

Abstract
Rationale
Notch signaling programs cardiac conduction during development, and in the adult ventricle, injury-induced Notch reactivation initiates global transcriptional and epigenetic changes.
Objective
To determine whether Notch reactivation may stably alter atrial ion channel gene expression and arrhythmia inducibility.
Methods and Results
To model an injury response and determine the effects of Notch signaling on atrial electrophysiology, we transiently activate Notch signaling within adult myocardium using a doxycycline-inducible genetic system (iNICD). Significant heart rate slowing and frequent sinus pauses are observed in iNICD mice when compared with controls. iNICD mice have structurally normal atria and preserved sinus node architecture, but expression of key transcriptional regulators of sinus node and atrial conduction, including Nkx2-5, Tbx3 and Tbx5 are dysregulated. To determine whether the induced electrical changes are stable, we transiently activated Notch followed by a prolonged washout period and observed that, in addition to decreased heart rate, atrial conduction velocity is persistently slower than control. Consistent with conduction slowing, genes encoding molecular determinants of atrial conduction velocity, including Scn5a (Nav1.5) and Gja5 (connexin 40), are persistently down-regulated long after a transient Notch pulse. Consistent with the reduction in Scn5a transcript, Notch induces global changes in the atrial action potential, including a reduced dVm/dtmax. In addition, programmed electrical stimulation near the murine pulmonary vein demonstrates increased susceptibility to atrial arrhythmias in mice where Notch has been transiently activated. Taken together, these results suggest that transient Notch activation persistently alters ion channel gene expression and atrial electrophysiology, and predisposes to an arrhythmogenic substrate.
Conclusions
Our data provide evidence that Notch signaling regulates transcription factor and ion channel gene expression within adult atrial myocardium. Notch reactivation induces electrical changes resulting in sinus bradycardia, sinus pauses and a susceptibility to atrial arrhythmias, which contribute to a phenotype resembling sick sinus syndrome.
Keywords: Sick sinus syndrome, Notch, atrial arrhythmia, ion channels, electrophysiology
Subject Terms: Electrophysiology, Ion Channels, Gene Expression and Regulation, Cell Signaling
INTRODUCTION
Rhythmic activation of the heart is governed by an intricate network of distinct cardiomyocyte subtypes, each with unique electrical characteristics due in part to the combination of ionic currents. Nodal cells express higher levels of subunits encoding the membrane voltage and Ca2+ clocks which contribute to spontaneous diastolic depolarization during phase 4 of the action potential. In contrast, atrial and ventricular cardiomyocytes display a more hyperpolarized and more stable resting membrane potential and less diastolic depolarization. Atrial and ventricular cells have a high density of the Na+ current INa and a fast upstroke velocity, while nodal cells exhibit a slow upstroke velocity primarily driven by the L-type Ca2+ current, ICa,L. There is an emerging paradigm dictating that many genetic pathways guiding morphologic patterning of the heart also program myocardial electrical properties1–5. A better understanding of the transcriptional networks and epigenetic regulators that program cardiomyocyte electrical properties and maintain expression patterns in the adult may further our understanding of arrhythmias, a significant cause of morbidity and mortality.
Genome-wide association studies have linked variation in baseline heart rate (HR) with polymorphisms located near ion channel and transcription factor genes, and many of these genetic variants also predispose to atrial arrhythmias6. For example, the transcription factor NKX2-5 regulates gene networks within the heart, and polymorphisms located near this locus have been linked with both HR variability and increased susceptibility to atrial fibrillation (AF)6, 7. Cardiomyocyte-specific loss of Nkx2-5 results in sinus bradycardia, increased arrhythmia susceptibility, and increased Notch activity8. The Notch signaling pathway regulates many aspects of cardiac morphology and electrical programming in the ventricles, however, the role of Notch signaling in regulation of atrial gene expression and impulse propagation is currently unknown3, 9–11.
While Notch signaling is quiescent in adult cardiomyocytes under homeostatic conditions, Notch is transiently activated within cardiomyocytes in response to injury11–15. We previously demonstrated that injury-induced Notch reactivation in ventricular cardiomyocytes leads them to adopt an electrical phenotype resembling Purkinje-like cells9, 10. This is due in part to decreased expression of the subunits encoding voltage-sensitive K+ channels11. Interestingly, we found significantly different effects of Notch signaling within left and right ventricular myocardium, due in part to distinct transcriptional responses11. Utilizing a genetic mouse model to transiently activate Notch signaling within adult atrial myocardium we found that, in contrast to its effects on ventricular myocardium, transient Notch activation in atrial myocardium reduces Scn5a expression, which encodes the major sodium channel in the heart. Notch activation alters fundamental conduction parameters including baseline HR and atrial conduction velocity (CV) without inducing structural alterations of the atria or sinoatrial node (SAN). Notch-induced electrical changes result in sinus bradycardia, sinus pauses, and increased susceptibility to atrial arrhythmias, and together this phenotype resembles Sick Sinus Syndrome (SSS). Interestingly, transient induction of Notch, which may occur in response to cardiac injury or elevated atrial pressure, induces long-term changes in conduction parameters, consistent with an “electrical scar”, suggesting that the Notch pathway may intersect with epigenetic regulators to control or modulate ion channel gene expression11.
METHODS
Expanded methods are presented in the Online Supplement available at http://circres.ahajournals.org
Animals
αMHC-rtTA63, tetO_NICD64, tdTomato65, N1IP::CreHI16, Cntn2-eGFP66, Hcn4CreERT228, and R26RNICD67 mice have been described previously, and were maintained on a mixed genetic background. For experiments involving conditional gene expression, induction of NICD expression was accomplished with doxycycline chow during the stated time points. αMHC-rtTA, αMHC-rtTA; Cntn2-eGFP, and tetO_NICD littermates fed doxycycline, or αMHC-rtTA; tetO_NICD without doxycycline, were used for comparison with αMHC-rtTA; tetO_NICD or αMHC-rtTA; tetO_NICD; Cntn2-eGFP fed doxycycline (iNICD) as noted in the figure legends.
Autonomic response drug studies
Drug responses were tested by intraperitoneal injection of isoproterenol (0.2 mg/kg), atropine (1mg/kg) and carbachol (0.3 mg/kg).
Histology and immunohistochemistry
Immunohistochemistry was performed on paraffin-embedded sections with antibodies recognizing Contactin-2 (1:25, AF4439, R&D Systems), connexin 40 (1:1000, CX40-A, Alpha Diagnostic International), Nkx-2.5 (1:25, N-19, sc-8697, Santa Cruz) and on frozen sections with antibodies recognizing Nav1.5 (1:25, ASC-005, Alomone Labs) and PECAM-1 (1:20, EIA-310, Dianova).
Optical mapping
Optical mapping experiments were performed to determine conduction velocity according to standard techniques on RA preparations performed as previously described, with minor modifications68. Isolated atria were prepared by removing the ventricles and making an incision along the atrial septum from the tricuspid valve to the superior vena cava. Changes in the membrane potential were detected by Di-4-ANEPPS and blebbistatin was used to eliminate motion artifacts in the recorded optical signal. For determination of arrhythmia susceptibility, a programmed pacing protocol that consists of single extrastimulus pacing with varying coupling intervals was performed on each whole heart.
Microelectrode recordings
Mouse hearts were Langendorff perfused and recorded while in sinus rhythm. Using glass sharp microelectrodes, single RA cardiomyocytes were sampled near the epicardial surface.
Statistical analysis
All data are expressed as means ± standard error of the mean. Statistical analyses were performed after assessing for normal distribution using unpaired t tests for comparison of 2 groups with a Welch’s correction, and one-way ANOVA followed by a Dunnett’s multiple comparisons test for comparison of 3 groups. Detection of genotype effect, drug effects, and interaction between drug and genotype for the autonomic innervation studies were tested with a repeated measures 2 way ANOVA. Post-hoc Sidak’s multiple comparisons test was used to compare each drug to baseline within genotypes as indicated in the table. Values of P<0.05 were considered statistically significant.
RESULTS
Notch signaling is reactivated in the atrium in models of increased right-sided pressure
Notch is a critical regulator of diverse developmental processes, including cardiac morphogenesis and cardiomyocyte electrical programming3, 9, 11. To demarcate cells where Notch1 signaling has been active during development, we performed lineage-tracing experiments in mice where the Notch1 activity-trap line is combined with the TdTomato reporter allele (N1IP::CreHI; TdTom, Supplemental Figure IA16). Clusters of atrial cardiomyocytes within right and left atria are labeled (Supplemental Figure IB–G), indicative of active myocardial Notch signaling, and consistent with previous reports delineating active Notch signaling within embryonic atrial cardiomyocytes using immunohistochemical methods to detect the Notch1 intracellular domain (NICD)15. While Notch signaling is normally quiescent in adult myocardium, it can be reactivated in response to injury11, 15. We previously demonstrated in a model of gradual and predictable progression of adverse left ventricular remodeling leading to heart failure (moderate transaortic constriction and small myocardial infarction within the same animal), that transcripts encoding several Notch pathway components are up-regulated within the left ventricle11. Since left heart failure can be associated with sinus bradycardia in mice, we asked whether the Notch pathway is reactivated in the right atrium (RA) in heart failure. Mice were sacrificed four weeks after surgery, at which time the direct Notch targets Hes1 and Nrarp are up-regulated within the RA (Figure IA, Supplemental Table I). To determine in which cells Notch activation occurs, we made use of a tamoxifen-inducible Notch1 activation-dependent reporter knock-in mouse line (NIP1::CreERT2; R26RTdTomato17, Supplemental Figure IIA). In this line, the intracellular domain of Notch1 was replaced with a complementary DNA encoding a 6× myc-tagged CreERT2 (6mtCreERT2). Binding of Notch ligands to the NIP1::CreERT2 triggers CreERT2 release from the membrane, which only enters the nucleus in the presence of tamoxifen. In the absence of tamoxifen no cells are labeled (data not shown), but sham surgical animals treated with tamoxifen display active Notch1 signaling within PECAM-1+ (platelet and endothelial cell adhesion molecule-1) endothelial cells, as expected, and labeling of α-actinin+ atrial cardiomyocytes was not detected (Figure 1B, top panel, Supplemental Figure IIB, top panel). In contrast, induction of left ventricular remodeling together with administration of tamoxifen results in Notch1 activation (red) in many α-actinin+ atrial cardiomyocytes (green) (Figure 1B, bottom panel) that are PECAM-1 negative (green) (Supplemental Figure IIB, bottom panel).
Figure 1. Notch signaling is reactivated in the atrium with increased right-sided pressure and results in sinus bradycardia.
