

Abstract
Background: Coronary heart disease (CHD) is characterized by arterial wall inflammation and matrix degradation. Matrix metalloproteinase (MMP)-22 and -29 and pro-inflammatory cytokine interleukin-18 (IL18) are present in human hearts. IL18 may regulate MMP-22 and -29 expression, which may correlate with CHD progression. Methods and results: Immunoblot analysis showed that IL18 induced MMP-22 expression in human aortic smooth muscle cells. The Mann Whitney test from a prospective study of 194 CHD patients and 68 non-CHD controls demonstrated higher plasma levels of IL18, MMP-22 and -29 in CHD patients than in the controls. A logistic regression test suggested that plasma IL18 (odds ratio (OR)=1.131, P=0.007), MMP-22 (OR=1.213, P=0.040), and MMP-29 (OR=1.198, P=0.033) were independent risk factors of CHD. Pearson’s correlation test showed that IL18 (coefficient (r)=0.214, P=0.045; r=0.246, P=0.031) and MMP-22 (r=0.273, P=0.006; r=0.286, P=0.012) were associated with the Gensini score before and after adjusting for potential confounding factors. The multivariate Pearson’s correlation test showed that plasma MMP-22 levels correlated positively with high-sensitive-C-reactive protein (hs-CRP) (r=0.167, P=0.023), and MMP-29 levels correlated negatively with triglyceride (r=−0.169, P=0.018). Spearman’s correlation test indicated that plasma IL18 levels associated positively with plasma MMP-22 (r=0.845, P<0.001) and MMP-29 (r=0.548, P<0.001). Conclusions: Our observations suggest that IL18, MMP-22 and -29 serve as biomarkers and independent risk factors of CHD. Increased systemic IL18 in CHD patients may contribute to elevated plasma MMP-22 and -29 levels in these patients.
Keywords: Interleukin-18, Matrix metalloproteinase (MMP)-22, MMP-29, Coronary heart disease, Risk factor
1. Introduction
Coronary heart disease (CHD) remains the leading cause of death worldwide (Pagidipati and Gaziano, 2013). Recent studies have underscored the importance of detecting, monitoring, and controlling the vulnerability of atherosclerotic plaques to reduce the mortality rate among CHD patients (Cheng et al., 2006; Libby et al., 2009; Finn et al., 2010; Arbab-Zadeh and Fuster, 2015). Inflammatory cell accumulation and production of inflammatory cytokines are key processes that contribute to the initiation, progression, and ultimately the rupture and thrombosis of atherosclerotic plaques (Ross, 1999; Hansson, 2005; Libby et al., 2009). Arterial wall extracellular matrix (ECM) protein degradation is one of the most prominent mechanisms of aortic atherosclerotic lesion growth and rupture, in which matrix metalloproteinases (MMPs) are among the common proteases (Herman et al., 2001; Katsuda and Kaji, 2003; Newby, 2005; Siasos et al., 2012).
Previous studies showed significantly elevated plasma levels of pro-inflammatory cytokine interleukin-18 (IL18) in patients who developed coronary events. Plasma IL18 has been considered as an inflammatory risk factor of human CHD (Blankenberg et al., 2002; 2003; Evans et al., 2007; Jefferis et al., 2011; 2013). In addition to its systemic increases, IL18 expression was also found increased in human and experimental atherosclerotic lesions (Mallat et al., 2001; Gerdes et al., 2002; Wang et al., 2015). In mouse experimental models, deficiency of IL18 or IL18 receptors protected mice from diet-induced atherosclerosis (Elhage et al., 2003; Wang et al., 2015). Intraperitoneal administration of recombinant IL18 accelerated atherogenesis in mice (Tenger et al., 2005). Our recent study suggested that IL18 contributes to atherogenesis by inducing inflammatory cell (e.g. macrophages and T cells) production of inflammatory cytokines and chemokines, such as IL6, interferon-γ (IFN-γ), and monocyte chemoattractant protein-1 (MCP-1) (Wang et al., 2015). IL18 is also a known inducer of MMP expression from inflammatory cells, such as natural killer (NK) cell (Ishida et al., 2004), monocytes and macrophages (Quiding-Jarbrink et al., 2001; Abraham et al., 2002; Gerdes et al., 2002), and cardiomyocytes (Reddy et al., 2010).
MMPs are highly expressed in various pathological processes, such as inflammation, myocardial injury, and vascular aneurysms and remodeling (Creemers et al., 2001). Several major MMPs, such as MMP-1, -2, -7, -9, and -12 have been shown to be increased in the plasma or atherosclerotic lesions from CHD patients (Loftus et al., 2001; Nilsson et al., 2006; Lehrke et al., 2009; Ma et al., 2014; Goncalves et al., 2015). Genetic depletion or pharmacological inhibition of these MMPs protected mice from experimental atherosclerosis (Luttun et al., 2004; Kuzuya et al., 2006; Johnson et al., 2011; Hu et al., 2015). Human MMP-22 was first identified from human testis and heart. Northern blot detection showed its dominant expression in the human heart (Gururajan et al., 1998). Alternative splicing yielded three MMP-22 isoforms that differ in length of exon 4 or 5 (Gururajan et al., 1998). However, there is no further study to report its function or expression regulation in any tissue, cell type, or pathological condition. Human MMP-29 was also initially isolated from a human testis complementary DNA (cDNA) library, but it is more broadly expressed than MMP-22. Small intestine, liver, heart, kidney, and many other organs express this MMP (Wu et al., 2007). In addition to these initial expression profiles, there is no study to test the role of MMP-29 in the heart or vascular system. As atherosclerosis is a chronic inflammatory disease of the aortic artery and inflammation may induce the expression of these underestimated MMP members (Mallat et al., 2001; Blankenberg et al., 2002; 2003; Gerdes et al., 2002; Evans et al., 2007; Jefferis et al., 2011; 2013; Wang et al., 2015), we designed this study to test whether increased plasma and arterial wall inflammatory cytokine IL18 in CHD patient was correlated with or affected the expression of MMP-22 and -29, and whether plasma levels of these MMPs were also increased in CHD patients.
