

Abstract
Patients with Hermansky-Pudlak syndrome type 2 (HPS-2) have mutations in the β3A subunit of adaptor complex-3 (AP-3) and functional deficiency of this complex. AP-3 serves as a coat protein in the formation of new vesicles, including, apparently, the platelet's dense body and the melanocyte's melanosome. We used HPS-2 melanocytes in culture to determine the role of AP-3 in the trafficking of the melanogenic proteins tyrosinase and tyrosinase-related protein-1 (TRP-1). TRP-1 displayed a typical melanosomal pattern in both normal and HPS-2 melanocytes. In contrast, tyrosinase exhibited a melanosomal (i.e., perinuclear and dendritic) pattern in normal cells but only a perinuclear pattern in the HPS-2 melanocytes. In addition, tyrosinase exhibited a normal pattern of expression in HPS-2 melanocytes transfected with a cDNA encoding the β3A subunit of the AP-3 complex. This suggests a role for AP-3 in the normal trafficking of tyrosinase to premelanosomes, consistent with the presence of a dileucine recognition signal in the C-terminal portion of the tyrosinase molecule. In the AP-3–deficient cells, tyrosinase was also present in structures resembling late endosomes or multivesicular bodies; these vesicles contained exvaginations devoid of tyrosinase. This suggests that, under normal circumstances, AP-3 may act on multivesicular bodies to form tyrosinase-containing vesicles destined to fuse with premelanosomes. Finally, our studies demonstrate that tyrosinase and TRP-1 use different mechanisms to reach their premelanosomal destination.
INTRODUCTION
Adaptor protein complexes are components of organellar coats having the dual purpose of forming carrier vesicles and recruiting cargo to the newly created vesicles (Schekman and Orci, 1996; Robinson, 1997). Three adaptor complexes (AP-1, AP-2, and AP-3) have been recognized for several years and, recently, AP-4 has been identified (Dell'Angelica et al., 1999a; Hirst et al., 1999). Each of the four known adaptor complexes consists of four different subunits, and each subunit resembles in size and amino acid sequence its counterpart within the other adaptor complexes. One adaptor complex, AP-3, consists of a 160-kDa δ, a 120-kDa β, a 47-kDa μ, and a 22-kDa ς subunit. AP-1 is involved in sculpturing clathrin vesicles from the trans-Golgi network, or TGN, whereas AP-2 forms similar vesicles at the plasma membrane (Robinson, 1994). AP-4 appears to be associated with nonclathrin-coated vesicles at or near the TGN (Dell'Angelica et al., 1999a; Hirst et al., 1999). The function of AP-3 remains controversial. It has been suggested that AP-3 functions at the TGN (Simpson et al., 1996; Dell'Angelica et al., 1997a) and also within a late endosome-like compartment (Dell'Angelica et al., 1997a; Simpson et al., 1997). In addition, it has been demonstrated that AP-3 either does not (Simpson et al., 1996; Dell'Angelica et al., 1997b) or does (Dell'Angelica et al., 1998) interact with clathrin.
Whatever role AP-3 plays in generic cells, it appears to exert critical functions in pigment-producing cells. When the δ subunit of AP-3 is altered in Drosophila, the product is the mutant fly garnet (Ooi et al., 1997; Simpson et al., 1997), whose ommatidia contain pigment cells with abnormal or absent pigment granules. Similarly, mutations in μ3, ς3, and β3A create the carmine (Mullins et al., 1999), orange (Mullins et al., 1997), and ruby (Kretzschmar et al., 2000) eyes, respectively. In the mouse, mutation of δ produces a poorly balanced and hypopigmented animal called mocha (Kantheti et al., 1998). When the β3A subunit of AP-3 is mutated, the resulting mouse, called pearl, also lacks normal pigmentation (Feng et al., 1999). Moreover, both mocha and pearl lack the platelet dense bodies responsible for a normal secondary platelet aggregation response. The combination of a storage pool deficiency and hypopigmentation places mocha and pearl among the 14 known murine models (Swank et al., 1998) for a human disorder called Hermansky-Pudlak syndrome, or HPS (Hermansky and Pudlak, 1959; Shotelersuk and Gahl, 1998). This autosomal recessive disease consists of oculocutaneous albinism (Witkop et al., 1990; King et al., 1995; Gahl et al., 1998), a platelet storage pool deficiency due to absent dense bodies (Witkop et al., 1987), and accumulation of a lipid-protein complex called ceroid lipofuscin (Sakuma et al., 1995). In 1996, the gene HPS-1 was identified as a cause of HPS (Oh et al., 1996) and subsequently demonstrated to be responsible for the melanocyte dysfunction (Boissy et al., 1998), but the disorder is clearly genetically heterogeneous (Hazelwood et al., 1997; Oh et al., 1998). In fact, in 1999 two brothers with documented HPS were reported to express mutations in the β3A subunit of AP-3 (Dell'Angelica et al., 1999b; Shotelersuk et al., 2000); their disorder is called HPS-2.
These findings suggest that AP-3 functions not only in the formation of or recycling through late endosomes and platelet dense bodies, but also in the pigment-forming process that occurs within the melanosomes of melanocytes. The current dogma of melanogenesis holds that premelanosomes form from the smooth endoplasmic reticulum and later fuse with microvesicles containing tyrosinase, tyrosinase-related protein-1 (TRP-1), pmel17, and other components of the melanogenic pathway (Novikoff et al., 1968; Chakraborty et al., 1989; Zhou et al., 1993). The convergence of the premelanosomes and microvesicles permits the resulting melanosomes to progressively acquire melanin pigment as they mature and travel to melanocytic dendrites for transfer to keratinocytes (King et al., 1995). The microvesicles destined to fuse with premelanosomes are thought to bud from the TGN and possibly recycle through the late endosome (Setaluri, 2000). Because the tyrosinase molecule contains a dileucine-targeting signal motif specific for the μ3 subunit of AP-3 (Ohno et al., 1998), this enzyme may be cargo transported by an AP-3–mediated mechanism from the TGN/endosome into microvesicles (Höning et al., 1998). It has recently been demonstrated that this dileucine signal within the cytoplasmic tail of human tyrosinase can regulate its targeting to synapse-like microvesicles in a brefeldin A-sensitive AP3-dependent manner when transfected into PC12 cells (Blagoveshchenskaya et al., 1999).
