

Abstract
Relapses of aggressive B-cell lymphomas pose a higher risk to affected patients because of potential treatment resistance and usually rapid tumor growth. Recent advances, such as targeting Bruton tyrosine kinase, have provided promising results in small numbers of cases, but treatment for the majority of patients remains challenging and outcomes are generally poor. A number of recent studies have utilized state-of-the-art genomic technologies in an attempt to better understand tumor genome evolution during relapse and to identify relapse-specific genetic alterations. It has been found that in some settings (e.g. diffuse large B-cell lymphomas in immunocompromised patients, secondary diffuse large B-cell lymphomas as Richter transformations) a significant part of the recurrences are clonally-unrelated de novo neoplasms, which might have distinct genomic and drug sensitivity profiles as well as different prognoses. Similar to earlier findings in indolent lymphomas, genetic tumor evolution of clonally-related relapsing aggressive B-cell lymphomas is predominantly characterized by two patterns: early divergence from a common progenitor and late divergence/linear evolution from a primary tumor. The clinical implications of these distinct patterns are not yet clear and will require additional investigation; however, it is plausible that these two patterns of recurrence are linked to different treatment-resistance mechanisms. Attempts to identify drivers of relapses result in a very heterogeneous list of affected genes and pathways as well as epigenetic mechanisms and suggest many ways of how recurrent tumors can adapt to treatment and expand their malignant properties.
Introduction
Aggressive B-cell lymphomas are lymphoid neoplasms characterized by blastoid morphology and rapid tumor growth.1 As a whole, this group comprises multiple histologically and phenotypically defined diagnostic categories as well as “gray zone” categories. Despite their aggressive course, if properly diagnosed and treated, most aggressive B-cell lymphomas have favorable outcomes. For example, the most frequently occurring neoplasm within this category, diffuse large B-cell lymphoma (DLBCL), is treated by the now standard immunochemotherapy regimen, R-CHOP (rituximab, cyclophosphamide, hydroxydaunorubicin, oncovin, and prednisolone), and this treatment results in clinical remission or even cure of a large number of patients.2 However, approximately 30% to 40% of patients in whom initial therapy failed due to primary resistance (~15%) or relapse (~25%) have a poor prognosis and often succumb to disease.2,3 Treatments for such patients consist of more aggressive, high-dose chemotherapy regimens supported by autologous stem cell transplantation,4 which is associated with severe toxicity and risks of fatal infections. Therefore, only about 50% of relapsed patients can tolerate such intensive treatment, while others who are not eligible, because of advanced age or co-morbidities, are managed with experimental protocols or palliative care.5 Interestingly, while the addition of rituximab to first-line regimens improved therapy outcomes for the majority of DLBCL patients, its prior use is associated with worse outcomes after salvage chemotherapy and autologous stem cell transplantation in the subgroup of early relapsing patients.6
The majority of DLBCL recurrences arise during the first 3 years after initial therapy, although up to 10% of patients relapse >5 years after their initial diagnosis.7 It has been hypothesized that resistance/relapse occurs either due to intrinsic tumor heterogeneity, or as a consequence of ongoing genomic tumor evolution under the selective pressure of therapy, or both.8 An initial genetic heterogeneity of the tumor can confer a propensity to relapse due to usually minor tumor cell population(s) that have a pre-existing resistance to treatment (e.g. due to stemness properties9), and that can outgrow following extermination of the dominant non-resistant (and often more proliferative) clone.10 On the other hand, genomic tumor evolution can occur after treatment if the initial clone is not completely eradicated.11 This evolution can potentially be driven by intrinsic genomic/genetic instability of the tumor itself or by the genomic/genetic instability caused by alkylating- and DNA intercalating agents, which are part of the chemotherapy regimen (cyclophosphamide and hydroxydaunorubicin in CHOP) and, finally, consolidated by selective pressures exerted by the treatment.12
The group of refractory/relapsed aggressive B-cell lymphomas constitutes an unmet challenge in terms of delineating the underlying biological mechanisms, identifying prognostic markers to identify such patients before starting treatment and, most importantly, developing effective treatments capable of curing more patients. In this review, we summarize the latest findings on ancestry, genetic evolution and determinants of relapses of aggressive B-cell lymphomas.
