 progenitors (Figures 5F and 5G), prior to tie1 expression (Paffett-Lugassy et al., 2013), but no longer in tie1+ arch 3 angioblasts (Figures 5F and 5G). Finally, at 36 hpf, pSmad3 was observed in arch 5 progenitors (Figures 5H and 5I), prior to tie1 expression (Paffett-Lugassy et al., 2013), but no longer in tie1+ arch 4 angio-blasts (Figures 5H and 5I). These data uncover a cranial to caudal wave of TGF-β receptor activity in PAA progenitor cells that anticipates a subsequent wave of angioblast lineage commitment. The spatiotemporal dynamics of pSmad3 staining are consistent with TGF-β signaling playing a cell-autonomous role in initiating the PAA progenitor to angioblast transition. However, because additional pharyngeal lineages, including neural-crest-derived and endodermal cells, also stained positive for pSmad3 (Figures 5D–5J), we cannot rule out non-cell-autonomous functions for TGF-β in PAA morphogenesis. As anticipated, pSmad3 epitopes were essentially non-existent in LY-364947-treated embryos (Figures 5K–5N) confirming that the small molecule effectively antagonizes TGF-β signaling.
Figure 5. A Burst of TGF-β Signaling Precedes tie1+ PAA Angioblast Differentiation.

(A) Cartoon of zebrafish embryo highlighting the left-side pharyngeal arches 3–6 (boxed) analyzed at higher magnifications in (B)–(N). The spatial relationships between PAA progenitors (magenta), neural-crest-derived cells (green), and endodermal pouches (gray) are shown.
(B and C) Left-side dorsal views of 32 hr post-fertilization (hpf) Tg(sox10:GFP), Tg(nkx2.5:ZsYellow), or Tg(sox17:GFP),Tg(nkx2.5:ZsYellow) embryos immuno-stained for GFP and ZsYellow and imaged in the green and far red channels to visualize neural crest cells (pseudocolored green), endodermal pouches (pseudocolored gray), and PAA progenitor clusters (pseudocolored magenta). n = 5 for both groups.
(D–I) Spatiotemporal dynamics of TGF-β signaling during PAA angioblast differentiation. Dorsal views of Tg(nkx2.5:GFP) embryos at the indicated developmental stages immunostained for GFP and pSMAD3 imaged in the green and far red channels to visualize PAA progenitors (pseudocolored magenta) and cells experiencing active TGF-β signaling (pseudocolored yellow). 28 hpf, n = 3; 32 hpf, n = 4; 36 hpf, n = 6.
(J) Left-side dorsal views of Tg(sox17:GFP) embryos at 36 hpf immunostained for GFP and pSMAD3 imaged in the green and far red channels to visualize endodermal pouches (pseudocolored gray) and cells active for TGF-β signaling (pseudocolored yellow), n = 2.
(K–N) Left-side dorsal views of Tg(nkx2.5:GFP) embryos at 32 hpf immunostained for GFP (pseudocolored magenta) and pSMAD3 (pseudocolored yellow) following treatment from 20 hpf with DMSO (K and L; n = 3) or 50 μM LY-364947 (M and N; n = 8). Scale bars, 25 μm.
Premature TGF-β Signaling Is Sufficient to Stimulate Precocious Appearance of PAA Angioblasts
To test the sufficiency of TGF-β signaling for initiating the PAA progenitor to angioblast transition, we utilized a previously validated transgenic strain, Tg(hsp70:caALK5) (Zhou et al., 2011), to achieve heat-shock inducible expression of a constitutively active isoform of ALK5 beginning at 14 hpf, a time point that is 18 hr prior to angioblast emergence (Figure 6A). Embryos were analyzed for either etsrp+ or tie1+ PAA angioblast clusters at 24 or 28 hpf, respectively, a developmental stage when ~80% of control animals are completely devoid of angioblasts (Figures 6B, 6C, 6E, 6F, 6H, and 6I). Remarkably, 75% and 92% of embryos with premature TGF-β signaling contained one or two substantial etsrp+ and tie1+ angioblast clusters, respectively (Figures 6D, 6G, 6H, and 6I). These data demonstrate that genetic activation of TGF-β signaling is sufficient to initiate the PAA progenitor to angioblast transition. Because pharyngeal arches 5 and 6 are not seeded with nkx2.5+ progenitor cells until after 28 hpf (Paffett-Lugassy et al., 2013), the absence of pre-mature tie1 expression in these arches was not surprising. Together, our data demonstrate that TGF-β signaling is both necessary and sufficient to drive PAA angioblast lineage commitment.