We investigated the role of AP-3 in the process of melanogenesis by examining the localization of tyrosinase and TRP-1 in normal and HPS-2 melanocytes in culture. In normal cells, tyrosinase and TRP-1 colocalize in a granular pattern throughout the entire cell. In HPS-2 melanocytes, tyrosinase is confined to the perinuclear area of the melanocyte within structures resembling multivesicular bodies and/or late endosomes, whereas TRP-1 localization was normal. Transfection of HPS-2 cells with the cDNA encoding the β3A subunit of AP-3 normalized tyrosinase localization. Therefore, in HPS-2 melanocytes, a striking discordance between the localization of tyrosinase and TRP-1 was found, indicating that these proteins, each critical for melanin production, reach the melanosome by at least two different mechanisms.
MATERIALS AND METHODS
Patients
The patients involved in this study were enrolled in a protocol approved by the National Institute of Child Health and Human Development Institutional Review Board and provided written informed consent. HPS was diagnosed on the basis of oculocutaneous albinism and the absence of platelet dense bodies on electron microscopy. The clinical characteristics of the two patients with HPS-2, i.e., mutations in the β3A subunit of AP-3, have been previously described (Shotelersuk et al., 2000). In addition, melanocyte cultures developed from patients with oculocutaneous albinism type 1 and type 3, having mutations eliminating the expression of tyrosinase (Zhao and Boissy, 1994) and TRP-1 (Boissy et al., 1996), respectively, were utilized.
Cell Culture
Human melanocytes and fibroblasts were established from normal neonatal foreskins that were obtained from the nursery of University Hospital (Cincinnati, OH) after routine circumcision and from a 4-mm punch biopsy obtained from the two patients with HPS-2. Skins were incubated in 0.25% trypsin for 2 h at 37°C. The tissue was vortexed for 30 s to separate the dermis from the epidermal cell suspension. The dermis was placed in a 25-cm2 tissue culture flask with growth medium to establish fibroblasts. The growth medium consisted of minimal essential medium (GIBCO-BRL, Grand Island, NY) with 10% fetal bovine serum and 1% antibiotic/antimycotic solution. The epidermal cells were pelleted by centrifugation and resuspended in melanocyte growth medium. Melanocyte growth medium consisted of M154 basal medium (Cascade Biologicals, Portland, OR) supplemented with 4% heat-inactivated fetal bovine serum, 1% antibiotic/antimycotic solution (GIBCO-BRL), 1 μg/ml transferrin, 1 μg/ml vitamin E, 5 μg/ml insulin, 0.6 ng/ml human recombinant basic fibroblast growth factor, 10−9 M endothelin-1, and 10−8 M α-melanocyte stimulating hormone, from Sigma Chemical (St. Louis, MO) unless indicated differently. All cultures were maintained in a tissue culture incubator at 37°C with 5% CO2. Growth media were routinely changed twice each week.
Antisera
Mouse monoclonal antiserum to lysosome-associated membrane protein-3 (LAMP-3 or CD-63) and LAMP-1 were obtained from the Developmental Studies Hybridoma Bank (Iowa City, IA), mouse monoclonal antiserum to TRP-1 (or Mel-5) was purchased from Signet Laboratories (Dedham, MA), and mouse monoclonal antiserum to μ3 (p47A) and ς3 were obtained from Transduction Laboratories (Lexington, KY). Rabbit polyclonal antiserum to tyrosinase was generously provided by R. King and W.S. Oetting (University of Minnesota, Minneapolis). Antisera to the β3A and δ3 subunits of AP-3 and to the γ subunit of AP-1 were obtained from A. Theos and M.S. Robinson (Cambridge Institute for Medical Research, Cambridge, Great Britain).
Immunofluorescence Studies
Human fibroblast or melanocyte monolayers were seeded onto gelatin-coated eight-well Lab-Tek chamber slides (Nunc, Naperville, IL). After 24 h, cells were fixed for 10 min in 2% formaldehyde in phosphate-buffered saline (PBS).
Cells were incubated for 1 h at room temperature in a mixture of mouse monoclonal and rabbit polyclonal antibodies (diluted in PBS, 0.2% saponin [wt/vol], 0.1% bovine serum albumin [wt/vol]). After the incubation, unbound antibodies were removed by washing with PBS three times for 5 min. Cells were then incubated for 30 min with a mixture of secondary antibodies: Cy-2–conjugated donkey anti-mouse and Cy-3–conjugated donkey anti-rabbit (Jackson Immunoresearch, West Grove, PA). After washing the cells three times for 5 min in PBS, the cells were mounted onto glass slides with Fluoromont G (Southern Biotechnologies, Birmingham, AL).
Microscopy was performed on an Axioscop-20 or an LSM 510 upright confocal microscope (Zeiss, Oberkochen, Germany). In the latter case, the fluorochromes were excited with appropriate wavelength argon laser light. Emitted light was filtered with a 505-nm pass filter. The pixel dimensions were 0.17 × 0.17 mm, and the optical section thickness was 3.8 μm.
Western Blotting
Proteins in the cell extracts were separated by SDS-PAGE, with the use of 4–20% gradient precast gels (Novex, San Diego, CA), and electroblotted onto 0.2-μm nitrocellulose membranes (Schleicher & Schuell, Keene, NH). Nonspecific sites on the membrane were blocked by incubation in 5% (wt/vol) nonfat dry milk in PBS, 0.1% Tween. The membranes were incubated with primary antiserum diluted in PBS containing 0.1% Tween, followed by incubation with horseradish peroxidase-conjugated secondary antibodies (Amersham Pharmacia Biotech, Piscataway, NJ). Detection was performed with the enhanced chemiluminescence (ECL) system, according to the manufacturer's instructions (Amersham Pharmacia Biotech).
Electron Microscopy
Melanocytes were seeded onto gelatin-coated eight-well Lab-Tek chamber slides. After 24 h, cells were fixed in wells with half-strength Karnovsky's fixative (Karnovsky, 1965) in 0.2 M sodium cacodylate buffer at pH 7.2 for 30 min at room temperature. The cells were washed three times in buffer and in some cases incubated in a 0.1% solution of dihydroxyphenylalanine (l-DOPA) for 2 h twice at 37°C for the histochemical identification of tyrosinase. Cells were then postfixed with 1% osmium tetroxide containing 1.5% potassium ferrocyanide (Karnovsky, 1971) for 30 min. The cells were washed, stained en bloc with 0.5% uranyl acetate for 30 min, dehydrated, and embedded in Eponate 12. Cells were sectioned on an RMC, Inc. (Tucson, AZ) MT 6000-XL ultramicrotome, stained with aqueous solutions of uranyl acetate (2%) and lead citrate (0.3%) for 15 min each, and then viewed and photographed in a JEM-100CX transmission electron microscope (JEOL, Tokyo, Japan). All tissue processing supplies were purchased from Ted Pella, Inc. (Tustin, CA).