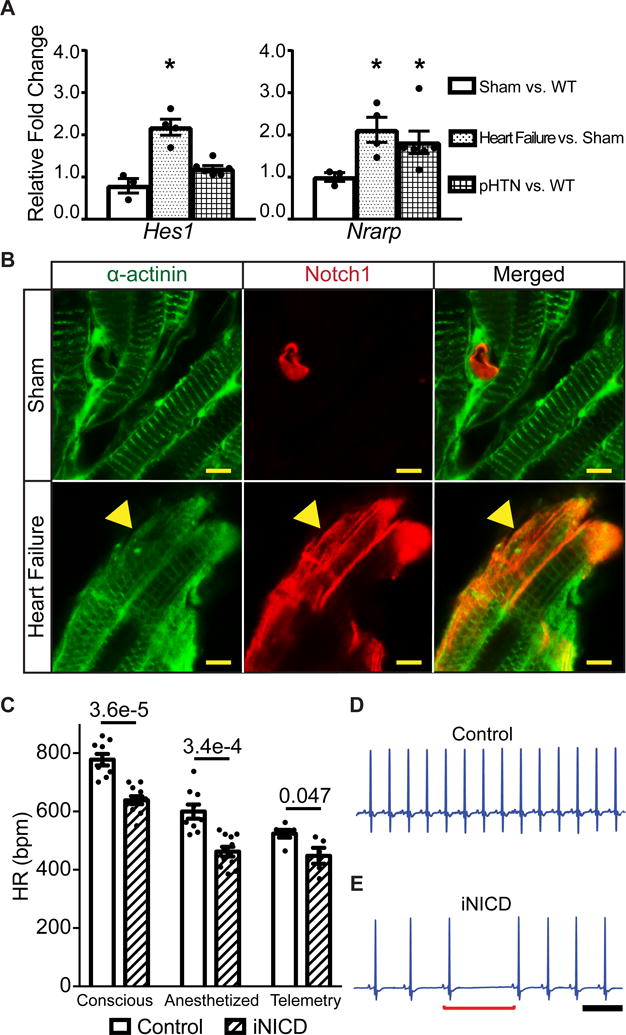
(A) We utilized a previously validated surgical approach that combines moderate transverse aortic constriction (TAC) and distal left anterior coronary ligation (MI) to produce a gradual and predictable progression of adverse left ventricular (LV) remodeling that leads to heart failure. Mice were sacrificed 4 weeks after surgery and RT-qPCR was performed on RA samples to assess expression of Notch target genes. Gene expression is represented as fold change in sham CD-1 RA versus non-instrumented CD-1 mice as a negative control, or TAC + MI mice versus shams (CD-1 control: n=2 females, n=1 male; sham: n=2 female, n=1 male; heart failure: n=3 females, n=1 male). In this heart failure model, the direct Notch targets Hes1 and Nrarp are significantly up-regulated. To test for Notch activation in a model of pulmonary hypertension, wild type CD-1 mice (n=3 females, n=3 males) were subjected to normoxic (21% O2) or hypoxic (10% O2) conditions for four weeks in an in vivo cabinet. Right atrial pressures are increased in chronic hypoxia when compared with normoxia (Supplemental Table II) and the direct Notch target Nrarp is significantly up-regulated in chronic hypoxic versus normoxic mice. All fold changes are relative to β-actin. Unpaired t test with Welch’s correction was performed between sham versus control, heart failure versus sham, and chronic hypoxia versus control normoxia for each gene and values of *P<0.05 were considered statistically significant. (B) Heart failure was induced via TAC + MI in NIP1::CreERT2; R26RtdTomato mice, where the intracellular domain of Notch1 was replaced with a complementary DNA encoding a 6× myc-tagged CreERT2 (6mtCreERT2). In this line, the intracellular domain of Notch1 was replaced with a complementary DNA encoding a 6× myc-tagged CreERT2 (6mtCreERT2). Binding of Notch ligands to the NIP1::CreERT2 triggers CreERT2 release from the membrane, but it only enters the nucleus in the presence of tamoxifen. While in the absence of tamoxifen no cells are labeled (data not shown), sham surgical animals treated with tamoxifen display active Notch1 signaling within PECAM-1+ (platelet and endothelial cell adhesion molecule-1) endothelial cells, as expected, and labeling of α-actinin+ atrial cardiomyocytes was not detected (Figure 1B, Supplemental Figure II). In contrast, induction of left ventricular remodeling together with administration of tamoxifen results in Notch1 activation (red) in many α-actinin+ atrial cardiomyocytes (green) (yellow arrowhead) as well as in PECAM-1+ endothelial cells (Supplemental Figure II). (C) Notch activation in adult mice significantly slows HR as assessed by conscious EKG, light anesthesia with isoflurane, and during conscious telemetric monitoring. Conscious and anesthetized EKGs were performed on mice that were administered doxycycline chow at 2 months of age to activate Notch for 3 weeks, followed by a 1–2 month washout period. Controls were either tetO_NICD (n=4 males) or αMHC-rtTA (n=1 female, n=4 males) mice fed doxycycline chow. Experimental iNICD mice were αMHC-rtTA; tetO_NICD (n=6 females, n=5 males). For telemetry studies, mice were between 3–5 months of age when doxycycline was administered. Controls were αMHC-rtTA (n=2 females, n=4 males) fed doxycycline, and experimental iNICD mice were αMHC-rtTA; tetO_NICD (n=2 females, n=3 males). The HR was slower in iNICD mice during both low and high activity periods (Supplemental Figure III). iNICD mice exhibit an increased frequency of pauses during telemetric monitoring, as demonstrated by HR variability in Poincaré plots (Supplemental Figure IV), and representative tracings from a control (D) and iNICD mouse (E) showing a sinus pause (underlined by red bracket) and slower HR in iNICD mice. Scale bar=500 ms. Statistics were performed using an unpaired t test with Welch’s correction. P values comparing control with iNICD mice are indicated for each condition, values of P<0.05 were considered statistically significant.
To determine whether Notch signaling is activated in other murine models of increased atrial pressure, in which sinus bradycardia and supraventricular arrhythmias are often observed in humans, we utilized a chronic hypoxia pulmonary hypertension (pHTN) model. The incidence of supraventricular arrhythmias, including sinus bradycardia, atrioventricular block, atrial flutter and atrial fibrillation is high in patients with pHTN18,19. Wild type mice were subjected to continuously normoxic (21% O2) or hypoxic (10% O2) conditions for four weeks in an in vivo cabinet, as previously described20, 21. As expected, mean right atrial pressure is significantly increased in chronic hypoxia mice (Supplemental Table II) and expression of the direct Notch target Nrarp is significantly elevated in the pHTN model when compared with controls (Figure 1A, Supplemental Table I).
Transient Notch activation results in bradycardia
We previously showed that reactivation of Notch within adult ventricular myocardium occurs in the setting of adult injury and leads to reprogramming of ventricular cardiomyocytes toward a Purkinje-like phenotype3, 10, 11. To assess the physiologic consequences of to Notch activation in adult atria, we utilized the Tet-on transgenic system to transiently activate Notch signaling in the myocardium at 2 months of age, as previously described (iNICD, inducible Notch Intracellular Domain; αMHC-rtTA; tetO_NICD10, 11). A significantly slower HR is observed in iNICD mice than in control mice, using multiple methodologies for assessment, including conscious EKGs (778±20bpm versus 638±14bpm, p=3.6E-5), EKGs performed under light anesthesia (600±24bpm versus 462±17bpm, p=3.4E-4), and during conscious telemetric monitoring (524±13bpm versus 448±27bpm, p=0.047, Figure 1C). iNICD mice demonstrate lower average HRs during daytime low activity periods as well as nighttime high activity periods, suggesting the defect is not specific for HR acceleration, as is seen in some bradycardia models22 (Supplemental Figure III). Control mice display a stable HR with low beat-to-beat dispersion on Poincaré plots and infrequent sinus pauses, while iNICD mice have high beat-to-beat dispersion and a high frequency of sinus pauses typical of SAN dysfunction (Figure 1D–E, Supplemental Figure IV). To determine whether structural or functional changes contribute to bradycardia, we performed echocardiography on iNICD mice. iNICD mice have a significantly slower HR measured during echocardiography, however, no other measure of cardiac function is altered (Supplemental Table III). Given that patients with sinus bradycardia due to SSS often develop additional conduction abnormalities, we measured these parameters in isoflurane sedated iNICD mice. Similar to what can sometimes be observed in human SSS, the P wave duration is prolonged in iNICD mice when compared with littermate controls, suggestive of slowed atrial conduction (Table 1A, 9.35±0.53 msec versus 7.61±0.34 msec, p=0.047), while atrioventricular (AV) and intra-ventricular conduction are not significantly altered (Table 1A).
Table 1.
EKG intervals and Response to Autonomic Stimuli
| A | ||
|---|---|---|
|
| ||
| Control n=4 |
iNICD n=8 |
|
| Heart Rate | 634 ±45 | 473 ±21* |
| P wave duration (msec) | 7.61 ±0.34 | 9.35 ±0.53* |
| PR (msec) | 33.5 ±1.6 | 36.9 ±1.2 |
| QRS (msec) | 9.57 ±0.88 | 11.59 ±0.99 |
| B | ||
|---|---|---|
|
| ||
| Control n=11 |
iNICD n=14 |
|
| Baseline HR | 649 ±21 | 559 ±35 |
| Isoproterenol HR (% change) | 728 ±14* (12.1) | 649 ±26† (16.3) |
| Atropine HR (% change) | 742 ±6* (14.2) | 676 ±16‡ (21.1) |
| Carbachol HR (% change) | 230 ±25§ (64.6) | 227 ±13§ (59.4) |
A. Controls are littermate tetO_NICD mice (n=4 males) and iNICD are αMHC-rtTA; tetO_NICD mice (n=4 females, n=4 males). Both genotypes were fed doxycycline chow at 8 weeks of age for 3 weeks with no washout. EKGs were performed on mice during isoflurane sedation. Statistics were performed using unpaired t tests with Welch’s correction.
P<0.05, data are expressed as mean ± SEM.
B. Controls are littermate αMHC-rtTA mice on dox (n=3 females, n=8 males), and iNICD are experimental αMHC-rtTA; tetO_NICD on dox group (n=8 females, n=6 males). All mice were fed doxycycline chow starting between 2–3 months of age for 3 weeks, followed by a washout period of 2 weeks. EKGs were performed on conscious mice. Adrenergic stimulation with isoproterenol was done by intraperitoneal injection of isoproterenol (0.2 mg/kg), inhibition of parasympathetic activity with atropine (1mg/kg), and carbachol injection was given intraperitoneally (0.3 mg/kg) to simulate increased vagal nerve activity. With repeated measures 2 way ANOVA there is a statistically significant genotype effect F(1,23)=10.6, p=0.0039. There is also a statistically significant drug effect across both genotypes F(3,69)=233.2, P<0.001. However, there is not a significant interaction between drug and genotype F(3,69)=1.739, P=0.17.
P<0.05,
P<0.01,
P<0.001,
P<0.0001, data are expressed as mean ± SEM.
Mice with Notch activation have preserved response to autonomic stimuli
HR is regulated by factors intrinsic and extrinsic to the heart, including autonomic input. To determine if responses to sympathetic and parasympathetic stimuli are affected in iNICD mice, we tested their response to isoproterenol, atropine and carbachol during conscious EKGs. At baseline, HR is slowed in iNICD mice but HR increases in response to isoproterenol to a similar degree in iNICD and littermate control animals (16.3% increase in iNICD versus 12.1% increase in control, Table 1B). Injection of atropine to inhibit parasympathetic stimulation also results in a similar HR increase in both iNICD and littermate control animals (21.1% versus 14.2% respectively, Table 1B), while administration of carbachol to simulate high vagal nerve activity reduces the HR in both iNICD and control animals (59.4% versus 64.6% respectively, Table 1B). Taken together, these data suggest that impaired response to the autonomic nervous system does not account for the bradycardia observed in iNICD mice.
