

Abstract
With the increasing use of mycophenolic acid (MPA) as an immunosuppressant in solid organ transplantation and in treating autoimmune diseases such as systemic lupus erythematosus, the need for strategies to optimize therapy with this agent has become increasingly apparent. This need is largely based on MPA’s significant between-subject and between-occasion (within-subject) pharmacokinetic variability. While there is a strong relationship between MPA exposure and effect, the relationship between drug dose, plasma concentration and exposure (area under the concentration-time curve [AUC]) is very complex and remains to be completely defined. Population pharmacokinetic models using various approaches have been proposed over the past 10 years to further evaluate the pharmacokinetic and pharmacodynamic behaviour of MPA. These models have evolved from simple one-compartment linear iterations to complex multi-compartment versions that try to include various factors, which may influence MPA’s pharmacokinetic variability, such as enterohepatic recycling and pharmacogenetic polymorphisms.
There have been major advances in the understanding of the roles transport mechanisms, metabolizing and other enzymes, drug-drug interactions and pharmacogenetic polymorphisms play in MPA’s pharmacokinetic variability. Given these advances, the usefulness of empirical-based models and the limitations of nonlinear mixed-effects modelling in developing mechanism-based models need to be considered and discussed. If the goal is to individualize MPA dosing, it needs to be determined whether factors which may contribute significantly to variability can be utilized in the population pharmacokinetic models. Some pharmacokinetic models developed to date show promise in being able to describe the impact of physiological processes such as enterohepatic recycling.
Most studies have historically been based on retrospective data or poorly designed studies which do not take these factors into consideration. Modelling typically has been undertaken using non-controlled therapeutic drug monitoring data, which do not have the information content to support the development of complex mechanistic models. Only a few recent modelling approaches have moved away from empiricism and have included mechanisms considered important, such as enterohepatic recycling. It is recognized that well thought-out sampling schedules allow for better evaluation of the pharmacokinetic data. It is not possible to undertake complex absorption modelling with very few samples being obtained during the absorption phase (which has often been the case). It is important to utilize robust AUC monitoring which is now being propagated in the latest consensus guideline on MPA therapeutic drug monitoring.
This review aims to explore the biological factors that contribute to the clinical pharmacokinetics of MPA and how these have been introduced in the development of population pharmacokinetic models. An overview of the processes involved in the enterohepatic recycling of MPA will be provided. This will summarize the components that complicate absorption and recycling to influence MPA exposure such as biotransformation, transport, bile physiology and gut flora. Already published population pharmacokinetic models will be examined, and the evolution of these models away from empirical approaches to more mechanism-based models will be discussed.
Mycophenolic acid (MPA) was approved for transplant rejection prophylaxis in the US and Europe in the mid-1990s and is now a widely used immunosuppressive agent.[1] MPA is the active compound of the pre-systemically hydrolysed prodrug myco-phenolate mofetil (MMF, CellCept®)[2] and is also found in de-layed-release formulation, enteric-coated mycophenolate sodium (EC-MPS, Myfortic®). MPA is a potent, selective, non-competitive and reversible inhibitor of inosine monophosphate dehydrogenase. It inhibits the de novo synthesis pathway of guanosine nucleotides, which triggers a potent cytostatic effect on T and B lymphocytes, thereby inhibiting their proliferative response. MPA also inhibits the proliferation of B lymphocytes.[3]
With increased use of MPA, there has been interest in optimizing therapeutic drug monitoring (TDM) of MMF therapy. Low drug exposure or replacing MMF therapy due to drug related adverse events has been associated with increased risks of graft rejection.[4,5] Therefore, it would be ideal to titrate the dose to individual patients’ needs. Currently, there are no universal guidelines for TDM of MPA therapies.[6,7] Varying approaches such as measuring predose trough concentrations as is common practice for ciclosporin and tacrolimus and abbreviated area under the concentration-time curve (AUC) estimation have been proposed,[6,8–11] in a recent consensus report on TDM of MPA in solid organ transplantation. The goal of this report was to offer transplant practitioners information on clinically relevant pharmacokinetic characteristics of MPA in support of the currently advised target exposure ranges for MPA in different types of organ transplantation, and to summarize the available methods for application of MPA TDM in clinical practice.[7]
Strategies to improve the optimization of MMF therapy have involved many investigations into the clinical pharmacokinetics of MPA. Population pharmacokinetic approaches have been used to describe the complex behaviour of MPA in renal transplant recipients. Most published population pharmacokinetic studies are undertaken in renal transplant recipients with a limited number of studies conducted in liver transplantation and in patients treated with MPA for autoimmune diseases. The majority of studies have used nonlinear mixed-effects modelling techniques to provide mean population estimates for the pharmacokinetic parameters and to characterize between-subject (BSV) and between-occasion (within-subject) variability (BOV). The focus has primarily been on identifying the influence of demographic (e.g. body-weight, sex) and clinical factors (e.g. albumin concentrations) on pharmacokinetic variability.[12] The interactions of drugs such as ciclosporin, tacrolimus and corticosteroids with MPA have been extensively investigated.
1. General Features of Mycophenolic Acid (MPA) Pharmacokinetics
Investigations into the pharmacokinetics of MPA have shown the existence of a concentration-effect relationship. However, the relationship between dose, plasma concentrations and exposure (AUC) is difficult to predict, with a greater than 10-fold range in MPA dose-normalized AUC between patients.[13] In renal transplant patients, the AUC has been shown to increase over time as changing renal function, protein binding and corticosteroid tapering give rise to lower clearance.[12] The absorption of MMF is a complex process that involves dissolution, transport and metabolism. This can be observed in the MPA concentration-time profiles that show varying lag times, varying times to maximal MPA concentration and double peaks in the absorption and post-absorption phases.[14,15] The large BSV and BOV in MPA pharmacokinetics[16] has been attributed to differences in albumin concentrations, change of renal and hepatic function, bilirubin and haemoglobin concentrations, bodyweight, sex, concomitant medication[13] and race. In addition, the exposure to and disposition of MPA are influenced by multiple enzymes and various transporters in which several functional single nucleotide polymorphisms (SNPs) have been found.[17–27]
2. Metabolism
MPA has four main metabolites: 7-O-MPA-β-glucuronide (MPAG), MPA acyl-glucuronide (AcMPAG), catalysed by uridine 5ʹ-diphosphate glucuronosyltransferases (UGTs), 7-O-MPA glucoside through UGT, and trace amounts of 6-O-des-methyl-MPA (DM-MPA) via cytochrome P450.[28,29] The main metabolite, MPAG, is pharmacologically inactive but plays an important role in enterohepatic circulation (EHC). AcMPAG is a minor metabolite, and there is ongoing debate regarding its activity in vitro.[30–32]
The specific role of the different UGT isoforms is not completely known, but several in vitro studies have suggested UGT1A9 as the predominant isoform with 1A8 and 1A7 contributing to a minor extent to the transformation of MPA to MPAG and, via UGT2B7, to AcMPAG.[30,33–37]
3. Effect of Concomitant Medications
In transplantation, MPA is used in combination with other immunosuppressive drugs including calcineurin inhibitors (ciclosporin, tacrolimus), m-Tor inhibitors (sirolimus) and corticosteroids (prednisone/prednisolone).[38] Studies involving humans and animalshave demonstrated that the AUC and trough concentration of MPA are decreased when administered concomitantly with ciclosporin. This is due to the inhibitory effect of ciclosporin on the multidrug resistance-associated protein (MRP)-2 transporter that controls MPAG’s active transport into bile.[39,40] Recent evidence also suggests a contribution of ciclosporin-mediated inhibition on the organic anion transporting polypeptide (OATP) 1B3.[41] As a result, EHC of MPA is decreased or absent, resulting in less pronounced or no secondary peaks.[42] MPA and sirolimus combination studies have demonstrated drug disposition variation between paediatric and adult patients, most likely because of developmental changes related to biliary transporters and metabolic enzymes.[43]
Studies by Filler et al.[44] and Kiberd et al.[45] have outlined biliary excretion as a nonlinear process, suggesting that with higher MMF doses MPA will undergo additional enterohepatic recycling. Consequently, there is a more pronounced inhibitory effect by ciclosporin seen at higher MMF doses. Another drug-drug interaction relevant to MMF therapy is the concomitant use of corticosteroids. Cattaneo et al.[46] showed that patients receiving high doses of methylprednisone had lower exposure to MPA, when compared with those on lower methylprednisone maintenance doses. Tapering of corticosteroids resulted in increased MPA exposure. An induction of UGTs caused by cor-ticosteroids has been proposed to explain this change in MPA clearance and, consequently, exposure.[46–49] However, debate remains about the effect of corticosteroids, with Cattaneo et al.[46] reporting an influence on the bioavailibity of MPA in transplant patients. Recently, differences in the inductive effect of corticosteroids on UGT were reported during the initial post-transplant period compared to the stable phase, which is thought to be related to changes in MPA clearance.[41,46]
4. Summary of the Enterohepatic Recycling of MPA
Figure 1 provides an outline of MMF disposition, indicating the associated drug-metabolizing enzymes and transporters known or suspected of playing a role. Enteral absorption of MPA is influenced by individual characteristics of a patient’s gastrointestinal tract, such as luminal pH, gastric emptying time, intestinal transit time, intestinal surface area, presence of gastrointestinal disease and mesenteric blood flow. Other influences may include the presence of food and/or drugs, and gut metabolism and microflora characteristics.[50]
Fig. 1.
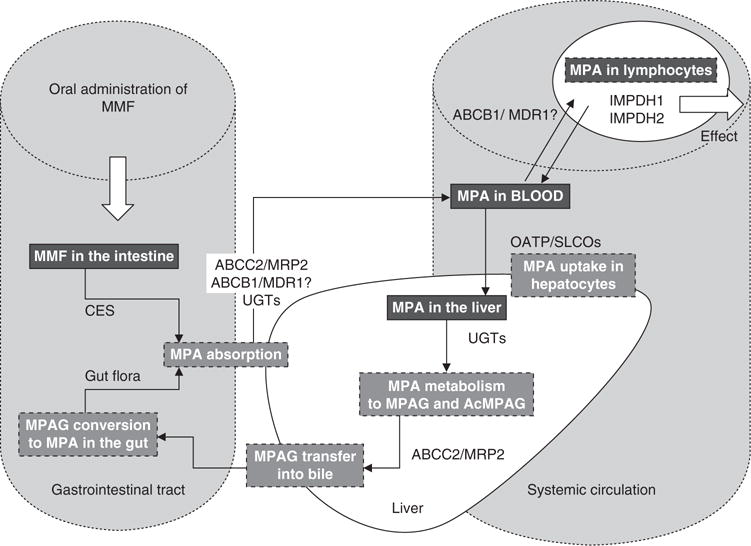
Outline of mycophenolate mofetil (MMF) absorption and dissolution in the body by known or proposed drug-metabolizing enzymes and transporters. ABCB1 (formally, MDR1 = multidrug resistance 1)=adenosine triphosphate-binding cassette, sub-family B, member 1; ABCC2 (formally, MRP2=multidrug resistance-associated protein 2) = adenosine triphosphate-binding cassette, sub-family C, member 2; AcMPAG = MPA acyl-glucuronide; CES =carboxyesterases; IMPDH=inosine monophosphate dehydrogenase; MPA=mycophenolic acid; MPAG=7-O-MPA-β-glucuronide; SLCO (formally, OATP=organic anion-transporting polypeptide)=solute carrier organic anion transporter family; UGT =uridine diphosphate glucuronosyltransferase.
After oral administration MMF is rapidly absorbed after being completely hydrolysed to its active form MPA by carboxyesterases found in the stomach, small intestine, blood and liver.[12] However, there is ongoing debate whether only MPA and/or MMF are absorbed across the gastrointestinal membrane. Studies undertaken by Lee et al.[51] and Bullingham et al.[52] suggest that MMF is also absorbed intact. Accordingly, MPA or a combination of both MMF and MPA is likely absorbed from the intestine.
Both MMF and MPA are classified as class II drugs under the Biopharmaceutics Drug Disposition Classification System predicting metabolism as the main route of elimination.[34,36,53] This is based on reasonable bioavailability, high permeability and low solubility (n-octanol-water partition coefficient of 238 at pH 7.4 and 570 at pH 2, for MMF and MPA, respectively).[51]
The transport of MPA across the membrane of enterocytes can be influenced by the affinity of the molecule to plasma membrane absorptive and efflux transporters.[53] Due to its high permeability, MPA can easily cross the gut membranes. Therefore, gut uptake transporters are unlikely to have a major clinical impact.[53,54] However, the low solubility of MMF and MPA limits the amount of drug entering the enterocytes and prevents saturation of efflux transporters. Generally, it is expected that gut efflux transporters have an effect on oral bioavailability. With respect to MMF, bioavailability is reasonable, suggesting the effects are most likely minor. Several studies have demonstrated that MPA is a substrate for the multidrug resistance transporters which are expressed in various tissues such as liver, brain, kidney and intestine.[55] These transporters are potential candidates to efflux MPA back into the intestinal lumen as both transporters are expressed on the membrane of enterocytes.[23,56–58]
In addition to hepatocytes, human intestinal microsomes are capable of forming the metabolites MPAG and AcM-PAG,[30,34] albeit with large interindividual variation in these formations. This may be related to the reduction in the amount and activity of enzymes found along the intestine.[59,60] Given that the intestine represents a large external surface and is highly exposed to MPA through enterohepatic recycling, it is probable that, despite low UGT activity in intestinal micro-somes, the latter may still contribute to the overall metabolism of MPA.