Immunocytochemistry/Electron Microscopy
Melanocytes were seeded onto gelatin-coated eight-well Lab-Tek chamber slides as described above. After 24 h, cells were washed in PBS for 5 min and then fixed in cold one-quarter-strength Karnovsky's fixative at 4°C for 5 min. Cells were then treated with 0.1% glycine in PBS for 5 min, permeablized with 0.02% Triton X-100 (in distilled water) for 1 min, and incubated for 30 min in 5% dry milk/1% bovine serum albumin A in distilled water. Cells were incubated in nonimmune blocking serum from the Vectastain ABC kit (Vector Laboratories, Burlingame, CA) for 30 min and then in Mel-5 (1:100) at room temperature for 1 h. Cells were processed with the Vectastain Elite ABC kit according to the manufacturer's instructions; the diaminobenzidine tetrahydrochloride incubation period lasted 10 min. Cells were then processed for electron microscopy as described above except that sections were stained with Reynold's alkaline lead only.
Transfection of HPS-2 Melanocytes
The complete cDNA encoding the β3A subunit of the human AP-3 complex was inserted into the EcoRV/XhoI site in pcDNA3.1(+) expression vector (Invitrogen, Carlsbad, CA). After confirmation of the presence of insert by restriction digest and sequencing, the plasmid was utilized for transfection into cultures of HPS-2 melanocytes. Briefly, confluent cultures of melanocytes were transfected with either vector alone (pcDNA3.1[+]) or the β3A subunit-containing construct (pcDNA[β3A]) with the use of Effectene reagent according to the manufacturer's protocols with modifications (Qiagen, Valencia, CA). The pcDNA(+) and pcDNA(β3A) were incubated with the appropriate reagents from the Effectene transfection kit and added to the melanocyte cultures. Selection with G418 (0.5 mg/ml) was started 72 h after initial incubation. After 3 d of selection, the medium was changed and cells allowed to recover for 2 d. The melanocyte cultures were maintained in growth medium without G418 for 5 d. To confirm successful transfection and protein expression, an aliquot of cells was used for immunohistochemical analysis.
RESULTS
In humans, deficiency of the β3A subunit of AP-3 has been demonstrated only in cultured fibroblasts (Dell'Angelica et al., 1999b). Yet, the phenotypic expression of the clinical disorder, HPS-2, involves dysfunction of platelets and melanocytes (Shotelersuk et al., 2000). Consequently, we investigated whether melanocytes also exhibit a deficiency of β3A. As previously demonstrated (Dell'Angelica et al., 1999b), immunofluorescence studies of normal fibroblasts revealed a typical lysosomal distribution for both LAMP-3 and the β3A subunit of AP-3 (Figure 1, top). Normal melanocytes also displayed a fluorescent signal for the LAMP-3 and β3A antibodies (Figure 1, middle and bottom). However, although HPS-2 fibroblasts and cultured melanocytes exhibited a normal amount and pattern of LAMP-3 expression, each of these cell types manifested a marked deficiency of β3A expression (Figure 1).
Figure 1.

LAMP-3 and β3A expression in normal and HPS-2 cells. Fibroblasts (A–D) and melanocytes (E–H) were fixed on coverslips and stained with mouse monoclonal antibodies to LAMP-3 (A, C, E, and G) or rabbit polyclonal antibodies to β3A (B, D, F, and H). In each normal cell type, the normal distribution patterns of LAMP-3 (green in A and E) and β3A (red in B and F) are shown. In HPS-2 cells, by comparison, the LAMP-3 pattern was normal (green in C and G) but β3A immunofluorescence was significantly reduced (red in D and H). Arrows in G and H denote the site of minimal β3A expression in the HPS-2 melanocytes.
In fibroblasts, deficiency of β3A was accompanied by deficiency of the other three AP-3 subunits, presumably because the AP-3 complex fails to form and stabilize its components (Dell'Angelica et al., 1999b). We now demonstrate that Western blots also showed markedly reduced levels of the other AP-3 subunits in cultured melanocytes from an HPS-2 patient that were compared with normal (Figure 2).
Figure 2.

Western blots of AP-3 subunits in melanocytes and fibroblasts. Protein extracts (25 mg) of cultured melanocytes and fibroblasts were electrophoresed on a 4–20% polyacrylamide gel, blotted onto nitrocellulose, and treated with antibodies to β3, μ3, ς3, δ3, and Lamp-1. N-1, N-2, normal cell extract from two different individuals; Pt, HPS-2 cell extract. Expression of β3, μ3, ς3, and δ3 was deficient in HPS-2 fibroblasts and melanocytes; Lamp-1 was normal for the patient and demonstrates a comparable amount of protein loaded in each lane.
These experiments confirm that HPS-2 melanocytes in culture express the β3A subunit deficiency, as expected, and can be used to study the effects of the absence of AP-3 upon protein trafficking. Subsequent investigations focused on two pivotal enzymes in melanogenesis, i.e., tyrosinase and TRP-1. These proteins are targeted to melanosomes and, indeed, tyrosinase and TRP-1 trafficked to the melanosomes in normal melanocytes. However, the relative amounts of tyrosinase/TRP-1 expression per melanocyte are not always equal, as illustrated by the immunofluorescence studies (Figure 3, left column). In HPS-2 melanocytes, TRP-1 displayed a normal, melanosomal distribution, with fluorescence appearing throughout the dendrites as well as in the perinuclear region (Figure 3, top, right). Normal localization of TRP-1 to the melanosomes in HPS-2 melanocytes was confirmed by immunocytochemical findings at the ultrastructural level (Figure 4). Tyrosinase, however, appeared mainly around the nucleus and was markedly absent from the cell periphery. Double-exposure fluorescence studies revealed the lack of colocalization of tyrosinase and TRP-1 in the HPS-2 melanocytes' dendrites, indicating that the absence of functional AP-3 prevented tyrosinase from entering and traveling with melanosomes.
Figure 3.

TRP-1 and tyrosinase distribution in normal and HPS-2 melanocytes. Melanocyte monolayers were fixed in 2% formaldehyde and stained with anti–TRP-1 monoclonal (green) and anti-tyrosinase polyclonal (red) antibodies. In normal melanocytes (A–C), TRP-1 (A) and tyrosinase (B) exhibited a similar distribution throughout the melanocyte (C = merged image). In HPS-2 cells (D–F), TRP-1 (D) had a normal distribution, but tyrosinase (E) was localized mainly in the perinuclear area and was markedly absent from the cell periphery (arrowheads) (F = merged image).
Figure 4.