Notch regulates Nkx2-5 expression within the sinus node
To further define the mechanism underlying bradycardia in iNICD mice, we examined the morphology of the SAN since significant structural alterations could lead to electrical source-sink mismatch. We did not detect changes in the size or architecture of the SAN in iNICD mice as assessed by comparison of Cntn2-eGFP expression in atrial preparations, or by immunohistochemical staining to delineate Contactin-2+/Connexin 40− (Cntn2+/Cx40−) nodal myocardium located at the lateral junction of the superior vena cava (SVC) and RA (Figure 2A, Supplemental Figures V and VI). We also did not detect excess fibrosis within the region of the SAN, which could have contributed to SSS (Figure 2B–D). Taken together, this suggests that sinus bradycardia in iNICD mice is likely a direct effect on ion channel gene expression and/or function, and not secondary to structural changes. Given that Notch reprograms AV myocardium to a chamber-like phenotype10, we probed for Notch-induced changes in several transcription factors and ion channels known to be important for normal SAN function. The T-box transcription factor Tbx3 is expressed within the SAN and is important for maintaining pacemaking activity23, 24. Tbx3 inhibits Nkx2-5 expression within the sinus node, while Nkx2-5 is highly expressed in atrial and ventricular cardiomyocytes25, 26. We found decreased expression of Tbx3 in the iNICD RA (Supplemental Figure VIIA), and consistent with this result, Nkx2-5 is ectopically mis-expressed within iNICD central nodal cells (Figure 2E–H). Expression of Cacna1g, which encodes Cav3.1 (T-type Ca2+ channel) and Cacna1d, which encodes Cav1.3 (L-type Ca2+ channel), which are responsible for currents important for pacemaker function are also significantly decreased (Supplemental Figure VIIA). Although expression of several transcription factors important for maintaining the nodal phenotype are altered in iNICD mice, expression of the transcription factor Shox2 remained unchanged. Shox2 plays a role in pacemaker development and function and antagonizes Nkx2-5 within the sinus node27. Since Shox2 expression remains unchanged in iNICD mice, this may at least in part explain the absence of changes in all nodal genes. For example, we did not detect ectopic Cx40 expression within the iNICD SAN (Figure 2E–H), and Hcn4 levels are unchanged (Supplemental Figure VIIB,C). Taken together, adult Notch activation dysregulates a subset of the nodal-specific gene expression program.
Figure 2. Nodal structure is preserved with mis-expression of Nkx2-5 in iNICD nodal myocardium.
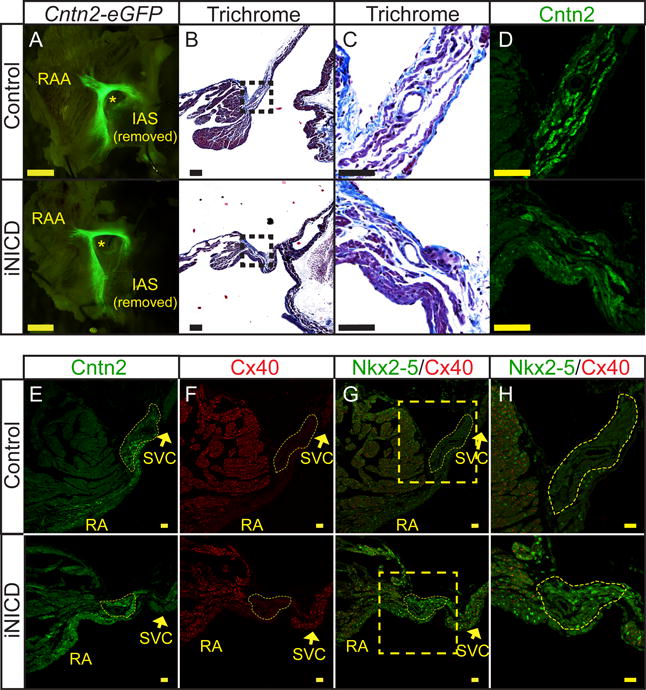
(A) Whole-mount pictures of the RA endocardial surface from a representative control (top, αMHC-rtTA; Cntn2-eGFP) and iNICD (bottom, αMHC-rtTA; tetO_NICD; Cntn2-eGFP) heart indicate no gross difference in Cntn2-eGFP expression delineating overall conduction system morphology (Scale bars=125 μM). Mice were between 5–6 months of age at the start of Notch induction for 3 weeks, followed by a 5-month washout. (B) Representative Masson’s Trichrome images from a control (top, αMHC-rtTA) and iNICD (bottom, αMHC-rtTA; tetO_NICD) SAN located at the junction of the SVC with the RA. Boxed regions in B (scale bars=100 μM) are enlarged in Panel C (scale bars=50 μM). (C) No excess fibrosis was detected within the iNICD SAN to account for the observed sinus bradycardia. (D) Serial sections to Panel C stained for Cntn2 further delineate the region of the SAN (scale bar=50 μM). (E,F) The SAN is histologically identified as Cntn2+ (E) and Cx40− (F) myocardium at the junction of the SVC and RA. Yellow dashed lines demarcate the compact SAN, which has a similar morphology between control (Supplemental Figure V) and iNICD mice (Supplemental Figure VI). Scale bars E,F=20 μM. (G,H) Nkx2-5 is mis-expressed within the compact SAN of iNICD mice. Yellow boxed region in G (scale bar=20 μM) is enlarged in Panel H (scale bar=50 μM) to better visualize the SAN. Mice were started on doxycycline chow between 2–3 months of age and remained on doxycycline chow for 4 weeks with no washout. IAS = inter-atrial septum, RAA = right atrial appendage, SVC = superior vena cava, RA = right atrium. * indicates lumen of SVC near region of the SAN.
To further delineate whether the nodal-specific changes are sufficient to induce bradycardia, we activated Notch specifically within the adult SAN using a tamoxifen inducible Cre (SAN-iNICD; Hcn4CreERT2; R26RNICD28). Lineage tracing in Hcn4CreERT2 mice demonstrates efficient recombination of the Rosa locus within the SAN (Supplemental Figure VIIIA). Interestingly, Notch activation within the SAN alone is not sufficient to cause bradycardia (Supplemental Figure VIIIB). Therefore, we hypothesize that changes in excitability within both the SAN and atrial tissue may play an important role in the etiology of bradycardia in iNICD mice.
Notch-induced electrophysiologic effects are stable after transient induction
Notch signaling is transiently activated during the response to injuries such as myocardial infarction with pressure overload11, 12, 29. To determine the persistence of the consequent Notch-induced electrical changes, we fed iNICD mice doxycycline chow for 3 weeks to activate Notch, followed by an 8-week washout period. NICD expression was up-regulated ~8-fold during doxycycline induction, and was associated with up-regulation of the Notch target genes Hrt1 and Hes1 (Figure 3A–B). As expected, NICD levels returned to baseline after the washout period (Figure 3C). Interestingly, Hes1 remained persistently elevated after cessation of Notch activation (Figure 3C). There was no evidence of structural changes (no increase in heart weight/tibia length, Figure 3D), but the HR also remained depressed in iNICD mice long after the washout period and cessation of Notch induction (Figure 3E). Even transient Notch activation for 12 hours resulted in long-term functional effects, as evidenced by a depressed HR that persists one year later (Figure 3E). This suggests that underlying ion channel gene expression may be stably altered after Notch induction, consistent with an epigenetic effect. To further characterize the persistence of the Notch-induced phenotype, subsequent experiments were performed after transient Notch activation followed by a washout period.
Figure 3. Transient Notch induction leads to prolonged sinus bradycardia.
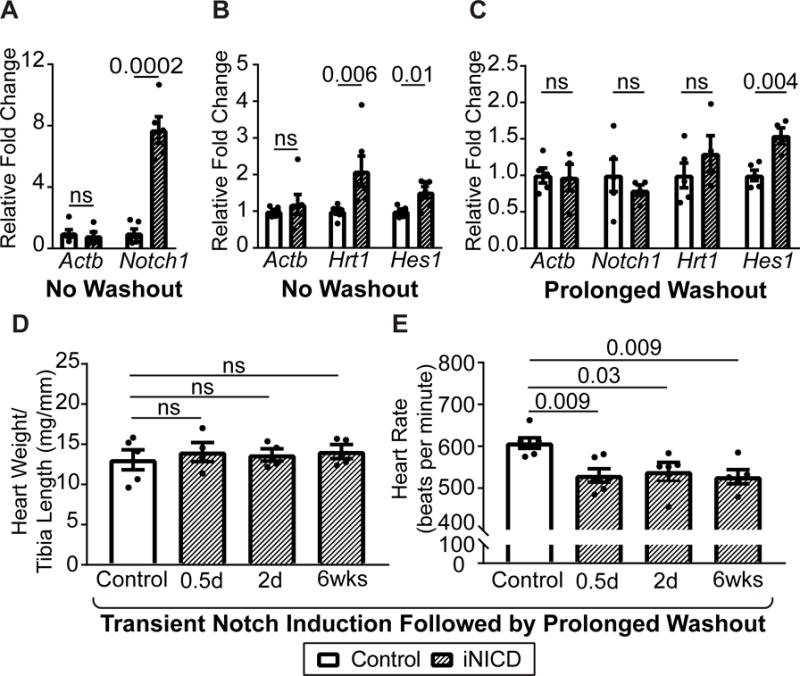
Quantification of N1ICD levels after 3-week doxycycline induction demonstrates approximately 8-fold activation (A), as well as up-regulation of known direct Notch targets Hrt1 and Hes1 (B). Mice were 8 weeks of age at the start of doxycycline administration, controls were αMHC-rtTA; tetO_NICD (n=2 females, n=4 males) never administered doxycycline, experimental mice were αMHC-rtTA; tetO_NICD iNICD (n=1 female, n=5 males) fed doxycycline chow. (C) N1ICD and Hrt1 levels return to baseline after 2 days of doxycycline induction followed by a 1-year washout period. Interestingly, Hes1 remains persistently up-regulated one year after the brief 2-day induction followed by a 1 year washout. (D) Heart weight/tibia length ratio of iNICD mice with varying lengths of Notch induction followed by a 1-year washout indicate no difference in heart size compared with controls. (E) The HR remains persistently depressed in iNICD mice after transient Notch induction for 0.5 days (n=3 females, n=3 males), 2 days (n=2 females, n=3 males), or 6 weeks (n=3 females, n=2 males) followed by a 40 week washout period and cessation of Notch induction. In C–E, control mice were αMHC-rtTA; tetO_NICD never administered doxycycline (n=3 females, n=3 males) and iNICD mice were αMHC-rtTA; tetO_NICD mice with periods of Notch induction beginning at 8 weeks of age for the length of time indicated within the figure. A–C statistics were performed using an unpaired t test with a Welch’s correction. D,E statistics were performed using a one-way ANOVA followed by a Dunnett’s multiple comparisons test. Values of P<0.05 were considered statistically significant. Relative fold changes were in comparison with α-actinin.