Uptake into the liver is of significant relevance as hepatic blood flow influences hepatic extraction, hepatocyte permeability and biliary or metabolic elimination.[50] Generally, drugs like MPA, which undergo enterohepatic recirculation, are transported as unchanged compounds or as solutes by different carrier-mediated systems. In humans, these systems include liver-specific transporters, OATPs/solute carrier organic anion transporters (SLCOs). There are also certain MRPs which are found at the plasma membrane.[50]
Like the transporters found in the gut, uptake and efflux transporters in the liver can also influence the disposition of drugs like MPA.[53,54] Uptake capacity of MPA may influence the rate of the metabolism.[24] The exact process by which a solute like MPA moves from the hepatocyte to reach the drug-metabolizing site in the endoplasmic reticulum or bile canaliculae is still unknown. Roberts et al.[50] state that most pharmacokinetic studies do not take into account the time between uptake from the blood and contact with the drug-metabolizing enzymes located in the endoplasmic reticulum or bile canaliculae. This may consequently bias estimates of clearance determined from these pharmacokinetic studies.
MPA is mainly metabolized by UGT1A9 and UGT2B7 in the liver as mentioned above.[28] Inhibition studies using mi-crosomes suggest UGT1A9 is responsible for 55% of MPAG formation in the liver.[34] MPAG is excreted into the bile by active transport facilitated by MRP2/adenosine triphosphate-binding cassette, sub-family C, member 2 (ABCC2).[23] Transport of drugs like MPA from the hepatocyte back into the sinusoidal blood is mediated by OATPs and can be bidirectional.[61] Although it has been suggested that the OATPs are driven by ion exchange, it is more likely that they predominantly act as uptake transporters.[62] The mechanisms involved in efflux of MPAG from the hepatocyte are still under-investigated. As there is large unexplained variability in MPAG plasma concentrations, further study accordingly appears warranted in this area.
A small fraction of bile is continuously excreted into the duodenum, with the majority stored in the gallbladder.[63] The sight, smell or ingestion of a meal results in the contraction of the gallbladder and relaxation of the sphincter of Oddi.[50,63] Emptying of the gallbladder into the duodenum is a sporadic process and is very difficult to predict. It has been demonstrated that secondary MPA absorption peaks correspond well to food intake times. However, it is difficult to obtain actual bile samples and information relating to biliary excretion or release into the gut of drugs and metabolites from the bile into the gut.[50,64]
The conversion of MPAG back to MPA occurs in the gut. Metabolism by the intestinal microflora can induce pharmacological changes, and the hydrolysis of biliary conjugates impacts on the EHC of these compounds.[50] Glucuronide conjugates such as MPAG are converted between the proximal and distal regions of the intestine by the enzyme β-glucuronidase.[65] There is large BSV related to activity of gut enzymes.[50] Further research is needed to describe the variability associated with differences in gut flora and MPA EHC.
5. Population Pharmacokinetic Modelling of MPA and Enterohepatic Recycling
The process of MPA enterohepatic recycling is complex, and the impact on the pharmacokinetics is not always predictable. The EHC comprises up to 60% (range 10–60%) of total MPA exposure and can be influenced by a range of factors such as co-medication, genetic variability and patient characteristics.[52] External factors can also influence MPA EHC including the type of food eaten and the time of food intake, which makes predicting MPA pharmacokinetics even more difficult.[50,66] To date there have only been a few population pharmacokinetic models[26,42,67–71] developed to try to accurately describe the impact of EHC on the variability in pharmacokinetic and total drug exposure.
5.1 Empirical Approaches/Compartmental Models
The first population pharmacokinetic models used to describe the pharmacokinetics of MPA were empirical models. The types of models developed were two- or three-compartment models of drug disposition with first-order absorption.[50] Other studies used two-compartment models, extended by a chain of compartments describing the transport of the drug from the gut to the central compartment. In some cases, more complex absorption models are utilized.[12] The web-based tool developed by the Limoges group[72] is one of the few models that uses a dual input function that works quite well as part of a Bayesian estimator.
5.2 Absorption Models
Modelling enteric drug absorption is complex, as the interplay between drug and patient specific variables needs to be capturedin a physiologically meaningful and mechanism-based fashion. Nevertheless, most absorption models are generally outlined by using either a first-order or zero-order absorption input. Other types of models have been used to describe atypical absorption profiles, including parallel first-order, mixed first-order and zero-order absorption or Weibull-type absorption models. It has been suggested that, although frequently described as first order, the real process is neither first nor zero order but predominantly time dependent.[73,74] The gamma distribution function is sometimes applied as a probability for waiting times to describe oral absorption.[75] However, the most common pharmacokinetic approach used to describe oral absorption delay is to use an absorption model with a lag time parameter. In general, this helps describe the delayed absorption profiles adequately.
5.2.1 Lag Time Models
Inclusion of lag time can improve estimations of the absorption processes and also allows for shifting of the time of dosing.[76] It has been suggested by inclusion of a lag time parameter that enterohepatic recycling models may account for the effect of gallbladder emptying.[50] Simple two-compartment models with a very prolonged lag time have been used to account for the occurrence of secondary peaks in plasma concentration-time profiles.[77] In relation to modelling enterohepatic recycling, lag times have also been used to describe the expulsion time of MPA from the gallbladder back into the gastrointestinal tract.[67,78]
5.2.2 Erlang Absorption Models and Transit Absorption Models
Erlang and transit absorption models are used to describe skewed and delayed absorption profiles. The Erlang absorption model is based on using analytical equations to define a chain of ‘n’ compartments between the depot and central compartment determined by serial addition.[79] The transfer rate between the compartments is similar for each step and uses a constant. The number of serial ‘n’ compartments is estimated by interposing and increasing the number of compartments until there is no further statistical improvement.[80,81] Transit compartment models describe drug absorption as a multiple-step process, thus mimicking the time dependency of the process. A chain of presystemic compartments is used to account for any delay in the passage of a drug.[82] Savic et al.[76] outlined a transit model where the number of transit compartments could be automatically estimated. These types of absorption models have been advocated as better reflecting physiological conditions, perhaps making them better suited to describe the complex absorption profiles associated with MPA.
6. Outline of Population Pharmacokinetic Modelling of MPA Published to Date
Over the last decade, various approaches have been pursued to develop (population) pharmacokinetic models that could adequately describe and predict MPA’s complex behaviour. Generally, the published pharmacokinetic models have used population nonlinear mixed-effects modelling (NONMEM). For this review, a literature search was performed using MEDLINE at Ovid. Abstracts were searched for the following keywords: ‘population pharmacokinetics’, ‘mycophenolic acid’, ‘mycophenolate mofetil’ and ‘model(l)ing’. Seventeen published papers relating to population pharmacokinetic modelling of MMF were found and reviewed. The specific details of each study are detailed in tables I and II.
Table I.
Description of published population pharmacokinetic (PK) modelling studies of mycophenolate mofetil (MMF) enterohepatic recirculation
| Study | Dose | Time post-transplant | No. of patients | Sample times | No. of samples | Concomitant medication | Adequate for EHCa | Description of concentration-time profile; evidence second peak | When second peak seen; absorption or post-absorption phaseb | Comments |
|---|---|---|---|---|---|---|---|---|---|---|
| Funaki (1999)[67] | 0.1–2.5 g MMFc | 140 adults | 0.5, 1, 2, 3, 4, 6, 8, 12 hours post-dose | 270 MPA | Maalox® | Yes | Yes | Absorption phase | Meta-analysisd study compared PK between different races | |
| Shum et al. (2003)[83] | 1g MMFe | Day 2, 5, 28 | 22 adults | Pre-dose, 0.25, 0.5, 0.75, 1, 1.25, 1.5, 2, 3, 4, 6, 8, 10, 12 hours post-dose | 557 MPA | Ciclosporin | Yes | Yes | Absorption and post-absorption phase | Evidence of lag time during absorption, complex absorption process and EHC |
| Le Guellec et al. (2004)[84] | 0.5–2g MMFe | Stable (>6 months) | 60 adults | Pre-dose, 0.33, 0.66, 1, 1.5, 2, 3, 4, 6, 9 hours post-dose | 300 MPA | Ciclosporin | Yes | Yes | Post-absorption phase | Dose administered under fasting conditions and meals given 0.5 and 4 hours after dose; Bayesian model for estimation individual MPA |
| Staatz et al. (2005)[85] | 1g MMFe | Day 3, 5, 7 | 117 adults | Pre-dose, 1, 2, 3, 4 hours post-dose | 1376 MPA | Ciclosporin, tacrolimus | No | Yes | Absorption phase | Evidence of lag time during absorption, complex absorption process and EHC |
| Van Hest et al. (2005)[86] | 0.45–1.7 g MMFe | Day 3, 7, 11, 21, 28, 56, 84, 112, 140 | 140 adults | Pre-dose, 0.33, 0.66, 1.25, 2, 6, 8, 12 hours post-dose | 6523 MPA | Ciclosporin, corticosteroids | Yes | No | No | PK data obtained from an RCCT |
| Payen et al. (2005)[68] | 600mg/m2 MMFe | 10–1538 days | 41 children (2–21 years) | Pre-dose, 1, 2, 4, 6, 8, 10, 12 hours post-dose | 449 MPA | Ciclosporin, tacrolimus, corticosteroids | No | No | No | Profiles taken median 183 (12–3754) days post-transplant, 40 (10–1538) days post-start of MMF |
| Premaud et al. (2005)[87] | 0.5–2g MMFe | Day 3, 7, 30 stable (>3 months) | 45 adults | Pre-dose, 0.33, 0.66, 1, 1.5, 2, 3, 4, 6, 9, 12 hours post-dose | Ciclosporin, corticosteroids | Yes | Yes | Absorption and post-absorption phase | Dose administered under fasting conditions and meals given 0.5 and 4 hours after dose | |
| Cremers et al. (2005)[42] | 0.5–1g MMFe | ≤1 year: week 2, 4, 6, 8, 10, 17, 21, 29, 39, 52 | 64 adults | Pre-dose, 1, 2, 3, 4, 6 hours post-dose | 2748 MPA, 2648 MPAG | Ciclosporin, tacrolimus | No | No | No | Modelled MPA and MPAG plasma concentrations simultaneously |
| Van Hest et al. (2007)[88] | 0.25–2.2 g MMFe | Day 1, 3, 4, 5, 7, 18, 21, month 1, 2, 3, 4, 5, 6, 7, 12 | 468 adults | Pre-dose, 0.33, 0.5, 0.67, 1, 1.25, 2, 3, 4, 6, 8, 12 hours post-dose | 1894 MPA | Ciclosporin, sirolimus, corticosteroids | Yes | No | No | Data pooled from 6 clinical studies in post-renal transplant subjects |
| Jiao et al. (2008)[26] | 0.5g MMFe | 42 adults | Pre-dose, 0.17, 0.25, 0.33, 0.5, 0.75, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 12, 24, 36, 48 hours post-dose | 590 MPA, 589 MPAG | Yes | Yes | Post-absorption phase | Healthy adult Chinese subjects enrolled in 2 open-label, randomized crossover design studies with a washout of 12 days; dose administered under fasting conditions and meals 4, 9.5/10, 24.25 hours after dose; modelled MPA and MPAG plasma concentrations simultaneously | ||
| de Winter et al. (2008)[89] | 0.25–2.2 g MMFe 0.72g EC-MPSe | Stable (4–257 months) | 259 adults | Pre-dose, 0.25, 0.33, 0.5, 0.66, 0.75, 1, 1.25, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 10, 12 hours post-dosef | 3764 MPA (208EC-MPS, 184 MMF profiles) | Ciclosporin, tacrolimus, everolimus, corticosteroids | Yes | Yes | Absorption and post-absorption phase | Pooled data from 7 clinical trials and 2 unpublished studies |
| Zahr et al. (2008)[90] | 0.5–1.5 g MMFe | 20 adults | Predose, 0.33, 0.66, 1, 1.5, 2, 3, 4, 6, 8, 12 hours post-dose | 220 MPA | Corticosteroids | Yes | Yes | Absorption and post-absorption phase | Patients with SLE on a stable MMF regimen for at least 10 weeks, and standard meals given 1, 5 and 10 hours after dose; EHC could not be described well in model | |
| Yau et al. (2009)[69] | 0.5–1g MMFe | >1 month | 14 adults | Pre-dose, 0.5, 1, 1.5, 2, 6, 12 hours post-dose | 98 MPA and MPAG | Ciclosporin, corticosteroids | Yes | Yes | Post-absorption phase | Open-label study in Asian patients; modelled MPA and MPAG plasma concentrations simultaneously |
| Sam et al. (2009)[70] | 0.36–0.72 g EC-MPSe | >1 month | 18 adults | Predose, 0.25, 0.5, 0.75, 1.5, 2, 3, 4, 5, 7, 9, 10, 12 hours post-dose | 232 MPA, MPAG and AcMPAG | Ciclosporin, tacrolimus | Yes | Yes | Post-absorption phase | Stable patients with diabetes mellitus post-renal transplant; modelled MPA, MPAG and AcMPAG plasma concentrations simultaneously |
| de Winter et al. (2009)[71] | 1g MMFe | 38 adults | Predose, 0.33, 0.67, 1, 1.5, 2, 3, 4, 6, 8, 12, 14, 24 hours post-dose | 492 MPA | Ciclosporin, corticosteroids | Yes | Yes | Post-absorption phase | Patients with autoimmune disease on a stable MMF regimen for at least 10 weeks | |
| Musuamba et al. (2009)[91] | 1g MMFe | 7, 9, 15 months | 40 adults | Predose, 0.33, 0.66, 1.25, 2, 4, 6, 8, 12 hours post-dose | 1035 MPA and MPAG | Ciclosporin, sirolimus, corticosteroids | Yes | Yes | Post-absorption phase | Modelled MPA and MPAG plasma concentrations simultaneously |
| de Winter et al. (2009)[92] | 1g MMFe | Day 3, 6, 7, 11, 21, 28, 49, 56, 84, 112, 140 | 75 adults | Predose, 0.33, 0.5, 0.66, 1, 1.25, 2, 6, 7, 12 hours post-dose (48 ciclosporin, 45 tacrolimus profiles) | 489 total and unbound MPA, 488 total MPAG, 210 unbound MPAG | Ciclosporin, tacrolimus, corticosteroids | Yes | Yes | Post-absorption phase | Data from 2 studies in de novo renal transplant patients; relationship between total MPA, unbound MPA, total MPAG and unbound MPAG |
Sampling determined to NOT be adequate for detecting EHC or second peak if no sampling time between 0 and 1 hours post-dosing.