TRP-1 is present within the melanosomes of HPS-2 melanocytes. Cultured HPS-2 (A), OCA-1 (B), and OCA-3 (C) melanocytes, containing predominantly hypomelanized melanosomes, were processed for the immunocytochemical localization of TRP-1 with the use (top row) or omission (bottom) of the MEL-5 primary antibody. Peroxidase reaction product occurred in melanosomes (arrows) of the HPS-2 melanocytes (A), null for β3A, and the OCA-1 melanocytes (B), null for tyrosinase. In contrast, peroxidase reaction product did not occur in melanosomes (arrowheads) of the OCA-3 melanocytes (C), null for TRP-1 and all melanocyte types not processed with Mel 5 primary antibody (bottom). Bar, 0.5 μm.
To further assess the distribution pattern of tyrosinase in HPS-2 melanocytes, we used DOPA histochemistry at the ultrastructural level. Tyrosinase activity was markedly reduced in most of the melanosomes throughout the cell body and dendrites of HPS-2 melanocytes (Figure 5). Approximately, 40–80% of the melanosomes within the dendrites of a melanocyte lacked tyrosinase and morphologically resembled stage I melanosomes. These organelles contained much floccular material and melanofilament-like structures. However, mature functional tyrosinase, as revealed by DOPA histochemistry, was normally trafficked through and exited from the Golgi apparatus (Figure 6). Reaction product in the trans-most cisternae and coated vesicles of the TGN appeared normal in HPS-2 melanocytes. Predominant in the AP-3–deficient HPS-2 melanocyte was the prevalence of multivesiculated bodies/late endosome-like structures that contained tyrosinase in unusual aggregates (Figure 7). These abundant structures contained multiple exvaginations of the limiting membrane lacking reaction product/tyrosinase.
Figure 5.

HPS-2 versus control melanocytes contain many melanosomes devoid of tyrosinase. Cultured HPS-2 (A) and control (B) melanocytes were processed for DOPA histochemistry to localize catalytically functional tyrosinase. Many of the melanosomes in the HPS-2 melanocytes remained amelanotic (arrowheads), indicating that they lacked tyrosinase, whereas those in the control melanocytes were heavily pigmented. However, in the HPS-2 melanocytes, reaction product and thus tyrosinase was apparent in some melanosomes (arrows) and in an occasional larger multivesiculated body/late endosome-like vesicles (open arrows). Bar, 2.0 μm.
Figure 6.

HPS-2 melanocytes exhibit a normal tyrosinase profile in the TGN. Cultured control (A) and HPS-2 (B) melanocytes were processed for DOPA histochemistry. In both cell types, reaction product was similarly present in the trans-most cisternae of the Golgi apparatus (arrows) and in coated vesicles budding off (arrowheads) and free in the vicinity of the TGN. Bar, 0.5 μm.
Figure 7.

Late endosome-like structures are prevalent in HPS-2 melanocytes. Cultured control (A) and HPS-2 (B and C) melanocytes were processed for DOPA histochemistry. (A) Multivesicular bodies with minimal (1) or no (2) reaction product were occasionally present in control melanocytes. (B) In contrast, multivesicular bodies with much reaction product (arrows) were abundant in HPS-2 melanocytes. (C) The HPS-2 multivesiculated bodies exhibited finger-like protrusions of their limiting membranes (1) and the DOPA positive reaction product appeared as aggregates (2), and/or within vesicles (3). Bars: A and B, 0.4 μm; C, 0.25 μm.
HPS-2 melanocytes were subsequently transfected with a cDNA encoding the β3A subunit of AP-3 (Figure 8). Tyrosinase localization in HPS-2 melanocytes transfected with mock cDNA remained restricted to the perinuclear area (Figure 8C). In contrast, tyrosinase acquired normal localization throughout the cell body and dendrites of HPS-2 melanocytes successfully transfected with the β3A (Figure 8D). In addition, the expression of the δ subunit of AP-3 was up-regulated in HPS-2 melanocytes transfected with β3A (Figure 8, E and F).
Figure 8.
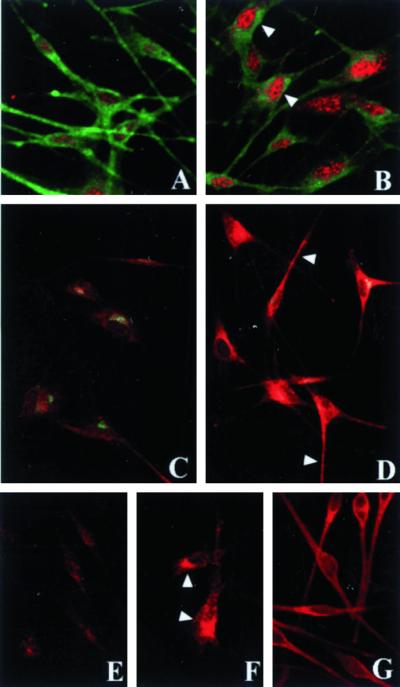
Tyrosinase localization on HPS-2 melanocytes transfected with the cDNA encoding the β subunit of AP-3. Cultured HPS-2 melanocytes were transfected with vector alone (A, C, and E) or the cDNA encoding the β subunit of AP-3 (B, D, and F). Selected transfectants were immunocytochemically processed for colocalization of the β3A subunit of AP-3 (red) and TRP-1 (green) (A and B); colocalization of the γ subunit of AP-1 (green) and tyrosinase (red) (C and D); and localization of the δ subunit of AP-3 (red) (E and F); G shows normal human melanocytes immunocytochemically processed for the localization of tyrosinase (red). Both the β3A (arrowheads in B) and the δ subunits of AP-3 (arrowheads in F) were up-regulated after transfection of the β3A subunit of AP-3. Concomitantly, expression of tyrosinase was normalized throughout the dendrites in the HPS-2 cells transfection with the β3A subunit of AP-3 (arrowheads in D) resembling tyrosinase expression in normal human melanocytes (G). Expression of both TRP-1 (green in A vs. B) and the γ subunit of AP-1 (green in C vs. D) were unaffected by transfection.