2. Materials and methods
2.1. Cell culture and Western blot
Human macrophages were prepared by differentiating Ficoll gradient-separated monocytes from human peripheral blood mononuclear cells in Roswell Park Memorial Institute 1640 (RPMI 1640) with 10% human serum (Gemini Bio-Products, Broderick, CA, USA) for 14 d on Falcon Primaria tissue culture dishes (Becton, Dickinson and Company, Franklin Lakes, NJ, USA). Human aortic smooth muscle cells (SMCs) were obtained from human donor aortas and subcultured at passages 2–5 in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS). Use of human primary cells or discarded tissues was pre-approved by the Partners Human Research Committee, Brigham and Women’s Hospital (Boston, MA, USA). For Western blot, human macrophages were starved for 24 h using serum-free RPMI 1640, followed by the addition of recombinant mouse IL18 (50 ng/ml, MBL International, Woburn, MA, USA) for 48 h. Human aortic SMCs were starved for 8 h using DMEM containing 1% FBS, followed by the same treatment as human macrophages for 48 h. Then cells were lysed in a lysis buffer containing 50 mmol/L Tris-HCl, pH 7.4, 150 mmol/L NaCl, 2 mmol/L ethylenediaminetetraacetic acid (EDTA), 1% Nonidet P-40 (NP-40), 0.1% (1 g/L) sodium dodecyl sulfate (SDS), 0.5% (5 g/L) sodium deoxycholate, and a protease inhibitor cocktail (Roche Diagnostics Corporation, Indianapolis, IN, USA). Cell lysates were centrifuged at 12 000 r/min for 20 min. Supernatant was collected for protein concentration determination using the BCA protein assay kit (Pierce, Rockford, IL, USA). For analysis, an equal amount of protein from each cell lysate was separated for Western blot analysis with rabbit anti-human MMP-22 polyclonal antibody (1:500 (v/v), ab87066, Abcam, Cambridge, MA, USA) and mouse anti-human β-actin monoclonal antibody (1:3000, sc-81178, Santa Cruz Biotechnology, Inc., Dallas, TX, USA).
2.2. Human study design
This prospective study was carried out in 194 consecutively enrolled CHD patients who had undergone coronary angiography (CAG) from October 2013 to October 2014 at the Department of Cardiology, the First Affiliated Hospital of Zhengzhou University, China. Patients were classified into two groups, including 75 patients with acute coronary syndrome (ACS) and 119 patients with stable angina pectoris (SAP). The ACS group consisted of 27 patients with acute myocardial infarction (AMI) and 48 patients with unstable angina pectoris (UAP). We selected 68 volunteers as controls from 158 consecutively enrolled individuals from the outpatient visits after excluding any of the following criteria: history of cardiovascular diseases; signs of ischemic heart disease; or electrocardiographic (ECG) criteria suggestive of CHD.
AMI was diagnosed with the presence of at least two of the following three characteristics from the universal definition of myocardial infarction (Thygesen et al., 2012): chest pain or equivalent symptoms; dynamic ECG changes consistent with AMI; and a typical serial increase (to at least three times the upper normal value) and decrease in cardiac necrosis markers such as creatine kinase-myocardial band isoenzyme (CK-MB) and/or troponin-I (Tn-I). UAP was defined by an ECG ST-segment depression of at least 0.1 mV or prominent T-wave inversion in two or more continuous ECG leads in an appropriate clinical setting (chest discomfort or anginal equivalent) without elevation of CK-MB or Tn-I (Anderson et al., 2013). SAP was diagnosed as chest pain of at least 6 months’ duration accompanied by evidence of severe coronary artery disease on CAG and by the absence of clinically evident ischemic episodes during the week preceding arteriography (Task Force et al., 2013). We also excluded patients with the following conditions to limit potential confounding effects: valvular heart disease, peripheral angiopathy, acute or chronic inflammatory diseases, immunologic diseases, respiratory insufficiency, renal or hepatic dysfunction, malignant neoplasia or terminal cachexia, and history or the presence of neoplastic diseases. The study was approved by the Ethics Committee of the First Affiliated Hospital of Zhengzhou University. All subjects were informed about the protocol at the time of enrollment and consented to study participation.
2.3. Clinical information and plasma IL18, MMP-22 and -29 measurement
Patient information, including age, sex, history of hypertension, diabetes mellitus, hyperlipidemia, cerebrovascular disease, CHD family history, and smoking (consuming tobacco for at least three years), was collected. Blood samples were collected from the peripheral vein into tubes containing EDTA after admission to hospital and 15 min of bed rest. Plasma was prepared by centrifugation at 3000 r/min for 10 min at 4 °C. All samples were stored at −80 °C until the assay. Plasma levels of total cholesterol (TC), triglyceride (TG), low-density lipoprotein (LDL), high-density lipoprotein (HDL), high-sensitive-C-reactive protein (hs-CRP), N-terminal pro-brain natriuretic peptide (NT-proBNP), and hemoglobin A1c (HbA1c) were determined using standard laboratory procedures at the clinical laboratory of the First Affiliated Hospital of Zhengzhou University, Henan, China. Plasma IL18 (JEB-10092, Nanjing Jin Yibai Biological Science and Technology Co., Ltd., Nanjing, China), MMP-22 (AE98622Hu, Shanghai Lianshuo Biological Technology Co., Ltd., Shanghai, China), and MMP-29 (JEB-12219, Nanjing Jin Yibai Biological Science and Technology Co., Ltd.) levels were determined using enzyme-linked immunosorbent assay (ELISA) kits in duplicates, according to the manufacturer’s instructions. All plasma sample aliquots were thawed only once.