Absorption phase – second peak observed during first 3 hours post-dosing or post-absorption phase – second peak observed between 6 and 12 hours post-dosing.
Once- or twice-daily dosing.
Study also included subjects with rheumatoid arthritis treated with MMF.
Twice-daily dosing.
Not all sample times were available for EC-MPS analysis.
AcMPAG=MPA acyl-glucuronide; EC-MPS=enteric-coated mycophenolate sodium; EHC=enterohepatic circulation; MPA=mycophenolic acid; MPAG=7-O-MPA-β-glucuronide; NONMEM=nonlinear mixed-effects modelling; RCCT= randomized controlled clinical trial; SLE = systemic lupus erythematosus.
Table II.
Summary of published pharmacokinetic (PK) models: parameter estimates, covariates, model variability and assessment
| Study | Outline of PK model | Software | EHC model | PK parameter estimates | Model variability | Covariates | Model assessment | Model schematic |
|---|---|---|---|---|---|---|---|---|
| Funaki (1999)[67] | 3-compartment based on a 1-compartment disposition modela | NONMEM | Yes | CL/F (L/h) 25–46b V1/F (L) 31.6–142c V1/F (L) 43–193.3a ke (h−1) 0.271–0.859 ka (h−1) 0.409–13.70 tlag (h) 0.412c tlag (h) 0.274b |
NS | Food, race on ka, Food, race on ke, Food, race, WT, Maalox®, CLCR on V1/F, Maalox® on tlag |
NS | Figure 2a |
| Shum et al. (2003)[83] | 2-compartment model with tlagd | NONMEM | Noe | CL/F (L/h) 27.1 [1.42] V1/F (L) 97.7 [12.6] ka (h−1) 2.27 [0.18] Q/F (L/h) 25.7 [3.45] V2/F (L) 206 [55.7] tlag (h) 0.145 [0.02] |
BSV CL/F 0.039 [0.024] V1/F 0.31 [0.18] V2/F 2.27 [2.22] BOV CL/F 0.017 [0.013] V1/F 0.9 [0.27] V2/F 0.26 [0.23] ka 3.4 [0.7] tlag 0.1 [0.36] RUV Exp 0.12 [0.076] Add (mg/L) 0.57 [0.5] |
NS | Diagnostic plots | |
| Le Guellec et al. (2004)[84] | 2-compartment model with zero-order absorption | NONMEM | Noe | CL/F (L/h) 15.7 [5]f V1/F (L) 36 [19] V2/F (L) 137 [17] Q/F (L/h) 25.9 [36] D1 (h) 0.69 [7] |
BSV CL/F (%) 28 V1/F (%) 63 Q/F (%) 45 D1 (%) 11 RUV Prop NS Add (mg/L) 2.04 |
WT on CL 0.246 | Diagnostic plots | |
| Staatz et al. (2005)[85] | Bi-exponential elimination model with first-order absorptiong | NONMEM | Noe | CL/F (L/h) 34.9h,i/25.4i,j V1/F (L) 65 [7] V2/F (L) 496 [20] Q/F (L/h) 30.7 [10] ka (h−1) 0.64 [14] |
BSV CL/F (%) 32 [29] Q/F (%) 78 [28] ka (%) 109 [21] BOV CL/F (%) 35 [14] RUV Prop (%) 41 [4] |
Alb and ciclosporin dose on CL | Diagnostic plots, bootstrap | |
| Van Hest et al. (2005)[86] | 2-compartment model, first-order absorption with lag timek | NONMEM | Noe | CL/F (L/h) 33 [5.4] V1/F (L) 91 [7.2] V2/F (L) 237 [10] Q/F (L/h) 35 [5.3] ka (h−1) 4.1 [6.8] tlag (h) 0.21 [1.3] |
BSV CL/F (%) 31 [15] V1/F (%) 91 [13] V2/F (%) 102 [25] ka (%) 111 [15] BOV CL/F (%) 20 [11] V1/F (%) 53 [17] ka (%) 116 [11] RUV Add (mg/L) 0.45 [2.3] |
CLCR on V1/F -0.62 [16] Alb on V1/F -1.13 [23] Sex on V1/F 1.11 [4.3] CLCR on CL/F -0.12 [30] Alb on CL/F -1.07 [11] Ciclosporin on CL/F 0.31 [11] |
Diagnostic plots, bootstrap | |
| Payen et al. (2005)[68] | 2-compartment model with zero-order absorption and lag timek | NONMEM | Noe | CL/F (L/h) 17 a (h−1) 7.5 b (h−1) 0.0072 [28.6] k21 (h−1) 0.017 [31.3] V1/F (L) 5 [17] ka (h−1) 0.63 [19.2] tlag (h) 0.69 [4.7] |
BSV CL/F (%) 50.8 V1/F (%) 35.1 β (%) 32.1 k21 (%) 22.4 ka (%) 44.1 tlag (%) 99.5 RUV Prop (%) 26.5 Add (mg/L) 0.57 |
WT on V1/F 4.75 [29.7] | Diagnostic plots | Figure 2b |
| Premaud et al. (2005)[87] | Double gamma absorption model, de novo patients (day 3, 7, 30)l | In-house software MMF® | Noe |
Day 3 a1 9.45 [6.25] b1 15.92 [8.97] a2 16.59 [9.79] b2 5.82 [4.47] A 1.76 [1.22] λ1 1.13 [0.62] MAT1 (h) 0.71 [0.39] MAT2 (h) 3.61 [1.86] CL/F (L/h) 40.16 [18.87] Day 7 a1 12.75 [8.4] b1 25.57 [12.92] a2 34.9 [15.15] b2 12.25 [5.41] A 1.49 [0.95] l1 1.03 [0.61] MAT1 (h) 0.56 [0.34] MAT2 (h) 3.38 [2.34] CL/F (L/h) 42.85 [15.21] Day 30 a1 13.43 [8.82] b1 21.79 [14.93] a2 15.38 [10.14] b2 5.46 [3.7] A 2.49 [1.44] λ1 1.95 [1.38] MAT1 (h) 0.69 [0.26] MAT2 (h) 3.01 [1.47] CL/F (L/h) 34.05 [13.8] |
NS | NS | Diagnostic plots | |
| Single gamma absorption model, stable (>3 months)l |
>3 months a 5.38 [2.5] b 10.39 [4.7] A 5.59 [5.1] λ1 7.36 [4.04] B 0.9 [0.6] λ2 0.69 [0.53] MAT (h) 0.53 [0.14] CL/F (L/h) 31.63 [15.38] |
|||||||
| Cremers et al. (2005)[42] | 4-compartment model with rate constant describing transfer from fourth to first compartment for EHCm | NONMEM | Yes | CL/F (L/h) 14.1h/11.9 [1.75]i Q/F (L/h) 20.1h/11.2i V1/F (L) 11.7h/10.3 [4.09]i V2/F (L) 465h/183 [75]i V4/F (L) MPAG 5.6h/8.9 [2.3]i k40(MPAG) (h−1) 0.16h/ 0.12 [0.03]i k41(MPAG) (h−1) 0.04 [0.02]i |
RUV PropMPA (%) 35i PropMPAG (%) 14i |
CLCR on k40 | Diagnostic plots | Figure 2c |
| Van Hest et al. (2007)[88] | 2-compartment model, first-order absorption with tlagk | NONMEM | Noe | CL/F (L/h) 23 [2] V1/F (L) 69 [6] V2/F (L) 298 [8] Q/F (L/h) 34 [7] ka (h−1) 4 [7] tlag (h) 0.24 [1] |
BSV CL/F (%) 36 [9] V1/F (%) 90 [16] Q/F (%) 60 [21] ka (%) 101 [14] BOV CL/F (%) 21 [10] V1/F (%) 71 [12] ka (%) 116 [10] Q/F (%) 41 [39] RUV Add (mg/L) 0.44 [2]n |
Ciclosporin on ka 9.8 × 10−4 [20] Alb on V1/F −1.2 [17] CLCR on V1/F −0.49 [10] Antacids on V1/F 1.4 [8] Ciclosporin on CL 4.8 × 10−4 [16] Alb on CL −0.72 [13] CLCR on CL −0.22 [7] Hb on CL −0.48 [16]° BSV CLCR on CL (%) 66 [29] Alb on CL (%) 112 [44] |
Bootstrap | |
| Jiao et al. (2008)[26] | 5-chain-compartment model with gallbladder compartmentg | NONMEM | Yes | CL/FMPA (L/h) 10.2 [5.7] CL/FMPAG (L/h) 1.38 [6.9] V2/F (L) 12.5 [8.3] V3/F (L) 213 [9.1] V4/F (L) 4.4 [6.4] Q/F (L/h) 16.1 [5.1] EHCP 29.1 [10.4] tlag (h) 0.096 [15.8] k12 (h−1) 3.53 [12.4] k51 (h−1) 67.5 [12.7] |
BSV CLMPA/F (%) 18.9 [35.6] V2/F (%) 34.5 [48.7] V3/F (%) 22.7 [39.2] V4/F (%) 23.1 [37.3] Q/F (%) 13.7 [48.9] EHCP (%) 29 [49.3] tlag (%) 57.3 [44.5] k12 (%) 60.3 [31.9] θ (%) 1.33 [27.2] RUV PropMPA (%) 45.3 [9.3] PropMPAG (%) 20.8 [16] |
WT onCLMPA,Q and V3 | Diagnostic plots, cross-method validation, VPC | Figure 2d |
| de Winter et al. (2008)[89] | 2-compartment model with first-order absorption and eliminationp | NONMEM | No | tlag(EC-MPS, M1) (h) 0.95 tlag(EC-MPS, M2) (h) 1.88 tlag(EC-MPS, M3) (h) 4.83 tlag(EC-MPS, EV) (h) 9.04 tlag(MMF) (h) 0.3 ka(EC-MPS) (h−1) 3.0 ka(MMF) (h−1) 4.1 V1/F (L) 40 CL/F (L/h) 16.0 V2/F (L) 518 Q/F (L/h) 22 POPtlag(EC-MPS, M1) 0.51 POP tlag(EC-MPS, M2) 0.32 POP tlag(EC-MPS, M3) 0.17 |
BSV tlag(EC-MPS, M) (%) 8.0 tlag(EC-MPS, EV) (%) 40 tlag(MMF) (%) 11 CL/F (%) 39 V1/F (%) 100 V2/F (%) 490 Q/F (%) 78 ka (%) 187 RUV Add (mg/L) 0.39 |
NS | Diagnostic plots, bootstrap, VPC | |
| Zahr et al. (2008)[90] | 1-compartment model with first-order elimination convoluted with a triple gamma distributionl | In-house software MMF® | No | CL/F (L/h) 40.3 [50.7] V1/F (L) 32.7 [18.6] |
NS | NS | Diagnostic plots, jack-knife method | |
| Yau et al. (2009)[69] | 5-compartment drug and metabolite model with EHCl | WinNonLin | Yes | CL/FMPA (L/h) 15 [5.6]q/ 18.2 [12.5]r CL/FMPAG (L/h) 0.85 [0.4]q/ 0.86 [0.3]r CLf/F (L/h) 13.2 [5.1]q/ 17.7 [11.9]r CL/FMPAG,bile (L/h) 0.4 [0.3]q/0.7 [0.1]r V1/F (L) 15.1 [10.9]q/7.6 [4.4]r V2/F (L) 188 [154]q/68 [39]r Vd/F (L) 203 [161]q/76 [43]r Vm/F (L) 3.74 [1.5]q/6.4 [4.2]r Q/F (L/h) 20.2 [11.9]q/ 9.7 [11.0]r ka (h−1) 1.57 [1.04]q/2.6 [0.39]r km (h−1) 1.69 [2.12]q/3.3 [3.45]r kbile (h−1) 0.12 [0.09]q/ 0.13 [0.06]r kr (h−1) 0.12 [0.1]q/0.11 [0.13]r kr,m (h−1) 0.13 [0.1]q/ 0.02 [0.01]r tlag (h) 0.13 [0.2]q/0.2 [0.3]r tbile (h) 9.4 [1.2]q/6 [2.1]r τgall (h) 1.4 [0.8]q/0.7 [0.3]r |
NS | Diagnostic plots | Figure 2e | |
| Sam et al. (2009)[70] | Initial 2-compartment model, linear elimination of MPAG and AcMPAG with EHC of MPAs | NONMEM | Yes |