In the center of the SAN, action potential propagation is slow and the central nodal cells express little or no Na+ current (INa). However, INa is expressed within SAN peripheral cells where it regulates the pacemaker rate through electrotonic interactions between peripheral and central nodal cells and by regulating impulse propagation from peripheral nodal cells to atrial myocardium. Polymorphisms within SCN5A result in distinct modes of SAN dysfunction, including abnormally slow pacemaker rate and sinus exit block, thought to result from insufficient inward current within peripheral nodal cells to drive the surrounding atrial muscle, or complete failure of the SAN to generate electrical impulses30. To verify that the resolution of our system could detect changes in the dominant pacemaker site, we blocked INa with tetrodotoxin, which results in sinus arrest at high doses and migration of the dominant pacemaker site distally along the crista terminalis at lower doses (Supplemental Figure IX). To determine the mechanism for the observed bradycardia in iNICD mice, we performed optical mapping and far-field EKG of the atria. Despite having a slower HR, the pacemaker location in iNICD hearts remains in the SAN region at the junction of the SVC and RA, similar to control hearts (Figure 4A). Impulse activation within the SAN region in iNICD mice corresponds 1:1 with activation of distant atrial myocardium (Figure 4A). In addition, the cycle length of the pauses during ambulatory telemetry recordings is not a multiple of the preceding cycle length, which is uncharacteristic of sinus exit block (Supplemental Figure X). Taken together, these results are most consistent with Notch activation causing slowing of the intrinsic sinus pacemaker rate.
Figure 4. Transient Notch activation results in stable slowing of atrial conduction velocity.
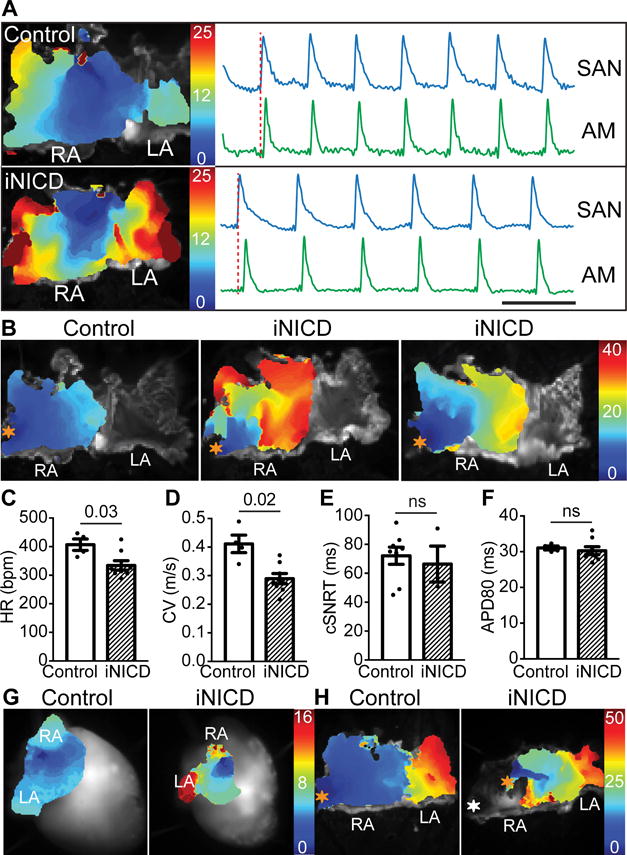
(A) Activation maps from isolated atria from a control and iNICD heart during sinus rhythm demonstrate that, despite a slower HR, the location of the dominant pacemaker remains at the junction of the SVC and RA in iNICD hearts. Optical action potentials of the SAN region and distant atrial myocardium demonstrate 1:1 activation in both control and iNICD atria. Together with RR interval plots of iNICD mice demonstrating that the pauses are not multiples of the preceding RR interval (Supplemental Figure X), the observed bradycardia is not likely due to exit block. Scale bar=0.2 seconds. (B) Activation maps of the RA obtained by pacing the RA appendage (orange asterisk) demonstrate overall slower conduction in iNICD hearts when compared with control. In addition, isochrones crowding and focal areas of conduction block were observed in iNICD atria. Under Langendorff perfusion, iNICD hearts exhibit slower HR (C). Optical mapping of isolated atria showed slower CV (D) in iNICD mice when compared with controls while the corrected SAN recovery time (cSNRT) (E) and APD80 (F) remain unchanged. In A–F, mice were fed doxycycline chow at 2 months of age for 3 weeks, followed by a washout period of 2–3 months. iNICD mice were αMHC-rtTA; tetO_NICD (n=4 females, n=4 males) and controls were littermate tetO_NICD (n=4 males). (G) Notch induction in juvenile mice (3 weeks of age) results in regional atrial inexcitability as evidenced by the absence of depolarization in a portion of the RA during spontaneous sinus rhythm (left), and an inability to capture with RA appendage pacing (H, white asterisk, right). iNICD were αMHC-rtTA; tetO_NICD and controls were littermate tetO_NICD. Statistics were performed using unpaired t tests with Welch’s correction. Values of P<0.05 were considered statistically significant. RA = right atrium, LA = left atrium, SAN = sinoatrial node, AM = atrial myocardium, ns = no statistical difference.
Notch activation slows atrial conduction velocity
To determine whether Notch affects atrial conduction, we performed programmed stimulation of isolated atrial tissue (Figure 4B). Isolated iNICD atrial preparations maintain a depressed HR when compared with control, demonstrating that the bradycardia observed above is not secondary to altered autonomic response (Figure 4C, Table 1B). iNICD mice have an ~30% reduction in RA CV (Figure 4D) while the corrected sinus node recovery time (cSNRT) and action potential duration at 80% repolarization (APD80) were unchanged (Figure 4E,F). This percent reduction in CV is comparable to that seen with complete loss of Cx40 within the atria, where CV is slowed 36% within the RA31. However, in contrast to Cx40−/− mice, in which the overall activation atrial pattern is not altered, iNICD activation maps obtained during RA pacing demonstrate areas of local isochrone crowding and occasional areas of focal conduction block (Figure 4B). Interestingly, when Notch signaling is induced in juvenile mice, we can detect regions of atrial inexcitability characterized by the absence of tissue depolarization during spontaneous sinus rhythm (Figure 4G) and an inability to capture these regions with pacing (Figure 4H). This phenomenon has previously been described in infants and children with congenital SSS due to loss of function mutations in SCN5A32.
Transient Notch activation strongly correlates with a transcriptional signature of atrial arrhythmias
Cardiac impulse propagation is determined by the rate of action potential depolarization (mostly defined by INa), intercellular coupling mediated by gap junctions, and aspects of tissue architecture such as cell shape and interstitial collagen content33. Conduction reserve is usually large, and this high safety factor protects against vulnerability to arrhythmias even when there is moderate impairment in one conduction parameter. Therefore, very large changes in a single conduction parameter, or modest changes in a combination of parameters, are needed to exceed the limits of conduction reserve and impair conduction. Previous animal models exhibiting massive atrial fibrosis have been associated with approximately 30% reduction in RA CV34, similar to the reduction observed in iNICD mice. iNICD mice exhibit a grossly normal atrial size, without increased fibrosis as assessed by Masson’s Trichrome staining (Figure 5A–B). In addition, iNICD mice have normal atrial cardiomyocyte cell size, and normal hydroxyproline levels, which approximate collagen content (Supplemental Figure XI). Immunohistochemical staining for Nav1.5 and Cx40 demonstrates normal localization of both at the sarcolemma in iNICD mice (Figure 5C and Supplemental Figure XII).
Figure 5. Transient Notch activation correlates with a transcriptional signature of atrial arrhythmias.
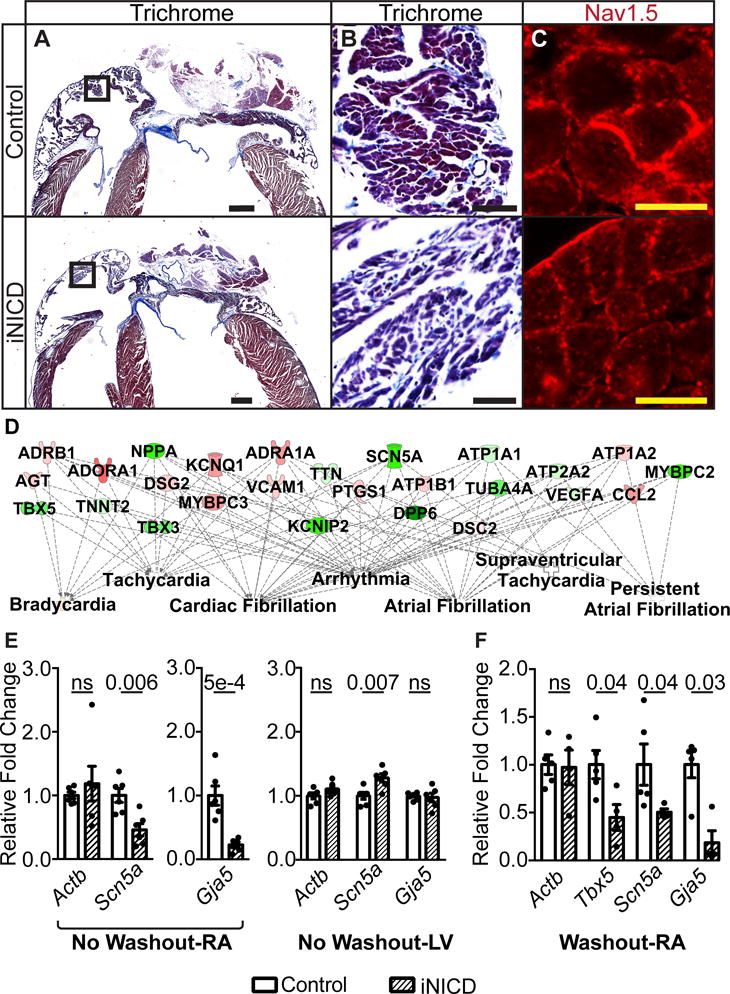
(A,B) Masson’s Trichrome staining demonstrates a grossly normal atrial size in iNICD mice. Boxed region within Panel A (scale bar=500 μM) is enlarged in Panel B (scale bar=50 μM) to reveal comparable levels of fibrosis in control and iNICD mice. Quantification of atrial cardiomyocyte cell size and collagen levels also revealed no differences (Supplemental Figure XI). (C) Nav1.5 immunostaining indicates proper localization to the sarcolemma in iNICD mice. Scale bar=10 μM. Representative images are from n=3 controls (αMHC-rtTA on doxycycline) and n=3 iNICD (αMHC-rtTA; tetO_NICD). In A–C, Notch was induced at 8 weeks of age for 3 weeks with no washout period. (D) RNA-sequencing was performed on 6 iNICD (α-MHC-rtTA; tetO_NICD, n=3 female, n=3 males) and 6 control (α-MHC-rtTA, n=4 female, n=2 males) RA from mice fed doxycycline for 3 weeks starting at 8 weeks of age with no washout period. The total number of differentially expressed transcripts is 910, and this set of differentially expressed genes was further analyzed using Ingenuity Pathway Analysis (IPA). Of the top 25 statistically significant Ingenuity Pathway generated disease or function network categories associated with this gene set, 10 of 25 are related to arrhythmias and 7 of 25 are related to atrial arrhythmias (Supplemental Table V). The FDR adjusted P value predicts existence of bias in gene regulation, with P values <0.05 considered statistically significant. Select disease categories are shown in a plot that also represents the key differentially expressed genes within the category. The green color represents genes that are down-regulated and red represents up-regulated genes, while a higher intensity of color indicates a higher fold change. RT-qPCR validation of genes that factor most prominently in the IPA analysis disease association is presented in Supplemental Table VI. (E) RT-qPCR of select genes demonstrates Scn5a and Gja5 down-regulation in iNICD RA after acute Notch activation. In contrast to the RA, RT-qPCR demonstrates Scn5a up-regulation in iNICD LV. iNICD (αMHC-rtTA; tetO_NICD, n=1 female, n=5 males) mice were fed doxycycline for 3 weeks starting at 8 weeks of age with no washout period. Controls were αMHC-rtTA; tetO_NICD mice never fed doxycycline chow (n=2 females, n=4 males). RA and LV gene expression were measured from the same cohort of mice. (F) RT-qPCR from RA where Notch was induced for 2 days followed by a prolonged 1-year washout period reveals persistent Tbx5, Scn5a, and Gja5 transcript down-regulation. iNICD are αMHC-rtTA; tetO_NICD (n=2 females, n=3 males) and controls are αMHC-rtTA; tetO_NICD mice that were never administered doxycycline (n=2 females, n=3 males). Relative fold changes were in comparison with α-actinin. Statistics were performed using unpaired t tests with Welch’s correction. Values of P<0.05 were considered statistically significant.