Figure 6. Premature TGF-β Signaling Induces Precocious Differentiation of tie1+ PAA Angioblasts.

(A) Experimental design for heat-shock-mediated (HS+) induction of TGF-β signaling in Tg(hsp70:caAlk5) embryos at 14 hpf (10 somites stage) followed by analysis of etsrp+ and tie1+ PAA angioblasts at 24 or 28 hr post-fertilization (hpf), respectively. Transgenic embryos not exposed to heat shock (HS–, Tg+) and heat shocked wild-type embryos (HS+, Tg–) were used as controls.
(B–G) Left lateral images of 24-hpf (B–D) or 28-hpf (E–G) control (B, C, E, and F) and experimental (D and G) embryos. Two large etsrp+ or tie1+ PAA angioblast clusters are visible in (D) and (G), respectively, whereas the majority of control embryos (B, C, E, and F) lacked PAA angioblasts at this stage. Scale bars, 200 μm.
(H and I) Bar graphs showing the average number of etsrp+ or tie1+ angioblast clusters ±SD within each experimental group (*p < 0.05; **p < 0.01). n = 40–50 embryos per group with a total of three biological replicates.
DISCUSSION
Our observations in zebrafish support a model in which nkx2.5+ PAA progenitor clusters receive a TGF-β signal that initiates their differentiation to the tie1+ angioblast lineage in a cranial to caudal sequence. Specifically, we witnessed a burst of active TGF-β signaling as detected by pSmad3 immunofluorescence in each nkx2.5+ PAA cluster just prior to angioblast lineage commitment. This differentiation event was sensitive to small-molecule inhibition of TGF-β signaling, demonstrating the necessity of this program for PAA morphogenesis. Most surprisingly, early activation of the ALK5 TGF-β receptor resulted in premature differentiation of PAA progenitor clusters, demonstrating the sufficiency of TGF-β signaling for driving the tie1+ angioblast fate. Because we observed pSmad3 epitopes broadly throughout the pharynx during angioblast emergence, tissue-specific requirements for TGF-β signaling in PAA establishment cannot be investigated until floxed alleles of TGF-β pathway components become available.
While TGF-β signaling has yet to be implicated in mammalian PAA establishment, it plays a clear role in PAA maintenance and remodeling. Specifically, mice deficient in TGF-β2 display IAA-B and ARSA that stem from abnormal regression of the fourth PAAs (Molin et al., 2002). Similarly, IAA-B and ARSA were observed in mice with neural-crest-specific deletions of the TGF-β type II receptor (TβRII) (Choudhary et al., 2006) or Smad4 (Nie et al., 2008). However, because mice that are homozygous for null alleles of TGF-β1 (Dickson et al., 1995), TβRI (Larsson et al., 2001), TβRII (Oshima et al., 1996), Smad2 (Nomura and Li, 1998; Weinstein et al., 1998), or Smad4 (Yang et al., 1998) die prior to PAA establishment, they could not be assessed for potential defects in PAA formation. Our zebrafish data bring up the possibility that bypassing this early embryonic lethality with more refined genetic tools might reveal PAA establishment phenotypes in mice. TGF-β signaling is essential for extra-embryonic vasculogenesis (Dickson et al., 1995; Oshima et al., 1996), and the TGF-β2 ligand was recently shown to be required and sufficient to promote endothelial cell differentiation from murine induced pluripotent stem cells (iPSCs) in culture (Di Bernardini et al., 2014). These observations, together with our zebrafish studies, provide a developmental context within which TGF-β might serve as a critical regulator of vasculogenesis in the mammalian embryo proper.
