

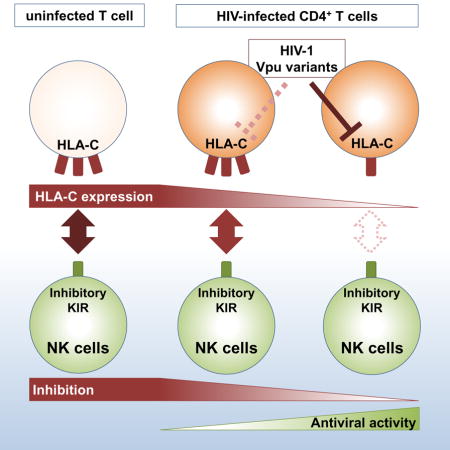
Summary
It was widely accepted that HIV-1 downregulates HLA-A/B to avoid CTL recognition while leaving HLA-C unaltered in order to prevent NK-cell activation by engaging inhibitory NK-cell receptors, but it was recently observed that most primary isolates of HIV-1 can mediate HLA-C downmodulation. Now we report that HIV-1-mediated downmodulation of HLA-C was associated with reduced binding to its respective inhibitory receptors. Despite this, HLA-C-licensed NK cells displayed reduced antiviral activity compared to their unlicensed counterparts, potentially due to residual binding to the respective inhibitory receptors. Nevertheless, NK cells were able to sense alterations of HLA-C expression demonstrated by increased antiviral activity when exposed to viral strains with differential abilities to downmodulate HLA-C. These results suggest that the capability of HLA-C-licensed NK cells to control HIV-1 replication is determined by the strength of KIR/HLA-C interactions and is thus dependent on both host genetics and the extent of virus-mediated HLA-C downregulation.
eTOC Blurb
HIV-1 modulates the expression of ligands for receptors expressed by natural killer (NK) cells on infected cells to evade NK-cell recognition. Körner et al. demonstrate that NK cells are able to sense HIV-1-mediated downmodulation of HLA-C through decreased binding of inhibitory KIR receptors to HLA-C.
Introduction
Natural killer (NK) cells represent an integral component of the innate immune system with a critical involvement in antiviral immunity (Jost and Altfeld, 2013). The initial control of viral infections by NK cells is mediated through direct cellular and antibody-mediated cytotoxicity and early production of pro-inflammatory cytokines, resulting in the elimination of virus-infected cells (Biron et al., 1999). Activation of NK cells is tightly regulated through a plethora of activating and inhibitory receptors balancing self-tolerance and effective responses against viral infections, ultimately defining the activation threshold of each NK cell (Long et al., 2013).
One important family of NK-cell receptors is the killer-cell immunoglobulin-like receptor (KIR) family, comprising 14 activating and inhibitory receptors. Although all KIR share common structural features, they exhibit differential characteristics in terms of expression, signaling pathways and ligand specificity. HLA class I molecules serve as natural ligands for KIR, however each KIR itself exhibits affinities for only specific types of HLA class I. For example, KIR3DL2 recognizes only a few HLA-A molecules, whereas KIR3DL1 only interacts with molecules carrying the serological Bw4 motif (Hansasuta et al., 2004; Lanier et al., 1995). In contrast, virtually all HLA-C molecules serve as ligands for members of the KIR2DL family but with varying affinities (Colonna et al., 1993). HLA-C molecules can be subdivided into two groups with distinct affinities for KIR2DL receptors (Biassoni et al., 1995). KIR2DL1 binds HLA-C group 2 molecules with high affinity, whereas KIR2DL3 recognizes predominantly HLA-C group 1 allotypes.
Engagement of inhibitory KIR via self HLA class I prevents auto-reactivity of NK cells but is additionally associated with the acquisition of functional competence during development, a process termed licensing (Elliott and Yokoyama, 2011). In this model, exposure to target-cells lacking self HLA class I results in increased response rates of NK cells expressing inhibitory KIR for self HLA class I whereas NK cells lacking self-inhibitory KIR remain hyporesponsive.
Multiple genetic association studies identified specific KIR/HLA haplotypes that influence the outcome of viral infections, most prominently HIV-1 infection (Khakoo, 2004; Martin et al., 2002, 2007). The inhibitory receptor KIR3DL1 and its activating counterpart KIR3DS1 were both associated with delayed progression to AIDS in combination with certain HLA-Bw4 alleles (Martin et al., 2002, 2007). While the protective effects of certain KIR3DL1/S1/HLA-Bw4 combinations in HIV-1 infection have been confirmed in additional experimental studies (Alter et al., 2007, 2009), accumulating evidence has drawn attention to KIR/HLA interactions between HLA-C and its corresponding KIR2DL receptors. In HIV-1-infected patients, specific KIR2DL2/3 genotypes were associated with HIV-1 sequence mutations indicating NK-cell-mediated immune pressure (Alter et al., 2011). In addition, it was shown that cell surface HLA-C expression levels were associated with protection against multiple outcomes of HIV-1 infection (Apps et al., 2013).
Until recently, HIV-1-mediated alterations of HLA class I molecules were thought to be limited to HLA-A and -B molecules, sparing HLA-C to evade NK-cell-mediated immune pressure by engaging inhibitory KIR2DL receptors (Cohen et al., 1999). A study recently refined this paradigm, showing that HIV-1 exhibits the ability to downmodulate HLA-C via the accessory protein Vpu (Apps et al., 2016). Little is known about the consequences for NK-cell-mediated control of HIV-1 infection and the potential contribution of NK cells expressing KIR2DL receptors. Therefore, we investigated the role of the inhibitory KIR2DL receptors in target-cell recognition and inhibition of viral replication in the context of HIV-1-mediated downmodulation of HLA-C.
Results
HIV-1 modulates HLA-C expression on infected CD4+ T cells
First, we investigated the effects of HIV-1 infection on the expression of HLA-C (Figure 1). Primary CD4+ T cells were infected with several primary clones and laboratory strains of HIV-1. Infection with the lab-adapted strain NL4-3 or the primary isolate CH293 were not associated with significant downmodulation of HLA-C on infected CD4+ T cells (Figure 1A). In contrast, CH077, CH198 as well as the lab-adapted strain JR-CSF were able to robustly downmodulate HLA-C (Figure 1B). Subsequent experiments were carried out using JR-CSF as the extent of HLA-C downmodulation was most pronounced with this strain. Since basal HLA-C expression levels have been associated with differential control of HIV-1 infection, we looked for a potential correlation between baseline HLA-C expression and the extent of HIV-1-mediated downmodulation. Independent of basal levels of HLA-C, infection with JR-CSF was associated with a consistent decrease of HLA-C surface levels (Figure 1C, p<0.0001). Overall, higher expression of HLA-C before HIV-1 infection resulted in higher expression after HIV-1 infection as indicated by the significant correlation between HLA-C surface density of uninfected and infected CD4+ T cells (Figure 1D; rs=0.91, p<0.0001). Interestingly, CD4+ T cells with high HLA-C surface levels were subject to a greater degree of HIV-1-mediated downmodulation of HLA-C (Figure 1E; rs=0.59, p=0.003). Taken together, our results showed that the ability and extent of HIV-mediated HLA-C downmodulation is not only strain-dependent as previously described but was additionally determined by baseline HLA-C surface expression.