2.4. Percutaneous coronary intervention, angiographic analysis, and echocardiographic study
CAG and percutaneous coronary intervention (PCI) were performed by the Judkins technique with standard equipment. Coronary angiograms were used for quantitative coronary angiographic analysis by two experienced investigators who were unaware of the clinical data. The severity of coronary artery stenosis was qualified by the Gensini score (Gensini, 1983).
Transthoracic two-dimensional echocardiography was performed by an experienced echocardiographer for all participants on Day 2 after admission, using a digital imaging system (Vivid-7; GE Medical System, Willoughby, Ohio, USA). The left ventricular end-systolic volume (LVESV) and end-diastolic volume (LVEDV) were measured using biplane Simpson’s method, and left ventricular ejection fraction (LVEF, %) was calculated by (LVEDV−LVESV)/LVEDV×100% from the apical four chambers position.
2.5. Statistical analysis
Mean levels of variables were compared across quartiles of IL18, MMP-22 and -29 levels by analysis of variance (ANOVA) for continuous variables and Mann Whitney U test was used to compare levels of these variables between different groups. Biomarker values were logarithmically transformed to reduce skewness. Logistic regression model was used to assess the relationship between CHD status and clinical risk factors. We used a forward selection model-building strategy and knowledge from a thorough literature review to select the covariates for further consideration in the multivariable analysis. Age, sex, smoking, diabetes, and hypertension were adjusted in the multivariable model. To analyze the correlation of plasma levels of IL18, MMP-22 and -29, we used Spearman’s correlation test. All analyses were performed by the SAS software (v.9.3, SAS Institute Cary, NC, USA), and P<0.05 was considered statistically significant.
3. Results
3.1. MMP-22 expression in human SMCs and macrophages
MMP-22 has been identified for over a decade, but limited information is available besides its known overall expression in the human heart (Anderson et al., 2013). It remains unknown whether cardiovascular or inflammatory cells express this protease differently under physiological or pathological conditions, how this protease expression is regulated, and whether its expression correlates with any pathological conditions of the heart. The availability of rabbit anti-human MMP-22 antibody allowed us to detect the expression of this MMP in human aortic SMCs and monocyte-derived macrophages. In human aortic SMCs, IL18 increased the expression of all three isoforms of MMP-22 (Fig. 1a), derived from alternative splicing (Gururajan et al., 1998). In contrast, IL18 did not affect MMP-22 expression in human monocyte-derived macrophages. Human macrophage expressed only the longest isoform of human MMP-22 (Fig. 1b). This observation suggests a role of IL18 in increasing the expression of MMP-22 in the vasculature of patients with CHD. Due to the lack of human MMP-29-specific antibody, we were unable to test whether IL18 affected MMP-29 expression in human aortic SMCs or macrophages. Therefore, increased plasma and arterial wall IL18 levels in CHD patients may lead to higher plasma and arterial wall expression of MMP-22 (Mallat et al., 2001; Blankenberg et al., 2002; 2003; Gerdes et al., 2002; Evans et al., 2007; Jefferis et al., 2011; 2013; Wang et al., 2015).
Fig. 1.
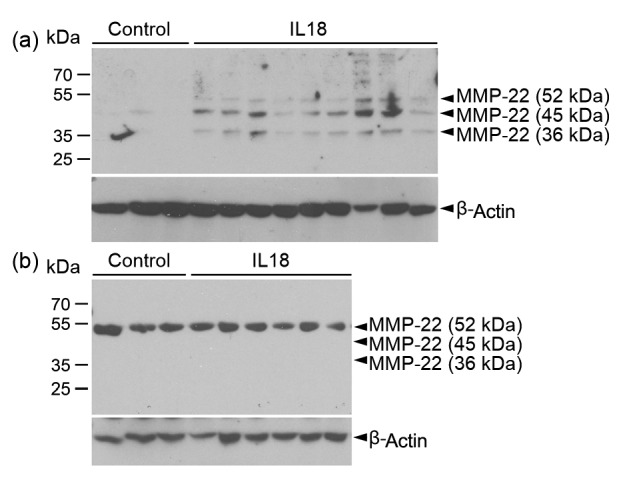
Immunoblot detection of MMP-22 expression in human aortic SMCs and monocyte-derived macrophages
(a) Human aortic SMCs were treated without (n=3) and with (n=9) recombinant human IL18, followed by MMP-22 immunoblot analysis. (b) Human monocyte-derived macrophages were treated without (n=3) and with (n=6) recombinant human IL18, followed by MMP-22 immunoblot analysis. The same blots were re-probed for β-actin to ensure equal protein loading. Arrowheads indicate all three isoforms of human MMP-22
3.2. General characteristics of CHD and non-CHD patients
A total of 194 patients with CHD (75 ACS patients and 119 SAP patients) and 68 non-CHD volunteers were included in the study. We examined the impact of sex, age, current smoking, diabetes mellitus, hypertension, hyperlipidemia, cerebrovascular disease, and CHD family history between CHD patients and non-CHD volunteers to the risk of CHD. Logistic regression showed that, among all tested common risk factors, sex (odds ratio (OR)=4.981, P=0.017) and age (OR=0.945, P=0.040) showed as significant risk factors to CHD in this population. Hypertension may also be a CHD risk factor, but statistical analysis showed borderline significance (OR=3.802, P=0.050) (Table 1).
Table 1.