Given the reduction of RA CV, we performed RNA sequencing on adult RA from iNICD and control mice after 3 weeks of doxycycline induction. This experiment yielded 910 differentially expressed transcripts of which 596 are up-regulated and 314 are down-regulated (Supplemental Table IV, NCBI GEO database). To enhance biological interpretation, this set of differentially expressed genes was further analyzed using Ingenuity Pathway Analysis (IPA). Of the top 25 statistically significant IPA disease or function network categories, 10 of 25 are related to arrhythmias and 7 of 25 are related to atrial arrhythmias (Supplemental Table V). Select disease categories relevant to atrial arrhythmias are shown in a plot that also represents the key differentially expressed genes within the category (Figure 5D), where green represents down-regulated and red represents up-regulated genes. RT-qPCR validation confirms expression changes for 24 of 26 atrial arrhythmia-associated genes (Supplemental Table VI).
Notch induces a distinct transcriptional response in atrial versus ventricular tissue
Further analysis to probe for changes that may contribute to conduction slowing reveals that Scn5a (which encodes Nav1.5) and Gja5 (which encodes Cx40) expression levels are significantly down-regulated within the RA (Figure 5E). Interestingly, acute Notch activation led to significant up-regulation of Scn5a within the left ventricle whereas Gja5 was not altered, highlighting important chamber-specific differences in the Notch-induced response (Figure 5E). We next assayed expression of select genes after a washout period to determine whether they may potentially be involved in the persistence of electrical changes. Tbx5 is an important transcription factor in the network of factors that play a critical role in maintenance of atrial rhythm35. Tbx5 levels are significantly decreased after a prolonged washout period (Figure 5F), suggesting that Notch may indirectly interact with this transcriptional network. Scn5a and Gja5 also remain persistently down-regulated, consistent with the persistent conduction velocity slowing (Figure 5F).
Notch activation reduces atrial cardiomyocyte excitability
To investigate whether Notch activation alters atrial cardiomyocyte electrophysiology at the single cell level, sharp microelectrode recordings were performed on Langendorff-perfused intact hearts. Representative traces recorded from a control and iNICD RA cardiomyocyte, as well as single representative action potentials, are shown (Supplemental Figure XIII, Figure 6A,B). When compared with littermate controls, iNICD RA cardiomyocytes exhibit decreased maximum upstroke velocity (dVm/dtmax, 206.9 ± 3.3 versus 164.8 ± 11.1 V/sec, p=0.02) and a trend toward decreased maximum action potential amplitude (APA) (96.6 ± 0.7 versus 89.0 ± 2.8, p=0.05), while other action potential characteristics, including resting membrane potential (RMP) and APD90 are not changed in response to Notch activation (Figure 6C–F, Supplemental Table VII). Together with reduced conduction velocity and areas of focal conduction block (Figure 4), these data are most consistent with decreased Na+ current causing reduced atrial cardiomyocyte excitability in iNICD mice.
Figure 6. Notch activation reduces atrial myocyte excitability.
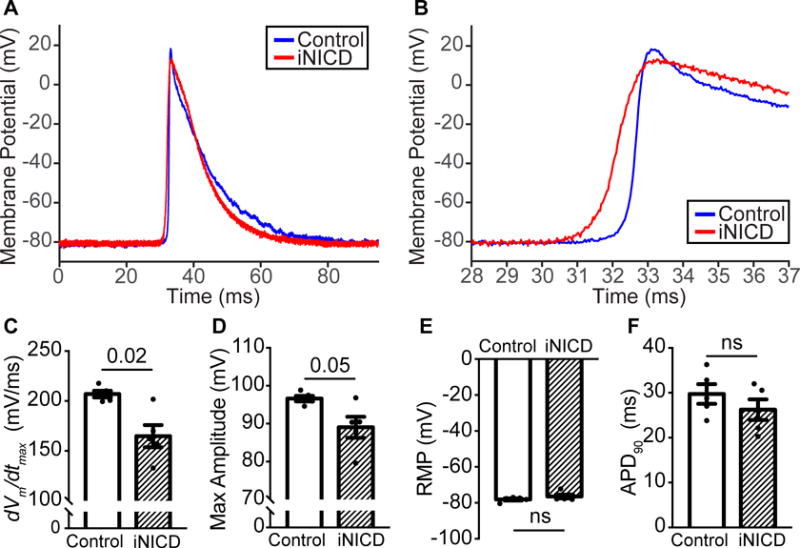
(A) Averaged individual action potentials from control (blue) and iNICD (red) RA cardiomyocytes recorded with sharp microelectrodes. (B) Zoom of a portion of the action potential including Phase 0 demonstrates a reduction in dVm/dtmax in iNICD mice. Action potential parameters that are highly dependent on Na+ current, including dVm/dtmax (C) and maximum action potential amplitude (APA, D) are most affected, while the RMP (E) and APD90 (F) are not significantly different. Mice were between 2–3 months of age when doxycycline was administered for 3 weeks, followed by a 2–4 month washout. Controls were αMHC-rtTA; Cntn2-eGFP (n=2 females, n=3 males) and experimental iNICD mice were αMHC-rtTA; tetO_NICD; Cntn2-eGFP (n=4 females, n=1 male). Statistics were performed using unpaired t tests with Welch’s correction. Values of P<0.05 were considered statistically significant.
Transient Notch activation predisposes to atrial arrhythmias
The wavelet theory provides an explanation for the mechanism of atrial arrhythmia maintenance36. According to this theory, the likelihood that a trigger initiates a sustained arrhythmia is influenced by the presence of an arrhythmic substrate, which is dependent on the wavelength of the wavelet, or the effective refractory period and the CV. Therefore, alterations in atrial CV, areas of severely slowed conduction, or conduction block, could provide a substrate for re-entry37. Given the small size of the mouse atria, severe disturbances in the conduction parameters are often needed to support an atrial arrhythmia. To investigate whether the iNICD substrate predisposes to atrial arrhythmias, we performed programmed electrical stimulation near the murine pulmonary vein. A programmed pacing protocol that consists of a drive train (S1) followed by a single extrastimulus (S2) with different coupling intervals was performed on each heart. iNICD mice demonstrate a significantly greater number of episodes of supraventricular tachycardias (SVTs) when compared with littermate controls as the S2 coupling interval decreased (Figure 7 and Supplemental Online Videos 1–3). The single extrastimulus pacing protocol was also used to measure the atrial effective refractory period (AERP). Control mice demonstrated an average AERP of 15ms, however, we were unable to measure the AERP in many iNICD mice due to the frequent episodes of SVT that occurred with short S2 coupling intervals. Taken together, we interpret the increased incidence of pacing-induced SVT together with the paucity of spontaneous atrial arrhythmia episodes during telemetric monitoring of iNICD mice to indicate that although an arrhythmic substrate is present, initiating triggers do not frequently occur in this model.
Figure 7. Transient Notch activation predisposes to atrial arrhythmias.
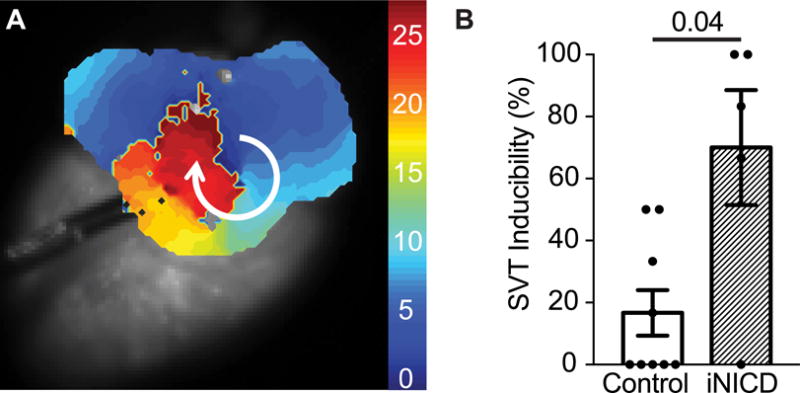
A programmed pacing protocol that consists of a drive train (S1) followed by a single extrastimulus (S2) with coupling intervals of 20ms and 15ms was performed on each heart. Stimulation was performed near the pulmonary vein to mimic the site of origin of triggers in human arrhythmias. An episode of supraventricular tachycardia (SVT) was defined as a reentrant and/or fast, abnormal ectopic activity lasting one second or longer. iNICD mice demonstrate a significantly greater number of episodes of SVTs when compared with littermate controls (example activation map during a macro-reentrant arrhythmia, A, Supplemental Videos I, II, and III) in response to programmed pacing (B, 16.7±7.4% vs. 70±18.6%, P=0.04). SVT inducibility is represented as the percentage of electrical stimulations that successfully induced an episode of SVT at S2 coupling intervals of 20 ms (each heart stimulated 3 times) and 15 ms (each heart stimulated 3 times). Doxycycline was administered at 8 weeks of age to control (αMHC-rtTA (n=3 females, n=2 males) and tetO_NICD controls (n=1 male)) and experimental iNICD mice (αMHC-rtTA; tetO_NICD (n=3 females, n=2 males)) followed by a 2–3 month washout. Statistics were performed using an unpaired t test with Welch’s correction. A value of P<0.05 was considered statistically significant.
DISCUSSION
Control of atrial electrical activity by Notch
There is emerging evidence that activation of a SAN-specific program and repression of a contractile cardiomyocyte program may regulate the developmental programming and maintenance of SAN structure and function4, 27, 38–40. Several transcriptional and epigenetic pathways cooperatively repress the contractile program within the node, typically acting through repression of Nkx2-5 within the adult SAN. For example, the Baf250a chromatin complex activates Tbx3, which acts together with histone deacetylase 3 to repress Nkx2-5 and hence repress components of the contractile gene expression program25. Baf250a knockout mice mis-express Nkx2-5 within the SAN resulting in sinus bradycardia25. In this manuscript, we implicate the Notch transcriptional network in HR regulation. Consistent with previous findings, we demonstrate Tbx3 down-regulation and ectopic Nkx2-5 mis-expression within nodal myocardium associated with sinus bradycardia in iNICD mice (Figure 2, Supplemental Figure VII). However, expression of many SAN-specific genes including Shox2 and Hcn4 remain unaffected in iNICD mice (Supplemental Table IV, Supplemental Figure VII).
