

Abstract
Background
Anorexia nervosa (AN) is a serious mental illness characterized by emaciation, an intense fear of gaining weight despite being underweight, and distorted body image. Few treatments reverse the core symptoms in AN such as profound aversion to food and food avoidance. Consequently, AN has a chronic and relapsing course and the highest mortality rate of any psychiatric illness. A more complete understanding of the disease pathogenesis is needed in order to develop better treatments and improve AN outcome. The pathogenesis and psychopathophysiology of AN can be better elucidated by combining longitudinal phenotyping with multiple “omics” techniques, including exon sequencing, proteomics, lipidomic, and metabolomics.
Design
This paper summarizes the key findings of a series of interrelated studies including new experimental data and previously published data, and describes our current initiatives and future directions.
Results
Exon sequencing data was analyzed in 1205 AN and 1948 controls. Targeted metabolomics, lipidomics, and proteomics data were collected in two independent convenience samples consisting of 75 subjects with eating disorders and 61 age-matched healthy controls. Study participants were female and the mean age was 22.9 (4.9 [SD]) years. Epoxide hydrolase 2 (EPHX2) genetic variations were significantly associated with AN and that activity of epoxide hydrolase (sEH) was elevated in AN compared to controls. The polyunsaturated fatty acid (PUFA) and eicosanoids data revealed that Cytochromes P450 pathway was implicated in AN, and AN displayed a dysregulated postprandial metabolism of PUFAs and sEH-dependent eicosanoids.
Implication and current Initiatives
Collectively, our data suggest that dietary factors may contribute to the burden of EPHX2-associated AN susceptibility and affect disease outcome. We are implementing new investigations using a longitudinal study design in order to validate and develop an EPHX2 multi-omics biomarker system. We will test whether sEH-associated postprandial metabolism increases AN risk and affects treatment outcome through an ω-6 rich breakfast challenge. Participants will include 100 ill AN patients, 100 recovered AN patients, and 100 age- and race-matched healthy women. These data will allow us to investigate 1) how genetic and dietary factors independently and synergistically contribute to AN risk and progression, and 2) if clinical severity and treatment response in AN are affected by sEH activity and eicosanoid dysregulation. Results of our study will 1) identify clinically relevant biomarkers, 2) unravel mechanistic functions of sEH, and 3) delineate contributory roles of dietary PUFAs and Cytochrome P450 pathway eicosanoids for the purpose of developing novel AN treatments and improving disease prognosis.
Keywords: Psychiatric metabolomics, Anorexia Nervosa, Eating disorder, Soluble epoxide hydrolase, EPHX2, Cytochrome P450 Pathway, Bioactive lipid mediators, Polyunsaturated fatty acids, Epoxides and diols, Metabolomics, Lipidomics, Genetic Polymorphism, Inflammation, Inflammatory biomarkers, Nutritional intervention, Personalized treatment development
INTRODUCTION
Anorexia Nervosa (AN) is a serious mental illness that often has an onset during adolescence. AN predominantly affects females [1] and is characterized by emaciation, an intense fear of gaining weight despite being underweight, and disturbed body image [2]. There are few effective treatments that reverse the core symptoms associated with AN. Consequently, AN often has a chronic and relapsing course, and as a result has the highest death rate of any psychiatric illness [3, 4].
Although social-cultural and psychosocial factors have been traditionally hypothesized as important contributory factors to AN [5], the pathophysiology and etiology of AN are unclear. More recently, significant familial risks reported in familial transmission studies [5] and high heritability (h2) [6] reveal that AN risk also has a significant genetic basis, supporting the “biopsychosocial entity” concept proposed for this psychiatric illness [7]. Almost two decades have passed since the first AN genetic association paper was published [8]. Nonetheless, data elucidating the etiology of AN remain sparse. While many associated candidate genes cannot be replicated across independent studies, identification of AN-associated genes has not successfully led to useful knowledge to guide new treatment development or improve maintenance strategies for this deadly disease [9].
Candidate gene [10–14] and whole genome [15–20] association studies have been employed to discover regions and specific genetic variants that contribute to AN risk, yet the results have been disappointing to date [9]. Utilizing the lead candidate genes identified in an earlier genome-wide association study [21], the use of an exon sequencing approach [14] coupled with rigorous association and replication analyses of 152 candidate genes led to the identification of soluble epoxide hydrolase 2 (EPHX2) as a novel AN gene [14]. EPHX2 is a gene known to be associated with subclinical cardiovascular disease [22, 23], stroke [24], insulin resistance [25], cholesterol phenotypes [26], and kidney functions [27]. The surprising discovery of its association to a major psychiatric illness opens up the possibility that researchers have been choosing an incomplete set of “candidate genes” due to insufficient understanding of genes’ biology. Related to this limitation, we may have overlooked or underestimated the functions and mechanisms of susceptibility genes in the context of disease development and progression, including mechanism-associated downstream biochemical and biological consequences.
The motivation of the series of studies described came from the need to elucidate mechanisms and consequences of epoxide hydrolase 2 gene (EPHX2) in AN. Specifically, to determine how EPHX2 genetic variation influence variability of soluble epoxide hydrolase (sEH) activity, thereby regulate bioactive metabolite derivatives of polyunsaturated fatty acids (PUFAs) to affect AN risk, progression, and outcome. EPHX2 codes sEH, a bifunctional enzyme that binds to specific lipid epoxides (metabolite derived from PUFAs) and converts them to the corresponding diols through the addition of a water molecule [28]. This catalytic function affects lipid signaling of a class of potent bioactive lipid mediators (termed “eicosanoid”) by converting epoxy fatty acids (e.g., EETs from arachidonic acid [ARA]) to diol fatty acids (e.g., DiHETs; Fig. 1). The sEH’s catalytic process contributes to diverse physiological and pathological processes by turning generally stable, anti-inflammatory eicosanoids to less stable, pro-inflammatory eicosanoids [29].
Figure 1.

Health Implications of Soluble Epoxide Hydrolase’s Catalytic Actions
The biosynthesis of eicosanoids involving sEH starts when these potent bioactive lipids are formed from several omega-3 and omega-6 polyunsaturated fatty acids (ω-3 and ω-6 PUFAs) by enzymes of the cytochrome P450 family. The precursor ω-3 and ω-6 PUFAs also undergo catalytic processing by enzymes of two other major metabolic enzyme families: cyclooxygenase (COX) and lipoxygenase (LOX). While the well-studied ω-6 PUFA ARA is metabolized in all three pathways that lead to production of a spectrum of important eicosanoids such as prostaglandins (by COX) [30], leukotrienes (LOX) [31], and epoxyeicosatrienoic acids (by CYP) [29], other major PUFAs including ω-6 linoleic acid (LA), ω-3 alpha-ainolenic acid (ALA), ω-3 eicosapentaenoic acid (EPA), and ω-3 docosahexaenoic acid (DHA) are also transformed by these enzymes. The sEH’s catalytic action takes place after the CYP enzymatic conversion of precursor PUFAs to their corresponding epoxygenated fatty acids (epoxy-fatty acids). These epoxy-fatty acids are then quickly inactivated by sEH to the diol-fatty acid products, leading to a generally more pro-inflammatory and pain-prong cellular state [32–34].