MPA ka (h−1) 0.67 [24.8] CL/F (L/h) 10.6 [11.1] V1/F (L) 25.9 [34.9] Q/F (L/h) 8.11 [24.2] V2/F (L) 39.6 [86.9] MPAG FMAG (L−1) 0.38 [27.3] k30(h−1): GFR <80 mL /min/ 1.73 m2 = a×(GFR/51.6)a, where a = 0.17 [34.7], b 0.33 [262] k30(hr−1): GFR >80 mL /min/ 1.73 m2 = 0.32 [28.4] k3G (hr−1) 0.15 [28.8] KGB (h−1) 0.007 [167] AcMPAG FMAC (L−1): GFR ≤60mL/ min/ 1.73 m2 = c×(GFR/45.3)e, where c 0.014 [17.5] d−1.95 [11.7] FMAC (L−1): GFR ≤60mL / min/ 1.73 m2 = 0.013 [15.1] k40 (h−1) 0.21 [13.8] k4G (h −1) 0.15 [26.5] |
BSV MPA CL/F (%) 21.4 [66.1] V1/F (%) 87.8 [32] V2/F (%) 239 [248] MPAG FMAG (%) 34.6 [85.8] a (%) 29.1 [199] k30 (%) 29.1 [199] kGB (%) 35.9 [260] AcMPAG c (%) 24.6 [68.7] FMAC (%) 24.6 [68.7] k40 (h−1) 48.8 [47.1] k4G (h−1) 67.3 [41.1] RUV MPA (%) 69.9 [15.3] MPAG (%) 19.4 [32.1] AcMPAG (%) 17.8 [23] |
GFR on k30 | Diagnostic plots, VPC | Figure 2f |
| de Winter et al. (2009)[71] | 2-compartment model, EHC of MPA with absorption of MPA described with 2 first-order processes (short and long lag time)k | NONMEM | Yes | Ffast 0.71 [9] tlag short (h) 0.287 [5] tlag long (h) 0.643 [3] ka (h−1) 6.2 [22] CL/F (L/h) 8.27 [5] V1/F (L) 52.4 [17] V2/F (L) 262 [5] Q/F (L/h) 16.2 [222] TGB1 (h) 6 TGB2 (h) TGB1+ 4 DGB (h) 0.1 EHCP 0.37 k63 (h−1) 1 |
BSV tlag short (%) 32 ka (%) 182 [40] CL/F (%) 34 [41] V1/F (%) 53 [48] TGB1 (%) 200 EHCP (%) 35 RUV (%) 0.414 [6] |
CLCR on CL/F 0.42 [26] | Diagnostic plots, bootstrap, VPC | Figure 2g |
| Musuamba et al. (2009)[91] | 2-compartment model with first-order absorption and elimination, with MPAG and EHC compartmentsk | NONMEM | Yest | tlag (h) 0.26 [7.4] k12 (h−1) 1.83 [32] V1/F (L) 14.7 [22] V2/F (L) 250 [32] V3/Fm (L) 6.31 [17] CL/F (L/h) 14.7 [11] k20 (h−1) 0.36 [23] Q/F (L/h) 21.1 [0.8] k20 (h−1) 0.36 [23] k40 0.12 [10.7] |
BSV V1/F (%) 3.2 [12] Q/F (%) 17 [8] k40 (%) 2 [16] BOV k12 (%) 62 [18] V1/F (%) 21 [31] CL/F (%) 13 [26] k40 (%) 5 [39] RUV PropMPA (%) 0.4 [59] PropMPAG (%) 0.2 [0.4] AddMPA (μg/mL) 0.2 [63] |
Sirolimus on k41 0.1 [14] GFR on k40 0.008 [41] AST/ALT on CL 3.1 [33] |
Diagnostic plots, bootstrap, cross-method validation, VPC | |
| de Winter et al. (2009)[92] | 2- and 1-compartment models for unbound MPA and unbound MPAG, with EHC compartment | NONMEM | Yes | tlag (h) 0.231 ka (h−1) 4.0 V1/F fmPA (L) 189 CL/F fmPA (L/h) 747 V2/F fmPA (L) 34300 Q/F fmPA (L/h) 2010 k24 (h−1 mmol−1) 0.153 Bmax (mmol) 35100 k42 (h−1) 169 V1/F fMPAG (L) 8.56 k56 (h−1 mmol−1) 0.0133 k65 (h−1) 93.1 CL/F fMPAG (L/h) 4.75 TGB (h) 7.9 DGB (h) 1 k72 (h−1) 10 k57 (h−1) 0.0796 |
BSV tlag (%) 161 V1/F fmPA (%) 116 CL/F fmPA (%) 97 Bmax (%) 48 CL/F fMPAG (%) 106 TGB (%) 141 k57 (%) 71 RUV tMPA (mmol/L) 0.52 fMPA (mmol/L) 0.993 tMPAG (mmol/L) 0.18 fMPAG (mmol/L) 0.55 |
CLCR on CL fMPAG 1.36 Alb on Bmax 1.39 Ciclosporin on k57 0.002 |
Diagnostic plots, VPC |
Values are range of mean PK parameters among races and fasted, after-meal and before-meal states.
It was unknown if Maalox® was taken.
No Maalox® taken.
Summary of results of the final base model, mean estimates [SE].
Considered but not in final model.
Based on median study weight (kg) with corticosteroid co-medication.
Population PK estimates given as mean [SE%].
Ciclosporin co-therapy.
Tacrolimus co-therapy.
Based on median study albumin 26g/L.
Final population PK model estimates given as mean [CV%].
PK parameters values are expressed as mean [SD].
Final population PK estimates given as mean [SE].
RUV is on a natural logarithmic-scale as data were logarithmically transformed.
During first 6 months post-transplant.
Final population PK model estimates expressed as mean.
Received MMF, ciclosporin and prednisolone.
Received MM F and prednisolone.
Parameter estimates of final population PK model expressed as mean [%RSE].
EHC model only developed in those patients receiving concomitant medication with sirolimus.
α=apparent rate constant of distribution; β=apparent rate constant of elimination; θ=covariate effects and correlation between CL/FMPAG and Q/F; λ1, λ2 = disposition rate constants of the 2 compartments; τgall=gallbladder emptying interval; a, b = gamma distribution parameters with single gamma absorption model; a1,b1, a2, b2=gamma distribution parameters with double gamma distribution model; A, B = disposition coefficient standardized per 100 mg dose; AcMPAG = MPA acyl-glucuronide; Add=additive error; Alb = plasma albumin concentration; bile=amount of MPAG in the gallbladder compartment; Bmax=maximum number of binding sites; BOV=between-occasion variability; BSV= between-subject variability; CL = apparent total clearance; CLCR=creatinine clearance; CL/F=apparent oral clearance; CLf=apparent formation clearance of MPA to MPAG; CV=coefficient of variation; D1,=duration of input (absorption); DGB=duration of gall bladder opening; EC-MPS=enteric-coated mycophenolate sodium; EHC=enterohepatic circulation; EHCP=% of MPA recycled into the body; EV=evening; Exp = exponential; Ffast=part of dose ending up in the fast absorption compartment; Fm=fraction of the MPA dose converted to MPAG; FMAG= ratio of the fraction of MPA metabolized to MPAG to the volume of distribution of MPAG; fMPA = unbound MPA; fMPAG = unbound MPAG; GFR=glomerular filtration rate; Hb = haemoglobin; kxy=transfer rate constant from compartment x to y; ka=absorption rate constant; kbile=biliary excretion rate constant of MPAG; ke = renal elimination constant; kGB = rate constant for the release of recirculated MPA from MPAG and AcM PAG into the depot compartment; km=formation rate constant of MPAG; kr = renal excretion rate constant of MPA; kr,m = renal excretion rate constant of MPAG; Laglong = lag time long absorption; Lagshort= lag time short absorption; M = morning; M1 =morning 1; M2 = morning 2; M3=morning 3; MAT=mean absorption time with single gamma absorption model; MAT1, MAT2 = mean absorption times with double gamma absorption model; MMF = mycophenolate mofetil; MPA=mycophenolic acid; MPAG=7-O-MPA-β-glucuronide; NONMEM = nonlinear mixed-effects modelling; NS = not stated; POP = part of the population; Prop = proportional error; Q/F = apparent intercompartmental clearance; RSE=relative standard error; RUV=residual variability; SD=standard deviation; SE=standard error; Tbile=time of bile release after dosing; TGB„=time of nth opening gallbladder compartment; tlag=lag time; tMPA=total MPA; tMPAG=total MPAG; V1/F = apparent volume of distribution in the central compartment after oral administration; V2/F=apparent volume of distribution of the peripheral compartment of MPA; V3/F = apparent volume of distribution of the central compartment of MPAG; V4/F = apparent volume of distribution of MPAG; Vd = apparent volume of distribution (V1/F + V2/F) for MPA; Vm=volume of central compartment for MPAG; VPC=visual predictive check; WT=bodyweight.
The studies mainly involved adults, with only one study using data collected from paediatric patients.[68] Studies involved healthy subjects, kidney transplant recipients and patients treated with MMF for autoimmune diseases. Approaches to modelling included first- and zero-order absorption models with and without lag time and more complex models used gamma distribution/absorption phases. Only a couple of the published papers included pharmacokinetic models developed to account for MPA enterohepatic recirculation (tables I and II).
One of the first population pharmacokinetic models for MPA that included an EHC component was published in 1999 by Funaki.[67] The model included a gallbladder compartment, which described the physiological process of enterohepatic recirculation and biliary excretion (tables I and II, figure 2a). The release of bile was assumed to occur as a bolus at time of gallbladder emptying, and was modelled by providing an expulsion time. Funaki’s model only described one single episode of gallbladder emptying. However, the fact that bile is continuously excreted and influenced by feeding habits, including difference between high- and low-fat meals, would mean the model has limitations.[50]
Fig. 2.