DISCUSSION
Adaptor protein complexes function to create new vesicles from extant membranes and to select cargo proteins that will be constituents of the newly formed vesicles. AP-3 was originally considered to serve this function at the TGN and to target proteins to the late endosome or lysosome (Simpson et al., 1996, 1997; Dell'Angelica et al., 1997a). However, AP-3 may also be localized to a more distal compartment, perhaps the late endosome (Dell'Angelica et al., 1997a; Simpson et al., 1997; Faundez and Kelly, 2000). In yeast, deficiency of any of the AP-3 subunits causes failure of alkaline phosphatase and Vam3p, a t-SNARE, to reach the vacuole (Cowles et al., 1997; Darsow et al., 1998). In mice, mutations in the β3A subunit of AP-3 produce the pearl mouse, with hypopigmentation and platelet storage pool deficiency (Feng et al., 1999; Zhen et al., 1999). The human correlate of pearl occurs in patients with HPS-2, a disorder manifest in two brothers having mild oculocutaneous albinism, absent platelet dense bodies, and neutropenia (Dell'Angelica et al., 1999b; Shotelersuk et al., 2000). These individuals have compound heterozygous mutations in the β3A subunit of AP-3, resulting in a 21-amino acid deletion (D390-410) and an amino acid substitution (L580R). Fibroblasts cultured from these patients have a reduction in all four AP-3 subunits and exhibit enhanced internalization of the lysosomal integral membrane proteins CD63, LAMP-1, and LAMP-2 at the plasma membrane (LeBorgne et al., 1998; Dell'Angelica et al., 1999b), indicating a role for AP-3 in the normal trafficking of these proteins. Nevertheless, the ultimate steady-state levels and distribution of an integral lysosomal membrane protein such as CD63 (LAMP-3) appears normal in AP-3–deficient cells (Figure 1), either because default trafficking by the plasma membrane (Dell'Angelica et al., 1999b) has delivered a normal contingent of LAMP-3 to the lysosomal membrane or because residual AP-3 has carried sufficient LAMP-3 to the lysosome.
Although AP-3–deficient fibroblasts express an abnormal phenotype in vitro, more striking in vivo manifestations of β3A subunit deficiency are caused by abnormal pigment production by melanocytes. This process depends on a series of melanogenetic enzymes whose trafficking to the melanosome can be assessed in melanocytes cultured from HPS-2 patients. Indeed, HPS-2 melanocytes, like fibroblasts, do exhibit a deficiency of the β3A subunit of AP-3 (Figures 1 and 8). Moreover, the μ, ς, and δ subunits of AP-3 are markedly deficient in HPS-2 melanocytes (Figures 2 and 8), presumably because they are not stabilized in a complete AP-3 complex because of absence of the β3A subunit.
The μ3 subunit of AP-3 is recognized to interact with cargo proteins by virtue of a dileucine motif in the cytoplasmic tail of such proteins. Tyrosinase has an appropriate dileucine motif, and studies with the use of yeast 2-hybrids (Dell'Angelica et al., 1997a) and surface plasmon resonance (Höning et al., 1998) have demonstrated that μ3A and tyrosinase interact with each other. However, it is not the dileucine residue alone that determines which proteins are recognized by the μ subunit of AP-3. In LIMP II, for example, the acidic amino acid residues Asp470 and Glu471, located four and five residues proximal to the dileucine motif, are critical for binding to AP-3 (Höning et al., 1998; Tabuchi et al., 2000), as well as for targeting to lysosomes (Pond et al., 1995). Tyrosinase and LIMP II share the same D(E)ERXP amino acid sequence preceding the dileucine signal, and the two acidic amino acids in this motif appear essential for binding to AP-3. Moreover, mutagenesis of two leucine residues within the dileucine-signaling motif of tyrosinase has been shown to prevent late endosomal localization in HeLa cells transfected with the construct (Calvo et al., 1999). Binding to AP-3 specifically has been abrogated in nature on at least two occasions. First, when the cytoplasmic tail is truncated at residue −10 in the murine platinum allele of tyrosinase, the dileucine motif is lost, tyrosinase is misrouted, and oculocutaneous albinism results (Beerman et al., 1995). Second, when β3A mutations result in deficiency of μ3 in humans with HPS-2, tyrosinase is misrouted in melanocytes (Figures 3 and 7), and an oculocutaneous albinism typical of HPS results (Dell'Angelica et al., 1999b; Shotelersuk et al., 2000). This misrouting of tyrosinase can be rescued with transfection of the β3A subunit (Figure 8).
The amino acid sequence of TRP-1 surrounding its dileucine signal differs from that of tyrosinase. Instead of having two acidic amino acids in the −5 and −4 positions relative to the dileucines, TRP-1 has a single acidic residue. This may account for the failure of AP-3 deficiency to disturb the normal distribution of TRP-1 in HPS-2 melanocytes (Figures 3 and 8), in marked contrast to the effects on tyrosinase. In fact, Höning et al. (1998) demonstrated that, when the −5 amino acid of LIMP II is substituted by an A, as occurs naturally in TRP-1, interaction with AP-3 is abrogated.
These studies of normal and HPS-2 melanocytes offer a fuller understanding of the movement of melanogenic proteins within these cells. Tyrosinase appears to move from the TGN to premelanosomes by a vesicle-mediated path (Maul, 1969; Chakraborty et al., 1989). Coated vesicles containing tyrosinase have been shown to form at the trans-most cisternea of the Golgi, and 50-nm vesicles containing tyrosinase eventually fuse with stage 1 premelanosomes. It remains unknown whether this route of translocation between the TGN and the premelanosome is direct or requires an intervening structure, such as the late endosome or multivesicular sorting body (Setaluri, 2000). The intervening structures through which tyrosinase could transit may also represent the recently described hybrid organelles (Luzio et al., 2000). Lysosomes can fuse directly with late endosomes forming a hybrid organelle from which lysosomes are then reformed by a process involving condensation of contents (Bright et al., 1997; Mullock et al., 1998). Structures morphologically resembling late endosome/multivesicular bodies, particularly those containing reaction product after DOPA histochemistry, are relatively rare in normal melanocytes (Figure 7). This suggests that, if tyrosinase is shuttled through an intervening structure, this transit occurs rapidly.
In the β3A-deficient HPS-2 melanocytes, tyrosinase was present in the trans-most cisternea of the Golgi as well as in nearby coated vesicles (Figure 6), indicating that a coat protein complex other than AP-3 functions in the formation of cargo vesicles at this site in the melanocyte. This is consistent with localization studies in normal melanocytes (Figure 1), which demonstrated a distribution pattern of AP-3 extending beyond the immediate perinuclear/Golgi area.
In contrast to its normal appearance in the TGN and coated vesicles of HPS-2 melanocytes, tyrosinase apparently does not efficiently reach premelanosomes (Figures 5 and 7). Instead, structures resembling late endosomes/multivesicular bodies are prevalent and contain abundant DOPA reaction product, i.e., tyrosinase. However, the structures also contain exvaginations lacking tyrosinase. In the absence of a functional AP-3 complex, these exvaginations may represent vesicles aborted or delayed in their formation and lacking both tyrosinase cargo and clathrin. Consequently, they would be unable to form a budding transport vesicle destined for the premelanosome. The exact role of AP-3 in this putative process has not been determined and requires further investigation.