Common food sources of PUFA found in a typical North American diet include a variety seeds and oil for ALA (e.g., flaxseed, canola oil), marine product for EPA and DHA (e.g., fish oil), and vegetable oils for LA (e.g., corn oil). Of interest, ω-6 ARA is primarily found in dietary products of animals such as meat and egg. Patients with AN are known to exhibit a chronic, pathological fear and avoidance of fatty foods [35], in particular animal-sourced fats [36]. Pre-meal anxiety in AN has been found to correlate with the consumption of high fat foods [37] suggesting that psychological “fear of weight gain” drives aversion to fatty foods and restrictive eating behavior. However, it has not been determined whether physiological responses from fatty foods consumption may also contribute to the persistent aversion and refusal of fatty foods. Pathological food avoidance is a serious treatment challenge, as the more severe the food restrictions patients exhibit, the more damage occurs in both their physical and neuronal health due to increased deficiency in macro and micro nutrition which is required to support a healthy cellular homeostasis. Therefore, better understandings of the physiological and biological impact of fatty foods in AN can open the door to identifying new treatments that ameliorate food aversion and food avoidance.
Echoing what Leonard Bernstein once said: “the best way to know a thing, is in the context of another discipline”. This multidisciplinary spirit has inspired a sequence of interrelated studies that investigated how an unsuspected lipid regulatory gene affects AN risk, how in vivo activity of this gene transforms key lipid substrates sourced primarily from foods, and whether inflammation consequences of this gene are relevant to AN pathophysiology. The purpose of this paper is to summarize and synthesize the key findings of our previously published studies [14, 38] with subsequently completed pilot studies to demonstrate that interactive relationships among dietary PUFAs, sEH, and individual genetic variability may play a key role in AN risk and outcome. Lastly, to describe our current initiatives, the study design, recruitment strategies, human subjects, methods for biomarker and postprandial metabolism investigations, and planned outcomes in our new multidisciplinary study are summarized.
MATERIALS AND METHODS
Subjects in published articles and new studies
All subjects included in the studies are female. The ill AN cases (IAN) in this study are defined as having ongoing ED symptoms and BMI of less than or equal to 17.5 kg/m2 in the past year; whereas the recovered AN cases (RecAN) are defined as having an absence of ED symptoms in the past year and maintaining a BMI of 18 kg/m2 or greater. The healthy controls were sex- and age-matched to AN cases. More detailed subject description has been published previously [14, 38–41]. In brief, candidate gene exon sequencing study was conducted in a total of 1205 female AN and 1948 female controls through multiple sample association and replication analyses [14]. Polyunsaturated fatty acid investigation was evaluated in 30 ill AN (age: 22.2 ± 4.9 [SD]), 30 recovered AN (age: 24.2 ± 5.2 [SD]), and 36 controls (age: 20.0 ± 1.6 [SD]) [39]. Metabolites (eicosanoids) selected from cytochrome p450, LOX, and COX pathways were measured in 10 ill (age: 19 ± 0.8 [SD]), 10 recovered AN (age: 20.4 ± 1.2 [SD]), and 38 controls (age: 21.6 ± 2.9 [SD]) in the first study [38]. 20 subjects including 15 recovered eating disorder subjects (age: 26.3 ± 4.5 [SD]) and 5 healthy controls (age: 30 ± 8.1 [SD]) were measured in the follow-up pilot study [41]. Ex vivo sEH activity was measured using buffy coats extracted from 12 recovered AN, 3 recovered bulimic nervosa subjects (age: 26.3 ± 4.5 [SD]), and 5 healthy control women (age: 30 ± 8.1 [SD]) [40]. Postprandial metabolism was studied in 6 recovered patients with eating disorders (5 AN and 1 bulimic nervosa) (age: 30.8 ± 8.1 [SD]) and 5 controls (age: 29.2 ± 4.5 [SD]) selected from the second metabolite study [41].
Biospecimens: plasma and buffy coats
Polyunsaturated fatty acid and eicosanoid (metabolites) assays were performed using banked plasma from convenience samples [42, 43]. Whole blood was collected in EDTA vacutainer tubes and plasma was separated by centrifugation at 1350 rpm for 10 minutes and removed using pipet. Ex vivo sEH activity was measured in buffy coats derived from whole blood sample of each subject by methods previously described [44].
Lipidomic analysis: polyunsaturated fatty acid analysis
Plasma non-esterified fatty acids were purified and enriched with the application of a bi-phasic solution of acidified methanol and isooctane. A set of deuterated fatty acids was added to the samples to serve as internal standards for quantitation and to compensate for any losses during the analytical procedure. Gas chromatography coupled with mass spectrometry (GC/MS) was used to quantify each fatty acid, as previously described [45]. The omega 6 (ω-6) and omega 3 (ω-3) fatty acids were quantified in ill AN, recovered AN, and control groups. Concentration of each fatty acid was measured in the unit of μmol/L and reported as a percentage of total plasma fatty acid.
Targeted metabolomic analysis
A targeted metabolomics assay was carried out in two independent samples. The first study utilized a cross-sectional study design to assess AN association. The second study was designed to explore if postprandial metabolism was associated with AN, and if PUFA eicosanoids were associated with key AN clinical phenotypes (such as anxiety). The targeted eicosanoid assay was designed based on the ω-3 and ω-6 PUFA pathway and analyzed using a targeted GC/MS and LC/MS/MS systems platform as described previously [46, 47]. In brief, these metabolite markers were prepared by solid phase extraction using Oasis HLB cartridges followed by reversed phase HPLC analysis utilizing C18 columns [48]. The analytes were quantified by tandem mass spectrometry in multiple-reaction monitoring mode that utilizes negative electrospray ionization for eicosanoid profiling. Absolute concentrations of eicosanoid are presented as nmol/L.
Proteomics
sEH activity was measured in blood-derived buffy coating that contains white blood cells known to reflect systemic sEH activity [49] in 15 recovered AN, 3 recovered women with bulimic nervosa, and 5 healthy controls. The assay used a well characterized surrogate substrate for sEH, trans-diphenyl-propene oxide (t-DPPO) for solution preparation, incubation, and reaction assay and protein quantification as described previously [44]. The absolute sEH activity was divided by protein level to derive the normalized sEH activity. The unit of sEH activity is presented as nmol/(min.mg).