Dichotomous and continuous variables between CHD patients and non-CHD controls of logistic regression analysis
| Variable | OR (95% CI) (CHD vs. non-CHD) | P |
| Sex | 4.981 (1.336, 18.568) | 0.017 |
| Age | 0.945 (0.896, 0.997) | 0.040 |
| Current smoking | 2.000 (0.425, 9.402) | 0.380 |
| Diabetes mellitus | 0.760 (0.196, 2.951) | 0.692 |
| Hypertension | 3.802 (1.021, 14.162) | 0.050 |
| Hyperlipidemia | 0.875 (0.100, 7.646) | 0.904 |
| Cerebrovascular disease | 0.345 (0.063, 1.889) | 0.220 |
| CHD family history | 0.532 (0.104, 2.732) | 0.450 |
3.3. Plasma IL18, MMP-22 and -29 are independent risk factors of CHD
The Mann Whitney U test demonstrated that plasma levels of IL18 were significantly higher in the ACS group than in the control group ((17.20±0.88) ng/dl vs. (8.19±3.16) ng/dl, mean±standard error of the mean (SEM); P=0.011; Fig. 2a). We also found significantly higher plasma levels of MMP-22 from patients with ACS than those from SAP patients ((2.42±0.14) ng/ml vs. (1.49±0.26) ng/ml, mean±SEM; P=0.034; Fig. 2b). Likewise, plasma MMP-29 levels were significantly higher in the ACS group ((205.29±24.64) ng/ml, mean±SEM) than in the SAP group ((156.48±18.26) ng/ml, mean±SEM; P=0.038) and control ((112.43±15.56) ng/ml, mean±SEM; P<0.001) groups (Fig. 2c).
Fig. 2.
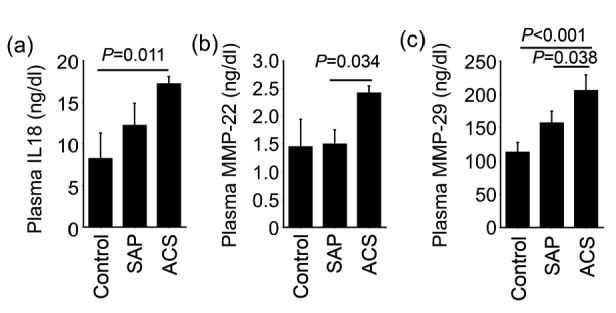
Plasma IL18 (a), MMP-22 (b), and MMP-29 (c) levels among ACS (n=75), SAP (n=119), and non-CHD control (n=68) groups
Data are expressed as mean±SEM. P<0.05 was considered as statistical significance, using Mann Whitney U test
Univariate logistic regression analysis showed that plasma IL18, MMP-22 and -29 levels were all significant risk factors of CHD with ORs at 1.131 (P=0.007), 1.213 (P=0.040), and 1.198 (P=0.033), respectively. After adjusting for smoking, diabetes, hypertension, age, and sex, these plasma cytokine and proteinases remained as significant risk factors of CHD (Table 2).
Table 2.
Plasma IL18, MMP-22, and MMP-29 are independent risk factors of human CHD
| Dependent variable | CHD status | Logarithmized data# | OR (95% CI) | Adjusted OR (95% CI) |
| ln(IL18) | CHD | 0.98 (0.81, 1.42) | 1.131 (1.027, 1.751) | 1.021 (1.001, 1.369) |
| Non-CHD | 0.84 (0.00, 1.00) | |||
| P | 0.002* | 0.007** | 0.005*** | |
| ln(MMP-22) | CHD | 0.15 (0.01, 0.51) | 1.213 (1.041, 2.529) | 1.092 (1.013, 1.777) |
| Non-CHD | 0.01 (0.00, 0.06) | |||
| P | 0.026* | 0.040** | 0.011*** | |
| ln(MMP-29) | CHD | 2.13 (2.01, 2.42) | 1.198 (1.039, 2.380) | 1.082 (1.011, 1.775) |
| Non-CHD | 2.04 (1.99, 2.10) | |||
| P | 0.009* | 0.033** | 0.012*** |
Data are expressed as ln(IL18) (ng/dl), ln(MMP-22) (ng/ml), and ln(MMP-29) (ng/ml) at percentile 50% (25%, 75%).
ANOVA test;
Logistic regression analysis;
Logistic regression analysis after adjusting for smoking, diabetes, hypertension, age, and sex
An ANOVA test of logarithmized data also indicated that the 194 CHD patients had significantly higher plasma ln(IL18) (0.98 (0.81, 1.42) vs. 0.84 (0.00, 1.00) ng/dl; P=0.002), ln(MMP-22) (0.15, (0.01, 0.51) vs. 0.01 (0.00, 0.06) ng/ml; P=0.026), and ln(MMP-29) (2.13 (2.01, 2.42) vs. 2.04 (1.99, 2.10) ng/ml; P=0.009) levels than those from the 68 non-CHD control patients (Table 2).
3.4. Correlations between plasma IL18, MMP-22 and -29 and CHD severity, heart functions, and common plasma CHD biomarkers
Univariate Pearson’s correlation analysis using logarithmized plasma IL18, MMP-22 and -29 levels demonstrated that plasma IL18 (coefficient (r)=0.214, P=0.045) and MMP-22 (r=0.273, P=0.006) levels were associated with the Gensini scores, as measure of CHD severity and extent. The multivariate Pearson’s correlation test showed that both IL18 (r=0.246, P=0.031) and MMP-22 (r=0.286, P=0.012) remained associated with the Gensini scores after adjusting for smoking, diabetes, hypertension, age, and sex (Table 3). However, the MMP-29 levels were not found to be associated with the Gensini scores under either univariate or multivariate Pearson’s correlation test (Table 3). Further, none of the tested plasma cytokine or proteinases (IL18, MMP-22 and -29) showed significant correlations with LVEF, LVEDV, or LVESV in this population, before and after adjusting for CHD risk factors including smoking, diabetes, hypertension, age, and sex (Table 3). Importantly, Spearman’s correlation test showed that the pro-inflammatory IL18 associated strongly and positively with both the MMP-22 (r=0.845, P<0.001; Fig. 3a) and MMP-29 (r=0.548, P<0.001; Fig. 3b) in all tested samples. These human data support our observations from in vitro cultured human SMCs (Fig. 1a) that IL18 induced MMP-22 expression in these cells. Increased systemic expression of IL18 from CHD patients may be responsible in part for increased plasma MMP-22 and possibly MMP-29.