In summary, our small-molecule screen represents the first unbiased approach to begin dissecting the molecular basis of PAA establishment, an understudied yet important line of inquiry. Identification of the genetic programs that regulate PAA development could be leveraged to inform the development of improved preventative and/or therapeutic approaches for treating congenital cardiovascular malformations involving the aortic arch and its branches.
EXPERIMENTAL PROCEDURES
Zebrafish Husbandry and Strains
Zebrafish were grown and maintained according to protocols approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee. The following strains were utilized: Tg(–4.9sox10:egfp)ba2(Carney et al., 2006), Tg(nkx2.5:GFP)el83(Choe et al., 2013), Tg(nkx2.5:ZsYellow)fb7(Zhou et al., 2011), Tg(sox17:GFP)s870 (Mizoguchi et al., 2008), Tg(kdrl:GFP)la116 (Jin et al., 2005), Tg(gata1:DsRed)sd2 (Traver et al., 2003), Tg(hsp70l:caALK5)fb6 (Zhou et al., 2011), Tg(nkx2.5:nZsYellow)fb17 (Paffett-Lugassy et al., 2013), and Tg(fli1a:nEGFP)y7 (Yaniv et al., 2006). Unless specified, live embryos were kept at 28.5°C and anesthetized in standard embryo media containing 0.4% tricaine (ethyl 3-aminobenzoate methanesulfonate, MS222; Sigma). Tg(hsp70:caAlk5) embryos were heat shocked at 37°C s for 1 hr and fixed at 24 hpf for etsrp and 28 hpf for tie1 expression analysis.
Chemical Exposures
For the small-molecule screen, zebrafish embryos were exposed to individual test compounds from tail-bud stage (10 hpf) until 36–38 hpf and evaluated by in situ hybridization for tie1 expression in PAA clusters. Fifteen to 20 wild-type and age-matched embryos per well were arrayed into 48-well plates and exposed to test compounds diluted to 10 μM. Embryos were transferred to 96-well MultiScreen-MESH Filter Plates (EMD Millipore), washed once in E3, and fixed in 4% paraformaldehyde/PBS (pH 7.4) (PFA). Dilute pronase was added overnight to facilitate removal of chorions prior to fixation. The NINDS Custom Collection 2 (320 compounds), BIOMOL ICCB Known Bioactives (415 compounds), Chembridge KinaCore (250 compounds), and Chembridge Nuclear Hormone Receptor Core (250 compounds) libraries were provided by the Institute of Chemistry and Chemical Biology (ICCB) at Harvard Medical School. Chembridge Library collection compound structures can be found using the following link and entering the compound ID number: https://www.hit2lead.com/search.asp?db=SC&SearchPage=id.
Dose-response curves were obtained by treating zebrafish embryos with 20–100 μM SB-505124 (Sigma) or 30–70 μM LY-364947 (Selleckchem) in the dark beginning at 20 hpf. In subsequent experiments, embryos were treated starting at indicated stages with 50 μM LY-364947. Control embryos were exposed concurrently to equivalent amounts of DMSO. After chemical exposure, embryos were washed three to five times in E3 solution and/or fixed with 4% PFA at the indicated stages.
Zebrafish Whole-Mount In Situ Hybridization
Single or double whole-mount in situ hybridizations were performed in glass vials as previously described (Paffett-Lugassy et al., 2013) or in 96-well mesh-bottom plates. Digoxygenin-labeled anti-sense RNA probes to nkx2.3, nkx2.5, tie1, etsrp, kdrl, tgfbr1a, tgfbr1b, tgfbr2a, tgfbr2b, tgfb1a, tgfb1b, tgfb2a, tgfb2b, tgfb3, and ltbp3 were synthesized using a DIG RNA Labeling Kit (SP6/T7/T3; Roche Applied Science). A blue (NBT/BCIP) chromogenic substrate was utilized (Promega). Double in situ hybridizations were performed using anti-sense fluorescein-labeled RNA probes to nkx2.3 that were synthesized using a Fluorescein RNA Labeling Kit (SP6/T7/T3; Roche Applied Science). An orange (INT/BCIP) chromogenic substrate was utilized (Roche Applied Science).