The precise mechanism(s) whereby Notch regulates gene expression in the SAN and atrial muscle remain to be elucidated. Many transcription factors, including Nkx2-5, Tbx3, and Tbx5, are continuously expressed and functionally active in adult cardiomyocytes. In contrast, Notch signaling is active during embryonic development, while full-length Notch receptors are expressed in the adult sarcolemma without evidence for Notch receptor cleavage and activation in healthy adult cardiomyocytes11. However, we have shown that Notch is transiently activated in a heart failure model, inducing a transcriptional and epigenetic response in left ventricular cardiomyocytes that results in partial reprogramming toward a Purkinje-like phenotype and down-regulation of voltage-gated K+ currents11, although Notch activation per se does not lead to ventricular systolic dysfunction (Supplemental Table III). Within the atrial myocardium, transient Notch activation mimicking pressure overload results in distinct transcriptional changes that have previously been associated with AF. Transcriptomic analysis of the RA appendage from humans in permanent AF demonstrates that the direct Notch target HEY1 (HRT1) is significantly upregulated41, similar to our iNICD model. Other similarities between these two models with respect to atrial fibrillation associated genes include dysregulation of genes involved in contractile remodeling (Tnnt2 down-regulation), action potential repolarization (down-regulation of Kcnip241 and Kcnq142–44), and Ca2+-handling (Atp2a2 and Cacna1g down-regulation). Transcriptional down-regulation of Tbx5, which regulates Scn5a expression, could result in decreased sodium current, reduced atrial cardiomyocyte excitibility (Figure 6) and reduced conduction velocity (Figure 4)7, 45 and contribute to an arrhythmic substrate. Indeed, when provided with a trigger near the pulmonary vein, iNICD mice have increased susceptibility to atrial arrhythmias (Figure 7). Interestingly, the Notch phenotype recapitulates many of the transcriptional and electrical changes seen in AF, but does not lead to the fibrosis. Taken together, this is suggestive that there may be a correlation between transient Notch activation and a substrate for AF, though future experiments will be required to further elucidate this connection. In addition, evidence is emerging that the outcome of Notch activation may depend on cell lineage-specific transcription factors and the chromatin context46. Indeed, we observe a distinct transcriptional output between LV versus RA in iNICD mice resulting in differential Scn5a regulation (Figure 5E).
Notch can confer cell fate decisions during development in part through its complex interactions with transcription factors and the epigenetic machinery46. Interestingly, Notch itself, as well as direct Notch targets, including Hrt1 and Hrt2, forms complexes with chromatin modifiers such as histone deacetylases and the SWI/SNF chromatin-modifying complex47, 48. In this way, Notch activation may confer long-term alteration of the cardiomyocyte transcriptional program in a cell-type specific manner to permanently alter ion channel homeostasis. One of the strongest clinical predictors for arrhythmias is a history of previous cardiac injury or myocardial infarction. In some cases, cardiac injury predisposes to structural changes that confer long-term arrhythmia risk. However, the degree to which Notch-induced chromatin changes may contribute to arrhythmia predisposition remains unclear.
iNICD as a model of Sick Sinus Syndrome
Our murine model of Notch activation resembles SSS, a common and debilitating syndrome encompassing a group of sinus rhythm disorders including inappropriate sinus bradycardia, frequent sinus pauses or arrest, and increased susceptibility to paroxysmal tachycardias such as atrial flutter and AF. SSS affects approximately 1 in 600 adults and is the most common indication for permanent pacemaker implantation49. The etiology is not well-understood, however it is common in the elderly, often occurring in individuals without preexisting cardiac history or signs of heart disease. SSS also occurs after operative procedures such as cardiac surgery for repair of congenital heart malformations involving the atria, where treatment typically requires lifelong pacemaker therapy50. Though there have been extensive efforts to define SSS in terms of abnormal automaticity, exit block, or impaired intra-atrial conduction and excitability, it remains largely an electrocardiographic diagnosis and very little is understood regarding the molecular etiology. Most known SSS-associated mutations involve genes encoding ion channels or structural proteins. Variants in Nav1.5, in the HCN channels that underlie If, and in the Ankyrin-B cytoskeletal adapter which targets ion channels to the membrane, as well as in desmosomal junctions, are all associated with familial SSS with similar phenotypes in mice and humans51, 52. Interestingly, some of these genes are primarily expressed within atrial myocardium, and not within the SAN itself53. This suggests that perhaps SSS can be triggered by distinct mechanisms, including primary abnormalities within the SAN and/or abnormalities within atrial myocardium that exert effects through electrotonic interactions. In support of this, an abnormal cSNRT is only 70% sensitive for SSS in humans54, while evidence of coexistent atrial disease, reflected in a prolonged P wave duration, often precedes symptomatic disease by several years55, 56. Atrial tachycardias coexist in about half of SSS patients, known as the “bradycardia-tachycardia syndrome”, but despite this clear association causal relationships are not well understood. In contrast to Hcn1 knockout mice, in which a prolonged cSNRT is seen57, we did not observe a prolonged cSNRT in the iNICD model (Figure 4E). Given that Notch activation in the SAN alone did not reproduce bradycardia (Supplemental Figure VIII), the effects on HR may reflect the effects of Notch activation in atrial myocytes more than a direct effect of Notch on the SAN. The mechanism in iNICD mice may be distinct from the Hcn1 loss-of-function model, which recapitulates the abnormal cSNRT often associated with SSS but may not recapitulate other aspects of SSS indicative of atrial pathology, such as atrial tachy-arrhythmias that are seen in the iNICD model (Figure 7). Hereditary Notch mutations can result in structural congenital heart defects including Tetralogy of Fallot, for which 20–30% of repaired patients and 50% of symptomatic patients have associated bradycardia58. There are no known human Notch mutations associated with SSS, although precise determination of the pathogenic initiators of SSS, including any role for Notch activation, may ultimately require analysis of primary human tissue from diseased and non-diseased atria.
In cases where conduction is primarily slowed as a result of reduced intercellular coupling, dVm/dtmax typically increases concomitant with an increased safety factor, such that conduction can reach very slow velocities as low as 1/200th of control before block occurs59. In contrast, reduced excitability decreases the safety factor such that the minimum velocity attainable before conduction block is only about one third of control59. Based on our observed decrease in dVm/dtmax and conduction block in iNICD atria (Figures 4,6), we expect that iNICD mice have reduced excitability through decreased sodium current. Expression levels of Scn5a in iNICD atria are reduced ~50% (Figure 5E). Given that 50% reduction in Scn5a expression results in only modest conduction slowing60, the iNICD model likely represents changes in both excitability and coupling, consistent with the observed decrease in expression levels of both Scn5a and Gja5 (Figure 5E). Indeed, combined deficiencies of sodium channels and Cx40 have been described in familial atrial standstill, similar to the partially inexcitable right atrial phenotype we observe when Notch is activated in juvenile mice (Figure 4G)61.
Conclusions and perspectives
Two disease mouse models, one of left heart failure as a result of TAC and MI and one of pulmonary hypertension due to hypoxia, lead to Notch activation primarily based on gene expression. We observe that chronic Notch activation predisposes to atrial arrhythmias when provided with a trigger (Figure 7). A limitation of the current study is that we did not attempt to recapitulate Notch overexpression-mediated arrhythmias through programmed stimulation in these disease models, so future experiments will be required to directly associate Notch activation with AF. Although arrhythmia induction is affected by parameters that differ significantly between human and murine hearts, including differences in atrial size, geometry, and distinct currents underlying repolarization, underlying principles may be similar and there may be conservation at the level of signaling pathways that predispose to an arrhythmogenic substrate. Genome-wide association studies have linked common variants near TBX5 and PITX2 with AF in humans, and perturbation of these gene regulatory networks can lead to atrial arrhythmias in mice35, 62. We hypothesize that the Notch receptor may be poised for cleavage in response to cardiac injury or stress, thereby activating an atrial gene regulatory cascade involving Tbx5. Given that even brief pulses of Notch activation can alter the HR, atrial conduction and arrhythmia predisposition many months later (Figure 3E), it will be important in the future to focus on how the Notch signal is reinforced over time to result in an “electrical scar”. Since it is well described clinically that “atrial fibrillation begets atrial fibrillation”, perhaps a better understanding of the transcriptional and epigenetic cascades activated downstream of injury and Notch activation could lead to a better understanding of the transition from paroxysmal to persistent to permanent AF.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Genome-wide association studies have linked common variants near TBX5 and PITX2 with atrial fibrillation in humans, and perturbation of these gene regulatory networks can lead to atrial arrhythmias in mice.
Notch signaling is normally quiescent in adult myocardium, and is reactivated in adult murine ventricular myocardium in heart failure.
Notch activation in ventricular myocardium partially reprograms ion channel gene expression toward a Purkinje-like phenotype and down-regulates voltage-gated K+ currents.
What New Information Does This Article Contribute?
Notch is reactivated in murine atrial myocardium in response to pressure overload.
The effects of transient Notch activation on atrial gene expression and atrial electrophysiology persist long after the transient pulse and results in a phenotype resembling sick sinus syndrome.
Notch-associated transcriptional and electrical changes are similar to those associated with atrial fibrillation, and transient Notch activation predisposes to murine atrial arrhythmias.
Notch signaling is normally quiescent in adult cardiac myocytes and is activated in response to pathological stimuli such as injury or elevated pressure. Here, we demonstrate atrial Notch activation in two disease models of increased atrial pressure, left heart failure and pulmonary hypertension. Transient activation of Notch signaling within adult myocardium using a genetic system induces distinct transcriptional changes in the right atrium when compared with changes in left ventricular myocardium, and leads to a phenotype resembling sick sinus syndrome. Atrial tachycardias occur in about half of sick sinus syndrome patients and may reflect coexistent atrial disease. Consistent with this notion, global Notch-induced transcriptional changes are similar to those previously associated with atrial fibrillation. In addition, Notch activation decreases atrial cardiomyocyte excitability and reduces conduction velocity, thereby providing a substrate for atrial arrhythmias in response to pulmonary vein triggers that persists long after the transient Notch pulse. Since cardiac injuries and pressure overload in humans can predispose to atrial arrhythmias including sick sinus syndrome and atrial fibrillation, a better mechanistic understanding of the cascade downstream of injury leading to Notch activation and culminating in persistent atrial transcriptional and electrical reprogramming could provide insights into novel atrial-selective therapeutic targets.
Acknowledgments
We would like to acknowledge Carla Weinheimer, Carrie Gierasch, and Attila Kovacs in the Mouse Cardiovascular Phenotyping Core in the Center for Cardiovascular Research for their assistance in performing invasive telemetry studies and echocardiograms, and the Developmental Biology Histology & Microscopy Core at Washington University for assistance with histology. We thank the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for help with genomic analysis.
SOURCES OF FUNDING
This work was supported by T32 GM007067 (CL), R01 HL130212 (SR), K08 HL107449 (SR), AHA Grant in Aid 14GRNT19510011 (SR), Center for the Investigation of Membrane Excitability Diseases (SR), and Department of Medicine funds from Washington University (SR). Dr. Rentschler holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund.