However, some tyrosinase successfully reaches the melanosome, as demonstrated by the few melanized melanosomes present in HPS melanocytes. This could result from the inadvertent fusion and incorporation of Golgi-derived vesicles containing tyrosinase with premelanosomes in the vicinity. It is also possible that this results from an alternate pathway for trafficking tyrosinase that is not AP-3 dependent. This could parallel the situation of the trafficking of synaptic vesicle proteins, which can apparently arrive into the synaptic vesicle via the plasma membrane (AP-2–mediated, brefeldin A-insensitive route) or the endosome (AP-3-mediated, brefeldin A-sensitive route) (Blagoveshchenskaya and Cutler, 2000). Similarly, it has been demonstrated that lysosomal enzyme transport from the Golgi to the lysosome can occur via the cell surface (Braun et al., 1989). This is conceivable in light of the fact that melanosomes and lysosomes appear to have a common lineage (Orlow, 1995). An alternative route of tyrosinase trafficking would also be consistent with observations made in melanocytes from HPS1 (Boissy et al., 1998) and Chediak-Higashi syndrome (Zhao et al., 1994), in which some tyrosinase reaches the melanosome whereas most is targeted away from the melanosome or secreted from the melanocyte, respectively.
We conclude that the AP-3 complex mediates tyrosinase trafficking in human melanocytes, that the location of this process is distal to the TGN and coated vesicle formation, and that TRP-1 reaches the premelanosome by a process not requiring functional AP-3. We speculate that AP-3 acts on late endosomes or multivesicular bodies to form tyrosinase-containing vesicles which subsequently fuse with premelanosomes in the melanogenic pathway.
ACKNOWLEDGMENTS
This work was supported in part by Grant AR-45429 fro the National Institutes of Health (to R.E.B.).
REFERENCES
- Beerman F, Orlow SJ, Boissy RE, Schmidt A, Boissy YL, Lamoreux ML. Misrouting of tyrosinase with a truncated cytoplasmic tail as a result of the murine platinum cp mutation. Exp Eye Res. 1995;61:599–607. doi: 10.1016/s0014-4835(05)80053-3. [DOI] [PubMed] [Google Scholar]
- Blagoveshchenskaya AD, Cutler DF. Sorting to synaptic-like microvesicles from early, and late endosomes requires overlapping but not identical targeting signals. Mol Biol Cell. 2000;11:1801–1814. doi: 10.1091/mbc.11.5.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagoveshchenskaya AD, Hewitt EW, Cutler DF. Di-leucine signals mediate targeting of tyrosinase and synaptotagmin to synaptic-like microvesicles with PC12 cells. Mol Biol Cell. 1999;10:3979–3990. doi: 10.1091/mbc.10.11.3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissy RE, Zhao H, Oetting WS, Austin LM, Wildenberg SC, Boissy YL, Zhao Y, Sturm RA, Hearing VJ, King RA, Nordlund JJ. Mutation in and lack of expression of tyrosinase related protein-1 (TRP-1) in melanocytes from an individual with brown oculocutaneous albinism: a new subtype of albinism classified as OCA3. Am J Hum Genet. 1996;58:1145–1156. [PMC free article] [PubMed] [Google Scholar]
- Boissy RE, Zhao Y, Gahl WA. Altered protein localization in melanocytes from Hermansky-Pudlak syndrome: support for the role of the HPS gene product in intracellular trafficking. Lab Invest. 1998;78:1037–1048. [PubMed] [Google Scholar]
- Braun M, Wahead A, von Figura K. Lysosomal acid phosphatase is transported to lysosomes via the cell surface. EMBO J. 1989;8:3633–3640. doi: 10.1002/j.1460-2075.1989.tb08537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright NA, Reaves BJ, Mullock BN, Luzio JP. Dense core lysosomes can fuse with late endosomes and are reformed from the resultant hybrid organelles. J Cell Sci. 1997;110:2027–2040. doi: 10.1242/jcs.110.17.2027. [DOI] [PubMed] [Google Scholar]
- Calvo PA, Frank DW, Bieler BM, Berson JF, Marks MS. A cytoplasmic sequence in human tyrosinase defines a second class of di-leucine-based sorting signals for late endosomal and lysosomal delivery. J Biol Chem. 1999;274:12780–12789. doi: 10.1074/jbc.274.18.12780. [DOI] [PubMed] [Google Scholar]
- Chakraborty AK, Mishima Y, Inazu M, Hatta S, Ichihashi M. Melanogenic regulatory factors in coated vesicles from melanoma cells. J Invest Dermatol. 1989;93:616–620. doi: 10.1111/1523-1747.ep12319736. [DOI] [PubMed] [Google Scholar]
- Cowles CR, Odorizzi G, Payne GS, Emr SD. The AP-3 adaptor complex is essential for cargo-selective transport to the yeast vacuole. Cell. 1997;91:109–118. doi: 10.1016/s0092-8674(01)80013-1. [DOI] [PubMed] [Google Scholar]
- Darsow T, Burd CG, Emr SC. Acidic di-leucine motif essential for AP-3-dependent sorting and restriction of the functional specificity of the Vam3p vacuolar tSNARE. J Cell Biol. 1998;142:913–922. doi: 10.1083/jcb.142.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Angelica EC, Klumperman J, Stoorvogel W, Bonifacino JS. Association of the AP-3 adaptor complex with clathrin. Science. 1998;280:431–434. doi: 10.1126/science.280.5362.431. [DOI] [PubMed] [Google Scholar]
- Dell'Angelica EC, Mullins C, Bonifacino JS. AP-4, a novel protein complex related to clathrin adaptors. J Biol Chem. 1999a;274:7278–7285. doi: 10.1074/jbc.274.11.7278. [DOI] [PubMed] [Google Scholar]
- Dell'Angelica EC, Ohno H, Ooi CE, Rabinovich E, Roche KW. AP-3: an adaptor-like protein complex with ubiquitous expression. EMBO J. 1997a;16:917–928. doi: 10.1093/emboj/16.5.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Angelica EC, Ooi CE, Bonifacino JS. β3A-adaptin, a subunit of the adaptor-like complex AP-3. J Biol Chem. 1997b;272:15078–15084. doi: 10.1074/jbc.272.24.15078. [DOI] [PubMed] [Google Scholar]