Zebrafish Immunohistochemistry
Manipulations were performed at room temperature unless otherwise stated. Embryos were fixed overnight in 4% PFA at 4°C and then washed at room temperature with PBS (pH 7.4) 0.1% Tween 20 (PBST) four times for 10 min. Embryos were incubated in a bleach solution (0.5% potassium hydroxide [KOH]/3% H2O2/0.1% Tween 20) in the dark until pigment was removed and washed four times for 10 min with PBST. Permeabilization was achieved by incubating embryos in PBST/0.5% Triton X-100 for 2 hr followed by incubation in block solution (5% BSA/5% goat serum in PBST) for 2 hr. Embryos were incubated with primary antibodies diluted in block solution overnight at 4°C. Embryos were washed six times for 20 min in PBST and incubated with secondary antibodies diluted in block overnight at 4°C. Secondary antibodies were removed by washing with PBST six times for 20 min prior to imaging or storage at 4°C in PBST. Primary antibodies specific for GFP (B-2 mouse monoclonal catalog number sc-9996; Santa Cruz Biotechnology), pSMAD3 (Smad3 [phospho S423 + S425] rabbit monoclonal antibody, catalog number ab52903, AbCam), and ZsYellow (RCFP rabbit polyclonal antibody; Clontech) were used at 1:50 dilutions. Secondary antibodies (Alexa Fluor 488 goat α-mouse immunoglobulin G [IgG], Alexa Fluor 555 goat α-rabbit IgG, Alexa Fluor 647 goat α-rabbit IgG [Invitrogen]) were used at 1:500 dilutions.
Microscopy
Live or processed embryos were imaged on a Nikon 80i compound microscope (Nikon) with Retiga 2000R high-speed charge-coupled device (CCD) camera (QImaging) and NIS-Elements advanced research image acquisition and analysis system (Nikon Instruments). Embryos processed for in situ hybridization were mounted on glass slides in 100% glycerol. Images of embryos processed by in situ hybridization were captured using a mounted Excelis AU600HDS HD Camera (Accu-Scope), and the resulting images were focus-stacked with Zerene Stacker Software (Build T201412212230). Confocal microscopy was performed using either a Zeiss LSM5 Pascal Laser Scanning Microscope (Carl Zeiss MicroImaging) or a Nikon A1SiR Confocal Microscope (Nikon). Live and fixed fluorescent embryos were mounted in 0.9% lo-melt agarose in glass-bottom dishes (MatTek) and covered with embryo media containing 0.4% tricaine or PBST. Live Tg(kdrl:GFP)la116;Tg(gata1:DsRed)sd2 embryos were imaged and analyzed for numbers of patent PAAs in z stack confocal images using ImageJ software (Schneider et al., 2012). GFP+ and ZsYellow+ nuclei in Tg(nkx2.5:nZsYellow)fb17; Tg(flia:nEGFP)y7 embryos were counted in z stack confocal images using ImageJ software (Schneider et al., 2012). Tg(nkx2.5:GFP)el83 and Tg(sox17:GFP)s870 embryos were fixed and immunostained for GFP and pSMAD3. z stack images acquired by confocal microscopy and analyzed with ImageJ software (Schneider et al., 2012).