Clonally-unrelated “relapse”
All B-cell lymphomas are clonal diseases resulting from the proliferation of a single B-cell (clone) that gives rise to a tumor. In fact, monoclonality is an important criterion to qualify a B-cell proliferation as a lymphoid neoplasm.13 While the definition of a cancer cell clone is a matter of ongoing debate in the field of solid tumors,14 it is widely accepted that lymphoid neoplasms sharing identical antigen-receptor gene rearrangements are clonally-related. This is because IG V(D)J recombination, generating a unique receptor gene sequence, occurs at an early stage of B-cell development, arguably earlier than genetic aberrations that initiate malignant proliferation appear, and is, therefore, irreversibly imprinted in the cell’s progeny. Thus, antigen receptor gene rearrangement represents a physiological marker of common origin,15 a feature that has been widely exploited in research and clinical settings to develop a broad variety of assay designs.16
Traditionally, second and subsequent occurrences of lymphoid neoplasms in the same patient after a period of complete remission are regarded as (clonally-related) outgrowths of the primary tumor.17–19 However, this concept has been challenged by studies of multiple subtypes of lymphomas showing that a significant percentage of secondary occurrences are clonally-unrelated neoplasms (summarized in Table 1). Although it has been suggested that clonally-unrelated relapses occur with different frequencies in different lymphoma subtypes,20 precise numbers are lacking. This can be attributed to small study cohorts, discrepancies in study designs and applied technologies, selection biases or any combination thereof. Several cohort-based studies showed that clonally-unrelated “relapses” occur in approximately 15–20% of DLBCL patients.11,21–23 Moreover, about 20% of DLBCL relapses emerging as Richter transformations had distinct IGH rearrangements compared to the initial chronic lymphocytic leukemia.24,25 In classical Hodgkin lymphoma, a higher frequency of approximately 35% was reported.26 Conversely, relapses of follicular lymphoma as well as their transformations to DLBCL are almost always clonally-related to the primary tumor.27–29
Table 1.
Summary of studies examining clonal relationships in lymphomas.
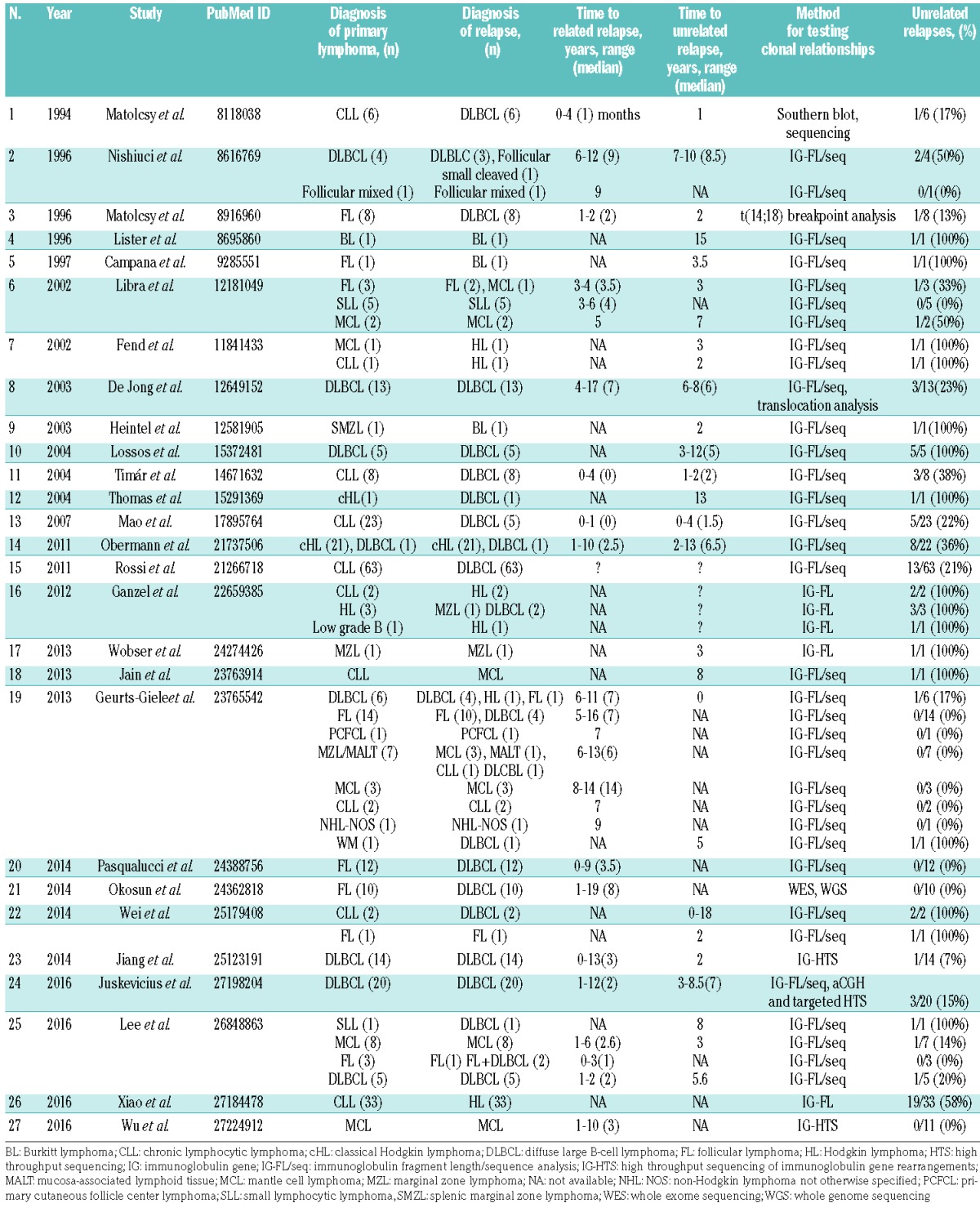
Collectively, these results show that the occurrence of clonally-unrelated lymphoma “relapses” at certain frequencies is specific to certain subtypes of B-cell neoplasms. This raises the following questions. (i) What are the factors that predispose individuals to develop a second independent lymphoid neoplasm? (ii) What are the clinical implications of such relapses? (iii) Can clonally-unrelated recurrences be predicted and, if so, how can current diagnostic procedures be adapted to facilitate their detection?