Figure 1. HIV-1 clones differentially modulate HLA-C expression after in vitro infection.

Effects of HIV-1 infection on HLA-C surface expression on primary CD4+ T cells as determined by flow cytometry. (A) HLA-C levels of infected and uninfected cells after infection with NL4-3 and JR-CSF. (B) Extent of HIV-1-mediated HLA-C downmodulation across various HIV-1 clones as determined by the ratio between HLA-C expression (median fluorescence intensity, MFI) of uninfected and infected cells (n=6). Box plots define median and IQR. (C) Baseline HLA-C expression of uninfected CD4+ T cells (clear) and after infection with JR-CSF (red) (n=24). Black bars represent the median. (D) Correlation analysis of HLA-C levels (MFI) between uninfected and JR-CSF-infected CD4+ T cells. (E) Correlation analysis between baseline HLA-C expression levels of uninfected CD4+ T cells and the extent of HIV-1-mediated HLA-C downmodulation. Statistical analyses: Wilcoxon matched-pairs signed rank test and Spearman rank correlation. See also Figure S3.
Decreased KIR binding through HIV-1-mediated downmodulation of HLA-C
HLA-C serves as the natural ligand for the inhibitory receptors KIR2DL1 and KIR2DL3. Therefore, HLA-C downmodulation may impact the ability of the respective inhibitory KIR to bind to infected CD4+ T cells. In order to study the potential effects on KIR binding, KIR-IgG fusion proteins of KIR2DL1 and KIR2DL3 were used to assess binding to infected and uninfected CD4+ T cells (Figure 2). As KIR2DL1 and KIR2DL3 display distinct affinities for HLA-C allotypes carrying either the C1 or C2 epitope, subjects were stratified according to their HLA-C group haplotype (C1Hom, C1/C2 and C2Hom).
Figure 2. HIV-1 infection alters binding of KIR2DL1 and KIR2DL3 to HLA-C.

KIR binding to HIV-1-infected and uninfected CD4+ T cells measured by flow cytometry and stratified by donor HLA-C haplotypes (C2Hom: n=9; C1/2: n=11; C1Hom: n=12). (A and B) KIR binding to infected cells after infection with JR-CSF (KIR2DL1: blue; KIR2DL3: yellow) as compared to uninfected cells (clear) displayed as relative fluorescence intensity. Black bars represent the median. (C) Correlation analysis between HIV-1-associated reduction of HLA-C expression and KIR binding on infected cells given as fold change reduction compared to uninfected cells. KIR2DL1 binding to cells from C2Hom donors (n=9) are displayed in blue, KIR2DL3 binding to cells from C1Hom donors (n=12) in yellow. Statistical analyses: Wilcoxon matched-pairs signed rank test and Spearman rank correlation. See also Figure S1 and S3.
Whereas KIR2DL1 bound to CD4+ T cells from HLA-C2+ donors, virtually no binding was observed to HLA-C1Hom cells (Figure 2A; p<0.0001). HIV-1 infection resulted in a significant decrease in KIR2DL1 binding to infected CD4+ T cells from HLA-C2+ donors (C2Hom: p=0.004; C1/C2: p=0.001). KIR2DL1 binding to infected cells from HLA-C2Hom donors remained detectable, whereas KIR2DL1 binding to infected cells from heterozygous donors was reduced to a greater extent and nearly abrogated in some cases. Similar findings were observed for KIR2DL3 (Figure 2B). Whereas CD4+ T cells from HLA-C2Hom donors did not allow for KIR2DL3 binding, binding of KIR2DL3 was detectable for cells from HLA-C1+ subjects. Of note, overall levels of KIR binding were significantly lower for KIR2DL3 than for KIR2DL1 (p<0.0001, C1Hom vs. C2Hom; p<0.0002, C1/C2), potentially reflecting reduced avidity of KIR2DL3 to HLA-C1 and/or overall lower expression levels of HLA-C1 than HLA-C2 molecules. Nevertheless, HIV-1 infection was associated with significantly reduced KIR2DL3 binding (C1Hom: p=0.0005; C1/C2: p=0.004). KIR binding was almost abrogated for both groups expressing either one or two HLA-C group 1 alleles. Finally, potential associations between HLA-C downmodulation and reduced KIR binding were investigated. Relative reduction in KIR binding strongly correlated with the relative reduction of HLA-C expression (Figure 2C; C1Hom: rs=0.78, p=0.004; C2Hom: rs=0.78, p=0.017). Of note, infection with NL4-3, a strain unable to downmodulate HLA-C, was not associated with decreased KIR binding to infected cells (Figure S1). Altogether, our results demonstrate that reduced KIR binding to HIV-1-infected CD4+ T cells was predominantly caused by HIV-1-mediated downmodulation of HLA-C.
Impaired antiviral activity of HLA-C-licensed KIR2DL+ NK cells
The function of NK cells is tuned by the interaction of inhibitory receptors and their cognate HLA class I ligands (licensing). KIR2DL1+ and KIR2DL3+ NK cells gain functional competence in the presence of the respective HLA-C group ligands (2DL+ Self) whereas NK cells remain hyporesponsive in the absence of a cognate ligand (2DL+ Nonself). To determine the licensing status of NK-cell subsets, levels of degranulation (CD107a) and cytokine production (TNF-α) were assessed after exposure to the MHC class I devoid cell line 721.221 (Figure 3A). NK cells were stratified based on the expression of KIR2DL1 and KIR2DL3 and their underlying donor HLA-C group haplotype (2DL+ Self; 2DL+ Nonself). Bulk NK cells and NK cells lacking both KIR2DL1 and KIR2DL3 served as internal controls (Bulk; 2DL(−)). Licensed KIR2DL+ NK cells displayed higher frequencies of CD107a+ cells as compared to unlicensed KIR2DL+ NK cells (Figure 3A; p=0.0005, Self vs. Nonself). Similar results were obtained with respect to the frequency of TNF-α-producing NK cells (p=0.01, Self vs. Nonself). The general functional pattern of licensed and unlicensed KIR2DL+ NK cells was not altered when including C1/C2 heterozygous donors and disregarding alternative licensing mediated through HLA-E/NKG2A and HLA-Bw4/KIR3DL1 (Figure S2).