Data analysis and statistics
Lipidomic (PUFA) and metabolomics (eicosanoid) data were assessed for normality with histograms. When non-normality was detected, values were log transformed prior to analysis. Significance was set at P<0.05 in publications; no adjustments were made for multiple testing due to the exploratory and hypothesis-driven nature of the analyses. PUFA and eicosanoids with >30% missing observations were excluded. Tests of association were conducted using ANOVA using the statistical analysis tools in R (version 2.14.2). Postprandial metabolism was first calculated using the difference between fasting-state and fed-state eicosanoid measurements to derive proxy markers of meal-induced “postprandial eicosanoid shift”. The comparison of postprandial “pro-inflammatory” diol-fatty acid markers between cases and controls was carried out in generalized linear models that included upstream epoxy-fatty acid level and age as covariates.
RESULTS
Investigations of AN genetic susceptibility
Through a series of complementary genomic/genetic study approaches and designs involving GWAS [21], candidate gene association study [50], and exon-based sequencing and single-locus association and replication studies [14], EPHX2 was found to harbor significantly more clusters of rare, likely deleterious genetic variants within sequenced region (14th intron down to 3′-UTR end) compared to healthy controls (p=0.0004 to 0.00000016) [14]. Common variants, including 3′-UTR polymorphisms rs1042032 and rs1042064, were also associated with AN risk [14]. To validate the biological link between EPHX2 and AN and to further understand how EPHX2 functionally affect AN, a series of targeted metabolomic, lipidomic, and enzymatic analyses were conducted.
Polyunsaturated fatty acid profile in AN and healthy controls, phenotype associations
We recently published a paper showing a significant difference in the concentrations of three ω-6 (DGLA, ARA and OBA) and four ω-3 (ALA, SDA, EPA and DHA) PUFAs between AN and controls [38]. Comparison of ill AN to controls revealed that ω-3 ALA and EPA were 2 and 2.5 times more concentrated respectively in AN(p-values=0.007 and <0.001, Table 1). The two major ω-6 to ω-3 ratios, LA:ALA and ARA:EPA, were significantly lower in ill AN compared to controls (p-values=0.001 and <0.001, Table 1). The ARA:EPA and LA:ALA were inversely associated with state anxiety (r= −0.261 and −0.268, p-values=0.04 and 0.04, respectively) in AN but not in controls. ARA:EPA ratio was significantly correlated with BMI (r= 0.415, p-value=0.0009) also in AN only. Multivariate PCA analysis of all PUFA markers revealed distinct patterns in ill AN, recovered AN, and controls. Consistent with the individual PUFA marker results, ill AN and controls were clustered with clear separation, whereas recovered AN showed overlap with both ill AN and controls, suggesting a normalization of PUFA profile in AN when their weight is recovered for at least a year.
Table 1.
Concentration of polyunsaturated fatty acids in anorexia nervosa and healthy controls
| ω-6: ω-3 (ratio) | AN mean |
Controls mean |
Statistics: IAN to Controls |
|---|---|---|---|
|
| |||
| LA/ALA | 126.7 | 195.7 | 0.001 |
| ARA/EPA | 7.9 | 17.1 | 1.82E-09 |
| ARA/DPA | 9.5 | 7.8 | 0.183 |
| ARA/DHA | 1.44 | 1.53 | 0.727 |
|
| |||
| Individual Non-esterified Fatty Acids (nM) | |||
|
| |||
| ω-6 Linoleic acid (LA) | 19.36 | 17.99 | 0.558 |
| ω-6 Arachidonic acid (ARA) | 1.06 | 0.95 | 0.208 |
|
| |||
| ω-3 Alpha-linolenic acid (ALA) | 0.22 | 0.11 | 0.007 |
| ω-3 Eicosapentaenoic acid (EPA) | 0.15 | 0.06 | 3.76E-06 |
| ω-3 Docosapentaenoic acid (DPA) | 0.13 | 0.15 | 0.625 |
| ω-3 Docosahexaenoic acid (DHA) | 0.92 | 0.76 | 0.069 |
Note: Modified from Shih et al, 2016 (Mol Psychiatry). The mean of ω-6 to ω-3 ratios (upper) and the mean of individual fatty acids (lower). Concentration of each fatty acid was measured in the unit of μmol/L and reported as percentage of total plasma fatty acid. Statistical comparisons were tested by age-adjusted ANOVA. AN: Ill anorexia nervosa with BMI<=17.5
Dysregulation of plasma eicosanoids in Cytochrome P450 pathway
The targeted metabolomics assay captured 80 individual eicosanoids, including direct substrates of sEH (e.g., EETs [EpETrEs] from ARA) and catalyzed products of sEH (e.g., DHETs [DiHETrEs] from ARA) of cytochrome P450 (CYP) enzyme pathway. The remaining eicosanoids are metabolites derived from related enzyme families, including the cyclooxygenase (COX) and lipoxygenase (LOX). Association analysis for each individual eicosanoid in the published paper [38] revealed that the eicosanoids significantly associated with AN were all metabolites within CYP pathway. The individual eicosanoids associated with AN risk (IAN compared to controls) were DHA metabolites 10.11.EpDPE (p-value = 0.049), 13.14.EpDPE (p-value = 0.039) and ARA’s metabolite 9.10.EpOME (p-value = 0.05) [38]. In the subsequent eicosanoid pilot study [41], associations with body weight (a measure of AN severity) and temperament traits that are clinically relevant in AN risk and disease prognosis (state and trait anxiety, harm avoidance, perfectionism, novelty seeking) were tested in 15 patients with remitted eating disorders [41]. The 9(10)-DiHOME from ω-6 LA was inversely associated with body mass index, whereas 13(14)- DiHDPE from ω-3 DHA, was positively associated with state anxiety [41].
Postprandial metabolism assessment
Postprandial metabolism assessment was conducted in 6 remitted patients (5 AN and 1 bulimic nervosa) and 5 healthy control women by the measurement of eicosanoids at two time points in each subject - once during fasting state, and again during fed state (after lunch). Significant variability was observed in individual eicosanoid levels between subjects and within subjects (fasting versus fed state comparison) [41]. The meal-induced postprandial eicosanoid shift was inferred by taking the difference between the fasting- and fed- state eicosanoid levels. sEH is the enzyme that converts epoxy-fatty acid to diol-fatty acid, turning eicosanoids to “pro-inflammatory state” in the diol form. After accounting for the variance of epoxy-fatty acid and age, the postprandial shift of pro-inflammatory diol-fatty acid of ω-6 ARA, 5.6.DHET, was increased in patients but decreased in controls (p=0.09), whereas no difference in the direction of postprandial shift in ω-3 DHA diol-fatty acid was observed between case and control groups [41].