A clonally-unrelated relapse, i.e. a second de novo lymphoid neoplasm in the same patient, cannot be explained by pure randomness, since the probability of this type of tumor is quite low in the general population (at an incidence of 19.5/100,000/year30 the probability of developing, e.g., a DLBCL twice within 15 years by chance is approximately 0.0009%). There must, therefore, be predisposing factors that increase the general risk of lymphoma in certain patients. Indeed, several such factors with various degrees of penetrance and incidence have been described, including inborn genetic properties,31,32 viral infections,33 immune suppression34,35 and exogenous exposure to mutagens.36–38
It is known that immunocompromised patients are more susceptible to lymphoma (e.g. post-transplant lymphoproliferative diseases in grafted patients, primary effusion lymphomas in patients with acquired immunodeficiency syndrome).39,40 Indeed, we and others have shown that clonally-unrelated DLBCL relapses can occur in patients with iatrogenic immunodeficiency due to transplantation.21,41 Additionally, some second de novo lymphoma recurrences in immunoproficient patients are frequently localized in immunologically-privileged anatomical sites (central nervous system, testes).21,42 In such cases, lack of immunosurveillance due to anatomical barriers could provide similarly favorable conditions for tumor development as acquired immunodeficiency; indeed, immunodeficiency can be reflected in the mutational background of DLBCL, as exemplified in a recent study showing that post-transplant DLBCL have fewer mutations in general, and do not harbor B2M mutations43 in particular, while B2M is mutated in approximately 12% of DLBCL occurring in immunocompetent patients.44
In addition to immunodeficiency, inherited or acquired genetic defects can further increase the risk of a second de novo lymphoma.31,45 It has recently been shown that somatic mutations of genes implicated in leukemo- and lymphomagenesis can be detected in the hematopoietic systems of healthy middle-aged and elderly individuals.46 This phenomenon was termed clonal hematopoiesis of indeterminate potential and was shown to increase the risk of hematopoietic cancers.46 Furthermore, it has been demonstrated that mice transplanted with hematopoietic stem cells purified from the bone marrow of patients with chronic lymphocytic leukemia developed monoclonal or oligoclonal B-cell proliferations that displayed IGH rearrangements different from those of the original chronic lymphocytic leukemia clone.47 These data suggest that predisposing mutations may be present in precursor cells at a stage prior to the antigen-receptor gene rearrangements and terminal differentiation.