Table 3.
Univariate and multivariate Pearson’s correlation analyses of plasma IL18, MMP-22 and -29 and Gensini score and echocardiographic parameters
| Variable | Correlation analysis | ln(IL18) |
ln(MMP-22) |
ln(MMP-29) |
|||
| r | P | r | P | r | P | ||
| Gensini score | Univariate | 0.214 | 0.045 | 0.273 | 0.006 | 0.177 | 0.078 |
| Multivariate* | 0.246 | 0.031 | 0.286 | 0.012 | 0.178 | 0.121 | |
| LVEDV | Univariate | −0.097 | 0.186 | −0.068 | 0.325 | 0.031 | 0.666 |
| Multivariate* | −0.090 | 0.249 | −0.063 | 0.416 | 0.009 | 0.911 | |
| LVESV | Univariate | −0.073 | 0.321 | −0.110 | 0.109 | 0.037 | 0.602 |
| Multivariate* | −0.054 | 0.491 | −0.080 | 0.307 | 0.060 | 0.440 | |
| LVEF | Univariate | 0.024 | 0.742 | 0.085 | 0.214 | −0.019 | 0.784 |
| Multivariate* | −0.002 | 0.978 | 0.014 | 0.862 | −0.051 | 0.515 | |
LVEDV, left ventricular end-diastolic volume; LVESV, left ventricular end-systolic volume; LVEF, left ventricular ejection fraction. * Adjusted for smoking, diabetes, hypertension, age, and sex
Fig. 3.
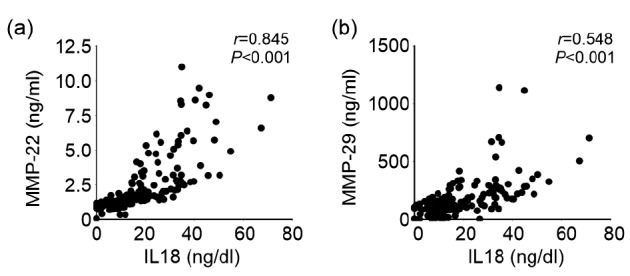
Spearman’s correlation test of plasma IL18 levels versus plasma MMP-22 (a) and -29 (b) levels in CHD patients (n=194) and non-CHD controls (n=68)
Both correlation coefficient (r) and P values are indicated for each analysis
Multivariate Pearson’s correlation analysis showed that plasma MMP-22 was associated significantly with plasma hs-CRP among all other common CHD risk factors, including NT-proBNP, HbA1C, TC, TG, HDL cholesterol (HDL-C), and LDL cholesterol (LDL-C), after adjusting for CHD risk factors smoking, diabetes, hypertension, age, and sex (r=0.167, P=0.023; Table 4). Plasma MMP-29 levels were associated significantly, but negatively, with plasma TG in both univariate (r=−0.165, P=0.012) and multivariate (r=−0.169, P=0.018) Pearson’s correlation analyses (Table 4). These observations support the hypothesis that elevated inflammation in CHD patients, as reflected by high plasma hs-CRP and IL18 levels, leads to increased protease expression, such as MMP-22 and many other MMPs and cathepsins and consequent arterial wall remodeling.
Table 4.
Univariate and multivariate Pearson’s correlation analyses of plasma IL18, MMP-22 and -29 and laboratory data
| Variable | Correlation analysis | ln(IL18) |
ln(MMP-22) |
ln(MMP-29) |
|||
| r | P | r | P | r | P | ||
| hs-CRP (mg/L) | Univariate | 0.112 | 0.110 | 0.112 | 0.091 | 0.012 | 0.860 |
| Multivariate* | 0.125 | 0.090 | 0.167 | 0.023 | 0.025 | 0.731 | |
| NT-proBNP (pg/ml) | Univariate | 0.018 | 0.807 | −0.004 | 0.959 | 0.026 | 0.707 |
| Multivariate* | 0.033 | 0.672 | 0.012 | 0.880 | −0.003 | 0.968 | |
| HbA1C (%) | Univariate | −0.116 | 0.343 | −0.017 | 0.879 | 0.009 | 0.938 |
| Multivariate* | −0.110 | 0.422 | −0.026 | 0.850 | −0.061 | 0.658 | |
| TC (mmol/L) | Univariate | 0.069 | 0.311 | 0.000 | 0.997 | 0.000 | 0.996 |
| Multivariate* | 0.074 | 0.307 | 0.076 | 0.291 | 0.007 | 0.920 | |
| TG (mmol/L) | Univariate | −0.056 | 0.414 | −0.058 | 0.366 | −0.165 | 0.012 |
| Multivariate* | −0.069 | 0.339 | 0.045 | 0.536 | −0.169 | 0.018 | |
| HDL-C (mmol/L) | Univariate | 0.035 | 0.605 | 0.068 | 0.295 | 0.130 | 0.048 |
| Multivariate* | 0.035 | 0.625 | −0.025 | 0.732 | 0.103 | 0.152 | |
| LDL-C (mmol/L) | Univariate | 0.077 | 0.258 | 0.036 | 0.578 | 0.036 | 0.582 |
| Multivariate* | 0.073 | 0.309 | 0.047 | 0.516 | 0.014 | 0.849 | |
hs-CRP, high-sensitivity C-reactive protein; NT-proBNP, N-terminal pro-brain natriuretic peptide; HbA1C, hemoglobin A1C; TC, total cholesterol; TG, triglyceride; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol. * Adjusted for smoking, diabetes, hypertension, age, and sex
4. Discussion
MMPs are among the most common proteases from the arterial wall in patients and experimental animals with atherosclerosis (Loftus et al., 2001; Nilsson et al., 2006; Lehrke et al., 2009; Ma et al., 2014; Goncalves et al., 2015). However, there has been limited information on MMP-22 and -29 since their initial identification (Gururajan et al., 1998; Wu et al., 2007). For unknown reasons, there is no study to suggest their matrix substrates, other intracellular and extracellular targets, or action mechanisms, and there is no information of how these proteases are regulated, although both MMPs are expressed in the human heart. Indeed, MMP-22 is expressed predominantly in the human heart (Gururajan et al., 1998). There must be a role for this protease to affect the heart functions, by acting on arterial wall matrix proteins or cardiac tissues, or by activating other tissue remodeling proteases, all which remain untested. Alternative splicing leads to three isoforms of human MMP-22 with size differences in either exon 4 or exon 5 (Gururajan et al., 1998). Here we showed that IL18 increased the expression of all three isoforms of MMP-22 in human SMCs (Fig. 1a). In contrast, human macrophages contained only the longest form of MMP-22 with extended exon 