Statistical Analysis
Statistical analysis was performed with GraphPad Prism software version 7.00 for Macintosh (GraphPad). Pharyngeal angioblast cluster numbers were counted bilaterally for each embryo. The larger number was recorded and used to calculate the average number of clusters per embryo per test condition. All results are expressed as mean ± SD. Differences between control and treated groups were assessed by one-way ANOVA followed by either Dunnett’s or Sidak’s multiple comparisons test. The number of PAAs supporting blood flow was determined per left side of each embryo, and a chi-square test was performed using GraphPad Prism Software. The number of ZsYellow+ nuclei was quantified per left side of each embryo. Results are expressed as mean ± SD. An unpaired t test was performed using GraphPad Prism software. In all cases, the investigators were blinded to cohort when quantifying pharyngeal cluster numbers and patent PAA formation. All embryos were included in the assessments.
Supplementary Material
Highlights.
Small-molecule screening implicates TGF-β signaling in PAA morphogenesis
TGF-β inhibition impairs nkx2.5+ PAA progenitor differentiation into angioblasts
A burst of TGF-β signaling activity precedes PAA angioblast differentiation
Premature TGF-β signaling induction promotes precocious angioblast differentiation
Acknowledgments
We thank Kimara Targoff for providing the nkx2.3 plasmid. We thank Dr. Caro-line Shamu and staff at the Institute of Chemistry and Cell Biology (ICCB) Long-wood Screening Facility, Harvard Medical School for helpful discussions throughout the small-molecule screen. M.A. received support from a Servier Institute International Mobility Help Grant and a Victor A. McKusick Fellowship from the Marfan Foundation (2016D001458). N.P.L. was supported by the Har-vard Stem Cell Institute Training Grant (5HL087735), a National Institutes of Health (NIH) National Research Service Award (1F32HL112579), and an MGH Tosteson and Fund for Medical Discovery Award. Creation of the Tg(hsp70:caALK5) zebrafish line was supported by an R01 award to C.G.B. (5R01HL096816) from the National Institutes of Health. This work was supported by an R35 award to C.E.B. (R35HL135831) from the National Institutes of Health. C.E.B. is also supported by an MGH Hassenfeld Cardiovascular Scholar Award and the d’Arbeloff MGH Research Scholar Award.
Footnotes
AUTHOR CONTRIBUTIONS
M.A. and N.P.-L. designed, performed, and interpreted the majority of experiments with support from S.J. and D.J. The small-molecule screen was performed by N.P.-L. with assistance from E.O. and C.J.F. C.G.B. and C.E.B. conceived the study, designed experiments, and interpreted data. M.A., C.G.B., and C.E.B. wrote the manuscript with input from all authors.
Supplemental Information includes four figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2017.07.002.
References
- Anderson MJ, Pham VN, Vogel AM, Weinstein BM, Roman BL. Loss of unc45a precipitates arteriovenous shunting in the aortic arches. Dev Biol. 2008;318:258–267. doi: 10.1016/j.ydbio.2008.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson ME, Hsu M, Conboy IM. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature. 2008;454:528–532. doi: 10.1038/nature07034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney TJ, Dutton KA, Greenhill E, Delfino-Machín M, Dufourcq P, Blader P, Kelsh RN. A direct role for Sox10 in specification of neural crest-derived sensory neurons. Development. 2006;133:4619–4630. doi: 10.1242/dev.02668. [DOI] [PubMed] [Google Scholar]