The aggressive disease course and lack of efficacious treatments in relapses of lymphoid neoplasms result in high rates of morbidity and mortality. The clinical behavior of clonally-unrelated relapses remains controversial as different studies report contradictory outcomes. In our previous study, two of three patients with second de novo DLBCL were treated successfully and had long-lasting clinical remissions.21 In contrast, Lee et al. reported dismal outcomes in two cases of clonally-unrelated DLBCL relapses.23 Rossi et al. reported that 12 patients who developed DLBCL, via Richter transformation, which was clonally-unrelated to the primary chronic lymphocytic leukemia had better survival than 49 patients with clonally-related transformations.25 However, an earlier, smaller study found no difference in survival between 19 patients with clonally-related and five with clonally-unrelated Richter transformation.24
Currently, if the patient’s health condition allows, aggressive B-cell lymphoma recurrences are treated with aggressive secondary chemotherapy regimens and autologous stem cell transplantation because acquired tumor resistance is anticipated. However, clonally-unrelated relapses, effectively being de novo neoplasms, could be sensitive to standard first-line treatments since the clone that gave rise to the second neoplasm was potentially not yet present during the initial therapy, and, therefore, had neither evolved under selective treatment pressure nor developed therapy resistance. If that is the case, then the hazardous side effects of aggressive therapies could be avoided. Additionally, patients who are not eligible for second-line treatment and are diverted to palliative care could be eligible for milder treatment regimens with curative intent. This possibility of treating clonally-unrelated relapses with first-line therapies must be evaluated by specifically designed prospective clinical trials, and the cumulative data reviewed here suggest the rationale for such investigations.
For the reasons described above, it would be useful to know which recurrences are most likely to be clonally-unrelated secondary de novo neoplasms. Time to relapse could be a useful indicator since unrelated relapses more often present as late recurrences (5 years or later after the initial disease).20,22 However, clonally-related relapses are also reported to occur long after the diagnosis of the primary tumor (up to 17 years later).22 In contrast, changes in morphology, phenotypic cell-of-origin conversions or viral infection status did not prove to be useful predictors of clonally-unrelated “relapses”.48 Ultimately, if therapy changes are envisioned for clonally-unrelated relapses, given their significant frequencies in some lymphoma subtypes (e.g. DLBCL, classical Hodgkin lymphoma), under certain circumstances (long interval, history of immunodeficiency or relapse in immunologically privileged site), determination of clonal relationship by genetic testing would be the most reliable solution.