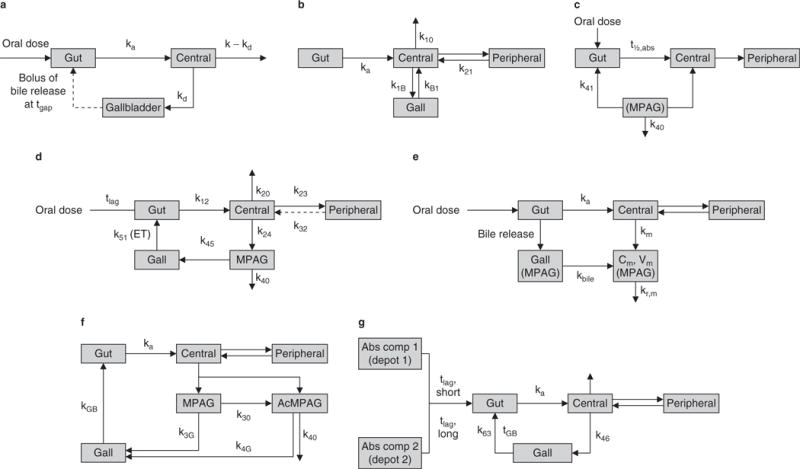
Schematicsof mycophenolic acid (MPA) pharmacokinetic models describing enterohepatic circulation (EHC). (a) 3-compartment EHC model based on a 1-compartment disposition model (reproduced from Funaki,[67] with permission). (b) Proposed EHC, 2-compartment structural model with first-order absorption with a lag time (tlag) [reproduced from Payen et al.,[68] with permission]. (c) 4-compartment model, with rate constant describing transfer from fourth to first compartment [enterohepatic recycling] (reproduced from Cremers et al.,[42] with permission). (d) Chain compartment model (intestinal, gallbladder, central and peripheral compartments for MPA and central compartment for 7-O-MPA-β-glucuronide (MPAG) [reproduced from Jiao et al.,[26] with permission]. (e) 5-compartment drug and metabolite EHC model with MPA and MPAG plasma concentrations simultaneously (reproduced from Yau et al.,[69] with permission). (f) 2-compartment model with linear elimination, with MPAG and MPA acyl-glucuronide (AcMPAG) produced from the central compartment with EHC of MPA via the two metabolites (reproduced from Sam et al.,[70] with permission). (g) 2-compartment model accounts for the enterohepatic recirculation of MPA. The absorption of MPA was described with two first-order processes with a short and a long tlag and subsequent first-order elimination (reproduced from de Winter et al.,[71] with permission). Abs comp=absorption compartment; Cm = concentration of MPAG in central compartment; ET = gallbladder emptying time; Gall = amount of MPAG in gallbladder compartment; Gut= amount of MPA in gut compartment; k=first-order rate constant; kxy = transfer rate constant from compartment x to y; ka= absorption rate constant; kbile= biliary excretion rate; kd= excretion rate constant into gallbladder; kGB = rate constant for the release of recirculated MPA from MPAG and AcMPAG; km = formation rate; kr,m = renal excretion rate of MPAG; t½,abs = absorption half-life; tgap = expulsion time of gallbladder; tGB = time of gallbladder compartment opening; Vm= volume of MPAG in central compartment.
In 2003, Shum et al.[83] described a two-compartment model with a lag time. They considered modelling enterohepatic recirculation of MPA but concluded that their data did not statistically support that type of model. They also investigated various other approaches, including time-dependent absorption models, maximum effect (Emax), Weibull and a dual sequential first-order absorption model. They found that none of these models improved the fit statistically as much as the addition of lag time using their available data. The same research group in 2005 developed a bi-exponential elimination model with first-order absorption.[85] They also described a single-exponential elimination model with first-order absorption, but this did not improve the model fit. Moreover, the addition of lag time to describe absorption did not improve the fit statistically, either (tables I and II).
Le Guellec et al.[84] developed a two-compartment model with zero-order absorption (tables I and II). They also investigated one- and two-compartment first-order absorption models. However, these models did not provide a good statistical fit. A model describing enterohepatic recirculation was considered but not used as the observed secondary peaks in the study patients were considered relatively small.
In one of the only published population pharmacokinetic models to include children, Payen et al.,[68] determined that a two-compartment model with first-order absorption and a lag time produced the best fit for their data. They also investigated a two-compartment model with enterohepatic recycling and first-order absorption and a lag time (tables I and II, figure 2b). They determined that there was no statistical improvement seen when applying that model to the data compared to the model without enterohepatic recycling.
The pharmacokinetic models outlined by van Hest et al.[86,88] both used a two-compartment model with time-lagged first-order absorption. Enterohepatic recirculation was not included in the models, as it was not considered to have a significant influence on the pharmacokinetics of MPA in this renal transplant cohort. Other models investigated included one-, two- and three-compartment models with and without lag time and with first- and zero-order absorption processes (tables I and II).
In the first study to simultaneously model MPA and MPAG concentrations, Cremers et al.,[42] used a four-compartment model describing the transfer of MPAG from the fourth and first compartments to simulate enterohepatic recycling (tables I and II, figure 2c). Due to the relatively sparse available data, secondary peaks were not observed in the population. Therefore, enterohepatic recycling was modelled by adding a rate constant between MPAG in the fourth compartment and the gut compartment. Similar to the model developed by Funaki,[67] this model assumed enterohepatic cycling as a constant process, when in actuality gallbladder emptying is both constant and sporadic.
Most of the studies analysed used a population approach (NONMEM) for the pharmacokinetic analysis. Only the study by Premaud et al.[87] used in-house software (MMF®) to perform individual Bayesian pharmacokinetic modelling in renal transplant recipients. They developed a double gamma absorption model in patients 3 months post-transplant and a one-compartment model with single gamma absorption phase for stable patients to successfully predict the contribution of MPA recycling (tables I and II). The models outlined by Premaud et al.[87] were able to accurately fit the varying concentration-time profiles observed between those patients who had recently received a renal transplant (<30 days) compared to those who were considered stable (>3 months).
The gamma distribution model as part of the MMF® algorithm was also used by Zahr et al.[90] to describe the absorption profile of MPA in patients with systemic lupus erythematosus (SLE). The application of Bayesian estimator[87] in a prospective concentration-controlled study in adult renal transplant patients showed good predictive performance resulting in significantly improved outcomes.[72] Zahr et al.[90] determined that a triple gamma distribution model provided the best fit to the third peak in addition to the first and second absorption peaks in these patients. The later peak or third peak is also assumed to be due to enterohepatic recycling of MPAG and subsequent re-conversion to MPA, representing, at least in the SLE study population, a significant proportion of the AUC. Recently, the same software (MMF®) was utilized in the pharmacokinetic modelling and development of a Bayesian estimator in haematopoietic stem-cell transplantation.[93]
The 2008 study undertaken by de Winter et al.[89] pooled data from patients treated with EC-MPS and MMF. As expected, the study determined that absorption of MPA was more delayed in those receiving EC-MPS compared to MMF. The model was also extended to include a bile compartment to describe enterohepatic recirculation, but this did not improve the fit of the model. The authors also investigated other modelling approaches, including two-compartment models with zero-order, with and without lag time, Weibull absorption and transit compartment models. A mixture model was also used to try to estimate time lag with an approximate longer lag time in the evening compared to the morning to simulate gastric emptying. The model estimated that gastric emptying was more delayed in the evening. This result is in keeping with previous observations of MPA absorption being more delayed at night in renal transplant patients receiving MMF.[94,95]
A chain compartment model simultaneously modelled MPA and MPAG in healthy subjects (tables I and II, figure 2d).[26] As this study involved healthy subjects, predictions for gallbladder emptying were set to meal times.[26] The investigators reported that they could not detect the effect of UGT1A9 polymorphisms on the pharmacokinetics of MPA and MPAG. However, this was probably related to the exclusion of subjects with functional SNPs such as UGT1A9−275 and −331/−440[96] from the study analysis. WinNonLin® was used by Yau et al.[69] to produce a five-compartment parent drug and metabolite EHC model (tables I and II, figure 2e). Simulations were used to investigate the influence of the time of bile release after dosing and the gallbladder emptying interval on enterohepatic recycling of MPA and MPAG. The model developed by Sam et al.[70] is the only population pharmacokinetic model to include MPA, MPAG and AcMPAG. Unlike the majority of other published studies, patients received EC-MPS rather than MMF (tables I and II, figure 2f).
In one of their recent studies, de Winter et al.[71] described MPA plasma concentrations in adults treated for autoimmune disease. EHC was included by using a rate constant to continuously fill the gallbladder from the central compartment. Emptying of the gallbladder into the gastrointestinal compartment was defined by two time points using rate and duration (tables I and II, figure 2g). This model appears to build on the group’s previously published model[89] by including a short and a long lag time. This concept is supported by the physiological mechanisms associated with gastric emptying, which have been shown to have variation related to the body's natural circadian rhythm.[97] It represents a more mechanism-based approach compared to earlier published MPA pharmacokinetic models. In contrast to other published pharmacokinetic models, this model was developed using data from patients treated with MMF for autoimmune disease. This model is similar to that described previously by Premaud et al.,[87] Zahr et al.[90] and Saint-Marcoux et al.,[93] who used different sets of gamma parameters to describe absorption rather than short and long lag time.
The latest published studies investigating population pharmacokinetics of MPA and including a mechanism-based model for EHC are those by Musuamba et al.[91] and de Winter et al.[92] Musuamba et al.[91] developed a model with a compartment to describe EHC in stable renal transplant recipients who were co-medicated with sirolimus (tables I and II). The application of Bayesian estimation allowed for accurate prediction of MPA AUC from 0 to 12 hours (AUC12) and individualized dosing. A complex mechanism-based model describing the pharmacokinetic role of protein binding of MPA was described recently by de Winter et al.[92] The model characterizes the relationship between total and unbound MPA and MPAG. From within the renal transplant recipient population, the correlation between the pharmacokinetic parameters, renal function, plasma albumin concentrations and co-medication with ciclosporin were included in the model. The process of EHC describing the reconversion of unbound MPAG to unbound MPA was included by using a gallbladder compartment that emptied into the central compartment at defined time intervals post-MMF dose. The model adequately described these complex relationships and allowed for the influences of renal function, plasma albumin and co-medication with ciclosporin to be quantified.
The estimation of pharmacokinetic parameters has been shown to moderately predict the outcome of MPA therapy.[16] The concentration-controlled study by LeMeur et al.[72] shows significantly improved outcomes. This study by the Limoges group was the first to report that TDM using a limited sampling strategy could be used to reduce the risk of treatment failure in renal transplant recipients. The Bayesian estimator was successfully used to adjust MMF doses in response to calculated MPA AUC. Yet, there are also conflicting results from some preliminary intervention studies.[98] In general, the individual modelling of MPA concentration-time profiles has been shown to predict MPA exposure expressed as an AUC12 with acceptable precision using a compartmental model approach. There is obvious debate about the inclusion or exclusion of enterohepatic recycling in a model. Currently, this appears to be data-driven but may be a source of error if not included, leading to an underestimation of clearance as well as exposure (AUC). Although the issue is complex, some empirical models that do not account for EHC per se but use a double gamma input are able to predict double peaks resulting in clinically acceptable AUC estimates. These models also allow capturing of very early secondary peaks or those that are higher than the initial peak.[52]
In summary, the review of the published papers found 12 studies[26,42,67,68,70,71,83–89,91,92] using NONMEM to describe the pharmacokinetics of MPA in renal transplant patients. Only one study included children[68] and one study used Win-NonLin.[69] Two studies[87] used MMF® software, with one of those studies investigating MPA pharmacokinetics in SLE patients.[90] In total, only three published papers involved non-transplant patients.[26,71,90] with two studies including patients treated with the EC-MPS formulation.[70,89] In the renal-transplant studies, time post-transplant ranged from 1 to 1538 days. Five studies[26,42,69,70,91] simultaneously modelled MPA and MPAG, with one study undertaken in healthy subjects.[26] One study included AcMPAG[70] and one modelled total and unbound MPA and MPAG.[92] Fourteen of the studies undertook sampling that is considered adequate for seeing evidence of EHC in concentration-time profiles. The EHC peak was typically observed in the post-absorption phase (6–12 hours post-dosing). Eight of the studies[26,42,67,69–71,91,92] included an EHC function in the final model, although most papers considered the inclusion of an EHC model during model development (tables I and II).
Importantly, in 15 of the studies, ciclosporin was used as co-therapy with MPA. In addition, tacrolimus (six stud-ies),[42,68,70,85,89,92] sirolimus (two studies)[88,91] and everolimus (one study)[89] were reported as concomitant therapies. Corti-costeroid co-therapy was reported in nine studies (table I). The estimated MPA apparent total oral clearance (CL/F) in adults post-renal transplant receiving MMF concomitant therapy with ciclosporin without corticosteroid therapy was 14.1–34.9 L/h[42,83–85] and with corticosteroid co-therapy was 15–42.85 L/h.[69,86–88] In those receiving tacrolimus the mean CL/F estimates ranged from 11.9 to 25.4 L/h without corticosteroid co-therapy.[42,85] In healthy subjects, MPA CL/F was reported to be 10.2 L/h[26] and in patients with SLE receiving corticosteroid co-therapy was 40.3 L/h.[90] In the non-transplant recipient studies, the covariates identified most frequently were bodyweight and creatinine clearance (CLCR) as influencing CL/F. In comparison, in the MPA renal transplant studies, serum albumin concentrations, bodyweight and ciclosporin co-medication were reported to have significant effects on CL/F and apparent volume of distribution in the central compartment after oral administration (V1/F) [table II].