Soluble epoxide hydrolase activity: in vivo markers
The ratios of sEH-associated eicosanoids (substrate/metabolite [product], or epoxy fatty acid/diol fatty acid) have been used by several groups as proxy markers of in vivo sEH activity to monitor effectiveness of sEH inhibition [51, 52]. In our published study [38], we adapted the reverse ratio (metabolite [product]/substrate, or diol fatty acid/epoxy fatty acid) to infer in vivo activity of sEH. Several eicosanoid ratios, including 15.16.DiHODE;15.16.EpODE of ALA and 19.20.DiHDPE;19.20.EpDPE of DHA, were significantly different among three groups (ill AN, recovered AN, healthy controls) and were consistently higher in AN compared to controls (Figure 4), suggesting a higher in vivo sEH activity, concentration, or efficiency in AN.
Figure 4. Product-to-substrate ratios of eicosanoids (diol fatty acid:epoxy fatty acid).
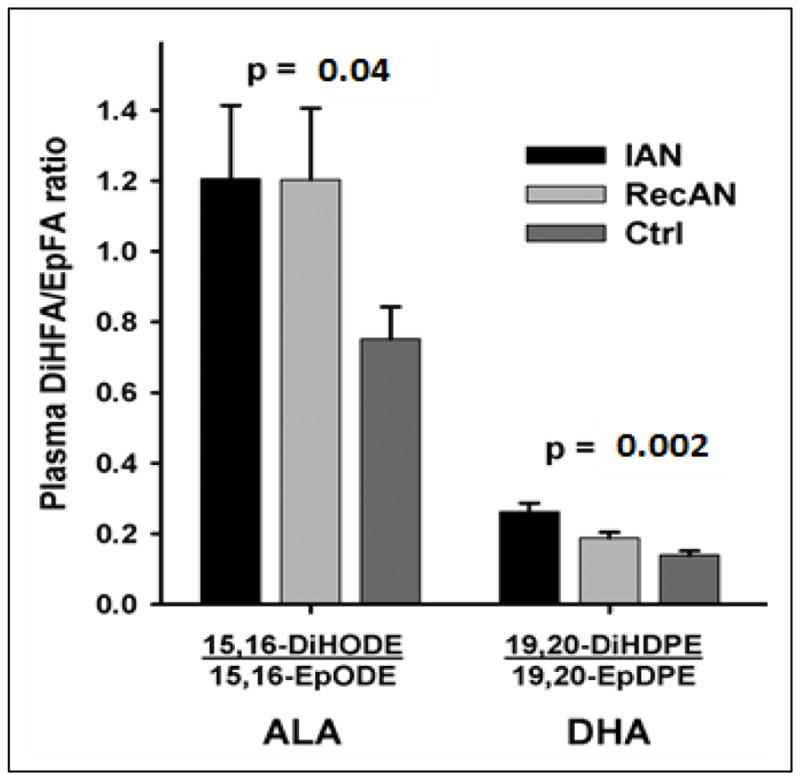
Adapted from Shih et al, 2016 (Molecular Psychiatry). Result columns are of the form eicosanoid ratios based on absolute eicosanoid value in nanomole (nM). All individual eicosanoid were log-transformed (natural-log) before ratio-transformation and inferential statistical analysis. Statistical comparisons among three groups (Ill AN n=10, recovered AN n=10, controls n=38) were tested by age-adjusted ANOVA.
Soluble epoxide hydrolase: ex vivo activity
Ex vivo soluble epoxide hydrolase (sEH) activity was measured in 20 convenient samples consisting of 15 remitted AN, 3 remitted bulimia nervosa (BN) subjects, and 5 healthy controls. There was no significant difference in ex vivo sEH between AN and BN (0.012 ± 0.002 and 0.012 ± 0.006 [mean ± SEM]). Combining both AN and BN, ex vivo sEH activity was found to be elevated in remitted eating disorder patients compared to controls (0.012 ± 0.002 and 0.007 ± 0.001 [mean ± SEM], p=0.05).
DISCUSSION
The results described in this paper were obtained following our original conceptual scheme (Fig. 2) that led to a series of interrelated, multi-disciplinary studies. Our data indicated that 1) Epoxide hydrolase 2 (EPHX2) genetic variation were significantly associated with AN, 2) activity of epoxide hydrolase (sEH) was elevated in AN compared to controls. 3) eicosanoids in the Cytochromes P450 pathway were implicated in AN, 4) AN displayed a dysregulated postprandial metabolism of PUFAs and sEH-dependent eicosanoids, and 5) PUFAs and sEH-associated eicosanoids are associated with not only AN risk, but also with a number of important clinical phenotypes. These data suggest that in addition to the documented EPHX2 association with AN [14], dietary factors also contribute to or modify the burden of EPHX2-associated AN susceptibility and affect outcome.
Figure 2.

Overarching EPHX2 Omics Hypothesis in AN
Anorexia nervosa: The need to develop better treatments and relapse prevention
AN is a serious health public issue not only because it has the highest death rate among all psychiatric illnesses [3], but also because its characteristics of extreme weight loss and preoccupation with body image and dieting often persist despite treatments, leading to chronicity. Despite the evolution of treatment strategies from the solo use of neuroleptics in the1950’s, to individual psychotherapy, to behavioral and cognitive interventions, family therapy, to better recognition of the promise of multifaceted treatment approach [4], personalized treatment strategies have yet to prove consistently successful due to heavy reliance on subjective patient report and physician observation. For example, a meta-analysis found that only 46.9% of patients met criteria of full recovery of core symptoms, 33.5% showed partial improvement, and 20.8% developed a chronic course of the disorder [4]. Specific clinical outcomes were equally disappointing as normalization of weight was achieved in 59.6% of the patients, while normalization of eating behavior was found only in 46.8% of subjects reviewed [4].
Multi-omics AN biomarkers
To overcome this treatment challenge, clinicians and patients need to have better tools than subjective assessment of symptoms. Tools such as biomarkers can provide objective indications of physiological state through accurate and reproducible measurements. Biomarkers also reflect objective and often quantifiable characteristics of biological processes involved in the disease/phenotype of interest, which enable treatment response monitoring and improve prognosis accuracy. To begin working towards improving treatment outcome of AN, a strategy of integrating EPHX2 genetic variation (static marker), sEH activity (dynamic proteomic marker), and eicosanoids (dynamic metabolomic marker) to develop a multi-domains biomarker system was implemented.
A synthesis of studies to date, limitations, and future directions
The completed studies illustrate the feasibility of developing objective, and biologically relevant biomarkers for AN. Although the sample sizes were modest in each individual study, our results showed multi-omics markers are associated not only with AN risk but also with “intermediate phenotypes” of AN. Phenotypes such as anxiety are “intermediate phenotypes” because they are intermediate in time and mechanism between gene action and onset of AN. Studying intermediate phenotype in biomarker development is a necessary strategy as these phenotypes have a logical involvement in the pathogenesis of AN, substantial heritability, and early penetrance. Therefore, intermediate phenotypes serve as superior phenotypic anchor points in the search for biological factors and biomarkers. In the following sections, I will synthesize each component of our “multi-omics” findings to date, address their limitations, and summarize the current initiatives of our work through a brief description of rationale, study design and methods, and anticipated outcomes for our newly funded longitudinal multi-omics study.