Figure 3. HLA-C-licensed KIR2DL+ NK cells display impaired antiviral activity.

(A) Assessment of HLA-C-mediated licensing of NK cells determined by frequency of CD107a expression and TNF-α production (left panel). Frequency of CD107a+ (middle panel) and TNF-α-producing cells (right panel) among different NK-cells subsets (n=12; C2Hom: n=4; C1Hom: n=8). (B) Antiviral capacity of NK-cell subsets. KIR2DL1+ and KIR2DL3+ NK cells were sorted by FACS (left panel). Viral loads in the presence or absence of bulk NK cells (middle panel). Level of NK-cell mediated inhibition of HIV-1 replication exhibited by NK-cell subsets as compared to CD4+ T cells alone (right panel). All donors were KIR haplotype A to avoid confounding effects of activating KIR and KIR2DL2 and homozygous for either HLA-C group (C1hom: n=6, C2hom: n=2). (A and B) Licensed (2DL+ Self) and unlicensed KIR2DL+ NK cells (2DL+ Nonself) are defined by the expression of KIR2DL1 and KIR2DL3 and the donor HLA-C haplotype (2DL+ Self: C2Hom: KIR2DL1+/L3(−), C1Hom: KIR2DL3+/L1(−); 2DL+ Nonself: C2Hom: KIR2DL1(−)/L3(+); C1Hom: KIR2DL3(−)/L1(+)). Bulk and KIR2DL1(−)/L3(−) NK cells (2DL(−)) served as internal controls. (C) Antiviral capacity of bulk NK cells in the presence or absence of KIR2DL antibodies (left panel; n=17: C1Hom: n=5, C1/C2: n=9, C2Hom: n=3). Viral loads in the presence or absence of NK cells (middle panel) or in the presence of KIR antibodies that do (α2DL Self) or do not (α2DL Nonself) bind to donor HLA-C ligands (right panel). α2DL Self indicates addition of α2DL1 to cells from HLA-C2+ donors or α2DL3 to cells from HLA-C1+ donors; α2DL Nonself indicates addition of α2DL1 to cells from HLA-C1Hom donors or α2DL3 to cells from HLA-C2Hom donors. Black bars represent the median. Statistical analyses: Wilcoxon matched-pairs signed rank test. See also Figure S2 and S3.
Next, we investigated whether HLA-C-licensed KIR2DL+ NK cells were able to exert superior antiviral activity in comparison to their hyporesponsive counterparts. NK cells were sorted based on the expression of the respective self-inhibitory KIR2DL receptors (Bulk, 2DL+ Self, 2DL+ Nonself, 2DL(−)) and co-cultured with autologous HIV-1-infected CD4+ T cells (Figure 3B). Antiviral activity was assessed by quantification of NK-cell-mediated inhibition of HIV-1 replication. Bulk NK cells from the majority of donors were able to inhibit HIV-1 replication (p=0.04); however, analysis of the sorted NK-cell subsets revealed differential antiviral activity. 2DL(−) and 2DL+ Nonself NK-cell subsets exerted comparable levels of anti-HIV-1 activity similar to the bulk population, whereas licensed KIR2DL+ NK cells (2DL+ Self) were unable to inhibit HIV-1 replication to the same degree (p=0.005 vs. 2DL+ Nonself). We sought to confirm that these results were due to the expression of self-inhibitory KIR2DL receptors. Therefore, bulk NK cells were co-cultured with autologous HIV-1-infected CD4+ T cells in the presence or absence of antibodies recognizing either KIR2DL1 or KIR2DL3 (Figure 3C). Presence of bulk NK cells was associated with reduced viral loads (p=0.0002). Addition of antibodies that block interaction between KIR2DL receptors and their HLA-C ligand led to additional reduction of viral loads as compared to NK cells alone indicating increased inhibition of viral replication (α2DL Self, p=0.0002). In contrast, no effects on HIV-1 replication were observed in the presence of αKIR2DL in co-cultures in which the targeted KIR2DL receptor had no corresponding HLA-C ligand (α2DL Nonself, p=0.8). Overall, our results indicate that HLA-C-licensed KIR2DL+ NK cells show impaired antiviral activity compared to their unlicensed counterparts due to the interaction between self-inhibitory KIR2DL receptors and their respective HLA-C ligands.
HLA-C downmodulation leads to improved antiviral activity of NK cells
Most primary HIV-1 clones are able to downmodulate HLA-C to various degrees, whereas infection with the lab-adapted strain NL4-3 did not result in reduced HLA-C surface density. Thus, we sought to compare HIV-1 strains with differential abilities to downmodulate HLA-C and to investigate the consequences for KIR binding and NK-cell-mediated antiviral activity (Figure 4). We demonstrated that the lab-adapted strain JR-CSF was able to robustly downmodulate HLA-C. Based on the HIV-1 JR-CSF full-length genome, a mutant strain (Vpu mut) was generated comprising amino acid residues derived from the Vpu sequence of NL4-3. Infection with wildtype JR-CSF resulted in downmodulation of HLA-C (Figure 4A). In contrast, Vpu mut was unable to reduce HLA-C surface expression to the same degree (p=0.008 vs. wt). As a result, decrease of KIR2DL binding to infected CD4+ T cells was far less pronounced when using Vpu mut as compared to wildtype JR-CSF (Figure 4B, p=0.001).
Figure 4. NK cells are able to sense HIV-mediated changes in HLA-C expression.