- Dell'Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the β3A subunit of the AP-3 adaptor. Mol Cell. 1999b;3:11–21. doi: 10.1016/s1097-2765(00)80170-7. [DOI] [PubMed] [Google Scholar]
- Faundez VV, Kelly RB. The AP-3 complex required for endosomal synaptic vesicle biogenesis is associated with a casein kinase Ia-like isoform. Mol Biol Cell. 2000;10:2591–2604. doi: 10.1091/mbc.11.8.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Seymour AB, Jiang S, To A, Peden AA, Novak EK, Zhen L, Rusiniak ME, Eicher EM, Robinson MS, Gorin MB, Swank RT. The β3A subunit gene (Ap3b1) of the AP-3 adaptor complex is altered in the mouse hypopigmentation mutant pearl, a model for Hermansky Pudlak syndrome and night blindness. Hum Mol Genet. 1999;8:323–330. doi: 10.1093/hmg/8.2.323. [DOI] [PubMed] [Google Scholar]
- Gahl WA, Brantly M, Kaiser-Kupfer MI, Iwata F, Hazelwood S, Shotelersuk V, Duffy LF, Kuehl EM, Troendle I, Bernardini I. Genetic defects and clinical characteristics of patients with a form of oculocutaneous albinism (Hermansky-Pudlak syndrome) N Engl J Med. 1998;338:1258–1264. doi: 10.1056/NEJM199804303381803. [DOI] [PubMed] [Google Scholar]
- Hazelwood S, Shotelesuk V, Wildenberg SC, Chen D, Iwata F, Kaiser-Kupfer MI, White JG, King RA, Gahl WA. Evidence for locus heterogeneity in Puerto Ricans with Hermansky-Pudlak syndrome. Am J Hum Genet. 1997;61:1088–1094. doi: 10.1086/301611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermansky F, Pudlak P. Albinism associated with hemorrhagic diathesis and unusual pigmented reticular cells in the bone marrow: report of two cases with histochemical studies. Blood. 1959;14:162–169. [PubMed] [Google Scholar]
- Hirst J, Bright NA, Rous B, Robinson MS. Characterization of a fourth adaptor-related protein complex. Mol Biol Cell. 1999;10:2787–2802. doi: 10.1091/mbc.10.8.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höning S, Sandoval IV, von Figura K. A di-leucine-based motif in the cytoplasmic tail of LIMP-II and tyrosinase mediates selective binding of AP-3. EMBO J. 1998;17:1304–1314. doi: 10.1093/emboj/17.5.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantheti P, Qiao X, Diaz M, Peden AA, Meyer GE, Carskadon SL, Kapfhamer D, Sufalko D, Robinson MS, Noebels JL, Burmeister M. Mutation in AP-3δ in the mocha mouse links endosomal transport to storage deficiency in platelets, melanosomes, and synaptic vesicles. Neuron. 1998;21:111–122. doi: 10.1016/s0896-6273(00)80519-x. [DOI] [PubMed] [Google Scholar]
- Karnovsky MJ. A formaldehyde-glutaraldehyde fixative of high osmolality for use in electron microscopy. J Cell Biol. 1965;27:137. (Abstract). [Google Scholar]
- Karnovsky MJ. Use of ferrocyanide-reduced oximum tetroxide in electron microscopy. J Cell Biol. 1971;51:146. (Abstract). [Google Scholar]
- King RA, Hearing VJ, Creel DJ, Oetting WS. Albinism. In: Scriver CR, Beaudet AL, Sly WS, Valle DV, editors. The Metabolic and Molecular Bases of Inherited Disease. 7th ed. New York: McGraw-Hill; 1995. pp. 4353–4392. [Google Scholar]
- Kretzschmar D, Poeck B, Roth H, Ernst R, Keller A, Porsch M, Stauss R, Pflugfelder GO. Defective pigment granule biogenesis, and aberrant behavior caused by mutations in the Drosophila AP-3 beta adaptin gene ruby. Genetics. 2000;155:213–223. doi: 10.1093/genetics/155.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBorgne R, Alconada A, Bauer U, Hoflack B. The mammalian AP-3 adaptor-like complex mediates the intracellular transport of lysosomal membrane glycoproteins. J Biol Chem. 1998;273:29451–29461. doi: 10.1074/jbc.273.45.29451. [DOI] [PubMed] [Google Scholar]
- Luzio JP, Rous BA, Bright NA, Pryer PR, Mullock BM, Piper RC. Lysosome-endosome fusion, and lysosome biogenesis. J Cell Sci. 2000;113:1515–1524. doi: 10.1242/jcs.113.9.1515. [DOI] [PubMed] [Google Scholar]
- Maul GG. Golgi-melanosome relationship in human melanoma in vitro. J Ultrastruct Res. 1969;26:163–176. doi: 10.1016/s0022-5320(69)90042-2. [DOI] [PubMed] [Google Scholar]
- Mullins C, Hartnell LM, Wassarman DA, Bonifacino JS. Mutations in subunits of the AP-3 adaptor complex result in defective pigment granule biogenesis in Drosophila melanogaster. Mol Biol Cell. 1997;10:223. (Abstract). [Google Scholar]
- Mullins C, Hartnell LM, Wassarman DA, Bonifacino JS. Defective expression of the μ3 subunit of the AP-3 adaptor complex in the Drosophila pigmentation mutant carmine. Mol Gen Genet. 1999;262:401–412. doi: 10.1007/s004380051099. [DOI] [PubMed] [Google Scholar]
- Mullock BM, Bright NA, Fearon CW, Gray SR, Luzio JP. Fusion of lysosomes with late endosomes produces a hybrid organelle of intermediate density and is NSF dependent. J Cell Biol. 1998;140:591–601. doi: 10.1083/jcb.140.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novikoff A, Albala A, Biempica L. Ultrastructural and cytochemical observations on B16 and Harding-Passey melanomas: the origin of premelanosomes and compound melanosomes. J Histochem Cytochem. 1968;16:299–319. doi: 10.1177/16.5.299. [DOI] [PubMed] [Google Scholar]
- Oh J, Bailin T, Fukai K, Feng GH, Ho L, Mao J, Frenk E, Tamura N, Spritz RA. Positional cloning of a gene for Hermansky-Pudlak syndrome, a disorder of cytoplasmic organelles. Nat Genet. 1996;14:300–306. doi: 10.1038/ng1196-300. [DOI] [PubMed] [Google Scholar]