Methodological considerations concerning clonality testing
The multiplex polymerase chain reaction approach introduced almost two decades ago remains the gold standard of clonality testing.49,50 With this technique, standardized sets of consensus primers are used to amplify rearranged IGH and IGL loci by polymerase chain reaction and, thereafter, lengths and sequences of the generated amplicons are investigated by capillary electrophoresis and Sanger sequencing.51 These methods provide conclusive results on clonality and/or clonal relationships in the vast majority of cases,49 but also have technical shortcomings that can lead to a lack of definitive output or misinterpretation.51,52
More recently, methods for high-throughput sequencing of B- and T-cell receptor genes have been developed, giving rise to a new field – immunogenomics.53–56 This new technique, also termed Ig-seq, allows qualitative and quantitative estimation of the entire immune repertoire of the investigated sample. In the context of clonality testing in lymphoid neoplasms this method can readily determine not only the sequence and the frequency of the dominant clone, but also provide information about less frequent clones “masked” in oligoclonal or polyclonal samples. This technology has already been advantageously used to provide answers about clonal relationships of recurrent tumors in cases in which results from traditional methods were hard to interpret and inconclusive.57 Ig-seq can also provide information about somatic hypermutations, which is useful in evolutionary analysis of emerging lymphoma populations.11,58 Furthermore, the high-throughput sequencing approach has been shown to be very sensitive, making it useful for monitoring minimal residual disease.59
Despite the aforementioned advantages, the introduction of Ig-seq into diagnostics and patient care has been slow. Like all current short read-based high-throughput sequencing techniques, Ig-seq is prone to sequencing mistakes, which can lead to an overestimation of receptor gene diversity or false interpretation of clonal relationship. Additionally, the nature of multiplex polymerase chain reactions can lead to amplification biases, with some rearrangements being preferred over others and causing incorrect estimation of clonotype frequencies. Multiple solutions have been suggested to deal with these shortcomings, including the use of unique molecular identifiers to label initial template molecules,60 synthetic spike-in controls to normalize biases and advanced sequencing library preparation methods.61,62 Data analysis represents an additional challenge that is slowing the broad implementation of this method. Ig-seq produces millions of reads, which have to be corrected for errors, aligned to reference sequences, normalized for biases and grouped into clonotypes according to the sequence similarity. To some avail, there are several publicly available software packages, which can handle these tasks.63–66 Analysis software is also offered by various commercial providers and can be purchased together with library preparation and sequencing kits.
Irrespectively of which technology is used to determine clonality, there are still potential problems that have to be taken into account. First, one drawback of IG-based clonality testing is that the analysis is based on very precise locations in the genome (IGK, IGH and IGL loci on chr2, chr14 and chr22, respectively), and if these are disturbed by somatic changes in lymphoma cells (e.g. chromosomal deletions or somatic hypermutations at the primer binding sites), only partial or no results will be obtained, which makes interpretation unreliable or impossible. In such cases, other clonality markers, such as the presence of a translocation with exactly identical junction regions in the primary and relapse tumors, can be utilized. However, recurring translocations only occur in some lymphomas (e.g. approximately 45% of DLBLC), and might not be applicable to the majority of samples. Alternatively, wide-scale genomic profiling would help to establish clonal relationship or lack thereof, as the presence or lack of shared copy number aberrations and/or somatic mutations between primary and relapse tumors are strongly indicative of clonally-related or -unrelated relapses, respectively, as previously shown in other types of cancers.67
Second, several studies have shown that genetic defects potentially increasing the risk of lymphomagenesis can occur in hematopoietic precursors prior to IG V(D)J recombination.21,47 This poses the following question: can subsequent lymphomas that bear distinct IG rearrangements but clearly share somatic mutations in genes implicated in lymphomagenesis be regarded as clonally related? In the sense of molecular oncology the answer is probably no, since lymphomagenesis is tightly associated with genetic events acquired due to aberrant effects of physiological mechanisms inherent to B-cell development that occur after IG rearrangements (“collateral damage” of somatic hypermutations, translocations related to class-switch recombination), and that shape clinically relevant tumor properties such as aggressiveness or treatment sensitivity. In the sense of pure tumor evolutionary biology, shared somatic aberrations, unless they occurred independently by chance, would be suggestive of a common lymphomagenesis and, even in the presence of independent IG rearrangements, bear witness to a common tumor derivation from a “primed” precursor cell pool.