(A) Comparison between JR-CSF wildtype (wt) and the Vpu mutant strain (Vpu mut) in terms of their ability to downmodulate HLA-C. Vpu mut contained the Vpu protein sequence of NL4-3 in position 4 and 5 (L4I+Q5del). Relative HLA-C expression of CD4+ T cells infected with either wt (clear) or Vpu mut (red) as compared to uninfected cells, given as fold change (n=8). HLA-C expression was quantified as median fluorescence intensity. (B) KIR2DL binding (KIR2DL1, KIR2DL3) to CD4+ T cells infected with either wt (clear) or Vpu mut (red) as compared to uninfected cells, given as fold change (n=11; KIR2DL1: n=6; KIR2DL3: n=5). KIR binding was quantified as relative fluorescence intensity. (C) Levels of NK-cell-mediated inhibition of HIV-1 replication after 7d co-culture with CD4+ T cells infected with either wt or Vpu mut (n=8). Inhibition of HIV-1 replication was calculated from viral loads (copies/ml) in the presence or absence of NK cells. (D) Frequency of CD107a+ NK cells after 6h co-incubation with various target cells. NK cells single positive (sp) for KIR2DL1, KIR2DL3, KIR3DL1 and NKG2A were identified by Boolean gating. Licensed KIR2DL+ NK cells were defined as KIR2DL1sp NK cells from C2+ donors (n=3) or KIR2DL3sp NK cells from C1+ donors (n=8) (grey box plots). Unlicensed NK cells lacked all mentioned inhibitory receptors (white box plots). (E) HIV-specific responses of licensed KIR2DL+ NK cells (n=11) after exposure to CD4+ T cells infected with either wildtype (clear) or Vpu mut (red). Absolute HIV-specific responses are given as % CD107a+ NK cells after adjustment for non-HIV-1-specific responses (NK cells alone & mock-infected CD4+ T cells). (F) Comparison of the relative HIV-specific responses of licensed KIR2DL+ NK cells after exposure to CD4+ T cells infected with either wildtype (clear) or Vpu mut (red). Relative HIV-1-specific responses are given as fold change compared to HIV-specific responses of unlicensed NK cells. Black bars represent the median. Box plots represent median and IQR. Statistical analyses: Wilcoxon matched-pairs signed rank test. See also Figure S3.
Next, potential effects on the antiviral activity of NK cells were tested (Figure 4C). The ability of bulk NK cells to inhibit HIV-1 replication was significantly stronger when exposed to CD4+ T cells infected with wildtype JR-CSF as compared to Vpu mut (p=0.016). We subsequently assessed HIV-1-specific responses of NK cells measuring CD107a expression after short-term exposure to CD4+ T cells infected with either JR-CSF wildtype or Vpu mut. Our results showed low levels of degranulation of both, licensed KIR2DL+ and unlicensed NK cells, either spontaneously or after exposure to mock-infected CD4+ T cells (Figure 4D). Co-incubation with HIV-1-infected CD4+ T cells or MHC class I devoid 721.221 was associated with increased CD107a expression frequencies. Importantly, exposure to wildtype-infected CD4+ T cells resulted in higher HIV-1-specific responses by KIR2DL+ NK cells than exposure to Vpu mutant-infected CD4+ T cells (p=0.02), whereas no differences were observed for licensed KIR3DL1+ (p=0.4, data not shown) or unlicensed NK cells (p=0.08, data not shown) (Figure 4E). This was also the case when HIV-1-specific responses were compared to the response rates of unlicensed NK cells of the same sample as an additional internal control (Figure 4F; KIR2DL+: p=0.005). Overall, licensed KIR2DL+ NK cells exhibited lower levels of CD107a compared to unlicensed NK cells in response to HIV-1. Taken together, the results indicate that NK cells expressing self-inhibitory KIR for HLA-C are able to sense HIV-associated changes in HLA-C expression.
Discussion
Genetic association studies have revealed an important role for HLA-C in the outcome of HIV-1 infection, raising the question of whether NK cells expressing receptors for HLA-C are involved in the underlying mechanisms (Apps et al., 2013). The recent observation that most primary isolates of HIV-1 can mediate downmodulation of HLA-C refined the widely accepted paradigm that HIV-1 specifically downregulates HLA-A/B to avoid CTL recognition while leaving HLA-C unaltered in order to prevent NK-cell activation by engaging inhibitory NK-cell receptors (Cohen et al., 1999).
Whereas the overall ability and degree of HLA-C downmodulation has been associated with sequence variations in the N-terminal part of the accessory protein Vpu (Apps et al., 2016), it remained unclear whether differential HLA-C surface densities in primary target cells may affect the magnitude of HLA-C reduction. Assessment of HLA-C expression in our cohort showed donor-specific differential expression levels confirming previous reports (Apps et al., 2013). Furthermore, HLA-C surface density of infected CD4+ T cells was strongly associated with its expression level on uninfected cells maintaining the gradient of expression levels along different HLA-C alleles. This indicates that patients carrying viral strains with the ability to downmodulate HLA-C may not impact the population level estimates of the overall protective effects of high HLA-C expression levels. Of note, the degree of HIV-1-mediated downmodulation of HLA-C varied between donors and correlated with baseline expression levels suggesting that HIV-1 aims to efficiently target HLA-C for downmodulation in order to evade CTL-driven immune pressure. In fact, high expression levels have been associated with greater frequencies of HLA-C-associated CTL responses and increased degree of viral mutations (Apps et al., 2013; Blais et al., 2012). Overall, we demonstrate that the extent of HIV-1-mediated HLA-C downmodulation is not only strain dependent but also determined by baseline HLA-C expression levels. HIV-1-mediated decrease of HLA-C expression on infected cells leads to impaired recognition by HLA-C-restricted CTL and subsequent increase in viral replication in vitro (Apps et al., 2016). However, it was not known whether HLA-C reduction impacts target-cell recognition and antiviral activity of NK cells. Our results demonstrate that reduction in HLA-C expression was associated with decreased KIR binding irrespective of the underlying HLA-C group. Furthermore, our data suggests that baseline HLA-C expression levels and the affinity of KIR2DL receptors to its respective HLA-C ligand determine residual KIR binding to HIV-1-infected cells. KIR binding was still detectable on HIV-1-infected cells reflecting residual HLA-C expression and thus potentially able to inhibit NK cells through engagement of inhibitory KIR2DL receptors.
Next, we investigated the contribution of HLA-C-mediated licensing to the antiviral activity of NK cells. Initially, we demonstrated that HLA-C-licensed KIR2DL+ NK cells exhibited superior functionality after exposure to MHC class I devoid target cells in comparison to NK cells lacking self-inhibitory receptors. In contrast, direct assessment of the antiviral activity revealed a diminished capacity of HLA-C-licensed KIR2DL+ NK cells to inhibit HIV-1 replication as compared to their unlicensed counterparts. These findings are most likely attributed to residual KIR binding to HLA-C leading to inhibition of licensed NK cells, which in turn can be overcome by blockade of self-inhibitory KIR resulting in an improved antiviral activity. Similarly, higher frequencies of CD107a have been observed in NK cells lacking self-inhibitory KIR2DL receptors after exposure to HIV-1-infected primary T cells as well as increased NK-cell-mediated cytotoxicity against HIV-1-infected cells after blockade of inhibitory receptors such as KIR2DL1/2/3 and NKG2A (Bonaparte, 2004; Davis et al., 2016).