The various published models vary not only in structure but also in utilization of varying populations for assessing MPA pharmacokinetics (table II). Overall model performance was highly variable with BSV mean estimates for MPA CL/F ranging from 28% to 50% (9–41% coefficient of variation [CV]) in adults receiving MMF post-renal transplant.[42,83–86,88,91] Five studies[83,85,86,88,91] reported estimates for BOV, with high variability associated with absorption rate constants. CL/F and V1/F estimates ranged from 13% to21% (10–26% CV) and from 53% to 71% (12–17% CV), respectively. Some models provided a good fit to the data but still had high residual variability, while other models reflected an adequate fit to the data with low residual variability; estimates ranged from 26.5% to 69.9% (4–15.3% CV).[42,83–86,88,91] All models utilized diagnostic plots to assess model fit. Other methods used for model evaluation included bootstrapping and visual predictive checks. Only two papers reported using a cross-validation method.[26,91]
7. Benefits of Modelling Enterohepatic Recycling
Few population pharmacokinetic analyses to date have used physiological or mechanism-based models to describe the EHC of MPA. In models derived in some populations, evidence of enterohepatic recycling – such as the characteristic secondary peak – are not as evident. Accordingly, EHC is not included in the model. The question whether all models developed for MPA should include enterohepatic recycling thus arises. On the other hand, inclusion of enterohepatic recycling may make the model too complex, and estimates produced with simpler models may still provide a reliable description of MPA pharmacokinetics. These are intriguing questions, especially as use of concomitant immunosuppressant therapy is known to decrease or inhibit enterohepatic recycling. As outlined previously, there are also other factorstoconsider that may increaseordecrease EHC and thus influence MPA pharmacokinetics. Can all these factors be included or estimated in a population pharmacokinetic model? Several parameters can be fixed based on prior information, but how much influence does this haveonthe estimates produced by the model?
MPA is quickly becoming standard therapy in solid-organ transplant patients and is increasingly being prescribed to those with autoimmune disorders. The MPA concentration-time profiles observed in kidney transplant patients include single-peak profiles, double-peak profiles with an early or later secondary peak, and atypical profiles.[87] The profiles from patients with SLE[99] or other autoimmune diseases[71,90] give a similar, if not more exaggerated, profile, especially in regard to the occurrence of the secondary peak. Transplant patients generally receive multiple drugs and experience post-surgical stress, further adding to the variability that exists between and within these patients. As time after organ transplantation lengthens, the proportion of patients with complex profiles tends to decrease. This may partly be due to tapering of corticosteroid and ciclosporin doses, progressive recovery of renal function and/or improvement in gastrointestinal motility after surgery. It is also thought that factors like absorption windows in the gastrointestinal tract, pre-systemic metabolism and efflux transports in mucosal epithelial cells may contribute to atypical drug absorption profiles.[100]
As outlined by van Gelder et al.,[6] optimal efficacy may require only a few dose adjustments. However, studies like the one undertaken by Le Guellec et al.[84] have used a population pharmacokinetic-based approach to demonstrate that Baye-sian estimation can accurately predict AUC. If EHC of MPA can truly account for up to 60% of exposure, it would be useful to be able to predict it[12] by the refinement of Bayesian en-terohepatic recycling models, which would allow further development of algorithms. This would facilitate the achievement of individualized MPA dosing.
Recently published pharmacokinetic models have advanced in their ability to mathematically describe the process of en-terohepatic recirculation of MPA. To date, the web-based Bayesian estimator developed by the Limoges group provides clinically robust results.[72] With incorporation of some genetic differences, e.g. UGT1A9 and MRP2, our ability to account for variability can be further improved.
8. Future Approaches
It is not easy to develop a mathematic model to fully describe the complex physiological processes that occur in relation to absorption of MPA. To date, there are only limited data available in regards to genetic differences in transporters and enzymes responsible for pre-systemic metabolism. As data for individual patient characteristics are slowly accumulated, these may further aid in describing the pharmacokinetics of MPA more accurately. However, there are still several problems that have to be resolved before the entire process of enterohepatic recycling of MPA will be able to be described in a model. NONMEM takes mainly an empirical approach to describe the multifaceted processes related to MPA pharmacokinetics, which has limitations. When using a compartment modelling approach it is difficult to isolate each physiological step into a compartment and to define the complex interactions between those compartments. Variability in model performance also exists, with some models able to accurately fit and describe data whether they include an EHC model or not, while other models contain high, unexplained residual variability even after modelling EHC. There is large variability observed even in estimating gastric emptying based on time of day as outlined by de Winter et al.,[89] and supported by results from other pharmacokinetic studies.[94,95] In addition, secondary peaks are not observed in all subjects and the number of sampling points around the secondary peak is usually limited. Given the complex physiology involved it makes sense to take a more physiologically and mechanism-based approach. This will be required if progress is to be made in explaining the large BSV and BOV observed in patients treated with MMF or EC-MPS.
Thus, it may be valuable to consider alternative, physiologically based approaches as offered by software packages such as Simcyp® (Simcyp, Sheffield, UK), GastroPlus™ (Simulation Plus, California, USA) and PKsim® (Bayer Technology Services, Leverkusen, Germany). These packages may be able to provide more insight into the mechanisms associated with the enterohepatic recycling of MPA. Literature is currently limited, as these packages have only gained wider application in the past 10 years. Simcyp® is a platform that provides whole-body physiologically based pharmacokinetic models that can incorporate enzyme kinetic data from routine in vitro studies.[101] Simcyp® not only allows typical modelling of absorption and distribution but also allows extensive simulation of metabolic drug-drug interactions and individual characteristics that determine the variability in drug exposure. PK-Sim® is a whole-body physiologically based pharmacokinetic simulation software, which derives models from parameters that are determined from a small set of physicochemical properties plus in vitro biochemical data, such as metabolic rates. It can be used to predict fraction absorbed, bioavailability and organ-specific pharmacokinetics.
GastroPlus™ combines the chemical properties (formulation, dosage and particle size), physiology (acid dissociation constant [pKa]) and kinetic information of a drug to simulate concentration-time profiles. By combining the physicochemical information about a drug and simulating a concentration-time profile, GastroPlus™ takes a more mechanism-based approach. As explained by Parrott et al.,[102] programs like GastroPlus™ can be used to predict the effect of food on the pharmacokinetics of a drug by using physiology-based absorption models.
9. Conclusions
Generally, the published population pharmacokinetic studies, which have used mainly empirical approaches, have not modelled MPA EHC physiology or have not had sufficient power to support such models. Conventional empirical models are useful but may be limited in their use to describe complex data that have multiple peaks. When evidence of EHC exists in the concentration-time profiles, it is important to attempt to include an EHC model, to aid in explaining high variability. It has become clear that, in order to describe the complex disposition of MMF, all relevant pharmacokinetic information should be used in building a model. Models need to not only include patient demographics but also other physiological factors, specifically those related to the biliary excretion of MPAG and to the overall EHC process. The influence of genetic differences and ontogeny in transporters and enzymes also needs to be considered as factors potentially influencing the disposition of MPA and affecting drug bioavailability and overall drug exposure as measured by the AUC.
Acknowledgments
The authors would like to acknowledge financial support from the following NIH grants: 5T32AR007594-15 (CMTS), 5K24HD050387-04 and 5U10HD037249-10 (TF, AAV). All authors have no conflicts of interest that are directly relevant to the content of this review.
References
- 1.Port FK, Dykstra DM, Merion RM, et al. Trends and results for organ donation and transplantation in the United States, 2004. Am J Transplant. 2005;5:843–9. doi: 10.1111/j.1600-6135.2005.00831.x. [DOI] [PubMed] [Google Scholar]
- 2.Cellcept®: US prescribing information. South San Francisco (CA): Genene-tech, Inc; 2010. Feb, [online]. Available from URL: http://www.gene.com/gene/products/information/cellcept/pdf/pi.pdf [Accessed 2010 Nov 5] [Google Scholar]
- 3.Allison AC. Mechanisms of action of mycophenolate mofetil. Lupus. 2005;14(Suppl. 1):s2–8. doi: 10.1191/0961203305lu2109oa. [DOI] [PubMed] [Google Scholar]
- 4.Knoll GA, MacDonald I, Khan A, et al. Mycophenolate mofetil dose reduction and the risk of acute rejection after renal transplantation. J Am Soc Nephrol. 2003;14:2381–6. doi: 10.1097/01.asn.0000079616.71891.f5. [DOI] [PubMed] [Google Scholar]
- 5.Pelletier RP, Akin B, Henry ML, et al. The impact of mycophenolate mofetil dosing patterns on clinical outcome after renal transplantation. Clin Transplant. 2003;17:200–5. doi: 10.1034/j.1399-0012.2003.00026.x. [DOI] [PubMed] [Google Scholar]
- 6.van Gelder T, Le Meur Y, Shaw LM, et al. Therapeutic drug monitoring of mycophenolate mofetil in transplantation. Ther Drug Monit. 2006;28:145–54. doi: 10.1097/01.ftd.0000199358.80013.bd. [DOI] [PubMed] [Google Scholar]
- 7.Kuypers DR, Le Meur Y, Cantarovich M, et al. Consensus report on therapeutic drug monitoring of mycophenolic acid in solid organ transplantation. Clin J Am Soc Nephrol. 2010;5:341–58. doi: 10.2215/CJN.07111009. [DOI] [PubMed] [Google Scholar]
- 8.Kuypers DR. Immunosuppressive drug monitoring: what to use in clinical practice today to improve renal graft outcome. Transpl Int. 2005;18:140–50. doi: 10.1111/j.1432-2277.2004.00041.x. [DOI] [PubMed] [Google Scholar]
- 9.Kaplan B. Mycophenolic acid trough level monitoring in solid organ transplant recipients treated with mycophenolate mofetil: association with clinical outcome. Curr Med Res Opin. 2006;22:2355–64. doi: 10.1185/030079906X148481. [DOI] [PubMed] [Google Scholar]
- 10.Nicholls AJ. Opportunities for therapeutic monitoring of mycophenolate mofetil dose in renal transplantation suggested by the pharmacokinetic/ pharmacodynamic relationship for mycophenolic acid and suppression of rejection. Clin Biochem. 1998;31:329–33. doi: 10.1016/s0009-9120(98)00056-3. [DOI] [PubMed] [Google Scholar]
- 11.Oellerich M, Shipkova M, Schutz E, et al. Pharmacokinetic and metabolic investigations of mycophenolic acid in pediatric patients after renal transplantation: implications for therapeutic drug monitoring. German Study Group on Mycophenolate Mofetil Therapy in Pediatric Renal Transplant Recipients. Ther Drug Monit. 2000;22:20–6. doi: 10.1097/00007691-200002000-00004. [DOI] [PubMed] [Google Scholar]