Nonstandard Abbreviations and Acronyms
- HR
Heart rate
- AF
Atrial fibrillation
- CV
Conduction velocity
- SAN
Sinoatrial node
- SSS
Sick sinus syndrome
- NICD
Notch intracellular domain
- RA
Right atrium
- PECAM-1
Platelet and endothelial cell adhesion molecule 1
- pHTN
Pulmonary hypertension
- iNICD
Inducible Notch intracellular domain
- AV
Atrioventricular
- Nkx2-5
NK2 homeobox 5
- Cntn2
Contactin-2
- Cx40
Connexin 40
- SVC
Superior vena cava
- cSNRT
Corrected sinus node recovery time
- APD
Action potential duration
- APA
Action potential amplitude
- dVm/dtmax
Maximum upstroke velocity
- SVT
Supraventricular tachycardia
- Hrt1
Hairy-related transcription factor 1
- Hes1
Hairy and Enhancer of Split-1
- Nrarp
Notch-Regulated Ankyrin Repeat Protein
- Shox2
Short Stature Homeobox 2
Footnotes
DISCLOSURES
None.
References
- 1.Bakker ML, Boink GJ, Boukens BJ, Verkerk AO, van den Boogaard M, den Haan AD, Hoogaars WM, Buermans HP, de Bakker JM, Seppen J, Tan HL, Moorman AF, t Hoen PA, Christoffels VM. T-box transcription factor TBX3 reprogrammes mature cardiac myocytes into pacemaker-like cells. Cardiovasc Res. 2012;94:439–49. doi: 10.1093/cvr/cvs120. [DOI] [PubMed] [Google Scholar]
- 2.Arnolds DE, Liu F, Fahrenbach JP, Kim GH, Schillinger KJ, Smemo S, McNally EM, Nobrega MA, Patel VV, Moskowitz IP. TBX5 drives Scn5a expression to regulate cardiac conduction system function. J Clin Invest. 2012;122:2509–18. doi: 10.1172/JCI62617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rentschler S, Yen AH, Lu J, Petrenko NB, Lu MM, Manderfield LJ, Patel VV, Fishman GI, Epstein JA. Myocardial Notch signaling reprograms cardiomyocytes to a conduction-like phenotype. Circulation. 2012;126:1058–66. doi: 10.1161/CIRCULATIONAHA.112.103390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bressan M, Liu G, Mikawa T. Early mesodermal cues assign avian cardiac pacemaker fate potential in a tertiary heart field. Science. 2013;340:744–8. doi: 10.1126/science.1232877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Briggs LE, Takeda M, Cuadra AE, Wakimoto H, Marks MH, Walker AJ, Seki T, Oh SP, Lu JT, Sumners C, Raizada MK, Horikoshi N, Weinberg EO, Yasui K, Ikeda Y, Chien KR, Kasahara H. Perinatal loss of Nkx2-5 results in rapid conduction and contraction defects. Circ Res. 2008;103:580–90. doi: 10.1161/CIRCRESAHA.108.171835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.den Hoed M, Eijgelsheim M, Esko T, et al. Identification of heart rate-associated loci and their effects on cardiac conduction and rhythm disorders. Nat Genet. 2013;45:621–31. doi: 10.1038/ng.2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang RT, Xue S, Xu YJ, Zhou M, Yang YQ. A novel NKX2.5 loss-of-function mutation responsible for familial atrial fibrillation. Int J Mol Med. 2013;31:1119–26. doi: 10.3892/ijmm.2013.1316. [DOI] [PubMed] [Google Scholar]
- 8.Nakashima Y, Yanez DA, Touma M, Nakano H, Jaroszewicz A, Jordan MC, Pellegrini M, Roos KP, Nakano A. Nkx2-5 suppresses the proliferation of atrial myocytes and conduction system. Circ Res. 2014;114:1103–13. doi: 10.1161/CIRCRESAHA.114.303219. [DOI] [PubMed] [Google Scholar]
- 9.Rentschler S, Harris BS, Kuznekoff L, Jain R, Manderfield L, Lu MM, Morley GE, Patel VV, Epstein JA. Notch signaling regulates murine atrioventricular conduction and the formation of accessory pathways. J Clin Invest. 2011;121:525–33. doi: 10.1172/JCI44470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gillers BS, Chiplunkar A, Aly H, Valenta T, Basler K, Christoffels VM, Efimov IR, Boukens BJ, Rentschler S. Canonical wnt signaling regulates atrioventricular junction programming and electrophysiological properties. Circ Res. 2015;116:398–406. doi: 10.1161/CIRCRESAHA.116.304731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khandekar A, Springer S, Wang W, Hicks S, Weinheimer C, Diaz-Trelles R, Nerbonne JM, Rentschler S. Notch-Mediated Epigenetic Regulation of Voltage-Gated Potassium Currents. Circulation Research. 2016;119:1324–1338. doi: 10.1161/CIRCRESAHA.116.309877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gude NA, Emmanuel G, Wu W, Cottage CT, Fischer K, Quijada P, Muraski JA, Alvarez R, Rubio M, Schaefer E, Sussman MA. Activation of Notch-mediated protective signaling in the myocardium. Circ Res. 2008;102:1025–35. doi: 10.1161/CIRCRESAHA.107.164749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang R, Han P, Yang H, Ouyang K, Lee D, Lin YF, Ocorr K, Kang G, Chen J, Stainier DY, Yelon D, Chi NC. In vivo cardiac reprogramming contributes to zebrafish heart regeneration. Nature. 2013;498:497–501. doi: 10.1038/nature12322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Campa VM, Gutierrez-Lanza R, Cerignoli F, Diaz-Trelles R, Nelson B, Tsuji T, Barcova M, Jiang W, Mercola M. Notch activates cell cycle reentry and progression in quiescent cardiomyocytes. J Cell Biol. 2008;183:129–41. doi: 10.1083/jcb.200806104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kratsios P, Catela C, Salimova E, Huth M, Berno V, Rosenthal N, Mourkioti F. Distinct roles for cell-autonomous Notch signaling in cardiomyocytes of the embryonic and adult heart. Circ Res. 2010;106:559–72. doi: 10.1161/CIRCRESAHA.109.203034. [DOI] [PubMed] [Google Scholar]
- 16.Liu Z, Brunskill E, Boyle S, Chen S, Turkoz M, Guo Y, Grant R, Kopan R. Second-generation Notch1 activity-trap mouse line (N1IP::CreHI) provides a more comprehensive map of cells experiencing Notch1 activity. Development. 2015;142:1193–202. doi: 10.1242/dev.119529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pellegrinet L, Rodilla V, Liu ZY, Chen SA, Koch U, Espinosa L, Kaestner KH, Kopan R, Lewis J, Radtke F. Dll1- and Dll4-Mediated Notch Signaling Are Required for Homeostasis of Intestinal Stem Cells. Gastroenterology. 2011;140:1230–+. doi: 10.1053/j.gastro.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanemoto N, Sasamoto H. Arrhythmias in Primary Pulmonary-Hypertension. Japanese Heart Journal. 1979;20:765–775. doi: 10.1536/ihj.20.765. [DOI] [PubMed] [Google Scholar]
- 19.Tongers J, Schwerdtfeger B, Klein G, Kempf T, Schaefer A, Knapp JM, Niehaus M, Korte T, Hoeper MM. Incidence and clinical relevance of supraventricular tachyarrhythmias in pulmonary hypertension. American Heart Journal. 2007;153:127–132. doi: 10.1016/j.ahj.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 20.Wanstall JC, Gambino A, Jeffery TK, Cahill MM, Bellomo D, Hayward NK, Kay GF. Vascular endothelial growth factor-B-deficient mice show impaired development of hypoxic pulmonary hypertension. Cardiovascular Research. 2002;55:361–368. doi: 10.1016/s0008-6363(02)00440-6. [DOI] [PubMed] [Google Scholar]
- 21.Patel D, Alhawaj R, Wolin MS. Exposure of mice to chronic hypoxia attenuates pulmonary arterial contractile responses to acute hypoxia by increases in extracellular hydrogen peroxide. American Journal of Physiology-Regulatory Integrative and Comparative Physiology. 2014;307:R426–R433. doi: 10.1152/ajpregu.00257.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Froese A, Breher SS, Waldeyer C, Schindler RF, Nikolaev VO, Rinne S, Wischmeyer E, Schlueter J, Becher J, Simrick S, Vauti F, Kuhtz J, Meister P, Kreissl S, Torlopp A, Liebig SK, Laakmann S, Muller TD, Neumann J, Stieber J, Ludwig A, Maier SK, Decher N, Arnold HH, Kirchhof P, Fabritz L, Brand T. Popeye domain containing proteins are essential for stress-mediated modulation of cardiac pacemaking in mice. J Clin Invest. 2012;122:1119–30. doi: 10.1172/JCI59410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frank DU, Carter KL, Thomas KR, Burr RM, Bakker ML, Coetzee WA, Tristani-Firouzi M, Bamshad MJ, Christoffels VM, Moon AM. Lethal arrhythmias in Tbx3-deficient mice reveal extreme dosage sensitivity of cardiac conduction system function and homeostasis. Proc Natl Acad Sci U S A. 2012;109:E154–63. doi: 10.1073/pnas.1115165109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mommersteeg MTM, Hoogaars WMH, Prall OWJ, de Gier-de Vries C, Wiese C, Clout DEW, Papaioannou VE, Brown NA, Harvey RP, Moorman AFM, Christoffels VM. Molecular pathway for the localized formation of the sinoatrial node. Circulation Research. 2007;100:354–362. doi: 10.1161/01.RES.0000258019.74591.b3. [DOI] [PubMed] [Google Scholar]
- 25.Wu M, Peng S, Yang J, Tu Z, Cai X, Cai CL, Wang Z, Zhao Y. Baf250a orchestrates an epigenetic pathway to repress the Nkx2.5-directed contractile cardiomyocyte program in the sinoatrial node. Cell Res. 2014;24:1201–13. doi: 10.1038/cr.2014.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Espinoza-Lewis RA, Liu H, Sun C, Chen C, Jiao K, Chen Y. Ectopic expression of Nkx2.5 suppresses the formation of the sinoatrial node in mice. Dev Biol. 2011;356:359–69. doi: 10.1016/j.ydbio.2011.05.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ye WD, Wang J, Song YN, Yu DK, Sun C, Liu C, Chen FD, Zhang YD, Wang F, Harvey RP, Schrader L, Martin JF, Chen YP. A common Shox2-Nkx2-5 antagonistic mechanism primes the pacemaker cell fate in the pulmonary vein myocardium and sinoatrial node. Development. 2015;142:2521–+. doi: 10.1242/dev.120220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang X, Wang G, Lin L, Lowe J, Zhang Q, Bu L, Chen Y, Chen J, Sun Y, Evans SM. HCN4 dynamically marks the first heart field and conduction system precursors. Circ Res. 2013;113:399–407. doi: 10.1161/CIRCRESAHA.113.301588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diaz-Trelles R, Scimia MC, Bushway P, Tran D, Monosov A, Monosov E, Peterson K, Rentschler S, Cabrales P, Ruiz-Lozano P, Mercola M. Notch-independent RBPJ controls angiogenesis in the adult heart. Nat Commun. 2016;7:12088. doi: 10.1038/ncomms12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Butters TD, Aslanidi OV, Inada S, Boyett MR, Hancox JC, Lei M, Zhang H. Mechanistic links between Na+ channel (SCN5A) mutations and impaired cardiac pacemaking in sick sinus syndrome. Circ Res. 2010;107:126–37. doi: 10.1161/CIRCRESAHA.110.219949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bagwe S, Berenfeld O, Vaidya D, Morley GE, Jalife J. Altered right atrial excitation and propagation in connexin40 knockout mice. Circulation. 