Insufficient lessons learned from genetic investigation without accounting for impacts of environmental factors
While genome wide association studies (GWAS) and candidate gene association studies have enabled both hypothesis-free and hypothesis-driven investigations, modest sample sizes, small genetic effect, and heterogeneous AN clinical presentation contributed to the difficulty in identifying AN genes. Despite the efforts of many [16, 20, 42, 53, 54], the community’s collective understanding of AN genetic susceptibility and its influence on prognosis remains limited. The phenotypic expression of a genotype is dependent on an array of host factors including genetic background, age and gender, developmental-, physiological- and pathological- conditions, as well as environmental factors such as dietary nutrition, exercise, and nicotine. Both acute and long-term environmental factors can interact with and influence an individual’s expression and functions of genes to affect AN pathophysiology. Careful consideration of impacts that environmental factors have on genetic susceptibility is warranted given that modification of environmental risk factors holds a great promise to mitigate disease risk and improve outcome.
Can nutritional modification be implemented to improve anorexia nervosa outcome?
The discovery that a gene integral to determining functional consequences of diet is a risk gene for AN [14] is significant for several reasons. First, while pathological eating behavior is well-established in AN, the malnutrition observed in patient population has traditionally been thought of solely as an outcome of restrictive eating. The finding that an AN-associated gene affects functional consequence of dietary-based PUFA, suggests that malnutrition may also be influenced by the type of the foods that patients restrict on, or by the host-specific energy metabolism of endogenous substrates. Secondly, a dysregulated metabolite cascade affects the balance of cellular inflammation which could lead to sensory abnormalities, pathological eating behavior, and other AN phenotypes. Our studies have shown evidence of differential plasma PUFA levels in ill AN compared to healthy controls, as well as partial normalization of PUFA levels in recovered AN. Despite the partial normalization of PUFA in remitted state, PUFA remained associated with key clinical symptoms: low BMI and high anxiety. These findings need to be confirmed in our new longitudinal study, and specific dietary or nutritional alteration that can effectively normalize PUFA dysregulation in AN need to be researched. We envision that a personalized PUFA modification approach will lead to an effective adjunctive therapy.
Promise of multi-omics biomarkers to assess risk, monitor disease course, and guide treatment strategy
Related to PUFA analysis which is a study of lipidomics, eicosanoids are potent bioactive metabolite derivatives of PUFAs that are analyzed in metabolomic assays. Eicosanoids are small molecules that are chemically transformed during metabolism of lipids. Because eicosanoids are more proximally correlated to phenotypes of interest, they serve as functional signatures of host cellular state with high precision. Our data have clearly demonstrated the feasibility of a targeted metabolomics approach to assess functional consequences of EPHX2 in AN. Human cells and tissues require not only ω-6 and ω-3 PUFAs but also their downstream corresponding eicosanoids for proper physiological functioning [55, 56]. A multi-disciplinary study which includes EPHX2 (genomic), sEH (proteomic), dietary PUFAs (lipidomic) and eicosanoids (metabolomic) data provides the best chance to uncover context-dependent mechanisms and consequences of EPHX2 in AN. The analysis of AN intermediate phenotype further demonstrated that ω-3 PUFA eicosanoids were associated with BMI and anxiety in patients, suggesting a potentially important role eicosanoids play in AN progression. The need to study eicosanoids (metabolomics) and their catalytic enzymes (proteomics) together and not in isolation comes from the established facts that enzymatic transformation of PUFAs play a significant role on eicosanoids. For example, CYP4A and CYP4F isoforms of Cytochrome P450 (CYP) enzymes are ARA ω/(ω-1)-hydroxylases producing 20-HETE, whereas the CYP2C and CYP2J subfamilies act as ARA epoxygenases producing sets of EETs [57], which later undergo catalytic conversion by sEH to become diol fatty acids (Fig. 3). The interactive relationship between enzymes and PUFA substrates allowed us to use the ratios of diol fatty acid to epoxy fatty acid to infer in vivo sEH activity. Genes coding for CYP enzymes have been found to associate with important intermediate and clinical phenotypes in eating disorders, including response to antidepressant [58], anxiety [59], and suicide attempts [60]. Just as important to consider is the contribution of diet. Our postprandial metabolism study showed a striking gene-by-PUFA interaction effect where the postprandial eicosanoid dysregulation in AN was observed only with ω-6 PUFA ARA but not with ω-3 DHA [41]. Taken together, a multi-omics research strategy showed great promise in establishing a novel biomarker system that can detect at-risk individuals, improve disease monitoring and nutritional assessment, and develop personalized treatment.
Figure 3. Postprandial shift of diol-fatty acids (eicosanoids).

Differential postprandial metabolism observed between AN patients and healthy controls in ω-6 PUFA ARA diol-fatty acid (upper) but not in ω-3 PUFA DHA diol-fatty acid(lower).
Implications and our work in progress - clarifying the effects of gene and diet in a longitudinal study
Collectively, the multi-omics data to date reveal EPHX2 affects AN risk through the regulation of sEH, that AN patients display a dysregulated profile both in upstream lipid substrates (PUFAs) and downstream metabolites (eicosanoid), and AN show postprandial inflammatory response in PUFA-dependent fashion. We have begun a new longitudinal study combining multiple “omics” techniques with a high-fat meal challenge protocol to better elucidate the molecular mechanisms underlying EPHX2-AN associations. We aim to test the following questions: 1) will EPHX2-associated metabolomic pathway be differentially altered in AN and controls upon the exposure to a high fat meal? 2) how do mechanisms underlying EPHX2-AN association and biochemical perturbation effect AN clinically? 3) can genetic variants, sEH activity, and eicosanoid markers be combined into a a clinical biomarker system? If successfully answered, we will 1) identify clinically relevant biomarkers, 2) unravel mechanistic functions of sEH, and 3) delineate contributory roles of dietary PUFAs and Cytochrome P450 pathway eicosanoids in AN pathogenesis,
Our 5-year study is a multi-disciplinary collaborative study conducted in University of California San Diego (PI institute), University of Toronto (Canada), and University of California, Davis. We will recruit and enroll a total of 300 subjects (100 ill AN women, 100 recovered AN women, and 100 age- and race-matched healthy control women) from the patients in the Program for Eating Disorders at Toronto General Hospital and from the city of Toronto and university campus. Ill AN will be recruited from the inpatient and day hospital programs to take advantage of the pre-existing meal serving infrastructure and to ensure patient safety. Multi-omics data and postprandial metabolism will be analyzed in all subjects through an ω-6 rich meal challenge protocol. Food aversion score, AN psychopathology traits (e.g., anxiety), and longitudinal clinical data will be applied to collect clinical and intermediate phenotypes, and to determine if sEH activity and postprandial metabolism affect disease progression and treatment outcomes. The strengths of our new study include the high likelihood of developing a reliable biomarker system that consists of both static biomarker (EPHX2 variants) and dynamic biomarkers (sEH activity, metabolites, PUFAs). Also, through an ω-6-rich meal challenge protocol, we are uniquely positioned to verify the mechanisms by which EPHX2 interacts with dietary substrates to affect AN. We anticipate the results of this study will lead to new insights on biological consequences of EPHX2, enable research and development of nutraceutical and pharmaceutical interventions and treatments, and further our knowledge on AN pathophysiology to improve disease prognosis.