4 and IL18 did not affect its expression in macrophages (Fig. 1b). In addition we do not know why macrophages contained only one MMP-22 isoform. We also do not know whether all three isoforms of MMP-22 are equally active against their substrates or what are the actual substrate(s) of these MMP-22 species. However, these biochemical data support the observations from human plasma samples that: IL18, MMP-22 and -29 from ACS patients all increased concurrently (Fig. 2); IL18 levels correlated positively with MMP-22 or -29 (Fig. 3); and hs-CRP levels correlated with MMP-22 in multivariate Pearson’s correlation analyses (Table 4). Together, these observations suggest that inflammation is involved in promoting the expression of MMP-22 and possibly MMP-29 during atherogenesis and that IL18 is one of those inflammatory regulators of these MMPs in CHD patients.
Prior studies demonstrated increased plasma IL18 levels in patients with stroke, myocardial infarction (MI), or CHD (Jefferis et al., 2011; 2013). Plasma IL18 levels were also higher in patients with heart failure than in those without (Mallat et al., 2004), and higher in in-hospital ACS patients with adverse events, including cardiac death, recurrence of unstable angina, re-infarction, life-threatening arrhythmias, and urgent revascularization during hospitalization, than in those without these adverse events (Chalikias et al., 2005). As a risk factor, plasma IL18 predicts CHD (Blankenberg et al., 2003) and death from cardiovascular causes in CHD patients, regardless of the patient clinical status at admission (Blankenberg et al., 2002). In a study of European men, plasma IL18 levels showed no correlations with diabetes, hypertension, smoking, or alcohol consumption, but correlated with TC:HDL ratio, CRP, fibrinogen, and IL6 (Blankenberg et al., 2003). Consistent with those prior studies, we also showed higher plasma IL18 in ACS patients (Fig. 2a) or CHD patients (Table 2) than non-CHD controls and demonstrated that IL18 is also an independent risk factor of CHD in this Chinese population (Table 2). Of note, among this population, we found that smoking, diabetes mellitus, and hyperlipidemia were not associated with plasma IL18 (Table 1), and IL18 levels did not associate with plasma cholesterol levels and diabetic markers (Tables 3 and 4). Similar inconsistent observations were obtained from prior studies. From a French population, plasma IL18 also did not associate with diabetes, smoking, or hypertension (Blankenberg et al., 2003), but plasma IL18 levels were found associated with smoking, plasma insulin, and systolic and diastolic blood pressure from the British Regional Heart Study (Jefferis et al., 2011). From a large population of British men, plasma IL18 levels were not associated with plasma insulin and glucose levels (Jefferis et al., 2013). In contrast, plasma IL18 levels were strongly associated with diabetes and blood pressure from a relatively small population of women from South Africa (Evans et al., 2007). Therefore, race and gender may affect the association between IL18 and smoking, diabetes, hypertension, and hyperlipidemia.
The mechanism by which IL18 participates in atherosclerosis has been tested in atherosclerosis prone apolipoprotein E-deficient mice. In the absence of T cells, intraperitoneal injection of IL18 increased atherosclerotic lesion expression of IFN-γ and chemokine (C-X-C motif) ligand 16 (chemokine CXCL16) (Tenger et al., 2005). Therefore, IL18 may contribute to atherogenesis independent of T cells. Similar to these findings, our prior study showed that IL18-induced expression of IFN-γ and IL6 in macrophages was hundreds of times higher than that from T cells (Wang et al., 2015). As an indirect mechanism, IL18-induced production of IFN-γ and IL6 from macrophages may be used to promote MMP-22 and -29 expression in other vascular and inflammatory cells. As a direct mechanism, IL18 may act on vascular cells (Fig. 1a) or other inflammatory cells, such as monocytes, NK cells, and even cardiomyocytes to induce their expression of MMP-22 and -29 (Quiding-Jarbrink et al., 2001; Abraham et al., 2002; Gerdes et al., 2002; Ishida et al., 2004; Reddy et al., 2010; Wang et al., 2015). A detailed expression profile of MMP-22 and -29 in inflammatory cells, vascular cells, and cardiac cells merits future investigation.
Footnotes
Project supported by the University of Science and Technology Innovation Team of Henan (No. 14IRTSTHN018), the Science and Technology Talents Team Construction Program of Zhengzhou City-Science and Technology Talents (No. 131PLJRC670), China, and the National Institutes of Health (Nos. HL60942 and HL123568), USA
Compliance with ethics guidelines: Dong-Yi JIN, Cong-Lin LIU, Jun-Nan TANG, Zhao-Zhong ZHU, Xue-Xi XUAN, Xiao-Dan ZHU, Yun-Zhe WANG, Tian-Xia ZHANG, De-Liang SHEN, Xiao-Fang WANG, Guo-Ping SHI, and Jin-Ying ZHANG declare that they have no conflict of interest.