- 12.Staatz CE, Tett SE. Clinical pharmacokinetics and pharmacodynamics of mycophenolate in solid organ transplant recipients. Clin Pharmacokinet. 2007;46:13–58. doi: 10.2165/00003088-200746010-00002. [DOI] [PubMed] [Google Scholar]
- 13.Shaw LM, Korecka M, Venkataramanan R, et al. Mycophenolic acid pharmacodynamics and pharmacokinetics provide a basis for rational monitoring strategies. Am J Transplant. 2003;3:534–42. doi: 10.1034/j.1600-6143.2003.00079.x. [DOI] [PubMed] [Google Scholar]
- 14.Armstrong VW, Tenderich G, Shipkova M, et al. Pharmacokinetics and bioavailability of mycophenolic acid after intravenous administration and oral administration of mycophenolate mofetil to heart transplant recipients. Ther Drug Monit. 2005;27:315–21. doi: 10.1097/01.ftd.0000163949.40686.0f. [DOI] [PubMed] [Google Scholar]
- 15.Bullingham R, Monroe S, Nicholls A, et al. Pharmacokinetics and bioavailability of mycophenolate mofetil in healthy subjects after single-dose oral and intravenous administration. J Clin Pharmacol. 1996;36:315–24. doi: 10.1002/j.1552-4604.1996.tb04207.x. [DOI] [PubMed] [Google Scholar]
- 16.Hale MD, Nicholls AJ, Bullingham RE, et al. The pharmacokinetic-pharmacodynamic relationship for mycophenolate mofetil in renal transplantation. Clin Pharmacol Ther. 1998;64:672–83. doi: 10.1016/S0009-9236(98)90058-3. [DOI] [PubMed] [Google Scholar]
- 17.Kuypers DR, Naesens M, Vermeire S, et al. The impact of uridine diphos-phate-glucuronosyltransferase 1A9 (UGT1A9) gene promoter region single-nucleotide polymorphisms T-275A and C-2152T on early mycophenolic acid dose-interval exposure in denovo renal allograft recipients. Clin Pharmacol Ther. 2005;78:351–61. doi: 10.1016/j.clpt.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 18.Baldelli S, Merlini S, Perico N, et al. C-440T/T-331C polymorphisms in the UGT1A9 gene affect the pharmacokinetics of mycophenolic acid in kidney transplantation. Pharmacogenomics. 2007;8:1127–41. doi: 10.2217/14622416.8.9.1127. [DOI] [PubMed] [Google Scholar]
- 19.Betonico GN, Abbud-Filho M, Goloni-Bertollo EM, et al. Influence of UDP-glucuronosyltransferase polymorphisms on mycophenolate mofetil-induced side effects in kidney transplant patients. Transplant Proc. 2008;40:708–10. doi: 10.1016/j.transproceed.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 20.Girard H, Court MH, Bernard O, et al. Identification of common polymorphisms in the promoter of the UGT1A9 gene: evidence that UGT1A9 protein and activity levels are strongly genetically controlled in the liver. Pharmacogenetics. 2004;14:501–15. doi: 10.1097/01.fpc.0000114754.08559.27. [DOI] [PubMed] [Google Scholar]
- 21.Kuypers DR, de Jonge H, Naesens M, et al. Current target ranges of mycophenolic acid exposure and drug-related adverse events: a 5-year, open-label, prospective, clinical follow-up study in renal allograft recipients. Clin Ther. 2008;30:673–83. doi: 10.1016/j.clinthera.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 22.Levesque E, Benoit-Biancamano MO, Delage R, et al. Pharmacokinetics of mycophenolate mofetil and its glucuronide metabolites in healthy volunteers. Pharmacogenomics. 2008;9:869–79. doi: 10.2217/14622416.9.7.869. [DOI] [PubMed] [Google Scholar]
- 23.Naesens M, Kuypers DR, Verbeke K, et al. Multidrug resistance protein 2 genetic polymorphisms influence mycophenolic acid exposure in renal allograft recipients. Transplantation. 2006;82:1074–84. doi: 10.1097/01.tp.0000235533.29300.e7. [DOI] [PubMed] [Google Scholar]
- 24.Miura M, Satoh S, Inoue K, et al. Influence of SLCO1B1, 1B3, 2B1 and ABCC2 genetic polymorphisms on mycophenolic acid pharmacokinetics in Japanese renal transplant recipients. Eur J Clin Pharmacol. 2007;63:1161–9. doi: 10.1007/s00228-007-0380-7. [DOI] [PubMed] [Google Scholar]
- 25.Wang J, Yang JW, Zeevi A, et al. IMPDH1 gene polymorphisms and association with acute rejectionin renal transplant patients. Clin Pharmacol Ther. 2008;83:711–7. doi: 10.1038/sj.clpt.6100347. [DOI] [PubMed] [Google Scholar]
- 26.Jiao Z, Ding JJ, Shen J, et al. Population pharmacokinetic modelling for enterohepatic circulation of mycophenolic acid in healthy Chinese and the influence of polymorphisms in UGT1A9. Br J Clin Pharmacol. 2008;65:893–907. doi: 10.1111/j.1365-2125.2008.03109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bernard O, Tojcic J, Journault K, et al. Influence of nonsynonymous polymorphisms of UGT1A8 and UGT2B7 metabolizing enzymes on the formation of phenolic and acyl glucuronidesofmycophenolic acid. Drug Metab Dispos. 2006;34:1539–45. doi: 10.1124/dmd.106.010553. [DOI] [PubMed] [Google Scholar]
- 28.Picard N, Cresteil T, Premaud A, et al. Characterization of a phase 1 meta-boliteofmycophenolic acid produced by CYP3A4/5. Ther Drug Monit. 2004;26:600–8. doi: 10.1097/00007691-200412000-00004. [DOI] [PubMed] [Google Scholar]
- 29.Shipkova M, Armstrong VW, Wieland E, et al. Identification of glucoside and carboxyl-linked glucuronide conjugates of mycophenolic acid in plasma of transplant recipients treated with mycophenolate mofetil. Br J Pharmacol. 1999;126:1075–82. doi: 10.1038/sj.bjp.0702399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shipkova M, Strassburg CP, Braun F, et al. Glucuronide and glucoside conjugation of mycophenolic acid by human liver, kidney and intestinal microsomes. Br J Pharmacol. 2001;132:1027–34. doi: 10.1038/sj.bjp.0703898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feichtiger H, Wieland E, Armstrong VW, et al. The acyl glucuronide metabolite of mycophenolic acid induces tubulin polymerization in vitro. Clin Biochem. 2010;43:208–13. doi: 10.1016/j.clinbiochem.2009.08.023. [DOI] [PubMed] [Google Scholar]
- 32.Gensburger O, Picard N, Marquet P. Effect of mycophenolate acyl-glucuronide on human recombinant type 2 inosine monophosphate dehy-drogenase. Clin Chem. 2009;55:986–93. doi: 10.1373/clinchem.2008.113936. [DOI] [PubMed] [Google Scholar]
- 33.Mackenzie PI. Identification of uridine diphosphate glucuronosyltransferases involved in the metabolism and clearance of mycophenolic acid. Ther Drug Monit. 2000;22:10–3. doi: 10.1097/00007691-200002000-00002. [DOI] [PubMed] [Google Scholar]
- 34.Picard N, Ratanasavanh D, Premaud A, et al. Identification of the UDP-glucuronosyltransferase isoforms involved in mycophenolic acid phase II metabolism. Drug Metab Dispos. 2005;33:139–46. doi: 10.1124/dmd.104.001651. [DOI] [PubMed] [Google Scholar]
- 35.Basu NK, Kole L, Kubota S, et al. Human UDP-glucuronosyltransferases show atypical metabolism of mycophenolic acid and inhibition by curcumin. Drug Metab Dispos. 2004;32:768–73. doi: 10.1124/dmd.32.7.768. [DOI] [PubMed] [Google Scholar]
- 36.Bowalgaha K, Miners JO. The glucuronidation of mycophenolic acid by human liver, kidney and jejunum microsomes. Br J Clin Pharmacol. 2001;52:605–9. doi: 10.1046/j.0306-5251.2001.01487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bernard O, Guillemette C. The main role of UGT1A9 in the hepatic metabolism of mycophenolic acid and the effects of naturally occurring variants. Drug Metab Dispos. 2004;32:775–8. doi: 10.1124/dmd.32.8.775. [DOI] [PubMed] [Google Scholar]
- 38.Filler G. Drug interactions between mycophenolate and cyclosporine. Pediatr Transplant. 2004;8:201–4. doi: 10.1111/j.1399-3046.2004.00194.x. [DOI] [PubMed] [Google Scholar]
- 39.Westley IS, Brogan LR, Morris RG, et al. Role of MRP2 in the hepatic disposition of mycophenolic acid and its glucuronide metabolites: effect of cyclosporine. Drug Metab Dispos. 2006;34:261–6. doi: 10.1124/dmd.105.006122. [DOI] [PubMed] [Google Scholar]
- 40.Kobayashi M, Saitoh H, Tadano K, et al. Cyclosporin A, but not tacrolimus, inhibits the biliary excretion of mycophenolic acid glucuronide possibly mediated by multidrug resistance-associated protein 2 in rats. J Pharmacol Exp Ther. 2004;309:1029–35. doi: 10.1124/jpet.103.063073. [DOI] [PubMed] [Google Scholar]
- 41.Picard N, Yee SW, Woillard JB, et al. The role of organic anion-transporting polypeptides and their common genetic variants in mycophenolic acid pharmacokinetics. Clin Pharmacol Ther. 2010;87:100–8. doi: 10.1038/clpt.2009.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cremers S, Schoemaker R, Scholten E, et al. Characterizing the role of enterohepatic recycling in the interactions between mycophenolate mofetil and calcineurin inhibitors in renal transplant patients by pharmacokinetic modelling. Br J Clin Pharmacol. 2005;60:249–56. doi: 10.1111/j.1365-2125.2005.02398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Filler G, Bendrick-Peart J, Christians U. Pharmacokinetics of mycophenolate mofetil and sirolimus in children. Ther Drug Monit. 2008;30:138–42. doi: 10.1097/FTD.0b013e31816ba73a. [DOI] [PubMed] [Google Scholar]
- 44.Filler G, Lepage N, Delisle B, et al. Effect of cyclosporine on mycophenolic acid area under the concentration-time curve in pediatric kidney transplant recipients. Ther Drug Monit. 2001;23:514–9. doi: 10.1097/00007691-200110000-00003. [DOI] [PubMed] [Google Scholar]
- 45.Kiberd BA, Puthenparumpil JJ, Fraser A, et al. Impact of mycophenolate mofetil loading on drug exposure in the early posttransplant period. Transplant Proc. 2005;37:2320–3. doi: 10.1016/j.transproceed.2005.03.059. [DOI] [PubMed] [Google Scholar]
- 46.Cattaneo D, Perico N, Gaspari F, et al. Glucocorticoids interfere with mycophenolate mofetil bioavailability in kidney transplantation. Kidney Int. 2002;62:1060–7. doi: 10.1046/j.1523-1755.2002.00531.x. [DOI] [PubMed] [Google Scholar]
- 47.Usui T, Kuno T, Mizutani T. Induction of human UDP-glucuronosyl-transferase 1A1 by cortisol-GR. Mol Biol Rep. 2006;33:91–6. doi: 10.1007/s11033-005-1750-9. [DOI] [PubMed] [Google Scholar]
- 48.Kanou M, Usui T, Ueyama H, et al. Stimulation of transcriptional expression of human UDP-glucuronosyltransferase 1A1 by dexamethasone. Mol Biol Rep. 2004;31:151–8. doi: 10.1023/b:mole.0000043582.35335.ff. [DOI] [PubMed] [Google Scholar]
- 49.Soars MG, Petullo DM, Eckstein JA, et al. An assessment of UDP-glucur-onosyltransferase induction using primary human hepatocytes. Drug Metab Dispos. 2004;32:140–8. doi: 10.1124/dmd.32.1.140. [DOI] [PubMed] [Google Scholar]
- 50.Roberts MS, Magnusson BM, Burczynski FJ, et al. Enterohepatic circulation: physiological, pharmacokinetic and clinical implications. Clin Pharmaco-kinet. 2002;41:751–90. doi: 10.2165/00003088-200241100-00005. [DOI] [PubMed] [Google Scholar]
- 51.Lee WA, Gu L, Miksztal AR, et al. Bioavailability improvement of mycophenolic acid through amino ester derivatization. Pharm Res. 1990;7:161–6. doi: 10.1023/a:1015828802490. [DOI] [PubMed] [Google Scholar]
- 52.Bullingham RES, Nicholls AJ, Kanmm BR. Clinical pharmacokinetics of mycophenolate mofetil. Clin Pharmacokinet. 1998;34:429–55. doi: 10.2165/00003088-199834060-00002. [DOI] [PubMed] [Google Scholar]
- 53.Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22:11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 54.Custodio JM, Wu CY, Benet LZ. Predicting drug disposition, absorption/ elimination/transporter interplay and the role of food on drug absorption. Adv Drug Deliv Rev. 2008;60:717–33. doi: 10.1016/j.addr.2007.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lecureur V, Courtois A, Payen L, et al. Expression and regulation of hepatic drug and bile acid transporters. Toxicology. 2000;153:203–19. doi: 10.1016/s0300-483x(00)00315-2. [DOI] [PubMed] [Google Scholar]
- 56.Miura M, Satoh S, Inoue K, et al. Influence of lansoprazole and rabeprazole on mycophenolic acid pharmacokinetics one year after renal transplantation. Ther Drug Monit. 2008;30:46–51. doi: 10.1097/FTD.0b013e31816337b7. [DOI] [PubMed] [Google Scholar]