Acknowledgments
This study was supported in part by NIH-NIDDK K01DK087813, NIH West Coast Metabolomics Center Pilot Award, Price Foundation of Switzerland, NIMH R01 092793, NIEHS R01 ES002710 and NIEHS P42 ES004699, and NARSAD Young Investigator Grant. The author is responsible for the design and conduct of the study, collection, management, analysis, and interpretation of the data, as well as preparation and submission of the manuscript. The author would like to thank all her study collaborators for their invaluable contributions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Micali N, Hagberg KW, Petersen I, Treasure JL. The incidence of eating disorders in the UK in 2000–2009: findings from the General Practice Research Database. BMJ open. 2013;3(5) doi: 10.1136/bmjopen-2013-002646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.American Psychiatric Association. American Psychiatric Association, Diagnostic and Statistical Manual of Mental Disorders. 5. Washington, DC: 2013. [Google Scholar]
- 3.Arcelus J, Mitchell AJ, Wales J, Nielsen S. Mortality rates in patients with anorexia nervosa and other eating disorders. A meta-analysis of 36 studies Archives of general psychiatry. 2011;68(7):724–731. doi: 10.1001/archgenpsychiatry.2011.74. [DOI] [PubMed] [Google Scholar]
- 4.Steinhausen HC. The outcome of anorexia nervosa in the 20th century. The American journal of psychiatry. 2002;159(8):1284–1293. doi: 10.1176/appi.ajp.159.8.1284. [DOI] [PubMed] [Google Scholar]
- 5.Strober M, Freeman R, Lampert C, Diamond J, Kaye W. Controlled family study of anorexia nervosa and bulimia nervosa: evidence of shared liability and transmission of partial syndromes. The American journal of psychiatry. 2000;157(3):393–401. doi: 10.1176/appi.ajp.157.3.393. [DOI] [PubMed] [Google Scholar]
- 6.Ben-Dor DH, Laufer N, Apter A, Frisch A, Weizman A. Heritability, genetics and association findings in anorexia nervosa. The Israel journal of psychiatry and related sciences. 2002;39(4):262–270. [PubMed] [Google Scholar]
- 7.Lucas AR. Toward the understanding of anorexia nervosa as a disease entity. Mayo Clin Proc. 1981;56(4):254–264. [PubMed] [Google Scholar]
- 8.Campbell DA, Sundaramurthy D, Markham AF, Pieri LF. Lack of association between 5-HT2A gene promoter polymorphism and susceptibility to anorexia nervosa. Lancet. 1998;351(9101):499. doi: 10.1016/S0140-6736(05)78688-8. [DOI] [PubMed] [Google Scholar]
- 9.Shih PA, Woodside DB. Contemporary views on the genetics of anorexia nervosa. Eur Neuropsychopharmacol. 2016;26(4):663–673. doi: 10.1016/j.euroneuro.2016.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bergen AW, van den Bree MB, Yeager M, Welch R, Ganjei JK, Haque K, Bacanu S, Berrettini WH, Grice DE, Goldman D, et al. Candidate genes for anorexia nervosa in the 1p33-36 linkage region: serotonin 1D and delta opioid receptor loci exhibit significant association to anorexia nervosa. Molecular psychiatry. 2003;8(4):397–406. doi: 10.1038/sj.mp.4001318. [DOI] [PubMed] [Google Scholar]
- 11.Bergen AW, Yeager M, Welch RA, Haque K, Ganjei JK, van den Bree MB, Mazzanti C, Nardi I, Fichter MM, Halmi KA, et al. Association of multiple DRD2 polymorphisms with anorexia nervosa. Neuropsychopharmacology. 2005;30(9):1703–1710. doi: 10.1038/sj.npp.1300719. [DOI] [PubMed] [Google Scholar]
- 12.Bergen AW, Yeager M, Welch R, Ganjei JK, Deep-Soboslay A, Haque K, van den Bree MB, Goldman D, Berrettini WH, Kaye WH, et al. Candidate gene analysis of the Price Foundation anorexia nervosa affected relative pair dataset. Current drug targets CNS and neurological disorders. 2003;2(1):41–51. doi: 10.2174/1568007033338760. [DOI] [PubMed] [Google Scholar]
- 13.Pinheiro AP, Bulik CM, Thornton LM, Sullivan PF, Root TL, Bloss CS, Berrettini WH, Schork NJ, Kaye WH, Bergen AW, et al. Association study of 182 candidate genes in anorexia nervosa. American journal of medical genetics Part B, Neuropsychiatric genetics: the official publication of the International Society of Psychiatric Genetics. 2010;153B(5):1070–1080. doi: 10.1002/ajmg.b.31082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scott-Van Zeeland AA, Bloss CS, Tewhey R, Bansal V, Torkamani A, Libiger O, Duvvuri V, Wineinger N, Galvez L, Darst BF, et al. Evidence for the role of EPHX2 gene variants in anorexia nervosa. Molecular psychiatry. 2014;19(6):724–732. doi: 10.1038/mp.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bacanu SA, Bulik CM, Klump KL, Fichter MM, Halmi KA, Keel P, Kaplan AS, Mitchell JE, Rotondo A, Strober M, et al. Linkage analysis of anorexia and bulimia nervosa cohorts using selected behavioral phenotypes as quantitative traits or covariates. American journal of medical genetics Part B, Neuropsychiatric genetics: the official publication of the International Society of Psychiatric Genetics. 2005;139B(1):61–68. doi: 10.1002/ajmg.b.30226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boraska V, Franklin CS, Floyd JA, Thornton LM, Huckins LM, Southam L, Rayner NW, Tachmazidou I, Klump KL, Treasure J, et al. A genome-wide association study of anorexia nervosa. Molecular psychiatry. 2014;19(10):1085–1094. doi: 10.1038/mp.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bulik CM, Bacanu SA, Klump KL, Fichter MM, Halmi KA, Keel P, Kaplan AS, Mitchell JE, Rotondo A, Strober M, et al. Selection of eating-disorder phenotypes for linkage analysis. American journal of medical genetics Part B, Neuropsychiatric genetics: the official publication of the International Society of Psychiatric Genetics. 2005;139B(1):81–87. doi: 10.1002/ajmg.b.30227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Devlin B, Bacanu SA, Klump KL, Bulik CM, Fichter MM, Halmi KA, Kaplan AS, Strober M, Treasure J, Woodside DB, et al. Linkage analysis of anorexia nervosa incorporating behavioral covariates. Hum Mol Genet. 2002;11(6):689–696. doi: 10.1093/hmg/11.6.689. [DOI] [PubMed] [Google Scholar]