- Oh J, Ho L, Ala-Mello S, Amato D, Armstrong L, Bellucci S, Carakushansky G, Ellis JP, Fong CT, Green JS, Heon E, Legius E, Levin AV, Nieuwenhius HK, Pinckers A, Tamura N, Whiteford ML, Yamasaki H, Spritz RA. Mutation analysis of patients with Hermansky-Pudlak syndrome: a frameshift hot spot in the HPS gene and apparent locus heterogeneity. Am J Hum Genet. 1998;62:593–598. doi: 10.1086/301757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno H, Aguilar RC, Yeh D, Taura D, Saito T, Bonifacino JS. The medium subunits of adaptor complexes recognize distinct but overlapping sets of tyrosine-based sorting signals. J Biol Chem. 1998;273:25915–25921. doi: 10.1074/jbc.273.40.25915. [DOI] [PubMed] [Google Scholar]
- Ooi CE, Moreira JE, Dell'Angelica EC, Poy G, Wassarman DA, Bonifacino JS. Altered expression of a novel adaptin leads to defective pigment granule biogenesis in the Drosophila eye color mutant garnet. EMBO J. 1997;16:4508–4518. doi: 10.1093/emboj/16.15.4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlow SJ. Melanosomes are specialized members of the lysosomal lineage of organelles. J Invest Dermatol. 1995;105:3–7. doi: 10.1111/1523-1747.ep12312291. [DOI] [PubMed] [Google Scholar]
- Pond L, Kuhn LA, Teyton L, Schutze MP, Tainer JA, Jackson MR, Peterson PA. A role for acidic residues in di-leucine motif-based targeting to the endocytic pathway. J Biol Chem. 1995;270:19989–19997. doi: 10.1074/jbc.270.34.19989. [DOI] [PubMed] [Google Scholar]
- Robinson MS. The role of clathrin, adaptors and dynamin in endocytosis. Curr Opin Cell Biol. 1994;6:538–544. doi: 10.1016/0955-0674(94)90074-4. [DOI] [PubMed] [Google Scholar]
- Robinson MS. Coats and vesicle budding. Trends Cell Biol. 1997;7:99. doi: 10.1016/S0962-8924(96)10048-9. [DOI] [PubMed] [Google Scholar]
- Sakuma T, Monma M, Satodate R, Satoh T, Takeda R, Kuriya S. Ceroid pigment deposition in circulating blood monocytes and T lymphocytes in Hermansky-Pudlak syndrome: an ultrastructural study. Pathol Int. 1995;45:866–870. doi: 10.1111/j.1440-1827.1995.tb03407.x. [DOI] [PubMed] [Google Scholar]
- Schekman R, Orci L. Coat proteins and vesicle budding. Science. 1996;271:1526–1539. doi: 10.1126/science.271.5255.1526. [DOI] [PubMed] [Google Scholar]
- Setaluri V. Sorting, and targeting of melanosomal membrane proteins: signals, pathways and mechanisms. Pigment Cell Res. 2000;13:128–134. doi: 10.1034/j.1600-0749.2000.130302.x. [DOI] [PubMed] [Google Scholar]
- Shotelersuk V, Dell'Angelica EC, Hartnell L, Bonifacino JS, Gahl WA. A new variant of Hermansky-Pudlak syndrome due to mutations in a gene responsible for vesicle formation. Am J Med. 2000;108:423–427. doi: 10.1016/s0002-9343(99)00436-2. [DOI] [PubMed] [Google Scholar]
- Shotelersuk V, Gahl WA. Hermansky-Pudlak syndrome: models for intracellular vesicle formation. Mol Genet Metab. 1998;65:85–96. doi: 10.1006/mgme.1998.2729. [DOI] [PubMed] [Google Scholar]
- Simpson F, Bright NA, West MA, Newman LS, Darnell RB, Robinson MS. A novel adaptor-related protein complex. J Cell Biol. 1996;133:749–760. doi: 10.1083/jcb.133.4.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson F, Peden AA, Christopoulou L, Robinson MS. Characterization of the adaptor-related protein complex, AP-3. J Cell Biol. 1997;137:835–845. doi: 10.1083/jcb.137.4.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swank RT, Novak EK, McGarry MP, Rusiniak ME, Feng L. Mouse models of Hermansky-Pudlak syndrome: a review. Pigment Cell Res. 1998;11:60–80. doi: 10.1111/j.1600-0749.1998.tb00713.x. [DOI] [PubMed] [Google Scholar]
- Tabuchi N, Akasaki K, Tsuji H. Two acidic amino acid residues, asp(470), and glu(471), contained in the carboxyl cytoplasmic tail of a major lysosomal membrane protein, LGP85/LIMP II, are important for its accumulation in secondary lysosomes. Biochem Biophys Res Commun. 2000;270:557–563. doi: 10.1006/bbrc.2000.2448. [DOI] [PubMed] [Google Scholar]
- Witkop CJJ, Krumwiede M, Sedano H, White JG. Reliability of absent platelet dense bodies as a diagnostic criterion for Hermansky-Pudlak syndrome. Am J Hematol. 1987;26:305–311. doi: 10.1002/ajh.2830260403. [DOI] [PubMed] [Google Scholar]
- Witkop CJJ, Nunez-Babcock M, Rao GH, Gaudier F, Summers CG, Shanahan F, Harmon KR, Townsend D, Sedano HO, King RA. Albinism and Hermansky Pudlak syndrome in Puerto Rico. Bol Asoc Med PR. 1990;82:333–339. [PubMed] [Google Scholar]
- Zhao H, Boissy RE. Distinguishing between the catalytic potential and apparent expression of tyrosinase activities. Am J Med Sci. 1994;308:322–330. doi: 10.1097/00000441-199412000-00003. [DOI] [PubMed] [Google Scholar]
- Zhao H, Boissy Y, Abdel-Malek Z, King RA, Nordlund JJ, Boissy RE. On the analysis of the pathophysiology of Chediak-Higashi syndrome: defects expressed by cultured melanocytes. Lab Invest. 1994;71:25–34. [PubMed] [Google Scholar]
- Zhen L, Jiang S, Feng L, Bright NA, Peden AA, Seymour AB, Novak EK, Elliott R, Gorin MB, Robinson MS, Swank RT. Abnormal expression and subcellular distribution of subunit proteins of the AP-3 adaptor complex lead to platelet storage pool deficiency in the pearl mouse. Blood. 1999;94:146–155. [PubMed] [Google Scholar]
- Zhou B-K, Boissy RE, Pifko-Hirst S, Moran DJ, Orlow SJ. Lysosome-associated membrane protein-1 (LAMP-1) is the melanocyte vesicular membrane glycoprotein, Band II. J Invest Dermatol. 1993;100:110–114. doi: 10.1111/1523-1747.ep12462775. [DOI] [PubMed] [Google Scholar]