In the context of murine cytomegalovirus infection (MCMV), control of MCMV infection was mainly driven by unlicensed NK cells rather than licensed NK cells (Orr et al., 2010). This study suggests that the presence of self MHC class I on the surface of virus-infected cells prevents activation of licensed NK cells and hinders their contribution to viral control. Our results clearly show that NK cells are able to respond to HIV-1-mediated modulation of HLA-C class I with increased antiviral activity. It is therefore possible that alterations of MHC class I expression in some viral infections such as HIV-1 infection may lead to the activation of licensed NK cells. This hypothesis is supported by increased frequencies and expansion of NK cells expressing self-inhibitory KIR in patients with acute and primary HIV-1 infection (Alter et al., 2009; Körner et al., 2014). Delayed progression to AIDS was observed in patients carrying a combination of certain subtypes of the inhibitory receptor KIR3DL1 and their cognate HLA-Bw4 ligands (Martin et al., 2007). Furthermore, HIV-1-mediated downregulation of HLA-Bw4 was associated with significant HIV-1-specific responses of licensed KIR3LD1+ NK cells and was predicted by the strength of NK cell education (Boudreau et al., 2016). These data suggest that both licensed and unlicensed NK cells may be involved in HIV-1 control. Nevertheless, their relative contribution to the control of viral replication will be strongly influenced by the degree of virus-mediated alterations of HLA class I expression and the strength of KIR/HLA interactions.
Potential contributions of KIR/HLA interactions to viral evolution have been previously suggested. Whereas CTL escape mutations were shown to abrogate KIR3DL1 binding to HLA-B*57, a HLA-C*03:04-restricted HIV-1 sequence variant was associated with viral escape from KIR2DL3+ NK cells (Fadda et al., 2011; Hölzemer et al., 2015). Finally, Vpu sequence polymorphisms associated with the presence of KIR2DL2 were identified in a cohort of chronically infected individuals leading to enhanced binding of KIR2DL2 to HIV-infected CD4+ T cells (Alter et al., 2011). It is therefore possible that immune pressure exerted by licensed KIR2DL+ NK cells may lead to the differential modulation of HLA-C expression by selecting Vpu sequence variations as an additional escape mechanism to balance between CTL- and NK-cell-mediated immune pressure.
Taken together, the comprehensive analyses of HLA-C expression, KIR binding and subsequent functional assessment of NK cells presented in this study revealed that NK cells are capable of sensing HIV-1-mediated downmodulation of HLA-C through inhibitory KIR2DL receptors. Target-cell recognition was determined by initial HLA-C expression levels and overall avidity of the respective inhibitory KIR to their cognate HLA-C ligands and is thus dependent on both host genetics and the extent of virus-mediated HLA-C downregulation.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Christian Körner (christian.koerner@hpi.uni-hamburg.de).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subjects
For this study a total of 72 HIV-1-negative healthy subjects were enrolled. Recruitment of study participants took place at the Massachusetts General Hospital, at the Brigham and Women’s Hospital in Boston, USA and at the Heinrich Pette Institute, Leibniz Institute for Experimental Virology, in Hamburg, Germany. The study was approved by the respective local Institutional Review Boards. All individuals gave written informed consent for participation in this study. Study participants have been randomly selected for the study and then grouped subsequently based on the underlying KIR/HLA haplotypes and the available blood volume for each individual experiment (Table S1). Information on sex and age was not available for all subjects, however was not relevant for the purpose of the study.
Cell lines
The cell line LCL 721.221 (Burlingham et al., 1989) was used to assess to NK-cell function. The cell line was maintained in complete medium (RPMI-1640 medium (Sigma) supplemented with 10% (v/v) fetal bovine serum (FBS, Sigma), 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (All from Corning Cellgro)). The sex of the cell is female.
The cell line HEK293T/17 (DSMZ) was used to produce replication-competent HIV-1 virions. The cell line was maintained in complete medium (DMEM (Sigma) supplemented with 10% (v/v) fetal bovine serum (FBS, Sigma), 100 U/ml penicillin, 100 μg/ml streptomycin (Corning Cellgro)). The sex of the cell is female.
METHOD DETAILS
Sample processing
ACD- or EDTA-treated venous peripheral blood was obtained through phlebotomy from all participants. PBMC were isolated by density-gradient centrifugation within 2 hours of sample collection, washed and subsequently resuspended in complete medium (RPMI-1640 medium (Sigma) supplemented with 10% (v/v) fetal bovine serum (FBS, Sigma), 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (All from Corning Cellgro)). PBMC were directly used for experiments or cryopreserved in FBS supplemented with 10% (v/v) dimethyl sulfoxide for future experimental applications.
Enrichment of CD4+ T cells and NK cells
CD4+ T cells and primary NK cells were enriched from PBMC through negative-selection strategy (Stemcell Technologies) according to the manufacturer’s protocol. Purity of the enriched cell populations was verified by flow cytometry using fluorochrome-conjugated antibodies against CD3, CD4, CD56, CD16 and CD8. CD4+ T cells were resuspended in complete media at 5*106 cells/ml and stimulated with 100 IU/ml human recombinant IL-2 (hrIL-2; NIH) and 1 μg/ml phytohaemagglutinin (PHA, Fisher) for up to 3 days. NK cells were resuspended in complete medium supplemented with 1 ng/ml human recombinant IL-15 (hrIL-15; R&D systems).
Primary Infectious Molecular Clones of HIV-1
The following reagents were obtained through the National Institutes of Health (NIH) AIDS Reagent Program, Division of AIDS, NIAID, NIH: pYK-JRCSF from Dr. Irvin SY Chen and Dr. Yoshio Koyanagi and pNL4-3 from Dr. Malcolm Martin (Adachi et al., 1986; Cann et al., 1990; Haltiner et al., 1985; Koyanagi et al., 1987). Parental versions of the TF HIV-1 strains CH077, CH198, CH293 were obtained from Beatrice Hahn (University of Pennsylvania) and previously described (Ochsenbauer et al., 2012; Parrish et al., 2013; Rücker et al., 2004). In addition, JR-CSF was modified using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent, Santa Clara, USA) to introduce mutations in HIV-1 Vpu (Vpu mut). First, Leucine (L) at amino acid position 4 was exchanged to Isoleucine (I). Consecutively Glutamine (Q) at position 5 was deleted. Mutagenesis was performed using the following 5′ oligonucleotide primers (all from Integrated DNA Technologies, Coralville, USA):
JR-CSF VPU L to I Fw (aagtagtgcatgtaatgcaacctatacaaatattagcaatagtagca),
JR-CSF VPU L to I RV (tgctactattgctaatatttgtataggttgcattacatgcactactt),
JR-CSF VPU Q5 del Fw (gtagtgcatgtaatgcaacctataatattagcaatagtagcattagta),
JR-CSF VPU Q5 del Rv (tactaatgctactattgctaatattataggttgcattacatgcactac)
Primer positions of the mutations are according to the wild type JR-CSF sequence (GenBank: M38429.1). Generated plasmids were sequenced by Sanger sequencing (Seqlab, Goettingen, Germany), in order to ensure correct insertion of the desired mutation and lack of any unwanted mutations. Infectious viral particles were produced in 293T/17 HEK cells using lipofectamine transfection (Lipofectamine 3000, Invitrogen).