- Choe CP, Collazo A, Trinh A, Pan L, Moens CB, Crump JG. Wnt-dependent epithelial transitions drive pharyngeal pouch formation. Dev Cell. 2013;24:296–309. doi: 10.1016/j.devcel.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary B, Ito Y, Makita T, Sasaki T, Chai Y, Sucov HM. Cardiovascular malformations with normal smooth muscle differentiation in neural crest-specific type II TGFbeta receptor (Tgfbr2) mutant mice. Dev Biol. 2006;289:420–429. doi: 10.1016/j.ydbio.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Di Bernardini E, Campagnolo P, Margariti A, Zampetaki A, Karamariti E, Hu Y, Xu Q. Endothelial lineage differentiation from induced pluripotent stem cells is regulated by microRNA-21 and transforming growth factor β2 (TGF-β2) pathways. J Biol Chem. 2014;289:3383–3393. doi: 10.1074/jbc.M113.495531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson MC, Martin JS, Cousins FM, Kulkarni AB, Karlsson S, Akhurst RJ. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development. 1995;121:1845–1854. doi: 10.1242/dev.121.6.1845. [DOI] [PubMed] [Google Scholar]
- Hiruma T, Nakajima Y, Nakamura H. Development of pharyngeal arch arteries in early mouse embryo. J Anat. 2002;201:15–29. doi: 10.1046/j.1469-7580.2002.00071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogai S, Horiguchi M, Weinstein BM. The vascular anatomy of the developing zebrafish: An atlas of embryonic and early larval development. Dev Biol. 2001;230:278–301. doi: 10.1006/dbio.2000.9995. [DOI] [PubMed] [Google Scholar]
- Jin SW, Beis D, Mitchell T, Chen JN, Stainier DYR. Cellular and molecular analyses of vascular tube and lumen formation in zebrafish. Development. 2005;132:5199–5209. doi: 10.1242/dev.02087. [DOI] [PubMed] [Google Scholar]
- Kameda Y. Hoxa3 and signaling molecules involved in aortic arch patterning and remodeling. Cell Tissue Res. 2009;336:165–178. doi: 10.1007/s00441-009-0760-7. [DOI] [PubMed] [Google Scholar]
- Larsson J, Goumans MJ, Sjöstrand LJ, van Rooijen MA, Ward D, Levéen P, Xu X, ten Dijke P, Mummery CL, Karlsson S. Abnormal angiogenesis but intact hematopoietic potential in TGF-beta type I receptor-deficient mice. EMBO J. 2001;20:1663–1673. doi: 10.1093/emboj/20.7.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HY, Wang Y, Heap CR, King CHR, Mundla SR, Voss M, Clawson DK, Yan L, Campbell RM, Anderson BD, et al. Dihydro-pyrrolopyrazole transforming growth factor-beta type I receptor kinase domain inhibitors: A novel benzimidazole series with selectivity versus transforming growth factor-beta type II receptor kinase and mixed lineage kinase-7. J Med Chem. 2006;49:2138–2142. doi: 10.1021/jm058209g. [DOI] [PubMed] [Google Scholar]
- Li P, Pashmforoush M, Sucov HM. Mesodermal retinoic acid signaling regulates endothelial cell coalescence in caudal pharyngeal arch artery vasculogenesis. Dev Biol. 2012;361:116–124. doi: 10.1016/j.ydbio.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng ZZ, Liu W, Xia Y, Yin HM, Zhang CY, Su D, Yan LF, Gu AH, Zhou Y. The pro-inflammatory signalling regulator Stat4 promotes vasculogenesis of great vessels derived from endothelial precursors. Nat Commun. 2017;8:14640. doi: 10.1038/ncomms14640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi T, Verkade H, Heath JK, Kuroiwa A, Kikuchi Y. Sdf1/Cxcr4 signaling controls the dorsal migration of endodermal cells during zebrafish gastrulation. Development. 2008;135:2521–2529. doi: 10.1242/dev.020107. [DOI] [PubMed] [Google Scholar]
- Molin DGM, DeRuiter MC, Wisse LJ, Azhar M, Doetschman T, Poelmann RE, Gittenberger-de Groot AC. Altered apoptosis pattern during pharyngeal arch artery remodelling is associated with aortic arch malformations in Tgfbeta2 knock-out mice. Cardiovasc Res. 2002;56:312–322. doi: 10.1016/s0008-6363(02)00542-4. [DOI] [PubMed] [Google Scholar]