- 19.Grice DE, Halmi KA, Fichter MM, Strober M, Woodside DB, Treasure JT, Kaplan AS, Magistretti PJ, Goldman D, Bulik CM, et al. Evidence for a susceptibility gene for anorexia nervosa on chromosome 1. Am J Hum Genet. 2002;70(3):787–792. doi: 10.1086/339250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakabayashi K, Komaki G, Tajima A, Ando T, Ishikawa M, Nomoto J, Hata K, Oka A, Inoko H, Sasazuki T, et al. Identification of novel candidate loci for anorexia nervosa at 1q41 and 11q22 in Japanese by a genome-wide association analysis with microsatellite markers. Journal of human genetics. 2009;54(9):531–537. doi: 10.1038/jhg.2009.74. [DOI] [PubMed] [Google Scholar]
- 21.Wang K, Zhang H, Bloss CS, Duvvuri V, Kaye W, Schork NJ, Berrettini W, Hakonarson H. A genome-wide association study on common SNPs and rare CNVs in anorexia nervosa. Molecular psychiatry. 2011;16(9):949–959. doi: 10.1038/mp.2010.107. [DOI] [PubMed] [Google Scholar]
- 22.Lee CR, North KE, Bray MS, Fornage M, Seubert JM, Newman JW, Hammock BD, Couper DJ, Heiss G, Zeldin DC. Genetic variation in soluble epoxide hydrolase (EPHX2) and risk of coronary heart disease: The Atherosclerosis Risk in Communities (ARIC) study. Hum Mol Genet. 2006;15(10):1640–1649. doi: 10.1093/hmg/ddl085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burdon KP, Lehtinen AB, Langefeld CD, Carr JJ, Rich SS, Freedman BI, Herrington D, Bowden DW. Genetic analysis of the soluble epoxide hydrolase gene, EPHX2, in subclinical cardiovascular disease in the Diabetes Heart Study. Diabetes & vascular disease research: official journal of the International Society of Diabetes and Vascular Disease. 2008;5(2):128–134. doi: 10.3132/dvdr.2008.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fava C, Montagnana M, Danese E, Almgren P, Hedblad B, Engstrom G, Berglund G, Minuz P, Melander O. Homozygosity for the EPHX2 K55R polymorphism increases the long-term risk of ischemic stroke in men: a study in Swedes. Pharmacogenet Genomics. 2010;20(2):94–103. doi: 10.1097/FPC.0b013e3283349ec9. [DOI] [PubMed] [Google Scholar]
- 25.Ramirez CE, Shuey MM, Milne GL, Gilbert K, Hui N, Yu C, Luther JM, Brown NJ. Arg287Gln variant of EPHX2 and epoxyeicosatrienoic acids are associated with insulin sensitivity in humans. Prostaglandins & other lipid mediators. 2014 doi: 10.1016/j.prostaglandins.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sato K, Emi M, Ezura Y, Fujita Y, Takada D, Ishigami T, Umemura S, Xin Y, Wu LL, Larrinaga-Shum S, et al. Soluble epoxide hydrolase variant (Glu287Arg) modifies plasma total cholesterol and triglyceride phenotype in familial hypercholesterolemia: intrafamilial association study in an eight-generation hyperlipidemic kindred. Journal of human genetics. 2004;49(1):29–34. doi: 10.1007/s10038-003-0103-6. [DOI] [PubMed] [Google Scholar]
- 27.Lee SH, Lee J, Cha R, Park MH, Ha JW, Kim S, Kim YS. Genetic variations in soluble epoxide hydrolase and graft function in kidney transplantation. Transplant Proc. 2008;40(5):1353–1356. doi: 10.1016/j.transproceed.2008.03.137. [DOI] [PubMed] [Google Scholar]
- 28.Morisseau C, Hammock BD. Epoxide hydrolases: mechanisms, inhibitor designs, and biological roles. Annu Rev Pharmacol Toxicol. 2005;45:311–333. doi: 10.1146/annurev.pharmtox.45.120403.095920. [DOI] [PubMed] [Google Scholar]
- 29.Fleming I. The pharmacology of the cytochrome P450 epoxygenase/soluble epoxide hydrolase axis in the vasculature and cardiovascular disease. Pharmacol Rev. 2014;66(4):1106–1140. doi: 10.1124/pr.113.007781. [DOI] [PubMed] [Google Scholar]
- 30.Motilva V, Alarcon de la Lastra C, Bruseghini L, Manuel Herrerias J, Sanchez-Fidalgo S. COX expression and PGE(2) and PGD(2) production in experimental acute and chronic gastric lesions. International immunopharmacology. 2005;5(2):369–379. doi: 10.1016/j.intimp.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 31.Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294(5548):1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 32.Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc Natl Acad Sci U S A. 2005;102(28):9772–9777. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Y, Dang H, Li D, Pang W, Hammock BD, Zhu Y. Inhibition of soluble epoxide hydrolase attenuates high-fat-diet-induced hepatic steatosis by reduced systemic inflammatory status in mice. PLoS One. 2012;7(6):e39165. doi: 10.1371/journal.pone.0039165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hammock BD, Wagner K, Inceoglu B. The soluble epoxide hydrolase as a pharmaceutical target for pain management. Pain management. 2011;1(5):383–386. doi: 10.2217/pmt.11.47. [DOI] [PubMed] [Google Scholar]
- 35.Drewnowski A, Pierce B, Halmi KA. Fat aversion in eating disorders. Appetite. 1988;10(2):119–131. doi: 10.1016/0195-6663(88)90063-3. [DOI] [PubMed] [Google Scholar]
- 36.Van Binsbergen CJ, Hulshof KF, Wedel M, Odink J, Coelingh Bennink HJ. Food preferences and aversions and dietary pattern in anorexia nervosa patients. Eur J Clin Nutr. 1988;42(8):671–678. [PubMed] [Google Scholar]
- 37.Steinglass JE, Sysko R, Mayer L, Berner LA, Schebendach J, Wang Y, Chen H, Albano AM, Simpson HB, Walsh BT. Pre-meal anxiety and food intake in anorexia nervosa. Appetite. 2010;55(2):214–218. doi: 10.1016/j.appet.2010.05.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shih PB, Yang J, Morisseau C, German JB, Zeeland AA, Armando AM, Quehenberger O, Bergen AW, Magistretti P, Berrettini W, et al. Dysregulation of soluble epoxide hydrolase and lipidomic profiles in anorexia nervosa. Molecular psychiatry. 2016;21(4):537–546. doi: 10.1038/mp.2015.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shih PA. Application of Sequencing, Fatty Acid Profiling, and Metabolomics Investigations to Explore Pathogenesis and Treatment Strategy for Anorexia Nervosa. Paper presented at the Hot Topics Session, American College of Neuropsychopharmacology Annual Meeting; Hollywood, FL. 2013. [Google Scholar]