Antibodies and flow cytometry
Multiparameter flow cytometry was used for phenotypical and functional characterisation of NK and CD4+ T cells as well as for cell sorting of NK-cell subsets according to their expression of KIR2DL1 and KIR2DL3. In that regard a variety of antibodies and reagents have been utilized to determine viability, purity, phenotype and function of the cells of interest. A comprehensive list of all antibodies and reagents is provided in Table S2. The respective gating strategies are displayed in Figure S3.
Flow cytometric assessment of HLA class I expression
Expression levels of HLA-C on HIV-1-infected and uninfected CD4+ T cells were determined using the commercially available antibody clone DT9 in a two-step staining. Cells were initially incubated with mouse anti-HLA-C, followed by anti-mouse IgG and a viability dye. Stained cells were further washed and then treated for intracellular detection of HIV-1 p24 gag using Cytofix/Cytoperm solution kit (BD Biosciences) and anti-HIV-1 core antigen. Mouse-derived antibodies for cell surface markers CD3 and CD4 were applied after washing and fixation together with the anti-HIV-1 core antigen. Uninfected cells and HIV-1-infected cells were discriminated based on the expression of CD4 and intracellular p24 Gag.
KIR binding assays
KIR2DL1 and KIR2DL3 IgG1 fusion proteins were used to assess the ability of cells to bind KIR molecules. In brief, CD4+ T cells were stained with 10μg/ml KIR-Fc for 45 min at 4°C, washed and then incubated for additional 45 min with anti-human IgG-PE, CD3, CD4 and LIVE/DEAD Blue viability dye at 4°C. Stained cells were further washed and then treated for the intracellular detection of HIV-1 p24 gag using Cytofix/Cytoperm solution kit and anti-HIV-1 core antigen. Uninfected cells and HIV-1-infected cells were discriminated based on the expression of CD4 and intracellular p24 Gag.
Enrichment of HIV-1-infected cells
Enrichment of HIV-1-infected cells was performed as previously described (Davis et al., 2011). Modification of the protocol entailed enrichment of CD45RO+ CD4+ T cells from CD4+ T cells through negative selection strategy (Miltenyi) according to the manufacturer’s protocol to increase frequency of CCR5+ CD4+ T cells before infection. In brief, PHA-stimulated CD4+ T cells CD45RO+ CD4+ T cells were infected with either JR-CSF wildtype or Vpu mut at an MOI of 0.1 through spinfection. After 7 day incubation infected cells were enriched by depletion of CD4+ T cells through positive selection strategy (Stemcell) according to the manufacturer’s protocol.
Assessment of NK-cell function
Levels of NK-cell activation and cytokine production were determined through expression of CD107a on the surface of NK cells and intra-cellular detection of TNF-α as previously described (Alter et al., 2004; Körner et al., 2014). PBMC were cultured in the presence or absence of MHC class I devoid target cell line LCL 721.221 (Burlingham et al., 1989) at an effector:target ratio of 5:1 for a total of 6 hours in the presence of 2.5 μg/mL anti-CD107a. Monensin and Brefeldin A were added one hour after setup of the co-culture followed by additional 5 hours of incubation. Subsequent intracellular detection of TNF-α was performed using Cytofix/Cytoperm solution kit and anti-TNF-α. For the assessment of HIV-1-specific responses enriched NK cells were co-incubated with HIV-1-infected CD4+ T cells for 6 hours at an effector:target ratio of 1:2 in the presence of 2.5 μg/mL anti-CD107a. Cells were stained for viability, expression of CD3, CD14, CD19, CD16 and CD56 as well as for the NK-cell receptors KIR2DL1, KIR2DL2, KIR2DL3, KIR3DL1 and NKG2A.
Quantification of NK cell-mediated inhibition of HIV-1 replication was conducted as previously described (He et al., 2016). In brief, overnight cultured CD4+ T cells were infected with the wildtype or Vpu mut of JR-CSF at a multiplicity of infection (MOI) of 0.01. CD4+ T cells were then plated in a 96 V-bottom well plate (5*104 cells/well) in the absence or presence of NK cells. Bulk NK cells were added at effector:target (E:T) ratios of 5:1. Previously sorted NK-cell subsets were added at E:T ratios of 1:1. HIV-1 replication was then assessed by Quantification of HIV-1 RNA by quantitative real-time reverse transcriptase polymerase chain reaction (RT-qPCR). Isolation of viral RNA from culture supernatants was performed using QIAamp Viral RNA Kits (Qiagen). Purified viral RNA was quantified through amplification of HIV-1 gag with a detection limit of 10 viral RNA copies per microliter of supernatant. RT-qPCR was performed with the QuantiFast SYBR Green RT-PCR Kit (Qiagen) according to manufacturer’s instructions in a 96 or 384 well plate on a Roche Lightcycler 480. The protocol utilizes a combination of gag “SK” primers that are components of the Amplicor HIV-1 Monitor viral load test:
SK145 primer (forward): AGTGGGGGGACATCAAGCAGCCATGCAAAT (30 bp; 72.5 Tm);
SK431 primer (reverse): TGCTATGTCACTTCCCCTTGGTTCTCT (27 bp; 61.3 Tm).
2 ul of sample was used and sampling was performed in duplicate with a standard deviation of 0.5 between crossing threshold (Ct) numbers as quality control cut-off. Concentrations were calculated from a 10,000 copy number standard of HIV-1 HxB2.
QUANTIFICATION AND STATISTICAL ANALYSIS
Acquisition of flow cytometric data was performed on a BD LSR Fortessa (BD Biosciences, Table S2) and further analysed using FlowJo software v10.1 (Tree Star, Inc.). Flow cytometric sorting of NK-cell subsets was performed using a BD FACS Aria II SORP (BD Biosciences, Table S2). GraphPad Prism 6.0 (GraphPad Software, Inc.) was used for statistical analyses and graphical display of the data. Non-parametric statistical tests were applied to test for differences between groups. Wilcoxon matched-pairs signed rank test was used for two groups with paired values, the Mann-Whitney U test for independent values. For comparisons between >2 groups Kruskal-Wallis test was applied for independent measures, Friedman test for paired values. Dunn’s post-test was used to correct for multiple comparisons. Spearman rank correlation was used to test for association between two parameters. Statistical parameters are stated in the results section as well as in the figure legends.