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2008 (5). Informed consent was obtained from all patients for being included in the study.
References
- 1.Abraham M, Shapiro S, Lahat N, et al. The role of IL-18 and IL-12 in the modulation of matrix metalloproteinases and their tissue inhibitors in monocytic cells. Int Immunol. 2002;14(12):1449–1457. doi: 10.1093/intimm/dxf108. [DOI] [PubMed] [Google Scholar]
- 2.Anderson JL, Adams CD, Antman EM, et al. 2012 ACCF/AHA focused update incorporated into the ACCF/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;61(23):e179–e347. doi: 10.1016/j.jacc.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 3.Arbab-Zadeh A, Fuster V. The myth of the “vulnerable plaque”: transitioning from a focus on individual lesions to atherosclerotic disease burden for coronary artery disease risk assessment. J Am Coll Cardiol. 2015;65(8):846–855. doi: 10.1016/j.jacc.2014.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blankenberg S, Tiret L, Bickel C, et al. Interleukin-18 is a strong predictor of cardiovascular death in stable and unstable angina. Circulation. 2002;106(1):24–30. doi: 10.1161/01.CIR.0000020546.30940.92. [DOI] [PubMed] [Google Scholar]
- 5.Blankenberg S, Luc G, Ducimetiere P, et al. Interleukin-18 and the risk of coronary heart disease in European men: the prospective epidemiological study of myocardial infarction (PRIME) Circulation. 2003;108(20):2453–2459. doi: 10.1161/01.CIR.0000099509.76044.A2. [DOI] [PubMed] [Google Scholar]
- 6.Chalikias GK, Tziakas DN, Kaski JC, et al. Interleukin-18:interleukin-10 ratio and in-hospital adverse events in patients with acute coronary syndrome. Atherosclerosis. 2005;182(1):135–143. doi: 10.1016/j.atherosclerosis.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Cheng C, Tempel D, van Haperen R, et al. Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation. 2006;113(23):2744–2753. doi: 10.1161/CIRCULATIONAHA.105.590018. [DOI] [PubMed] [Google Scholar]
- 8.Creemers EE, Cleutjens JP, Smits JF, et al. Matrix metalloproteinase inhibition after myocardial infarction: a new approach to prevent heart failure? Circ Res. 2001;89(3):201–210. doi: 10.1161/hh1501.094396. [DOI] [PubMed] [Google Scholar]
- 9.Elhage R, Jawien J, Rudling M, et al. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovasc Res. 2003;59(1):234–240. doi: 10.1016/S0008-6363(03)00343-2. [DOI] [PubMed] [Google Scholar]
- 10.Evans J, Collins M, Jennings C, et al. The association of interleukin-18 genotype and serum levels with metabolic risk factors for cardiovascular disease. Eur J Endocrinol. 2007;157(5):633–640. doi: 10.1530/EJE-07-0463. [DOI] [PubMed] [Google Scholar]
- 11.Finn AV, Nakano M, Narula J, et al. Concept of vulnerable/unstable plaque. Arterioscler Thromb Vasc Biol. 2010;30(7):1282–1292. doi: 10.1161/ATVBAHA.108.179739. [DOI] [PubMed] [Google Scholar]
- 12.Gensini GG. A more meaningful scoring system for determining the severity of coronary heart disease. Am J Cardiol. 1983;51(3):606. doi: 10.1016/S0002-9149(83)80105-2. [DOI] [PubMed] [Google Scholar]
- 13.Gerdes N, Sukhova GK, Libby P, et al. Expression of interleukin (IL)-18 and functional IL-18 receptor on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for atherogenesis. J Exp Med. 2002;195(2):245–257. doi: 10.1084/jem.20011022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goncalves I, Bengtsson E, Colhoun HM, et al. Elevated plasma levels of MMP-12 are associated with atherosclerotic burden and symptomatic cardiovascular disease in subjects with type 2 diabetes. Arterioscler Thromb Vasc Biol. 2015;35(7):1723–1731. doi: 10.1161/ATVBAHA.115.305631. [DOI] [PubMed] [Google Scholar]
- 15.Gururajan R, Grenet J, Lahti JM, et al. Isolation and characterization of two novel metalloproteinase genes linked to the Cdc2L locus on human chromosome 1p36.3. Genomics. 1998;52(1):101–106. doi: 10.1006/geno.1998.5401. [DOI] [PubMed] [Google Scholar]
- 16.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352(16):1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 17.Herman MP, Sukhova GK, Libby P, et al. Expression of neutrophil collagenase (matrix metalloproteinase-8) in human atheroma: a novel collagenolytic pathway suggested by transcriptional profiling. Circulation. 2001;104(16):1899–1904. doi: 10.1161/hc4101.097419. [DOI] [PubMed] [Google Scholar]
- 18.Hu JH, Touch P, Zhang J, et al. Reduction of mouse atherosclerosis by urokinase inhibition or with a limited-spectrum matrix metalloproteinase inhibitor. Cardiovasc Res. 2015;105(3):372–382. doi: 10.1093/cvr/cvv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishida Y, Migita K, Izumi Y, et al. The role of IL-18 in the modulation of matrix metalloproteinases and migration of human natural killer (NK) cells. FEBS Lett. 2004;569(1-3):156–160. doi: 10.1016/j.febslet.2004.05.039. [DOI] [PubMed] [Google Scholar]