- 40.Shih PA, Morisseau C, Yang J, Inceoglu AB, Bailer U, Van Zeeland A, Bergen A, Magistretti P, Berrettini W, Halmi K, et al. A Pilot Study of Soluble Epoxide Hydrolase Activity in Eating Disorders. Neuropsychopharmacology. 2014;39:S323–S324. [Google Scholar]
- 41.Yang J, Hammock BD, Halmi K, Woodside B, German B, Schork N, Bailer U, Kaye W, Morisseau C, Shih PB. Substrate-dependent Postprandial Oxylipin Responses Reveal the Potential Role of Nutrient-Gene Interaction in Anorexia Nervosa. In. Neuropsychopharmacology. 2015;40:S302. [Google Scholar]
- 42.Kaye WH, Bulik CM, Plotnicov K, Thornton L, Devlin B, Fichter MM, Treasure J, Kaplan A, Woodside DB, Johnson CL, et al. The genetics of anorexia nervosa collaborative study: methods and sample description. Int J Eat Disord. 2008;41(4):289–300. doi: 10.1002/eat.20509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wagner A, Simmons AN, Oberndorfer TA, Frank GK, McCurdy-McKinnon D, Fudge JL, Yang TT, Paulus MP, Kaye WH. Altered sensitization patterns to sweet food stimuli in patients recovered from anorexia and bulimia nervosa. Psychiatry Res. 2015;234(3):305–313. doi: 10.1016/j.pscychresns.2015.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morisseau C, Hammock B. Measurements of soluble epoxide hydrolase (sEH) activity. Curr Protoc Toxicol. 2007;33:4.23 1–18. doi: 10.1002/0471140856.tx0423s33. [DOI] [PubMed] [Google Scholar]
- 45.Quehenberger O, Armando AM, Dennis EA. High sensitivity quantitative lipidomics analysis of fatty acids in biological samples by gas chromatography-mass spectrometry. Biochimica et biophysica acta. 2011;1811(11):648–656. doi: 10.1016/j.bbalip.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zivkovic AM, Yang J, Georgi K, Hegedus C, Nording ML, O’Sullivan A, German JB, Hogg RJ, Weiss RH, Bay C, et al. Serum oxylipin profiles in IgA nephropathy patients reflect kidney functional alterations. Metabolomics: Official journal of the Metabolomic Society. 2012;8(6):1102–1113. doi: 10.1007/s11306-012-0417-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang J, Dong H, Hammock BD. Profiling the regulatory lipids: another systemic way to unveil the biological mystery. Curr Opin Lipidol. 2011;22(3):197–203. doi: 10.1097/MOL.0b013e3283468c10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang J, Schmelzer K, Georgi K, Hammock BD. Quantitative profiling method for oxylipin metabolome by liquid chromatography electrospray ionization tandem mass spectrometry. Analytical chemistry. 2009;81(19):8085–8093. doi: 10.1021/ac901282n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Newman JW, Morisseau C, Hammock BD. Epoxide hydrolases: their roles and interactions with lipid metabolism. Progress in lipid research. 2005;44(1):1–51. doi: 10.1016/j.plipres.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 50.Bloss CS, Berrettini W, Bergen AW, Magistretti P, Duvvuri V, Strober M, Brandt H, Crawford S, Crow S, Fichter MM, et al. Genetic association of recovery from eating disorders: the role of GABA receptor SNPs. Neuropsychopharmacology. 2011;36(11):2222–2232. doi: 10.1038/npp.2011.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee JP, Yang SH, Kim DK, Lee H, Kim B, Cho JY, Yu KS, Paik JH, Kim M, Lim CS, et al. In vivo activity of epoxide hydrolase according to sequence variation affects the progression of human IgA nephropathy. Am J Physiol Renal Physiol. 2011;300(6):F1283–1290. doi: 10.1152/ajprenal.00733.2010. [DOI] [PubMed] [Google Scholar]
- 52.Fife KL, Liu Y, Schmelzer KR, Tsai HJ, Kim IH, Morisseau C, Hammock BD, Kroetz DL. Inhibition of soluble epoxide hydrolase does not protect against endotoxin-mediated hepatic inflammation. The Journal of pharmacology and experimental therapeutics. 2008;327(3):707–715. doi: 10.1124/jpet.108.142398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wade TD, Gordon S, Medland S, Bulik CM, Heath AC, Montgomery GW, Martin NG. Genetic variants associated with disordered eating. Int J Eat Disord. 2013;46(6):594–608. doi: 10.1002/eat.22133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kaye WH, Lilenfeld LR, Berrettini WH, Strober M, Devlin B, Klump KL, Goldman D, Bulik CM, Halmi KA, Fichter MM, et al. A search for susceptibility loci for anorexia nervosa: methods and sample description. Biol Psychiatry. 2000;47(9):794–803. doi: 10.1016/s0006-3223(99)00240-1. [DOI] [PubMed] [Google Scholar]
- 55.Lands B. Consequences of essential fatty acids. Nutrients. 2012;4(9):1338–1357. doi: 10.3390/nu4091338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Buczynski MW, Dumlao DS, Dennis EA. Thematic Review Series: Proteomics. An integrated omics analysis of eicosanoid biology. J Lipid Res. 2009;50(6):1015–1038. doi: 10.1194/jlr.R900004-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arnold C, Markovic M, Blossey K, Wallukat G, Fischer R, Dechend R, Konkel A, von Schacky C, Luft FC, Muller DN, et al. Arachidonic acid-metabolizing cytochrome P450 enzymes are targets of {omega}-3 fatty acids. J Biol Chem. 2010;285(43):32720–32733. doi: 10.1074/jbc.M110.118406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Probst-Schendzielorz K, Viviani R, Stingl JC. Effect of Cytochrome P450 polymorphism on the action and metabolism of selective serotonin reuptake inhibitors. Expert Opin Drug Metab Toxicol. 2015;11(8):1219–1232. doi: 10.1517/17425255.2015.1052791. [DOI] [PubMed] [Google Scholar]
- 59.Persson A, Sim SC, Virding S, Onishchenko N, Schulte G, Ingelman-Sundberg M. Decreased hippocampal volume and increased anxiety in a transgenic mouse model expressing the human CYP2C19 gene. Molecular psychiatry. 2014;19(6):733–741. doi: 10.1038/mp.2013.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Penas-Lledo EM, Dorado P, Aguera Z, Gratacos M, Estivill X, Fernandez-Aranda F, Llerena A. High risk of lifetime history of suicide attempts among CYP2D6 ultrarapid metabolizers with eating disorders. Molecular psychiatry. 2011;16(7):691–692. doi: 10.1038/mp.2011.5. [DOI] [PubMed] [Google Scholar]