Supplementary Material
Highlights.
HIV-1 clones differentially modulate HLA-C expression after in vitro infection
HIV-1 infection alters binding of inhibitory NK receptors KIR2DL1 and KIR2DL3 to HLA-C
HLA-C-licensed KIR2DL+ NK cells display impaired antiviral activity
NK cells are able to sense HIV-1-mediated downmodulation of HLA-C expression
Acknowledgments
The following reagents were obtained through the National Institutes of Health (NIH) AIDS Reagent Program, Division of AIDS, NIAID, NIH: pYK-JRCSF from Dr. Irvin SY Chen and Dr. Yoshio Koyanagi and pNL4-3 from Dr. Malcolm Martin. The authors gratefully acknowledge the support of the Ragon Institute Flow Cytometry core and Virology platform. This study was supported by the NIH (R01-AI067031-08) and the Pathogenesis Program of the Heinrich Pette Institute, a Leibniz Institute for Experimental Virology. P.S., A.H and J.C. were supported by the German Center for Infection Research (TI07.001,TI07.002, and TI07.003). S.L. was supported by the German Research Foundation (DFG) through the SFB841. E.P.S. was supported by a grant from the Creative and Novel Ideas in HIV Research Program (CNIHR) through a supplement to the University of California at San Francisco (UCSF) Center for AIDS Research funding (P30 AI027763). S.J. was supported by the NIH (R01-AI116363-03). F.K. is supported by an ERC Advanced grant (Anti-Virome) and the DFG. This project has been funded in whole or in part with federal funds from the Frederick National Laboratory for Cancer Research, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This Research was supported in part by the Intramural Research Program of the NIH, Frederick National Lab, Center for Cancer Research.
The authors declare no competing financial interests.
Footnotes
Author Contributions
C.K., E.P.S., S.J. and M.A. designed the experiments. C.K, C.R.S., P.S., M.G., M.Z., A.H., S.L., J.C. and B.C. conducted the experiments. C.K., E.P.S., S.J., V.N. and M.C. analyzed the data. C.K. wrote the manuscript. F.K. provided resources. D.S.K. and M.A. provided funding. All authors critically reviewed the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods. 2004;294:15–22. doi: 10.1016/j.jim.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Alter G, Martin MP, Teigen N, Carr WH, Suscovich TJ, Schneidewind A, Streeck H, Waring M, Meier A, Brander C, et al. Differential natural killer cell mediated inhibition of HIV-1 replication based on distinct KIR/HLA subtypes. J Exp Med. 2007;204:3027–3036. doi: 10.1084/jem.20070695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alter G, Rihn S, Walter K, Nolting A, Martin M, Rosenberg ES, Miller JS, Carrington M, Altfeld M. HLA Class I Subtype-Dependent Expansion of KIR3DS1+ and KIR3DL1+ NK Cells during Acute Human Immunodeficiency Virus Type 1 Infection. J Virol. 2009;83:6798–6805. doi: 10.1128/JVI.00256-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alter G, Heckerman D, Schneidewind A, Fadda L, Kadie CM, Carlson JM, Oniangue-Ndza C, Martin M, Li B, Khakoo SI, et al. HIV-1 adaptation to NK-cell-mediated immune pressure. Nature. 2011;476:96–100. doi: 10.1038/nature10237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apps R, Qi Y, Carlson JM, Chen H, Gao X, Thomas R, Yuki Y, Del Prete GQ, Goulder P, Brumme ZL, et al. Influence of HLA-C Expression Level on HIV Control. Science. 2013;340:87–91. doi: 10.1126/science.1232685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apps R, Del Prete GQ, Chatterjee P, Lara A, Brumme ZL, Brockman MA, Neil S, Pickering S, Schneider DK, Piechocka-Trocha A, et al. HIV-1 Vpu Mediates HLA-C Downregulation. Cell Host Microbe. 2016;19:686–695. doi: 10.1016/j.chom.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biassoni R, Falco M, Cambiaggi A, Costa P, Verdiani S, Pende D, Conte R, Donato CD, Parham P, Moretta L. Amino acid substitutions can influence the natural killer (NK)-mediated recognition of HLA-C molecules. Role of serine-77 and lysine-80 in the target cell protection from lysis mediated by “group 2” or “group 1” NK clones. J Exp Med. 1995;182:605–609. doi: 10.1084/jem.182.2.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu Rev Immunol. 1999;17:189–220. doi: 10.1146/annurev.immunol.17.1.189. [DOI] [PubMed] [Google Scholar]
- Blais ME, Zhang Y, Rostron T, Griffin H, Taylor S, Xu K, Yan H, Wu H, James I, John M, et al. High Frequency of HIV Mutations Associated with HLA-C Suggests Enhanced HLA-C-Restricted CTL Selective Pressure Associated with an AIDS-Protective Polymorphism. J Immunol. 2012;188:4663–4670. doi: 10.4049/jimmunol.1103472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaparte MI. Killing of human immunodeficiency virus-infected primary T-cell blasts by autologous natural killer cells is dependent on the ability of the virus to alter the expression of major histocompatibility complex class I molecules. Blood. 2004;104:2087–2094. doi: 10.1182/blood-2004-02-0696. [DOI] [PubMed] [Google Scholar]
- Boudreau JE, Mulrooney TJ, Le Luduec J-B, Barker E, Hsu KC. KIR3DL1 and HLA-B Density and Binding Calibrate NK Education and Response to HIV. J Immunol Baltim Md. 2016 doi: 10.4049/jimmunol.1502469. 1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burlingham WJ, Ceman SS, DeMars R. Secretion and cell surface expression of IgG1 are impaired in human B lymphoblasts that lack HLA-A,-B, and-C antigens. Proc Natl Acad Sci. 1989;86:8005–8009. doi: 10.1073/pnas.86.20.8005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Cann AJ, Zack JA, Go AS, Arrigo SJ, Koyanagi Y, Green PL, Koyanagi Y, Pang S, Chen IS. Human immunodeficiency virus type 1 T-cell tropism is determined by events prior to provirus formation. J Virol. 1990;64:4735–4742. doi: 10.1128/jvi.64.10.4735-4742.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen GB, Gandhi RT, Davis DM, Mandelboim O, Chen BK, Strominger JL, Baltimore D. The Selective Downregulation of Class I Major Histocompatibility Complex Proteins by HIV-1 Protects HIV-Infected Cells from NK Cells. Immunity. 1999;10:661–671. doi: 10.1016/s1074-7613(00)80065-5. [DOI] [PubMed] [Google Scholar]