2005;112:2245–53. doi: 10.1161/CIRCULATIONAHA.104.527325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benson DW, Wang DW, Dyment M, Knilans TK, Fish FA, Strieper MJ, Rhodes TH, George AL., Jr Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A) J Clin Invest. 2003;112:1019–28. doi: 10.1172/JCI18062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.King JH, Huang CL, Fraser JA. Determinants of myocardial conduction velocity: implications for arrhythmogenesis. Front Physiol. 2013;4:154. doi: 10.3389/fphys.2013.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verheule S, Sato T, Everett TT, Engle SK, Otten D, Rubart-von der Lohe M, Nakajima HO, Nakajima H, Field LJ, Olgin JE. Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-beta1. Circ Res. 2004;94:1458–65. doi: 10.1161/01.RES.0000129579.59664.9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nadadur RD, Broman MT, Boukens B, Mazurek SR, Yang XA, van den Boogaard M, Bekeny J, Gadek M, Ward T, Zhang M, Qiao Y, Martin JF, Seidman CE, Seidman J, Christoffels V, Efimov IR, McNally EM, Weber CR, Moskowitz IP. Pitx2 modulates a Tbx5-dependent gene regulatory network to maintain atrial rhythm. Science Translational Medicine. 2016;8 doi: 10.1126/scitranslmed.aaf4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Panfilov AV. Is heart size a factor in ventricular fibrillation? Or how close are rabbit and human hearts? Heart Rhythm. 2006;3:862–864. doi: 10.1016/j.hrthm.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 37.Allessie MA. Atrial electrophysiologic remodeling: another vicious circle? J Cardiovasc Electrophysiol. 1998;9:1378–93. doi: 10.1111/j.1540-8167.1998.tb00114.x. [DOI] [PubMed] [Google Scholar]
- 38.Vedantham V, Galang G, Evangelista M, Deo RC, Srivastava D. RNA sequencing of mouse sinoatrial node reveals an upstream regulatory role for Islet-1 in cardiac pacemaker cells. Circ Res. 2015;116:797–803. doi: 10.1161/CIRCRESAHA.116.305913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wiese C, Grieskamp T, Airik R, Mommersteeg MT, Gardiwal A, de Gier-de Vries C, Schuster-Gossler K, Moorman AF, Kispert A, Christoffels VM. Formation of the sinus node head and differentiation of sinus node myocardium are independently regulated by Tbx18 and Tbx3. Circ Res. 2009;104:388–97. doi: 10.1161/CIRCRESAHA.108.187062. [DOI] [PubMed] [Google Scholar]
- 40.Liang XQ, Zhang QQ, Cattaneo P, Zhuang SW, Gong XH, Spann NJ, Jiang CZ, Cao XK, Zhao XD, Zhang XL, Bu L, Wang G, Chen HSV, Zhuang T, Van J, Geng P, Luo LN, Banerjee I, Chen YH, Glass CK, Zambon AC, Chen J, Sun YF, Evans SM. Transcription factor ISL1 is essential for pacemaker development and function. Journal of Clinical Investigation. 2015;125:3256–3268. doi: 10.1172/JCI68257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barth AS, Merk S, Arnoldi E, Zwermann L, Kloos P, Gebauer M, Steinmeyer K, Bleich M, Kaab S, Hinterseer M, Kartmann H, Kreuzer E, Dugas M, Steinbeck G, Nabauer M. Reprogramming of the human atrial transcriptome in permanent atrial fibrillation: expression of a ventricular-like genomic signature. Circ Res. 2005;96:1022–9. doi: 10.1161/01.RES.0000165480.82737.33. [DOI] [PubMed] [Google Scholar]
- 42.Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang Y, Xu WY, Jin HW, Sun H, Su XY, Zhuang QN, Yang YQ, Li YB, Liu Y, Xu HJ, Li XF, Ma N, Mou CP, Chen Z, Barhanin J, Huang W. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003;299:251–4. doi: 10.1126/science.1077771. [DOI] [PubMed] [Google Scholar]
- 43.Temple J, Frias P, Rottman J, Yang T, Wu Y, Verheijck EE, Zhang W, Siprachanh C, Kanki H, Atkinson JB, King P, Anderson ME, Kupershmidt S, Roden DM. Atrial fibrillation in KCNE1-null mice. Circ Res. 2005;97:62–9. doi: 10.1161/01.RES.0000173047.42236.88. [DOI] [PubMed] [Google Scholar]
- 44.Abraham RL, Yang T, Blair M, Roden DM, Darbar D. Augmented potassium current is a shared phenotype for two genetic defects associated with familial atrial fibrillation. J Mol Cell Cardiol. 2010;48:181–90. doi: 10.1016/j.yjmcc.2009.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sinner MF, Tucker NR, Lunetta KL, et al. Integrating genetic, transcriptional, and functional analyses to identify 5 novel genes for atrial fibrillation. Circulation. 2014;130:1225–35. doi: 10.1161/CIRCULATIONAHA.114.009892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bray SJ. Notch signalling in context. Nature Reviews Molecular Cell Biology. 2016;17:722–735. doi: 10.1038/nrm.2016.94. [DOI] [PubMed] [Google Scholar]
- 47.Stefanovic S, Barnett P, van Duijvenboden K, Weber D, Gessler M, Christoffels VM. GATA-dependent regulatory switches establish atrioventricular canal specificity during heart development. Nat Commun. 2014;5:3680. doi: 10.1038/ncomms4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schwanbeck R. The Role of Epigenetic Mechanisms in Notch Signaling During Development. Journal of Cellular Physiology. 2015;230:969–981. doi: 10.1002/jcp.24851. [DOI] [PubMed] [Google Scholar]
- 49.Kusumoto FM, Goldschlager N. Cardiac pacing. N Engl J Med. 1996;334:89–97. doi: 10.1056/NEJM199601113340206. [DOI] [PubMed] [Google Scholar]
- 50.Kugler JD, Gillette PC, Mullins CE, McNamara DG. Sinoatrial conduction in children: an index of sinoatrial node function. Circulation. 1979;59:1266–76. doi: 10.1161/01.cir.59.6.1266. [DOI] [PubMed] [Google Scholar]
- 51.Dobrzynski H, Boyett MR, Anderson RH. New insights into pacemaker activity: promoting understanding of sick sinus syndrome. Circulation. 2007;115:1921–32. doi: 10.1161/CIRCULATIONAHA.106.616011. [DOI] [PubMed] [Google Scholar]
- 52.Mezzano V, Liang Y, Wright AT, Lyon RC, Pfeiffer E, Song MY, Gu Y, Dalton ND, Scheinman M, Peterson KL, Evans SM, Fowler S, Cerrone M, McCulloch AD, Sheikh F. Desmosomal junctions are necessary for adult sinus node function. Cardiovasc Res. 2016;111:274–86. doi: 10.1093/cvr/cvw083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hagendorff A, Schumacher B, Kirchhoff S, Luderitz B, Willecke K. Conduction disturbances and increased atrial vulnerability in connexin40-deficient mice analyzed by transesophageal stimulation. Circulation. 1999;99:1508–1515. doi: 10.1161/01.cir.99.11.1508. [DOI] [PubMed] [Google Scholar]
- 54.Burnett D, Abi-Samra F, Vacek JL. Use of intravenous adenosine as a noninvasive diagnostic test for sick sinus syndrome. Am Heart J. 1999;137:435–8. doi: 10.1016/s0002-8703(99)70488-6. [DOI] [PubMed] [Google Scholar]
- 55.Ferrer MI. The sick sinus syndrome in atrial disease. JAMA. 1968;206:645–6. [PubMed] [Google Scholar]
- 56.De Sisti A, Leclercq JF, Fiorello P, Di Lorenzo M, Manot S, Halimi F, Attuel P. Sick sinus syndrome with and without atrial fibrillation: atrial refractoriness and conduction characteristics. Cardiologia. 1999;44:361–7. [PubMed] [Google Scholar]
- 57.Fenske S, Krause SC, Hassan SIH, Becirovic E, Auer F, Bernard R, Kupatt C, Lange P, Ziegler T, Wotjak CT, Zhang HG, Hammelmann V, Paparizos C, Biel M, Wahl-Schott CA. Sick Sinus Syndrome in HCN1-Deficient Mice. Circulation. 2013;128:2585–2594. doi: 10.1161/CIRCULATIONAHA.113.003712. [DOI] [PubMed] [Google Scholar]
- 58.Rooshesselink J, Perlroth MG, Mcghie J, Spitaels S. Atrial Arrhythmias in Adults after Repair of Tetralogy of Fallot - Correlations with Clinical, Exercise, and Echocardiographic Findings. Circulation. 1995;91:2214–2219. doi: 10.1161/01.cir.91.8.2214. [DOI] [PubMed] [Google Scholar]
- 59.Shaw RM, Rudy Y. Ionic mechanisms of propagation in cardiac tissue - Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circulation Research. 1997;81:727–741. doi: 10.1161/01.res.81.5.727. [DOI] [PubMed] [Google Scholar]
- 60.van Veen TA, Stein M, Royer A, Le Quang K, Charpentier F, Colledge WH, Huang CL, Wilders R, Grace AA, Escande D, de Bakker JM, van Rijen HV. Impaired impulse propagation in Scn5a-knockout mice: combined contribution of excitability, connexin expression, and tissue architecture in relation to aging. Circulation. 2005;112:1927–35. doi: 10.1161/CIRCULATIONAHA.105.539072. [DOI] [PubMed] [Google Scholar]
- 61.Groenewegen WA, Firouzi M, Bezzina CR, Vliex S, van Langen IM, Sandkuijl L, Smits JPP, Hulsbeek M, Rook MB, Jongsma HJ, Wilde AAM. A cardiac sodium channel mutation cosegregates with a rare connexin40 genotype in familial atrial standstill. Circulation Research. 2003;92:14–22. doi: 10.1161/01.res.0000050585.07097.d7. [DOI] [PubMed] [Google Scholar]
- 62.Wang J, Klysik E, Sood S, Johnson RL, Wehrens XHT, Martin JF. Pitx2 prevents susceptibility to atrial arrhythmias by inhibiting left-sided pacemaker specification. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:9753–9758. doi: 10.1073/pnas.0912585107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Valencik ML, McDonald JA. Codon optimization markedly improves doxycycline regulated gene expression in the mouse heart. Transgenic Res. 2001;10:269–75. doi: 10.1023/a:1016601928465. [DOI] [PubMed] [Google Scholar]
- 64.Stanger BZ, Datar R, Murtaugh LC, Melton DA. Direct regulation of intestinal fate by Notch. Proc Natl Acad Sci U S A. 2005;102:12443–8. doi: 10.1073/pnas.0505690102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–40. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pallante BA, Giovannone S, Fang-Yu L, Zhang J, Liu N, Kang G, Dun W, Boyden PA, Fishman GI. Contactin-2 expression in the cardiac Purkinje fiber network. Circ Arrhythm Electrophysiol. 3:186–94. doi: 10.1161/CIRCEP.109.928820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Murtaugh LC, Stanger BZ, Kwan KM, Melton DA. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci U S A. 2003;100:14920–5. doi: 10.1073/pnas.2436557100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Glukhov AV, Fedorov VV, Anderson ME, Mohler PJ, Efimov IR. Functional anatomy of the murine sinus node: high-resolution optical mapping of ankyrin-B heterozygous mice. American Journal of Physiology-Heart and Circulatory Physiology. 2010;299:H482–H491. doi: 10.1152/ajpheart.00756.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.