- Moore KL, Persaud TVN. The Developing Human. 5. Saunders; 1993. [Google Scholar]
- Nagelberg D, Wang J, Su R, Torres-Vázquez J, Targoff KL, Poss KD, Knaut H. Origin, Specification, and Plasticity of the Great Vessels of the Heart. Curr Biol. 2015;25:2099–2110. doi: 10.1016/j.cub.2015.06.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie X, Deng CX, Wang Q, Jiao K. Disruption of Smad4 in neural crest cells leads to mid-gestation death with pharyngeal arch, craniofacial and cardiac defects. Dev Biol. 2008;316:417–430. doi: 10.1016/j.ydbio.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura M, Li E. Smad2 role in mesoderm formation, left-right patterning and craniofacial development. Nature. 1998;393:786–790. doi: 10.1038/31693. [DOI] [PubMed] [Google Scholar]
- Oshima M, Oshima H, Taketo MM. TGF-beta receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev Biol. 1996;179:297–302. doi: 10.1006/dbio.1996.0259. [DOI] [PubMed] [Google Scholar]
- Paffett-Lugassy N, Singh R, Nevis KR, Guner-Ataman B, O’Loughlin E, Jahangiri L, Harvey RP, Burns CG, Burns CE. Heart field origin of great vessel precursors relies on nkx2.5-mediated vasculogenesis. Nat Cell Biol. 2013;15:1362–1369. doi: 10.1038/ncb2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotrowski T, Ahn DG, Schilling TF, Nair S, Ruvinsky I, Geisler R, Rauch GJ, Haffter P, Zon LI, Zhou Y, et al. The zebrafish van gogh mutation disrupts tbx1, which is involved in the DiGeorge deletion syndrome in humans. Development. 2003;130:5043–5052. doi: 10.1242/dev.00704. [DOI] [PubMed] [Google Scholar]
- Sadler TW. Langman’s Medical Embryology. 10. Lippincott; 2006. [Google Scholar]
- Schilling TF, Piotrowski T, Grandel H, Brand M, Heisenberg CP, Jiang YJ, Beuchle D, Hammerschmidt M, Kane DA, Mullins MC, et al. Jaw and branchial arch mutants in zebrafish I: Branchial arches. Development. 1996;123:329–344. doi: 10.1242/dev.123.1.329. [DOI] [PubMed] [Google Scholar]
- Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: Molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8:970–982. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Traver D, Paw BH, Poss KD, Penberthy WT, Lin S, Zon LI. Transplantation and in vivo imaging of multilineage engraftment in zebrafish bloodless mutants. Nat Immunol. 2003;4:1238–1246. doi: 10.1038/ni1007. [DOI] [PubMed] [Google Scholar]
- Vincent SD, Buckingham ME. How to make a heart: The origin and regulation of cardiac progenitor cells. Curr Top Dev Biol. 2010;90:1–41. doi: 10.1016/S0070-2153(10)90001-X. [DOI] [PubMed] [Google Scholar]
- Vogt J, Traynor R, Sapkota GP. The specificities of small molecule inhibitors of the TGFß and BMP pathways. Cell Signal. 2011;23:1831–1842. doi: 10.1016/j.cellsig.2011.06.019. [DOI] [PubMed] [Google Scholar]
- Weinstein M, Yang X, Li C, Xu X, Gotay J, Deng CX. Failure of egg cylinder elongation and mesoderm induction in mouse embryos lacking the tumor suppressor smad2. Proc Natl Acad Sci USA. 1998;95:9378–9383. doi: 10.1073/pnas.95.16.9378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Li C, Xu X, Deng C. The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc Natl Acad Sci USA. 1998;95:3667–3672. doi: 10.1073/pnas.95.7.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaniv K, Isogai S, Castranova D, Dye L, Hitomi J, Weinstein BM. Live imaging of lymphatic development in the zebrafish. Nat Med. 2006;12:711–716. doi: 10.1038/nm1427. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Cashman TJ, Nevis KR, Obregon P, Carney SA, Liu Y, Gu A, Mosimann C, Sondalle S, Peterson RE, et al. Latent TGF-β binding protein 3 identifies a second heart field in zebrafish. Nature. 2011;474:645–648. doi: 10.1038/nature10094. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.