- Colonna M, Borsellino G, Falco M, Ferrara GB, Strominger JL. HLA-C is the inhibitory ligand that determines dominant resistance to lysis by NK1- and NK2-specific natural killer cells. Proc Natl Acad Sci U S A. 1993;90:12000–12004. doi: 10.1073/pnas.90.24.12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis ZB, Ward JP, Barker E. Preparation and Use of HIV-1 Infected Primary CD4+ T-Cells as Target Cells in Natural Killer Cell Cytotoxic Assays. J Vis Exp. 2011 doi: 10.3791/2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis ZB, Cogswell A, Scott H, Mertsching A, Boucau J, Wambua D, Le Gall S, Planelles V, Campbell KS, Barker E. A Conserved HIV-1-Derived Peptide Presented by HLA-E Renders Infected T-cells Highly Susceptible to Attack by NKG2A/CD94-Bearing Natural Killer Cells. PLoS Pathog. 2016;12:e1005421. doi: 10.1371/journal.ppat.1005421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott JM, Yokoyama WM. Unifying concepts of MHC-dependent natural killer cell education. Trends Immunol. 2011;32:364–372. doi: 10.1016/j.it.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadda L, O’Connor GM, Kumar S, Piechocka-Trocha A, Gardiner CM, Carrington M, McVicar DW, Altfeld M. Common HIV-1 Peptide Variants Mediate Differential Binding of KIR3DL1 to HLA-Bw4 Molecules. J Virol. 2011;85:5970–5974. doi: 10.1128/JVI.00412-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haltiner M, Kempe T, Tjian R. A novel strategy for constructing clustered point mutations. Nucleic Acids Res. 1985;13:1015–1025. doi: 10.1093/nar/13.3.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansasuta P, Dong T, Thananchai H, Weekes M, Willberg C, Aldemir H, Rowland-Jones S, Braud VM. Recognition of HLA-A3 and HLA-A11 by KIR3DL2 is peptide-specific. Eur J Immunol. 2004;34:1673–1679. doi: 10.1002/eji.200425089. [DOI] [PubMed] [Google Scholar]
- He X, Simoneau CR, Granoff ME, Lunemann S, Dugast AS, Shao Y, Altfeld M, Körner C. Assessment of the antiviral capacity of primary natural killer cells by optimized in vitro quantification of HIV-1 replication. J Immunol Methods. 2016;434:53–60. doi: 10.1016/j.jim.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hölzemer A, Thobakgale CF, Jimenez Cruz CA, Garcia-Beltran WF, Carlson JM, van Teijlingen NH, Mann JK, Jaggernath M, Kang S, Körner C, et al. Selection of an HLA-C*03:04-Restricted HIV-1 p24 Gag Sequence Variant Is Associated with Viral Escape from KIR2DL3+ Natural Killer Cells: Data from an Observational Cohort in South Africa. PLoS Med. 2015;12:e1001900. doi: 10.1371/journal.pmed.1001900. discussion e1001900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost S, Altfeld M. Control of Human Viral Infections by Natural Killer Cells. Annu Rev Immunol. 2013;31:163–194. doi: 10.1146/annurev-immunol-032712-100001. [DOI] [PubMed] [Google Scholar]
- Khakoo SI. HLA and NK Cell Inhibitory Receptor Genes in Resolving Hepatitis C Virus Infection. Science. 2004;305:872–874. doi: 10.1126/science.1097670. [DOI] [PubMed] [Google Scholar]
- Körner C, Granoff ME, Amero MA, Sirignano MN, Vaidya SA, Jost S, Allen TM, Rosenberg ES, Altfeld M. Increased frequency and function of KIR2DL1-3 + NK cells in primary HIV-1 infection are determined by HLA-C group haplotypes: Innate immunity. Eur J Immunol. 2014;44:2938–2948. doi: 10.1002/eji.201444751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyanagi Y, Miles S, Mitsuyasu RT, Merrill JE, Vinters HV, Chen IS. Dual infection of the central nervous system by AIDS viruses with distinct cellular tropisms. Science. 1987;236:819–822. doi: 10.1126/science.3646751. [DOI] [PubMed] [Google Scholar]
- Lanier LL, Gumperz JE, Parham P, Melero I, López-Botet M, Phillips JH. The NKB1 and HP-3E4 NK cells receptors are structurally distinct glycoproteins and independently recognize polymorphic HLA-B and HLA-C molecules. J Immunol Baltim Md 1950. 1995;154:3320–3327. [PubMed] [Google Scholar]
- Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol. 2013;31:227–258. doi: 10.1146/annurev-immunol-020711-075005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MP, Gao X, Lee J-H, Nelson GW, Detels R, Goedert JJ, Buchbinder S, Hoots K, Vlahov D, Trowsdale J, et al. Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat Genet. 2002 doi: 10.1038/ng934. [DOI] [PubMed] [Google Scholar]
- Martin MP, Qi Y, Gao X, Yamada E, Martin JN, Pereyra F, Colombo S, Brown EE, Shupert WL, Phair J, et al. Innate partnership of HLA-B and KIR3DL1 subtypes against HIV-1. Nat Genet. 2007;39:733–740. doi: 10.1038/ng2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochsenbauer C, Edmonds TG, Ding H, Keele BF, Decker J, Salazar MG, Salazar-Gonzalez JF, Shattock R, Haynes BF, Shaw GM, et al. Generation of transmitted/founder HIV-1 infectious molecular clones and characterization of their replication capacity in CD4 T lymphocytes and monocyte-derived macrophages. J Virol. 2012;86:2715–2728. doi: 10.1128/JVI.06157-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr MT, Murphy WJ, Lanier LL. “Unlicensed” natural killer cells dominate the response to cytomegalovirus infection. Nat Immunol. 2010;11:321–327. doi: 10.1038/ni.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish NF, Gao F, Li H, Giorgi EE, Barbian HJ, Parrish EH, Zajic L, Iyer SS, Decker JM, Kumar A, et al. Phenotypic properties of transmitted founder HIV-1. Proc Natl Acad Sci U S A. 2013;110:6626–6633. doi: 10.1073/pnas.1304288110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rücker E, Grivel JC, Münch J, Kirchhoff F, Margolis L. Vpr and Vpu are important for efficient human immunodeficiency virus type 1 replication and CD4+ T-cell depletion in human lymphoid tissue ex vivo. J Virol. 2004;78:12689–12693. doi: 10.1128/JVI.78.22.12689-12693.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.