- 57.Wang J, Figurski M, Shaw LM, et al. The impact of P-glycoprotein and Mrp2 on mycophenolic acid levels in mice. Transpl Immunol. 2008;19(3–4):192–6. doi: 10.1016/j.trim.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 58.Sawamoto T, Van Gelder T, Christians U, et al. Membrane transport of mycophenolate mofetil and its active metabolite, mycophenolic acid in MDCK and MDR1-MDCK cell monolayers. J Heart Lung Transplant. 2001;20:234–5. doi: 10.1016/s1053-2498(00)00525-8. [DOI] [PubMed] [Google Scholar]
- 59.Strassburg CP, Kneip S, Topp J, et al. Polymorphic gene regulation and interindividual variation of UDP-glucuronosyltransferase activity in human small intestine. J Biol Chem. 2000;275:36164–71. doi: 10.1074/jbc.M002180200. [DOI] [PubMed] [Google Scholar]
- 60.Radominska-Pandya A, Little JM, Pandya JT, et al. UDP-glucuronosyl-transferases in human intestinal mucosa. Biochim Biophys Acta. 1998;1394:199–208. doi: 10.1016/s0005-2760(98)00115-5. [DOI] [PubMed] [Google Scholar]
- 61.Marzolini C, Tirona RG, Kim RB. Pharmacogenomics of the OATP and OAT families. Pharmacogenomics. 2004;5:273–82. doi: 10.1517/phgs.5.3.273.29831. [DOI] [PubMed] [Google Scholar]
- 62.Zamek-Gliszczynski MJ, Hoffmaster KA, Nezasa K, et al. Integration of hepatic drug transporters and phase II metabolizing enzymes: mechanisms of hepatic excretion of sulfate, glucuronide, and glutathione metabolites. Eur J Pharm Sci. 2006;27:447–86. doi: 10.1016/j.ejps.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 63.Ghibellini G, Leslie EM, Brouwer KL. Methods to evaluate biliary excretion of drugs in humans: an updated review. Mol Pharm. 2006;3:198–211. doi: 10.1021/mp060011k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cosson VF, Fuseau E. Mixed effect modeling of sumatriptan pharmacokinetics during drug development: II. From healthy subjects to phase 2 dose ranging in patients. J Pharmacokinet Biopharm. 1999;27:149–71. doi: 10.1023/a:1020601906027. [DOI] [PubMed] [Google Scholar]
- 65.Ilett KF, Tee LB, Reeves PT, et al. Metabolism of drugs and other xenobiotics in the gut lumen and wall. Pharmacol Ther. 1990;46:67–93. doi: 10.1016/0163-7258(90)90036-2. [DOI] [PubMed] [Google Scholar]
- 66.Bullingham R, Shah J, Goldblum R, et al. Effects of food and antacid on the pharmacokinetics of single doses of mycophenolate mofetil in rheumatoid arthritis patients. Br J Clin Pharmacol. 1996;41:513–6. doi: 10.1046/j.1365-2125.1996.03636.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Funaki T. Enterohepatic circulation model for population pharmacokinetic analysis. J Pharm Pharmacol. 1999;51:1143–8. doi: 10.1211/0022357991776831. [DOI] [PubMed] [Google Scholar]
- 68.Payen S, Zhang D, Maisin A, et al. Population pharmacokinetics of mycophenolic acid in kidney transplant pediatric and adolescent patients. Ther Drug Monit. 2005;27:378–88. doi: 10.1097/01.ftd.0000159784.25872.f6. [DOI] [PubMed] [Google Scholar]
- 69.Yau W-P, Vathsala A, Lou H-X, et al. Mechanism-based enterohepatic circulation model of mycophenolic acid and its glucuronide metabolite: assessment of impact of cyclosporine dose in Asian renal transplant patients. J Clin Pharmacol. 2009;49:684–99. doi: 10.1177/0091270009332813. [DOI] [PubMed] [Google Scholar]
- 70.Sam WJ, Akhlaghi F, Rosenbaum SE. Population pharmacokinetics of mycophenolic acid and its 2 glucuronidated metabolites in kidney transplant recipients. J Clin Pharmacol. 2009;49:185–95. doi: 10.1177/0091270008329558. [DOI] [PubMed] [Google Scholar]
- 71.de Winter B, Neumann I, van Hest RM, et al. Limited sampling strategies for therapeutic drug monitoring of mycophenolate mofetil therapy in patients with autoimmune disease. Ther Drug Monit. 2009;31:382–90. doi: 10.1097/FTD.0b013e3181a23f1a. [DOI] [PubMed] [Google Scholar]
- 72.Le Meur Y, Buchler M, Thierry A, et al. Individualized mycophenolate mofetil dosing based on drug exposure significantly improves patient outcomes after renal transplantation. Am J Transplant. 2007;7:2496–503. doi: 10.1111/j.1600-6143.2007.01983.x. [DOI] [PubMed] [Google Scholar]
- 73.Woltosz W. Weibull absorption models. PharmPK discussion [discussion list on the Internet] 2009 Jun 22; 21:00[online]. Available from URL: http://www.boomer.org/pkin/K09/K2009395.html [Accessed 2010 Oct 13]
- 74.Woltosz W. Weibull absorption models. PharmPK discussion [discussion list on the Internet] 2009 Jun 23; at 09:30 [online]. Available from URL: http://www.boomer.org/pkin/K09/K2009395.html [Accessed 2010 Oct 13]
- 75.Sun YN, Jusko WJ. Transit compartments versus gamma distribution function to model signal transduction processes in pharmacodynamics. J Pharm Sci. 1998;87:732–7. doi: 10.1021/js970414z. [DOI] [PubMed] [Google Scholar]
- 76.Savic RM, Jonker DM, Kerbusch T, et al. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J Pharmacokinet Pharmacodyn. 2007;34:711–26. doi: 10.1007/s10928-007-9066-0. [DOI] [PubMed] [Google Scholar]
- 77.Steimer JL, Plusquellec Y, Guillaume A, et al. A time-lag model for pharmacokinetics of drugs subjecttoenterohepatic circulation. J Pharm Sci. 1982;71:297–302. doi: 10.1002/jps.2600710308. [DOI] [PubMed] [Google Scholar]
- 78.Peris-Ribera JE, Torres-Molina F, Garcia-Carbonell MC, et al. General treatment of the enterohepatic recirculation of drugs and its influence on the area under the plasma level curves, bioavailability, and clearance. Pharm Res. 1992;9:1306–13. doi: 10.1023/a:1015861502354. [DOI] [PubMed] [Google Scholar]
- 79.Matis JH, Wehrly TE. Generalized stochastic compartmental models with Erlang transit times. J Pharmacokinet Biopharm. 1990;18:589–607. doi: 10.1007/BF01073940. [DOI] [PubMed] [Google Scholar]
- 80.Saint-Marcoux F, Marquet P, Jacqz-Aigrain E, et al. Patient characteristics influencing ciclosporin pharmacokinetics and accurate Bayesian estimation of ciclosporin exposure in heart, lung and kidney transplant patients. Clin Pharmacokinet. 2006;45:905–22. doi: 10.2165/00003088-200645090-00003. [DOI] [PubMed] [Google Scholar]
- 81.Rousseau A, Leger F, Le Meur Y, et al. Population pharmacokinetic modeling of oral cyclosporin using NONMEM: comparison of absorption pharmacokinetic models and design of a Bayesian estimator. Ther Drug Monit. 2004;26:23–30. doi: 10.1097/00007691-200402000-00006. [DOI] [PubMed] [Google Scholar]
- 82.Yu LX, Amidon GL. A compartmental absorption and transit model for estimating oral drug absorption. Int J Pharm. 1999;186:119–25. doi: 10.1016/s0378-5173(99)00147-7. [DOI] [PubMed] [Google Scholar]
- 83.Shum B, Duffull SB, Taylor PJ, et al. Population pharmacokinetic analysis of mycophenolic acid in renal transplant recipients following oral administration of mycophenolate mofetil. Br J Clin Pharmacol. 2003;56:188–97. doi: 10.1046/j.1365-2125.2003.01863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Le Guellec C, Bourgoin H, Buchler M, et al. Population pharmacokinetics and Bayesian estimation of mycophenolic acid concentrations in stable renal transplant patients. Clin Pharmacokinet. 2004;43:253–66. doi: 10.2165/00003088-200443040-00004. [DOI] [PubMed] [Google Scholar]
- 85.Staatz CE, Duffull SB, Kiberd B, et al. Population pharmacokinetics of mycophenolic acid during the first week after renal transplantation. Eur J Clin Pharmacol. 2005;61:507–16. doi: 10.1007/s00228-005-0927-4. [DOI] [PubMed] [Google Scholar]
- 86.van Hest RM, van Gelder T, Vulto AG, et al. Population pharmacokinetics of mycophenolic acid in renal transplant recipients. Clin Pharmacokinet. 2005;44:1083–96. doi: 10.2165/00003088-200544100-00006. [DOI] [PubMed] [Google Scholar]
- 87.Premaud A, Debord J, Rousseau A, et al. A double absorption-phase model adequately describes mycophenolic acid plasma profiles in de novo renal transplant recipients given oral mycophenolate mofetil. Clin Pharmacokinet. 2005;44:837–47. doi: 10.2165/00003088-200544080-00005. [DOI] [PubMed] [Google Scholar]
- 88.van Hest RM, van Gelder T, Bouw R, et al. Time-dependent clearance of mycophenolic acid in renal transplant recipients. Br J Clin Pharmacol. 2007;63:741–52. doi: 10.1111/j.1365-2125.2006.02841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.deWinter BC, van Gelder T, Glander P, et al. Population pharmacokinetics of mycophenolic acid: a comparison between enteric-coated mycophenolate sodium and mycophenolate mofetil in renal transplant recipients. Clin Pharmacokinet. 2008;47:827–38. doi: 10.2165/0003088-200847120-00007. [DOI] [PubMed] [Google Scholar]
- 90.Zahr N, Amoura Z, Debord J, et al. Pharmacokinetic study of mycophenolate mofetil in patients with systemic lupus erythematosus and design of Bayesian estimator using limited sampling strategies. Clin Pharmacokinet. 2008;47:277–84. doi: 10.2165/00003088-200847040-00005. [DOI] [PubMed] [Google Scholar]
- 91.Musuamba FT, Rousseau A, Bosmans JL, et al. Limited sampling models and Bayesian estimation for mycophenolic acid area under the curve prediction in stable renal transplant patients co-medicated with ciclosporin or siro-limus. Clin Pharmacokinet. 2009;48:745–58. doi: 10.2165/11318060-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 92.de Winter BC, van Gelder T, Sombogaard F, et al. Pharmacokinetic role of protein binding of mycophenolic acid and its glucuronide metabolite in renal transplant recipients. J Pharmacokinet Pharmacodyn. 2009;36:541–64. doi: 10.1007/s10928-009-9136-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Saint-Marcoux F, Royer B, Debord J, et al. Pharmacokinetic modelling and development of Bayesian estimators for therapeutic drug monitoring of mycophenolate mofetil in reduced-intensity haematopoietic stem cell transplantation. Clin Pharmacokinet. 2009;48:667–75. doi: 10.2165/11317140-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Satoh S, Tada H, Murakami M, et al. Circadian pharmacokinetics of mycophenolic acid and implication of genetic polymorphisms for early clinical events in renal transplant recipients. Transplantation. 2006;82:486–93. doi: 10.1097/01.tp.0000231874.53240.ba. [DOI] [PubMed] [Google Scholar]
- 95.Kagaya H, Inoue K, Miura M, et al. Influence of UGT1A8 and UGT2B7 genetic polymorphisms on mycophenolic acid pharmacokinetics in Japanese renal transplant recipients. Eur J Clin Pharmacol. 2007;63:279–88. doi: 10.1007/s00228-006-0248-2. [DOI] [PubMed] [Google Scholar]
- 96.Prausa SE, Fukuda T, Maseck D, et al. UGT genotype may contribute to adverse events following medication with mycophenolate mofetil inpediatric kidney transplant recipients. Clin Pharmacol Ther. 2009;85:495–500. doi: 10.1038/clpt.2009.3. [DOI] [PubMed] [Google Scholar]
- 97.Goo RH, Moore JG, Greenberg E, et al. Circadian variation in gastric emptying of meals in humans. Gastroenterology. 1987;93:515–8. doi: 10.1016/0016-5085(87)90913-9. [DOI] [PubMed] [Google Scholar]
- 98.de Winter BC, Mathot RA, van Hest RM, et al. Therapeutic drug monitoring of mycophenolic acid: does it improve patient outcome? Expert Opin Drug Metab Toxicol. 2007;3:251–61. doi: 10.1517/17425255.3.2.251. [DOI] [PubMed] [Google Scholar]
- 99.Zahir H, McCaughan G, Gleeson M, et al. Factors affecting variability in distribution of tacrolimus in liver transplant recipients. Br J Clin Pharmacol. 2004;57:298–309. doi: 10.1046/j.1365-2125.2003.02008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhou H. Pharmacokinetic strategies in deciphering atypical drug absorption profiles. J Clin Pharmacol. 2003;43:211–27. doi: 10.1177/0091270002250613. [DOI] [PubMed] [Google Scholar]
- 101.Jamei M, Dickinson GL, Rostami-Hodjegan A. A framework for assessing inter-individual variability in pharmacokinetics using virtual human populations and integrating general knowledge of physical chemistry, biology, anatomy, physiology and genetics: a tale of ‘bottom-up’ vs ‘top-down’ recognition of covariates. Drug Metab Pharmacokinet. 2009;24:53–75. doi: 10.2133/dmpk.24.53. [DOI] [PubMed] [Google Scholar]
- 102.Parrott N, Lukacova V, Fraczkiewicz G, et al. Predicting pharmacokinetics of drugs using physiologically based modeling: application to food effects. AAPS J. 2009;11:45–53. doi: 10.1208/s12248-008-9079-7. [DOI] [PMC free article] [PubMed] [Google Scholar]