- 20.Jefferis BJ, Papacosta O, Owen CG, et al. Interleukin 18 and coronary heart disease: prospective study and systematic review. Atherosclerosis. 2011;217(1):227–233. doi: 10.1016/j.atherosclerosis.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jefferis BJ, Whincup PH, Welsh P, et al. Prospective study of IL-18 and risk of MI and stroke in men and women aged 60–79 years: a nested case-control study. Cytokine. 2013;61(2):513–520. doi: 10.1016/j.cyto.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson JL, Devel L, Czarny B, et al. A selective matrix metalloproteinase-12 inhibitor retards atherosclerotic plaque development in apolipoprotein E-knockout mice. Arterioscler Thromb Vasc Biol. 2011;31(3):528–535. doi: 10.1161/ATVBAHA.110.219147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katsuda S, Kaji T. Atherosclerosis and extracellular matrix. J Atheroscler Thromb. 2003;10(5):267–274. doi: 10.5551/jat.10.267. [DOI] [PubMed] [Google Scholar]
- 24.Kuzuya M, Nakamura K, Sasaki T, et al. Effect of MMP-2 deficiency on atherosclerotic lesion formation in apoE-deficient mice. Arterioscler Thromb Vasc Biol. 2006;26(5):1120–1125. doi: 10.1161/01.ATV.0000218496.60097.e0. [DOI] [PubMed] [Google Scholar]
- 25.Lehrke M, Greif M, Broedl UC, et al. MMP-1 serum levels predict coronary atherosclerosis in humans. Cardiovasc Diabetol. 2009;8:50. doi: 10.1186/1475-2840-8-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Libby P, Ridker PM, Hansson GK, et al. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol. 2009;54(23):2129–2138. doi: 10.1016/j.jacc.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loftus IM, Naylor AR, Bell PR, et al. Plasma MMP-9–a marker of carotid plaque instability. Eur J Vasc Endovasc Surg. 2001;21(1):17–21. doi: 10.1053/ejvs.2000.1278. [DOI] [PubMed] [Google Scholar]
- 28.Luttun A, Lutgens E, Manderveld A, et al. Loss of matrix metalloproteinase-9 or matrix metalloproteinase-12 protects apolipoprotein E-deficient mice against atherosclerotic media destruction but differentially affects plaque growth. Circulation. 2004;109(11):1408–1414. doi: 10.1161/01.CIR.0000121728.14930.DE. [DOI] [PubMed] [Google Scholar]
- 29.Ma Y, Yabluchanskiy A, Hall ME, et al. Using plasma matrix metalloproteinase-9 and monocyte chemoattractant protein-1 to predict future cardiovascular events in subjects with carotid atherosclerosis. Atherosclerosis. 2014;232(1):231–233. doi: 10.1016/j.atherosclerosis.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mallat Z, Corbaz A, Scoazec A, et al. Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation. 2001;104(14):1598–1603. doi: 10.1161/hc3901.096721. [DOI] [PubMed] [Google Scholar]
- 31.Mallat Z, Heymes C, Corbaz A, et al. Evidence for altered interleukin (IL)-18 pathway in human heart failure. FASEB J. 2004;18(14):1752–1754. doi: 10.1096/fj.04-2426fje. [DOI] [PubMed] [Google Scholar]
- 32.Newby AC. Dual role of matrix metalloproteinases (matrixins) in in timal thickening and atherosclerotic plaque rupture. Physiol Rev. 2005;85(1):1–31. doi: 10.1152/physrev.00048.2003. [DOI] [PubMed] [Google Scholar]
- 33.Nilsson L, Jonasson L, Nijm J, et al. Increased plasma concentration of matrix metalloproteinase-7 in patients with coronary artery disease. Clin Chem. 2006;52(8):1522–1527. doi: 10.1373/clinchem.2006.067439. [DOI] [PubMed] [Google Scholar]
- 34.Pagidipati NJ, Gaziano TA. Estimating deaths from cardiovascular disease: a review of global methodologies of mortality measurement. Circulation. 2013;127(6):749–756. doi: 10.1161/circulationaha.112.128413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quiding-Jarbrink M, Smith DA, Bancroft GJ. Production of matrix metalloproteinases in response to mycobacterial infection. Infect Immun. 2001;69(9):5661–5670. doi: 10.1128/IAI.69.9.5661-5670.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reddy VS, Prabhu SD, Mummidi S, et al. Interleukin-18 induces EMMPRIN expression in primary cardiomyocytes via JNK/Sp1 signaling and MMP-9 in part via EMMPRIN and through AP-1 and NF-κB activation. Am J Physiol Heart Circ Physiol. 2010;299(4):H1242–H1254. doi: 10.1152/ajpheart.00451.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. 1999;340(2):115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 38.Siasos G, Tousoulis D, Kioufis S, et al. Inflammatory mechanisms in atherosclerosis: the impact of matrix metalloproteinases. Curr Top Med Chem. 2012;12(10):1132–1148. doi: 10.2174/1568026611208011132. [DOI] [PubMed] [Google Scholar]
- 39.Task Force M, Montalescot G, Sechtem U, et al. 2013 ESC guidelines on the management of stable coronary artery disease: the task force on the management of stable coronary artery disease of the European Society of Cardiology. Eur Heart J. 2013;34(38):2949–3003. doi: 10.1093/eurheartj/eht296. [DOI] [PubMed] [Google Scholar]
- 40.Tenger C, Sundborger A, Jawien J, et al. IL-18 accelerates atherosclerosis accompanied by elevation of IFN-γ and CXCL16 expression independently of T cells. Arterioscler Thromb Vasc Biol. 2005;25(4):791–796. doi: 10.1161/01.ATV.0000153516.02782.65. [DOI] [PubMed] [Google Scholar]
- 41.Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. J Am Coll Cardiol. 2012;60(16):1581–1598. doi: 10.1016/j.jacc.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 42.Wang J, Sun C, Gerdes N, et al. Interleukin 18 function in atherosclerosis is mediated by the interleukin 18 receptor and the Na-Cl co-transporter. Nat Med. 2015;21(7):820–826. doi: 10.1038/nm.3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu S, Chen J, Feder J, et al. 2007