Clonally-related relapses occur via two distinct genetic evolution patterns
Until recently, lymphoma relapses, even after prolonged remission, were regarded as direct outgrowths of the primary tumor, assuming that all genetic alterations present in the primary tumor were directly passed to the relapse. Therefore, a relapse was considered to be a more evolutionarily advanced stage of the primary tumor. This was supported by stable detection of some genetic aberrations (e.g. classical drivers of lymphomagenesis such as BCL2 or CCND1 translocations) throughout the entire history of the individual disease. The recent advent of high-throughput genome-wide technologies enabled a broader look at the genetic composition of recurring tumor samples. Analysis of matched primary-relapse pairs allowed identification of shared and unique features between matched tumors inferring genetic tumor evolution. Previous studies in other types of cancer have unequivocally shown that the majority of tumors are heterogeneous, and that tumor growth occurs via (sub-)clonal genome evolution.68 Multiple subsequent studies confirmed these findings in aggressive B-cell lymphomas and demonstrated in particular that relapses occur via at least two distinct patterns of clonal evolution.11,21,69–72 In one pattern, primary and relapse tumors share some common genetic alterations that attest to their clonal origin, but the majority of somatic mutations are unique to each tumor occurrence. This pattern suggests early divergence of tumor-founding clones and independent genetic evolution. In the other pattern, the relapsed clone bears the vast majority of genetic alterations of the primary tumor, but can additionally possess variable numbers of relapse-specific mutations. Importantly, in the majority of such cases, individual tumor occurrences have a low number of private, usually small scale mutations, which provide evidence against linear tumor evolution and instead suggest very late divergence of primary and relapse clones. Similar genetic evolution patterns have previously been shown in the progression of transformed follicular lymphoma.27,28,73,74
Currently it is difficult to estimate which pattern is more frequent due to conflicting reports that reflect the use of different techniques as well as differences between the various types of aggressive lymphomas. Analyzing somatic hypermutation patterns within rearranged IGH VDJ sequences by Ig-seq in matched primary-relapse pairs of DLBCL, Jiang et al. found that relapses in six patients followed early divergent, and in seven patients late divergent evolution scenarios.11 In our previous study using targeted high-throughput sequencing and array comparative genomic hybridization, we found that six of 17 DLBCL relapses diverged early from the common progenitor, while 11/17 diverged late.21 In a recent study investigating mantle cell lymphoma by whole exome sequencing, Wu et al. found that most relapses followed the early-divergent/branching evolution pattern.70 Conversely, cytogenetic karyotyping of Burkitt lymphoma pairs showed primarily late divergent or direct tumor evolution.72
Implications of distinct genetic patterns in diffuse large B-cell lymphoma relapse
The discovery of two distinct evolution patterns at relapse raises additional questions and provides the basis for new hypotheses concerning various aspects of lymphoma biology, such as considerations regarding tumor heterogeneity, timing of tumor growth and emergence of resistance. Testing these hypotheses could lead to new knowledge that would offer the potential for improved therapy.
Relapse patterns as markers of primary tumor genetic heterogeneity
It has previously been shown in chronic lymphocytic leukemia that intratumoral heterogeneity might increase the risk of relapse or disease progression.75 Although intra-tumoral heterogeneity has been documented in DLBCL,76 this connection was not confirmed by a recent study in DLBCL showing that genome-wide methylation heterogeneity rather than genetic heterogeneity increased the risk of relapse.77 Although not yet clearly demonstrated, it is likely that the evolutionary pattern at relapse could potentially correlate with the degree of intratumoral heterogeneity of a pre-diagnostic or diagnostic tumor. Primary tumors that relapse via the late-divergent mode of evolution would be expected to be more homogenous and dominated by one clone. In contrast, primary tumors in cases in which relapse occurs via late-divergent/branching evolution are likely to have a more complex subclonal structure, with a dominant clone accompanied by minor clones that are the product of branching tumor evolution during early tumor development.
Implications for mechanisms of therapy resistance
It is reasonable to speculate that the two different evolutionary patterns could represent two distinct mechanisms of therapy resistance (Figure 1). Late divergence could represent a spontaneous strategy of tumor survival under selective treatment pressure. In such case resistance is not pre-existing, but rather (rapidly) acquired by necessity. Mutagenic agents included in the chemotherapy regimen might create the genetic instability that is required for evolution to occur and to acquire resistance mutations essential for tumor survival. Already having all characteristics required for rapid tumor formation, such survivor clones would (more rapidly) replenish the tumor mass lost during initial treatment and form a relapse. In contrast, early divergence is obviously not occurring due to therapy pressure; rather it happens either due to limitations imposed by the surrounding microenvironment, such as the strength of immune response, the lack of nutrients, oxygen, etc., or due to intrinsic properties of the early tumor clone such as a higher degree of genomic instability due to defects in DNA repair, increased somatic hypermutation or aberrant mitotic mechanisms. The resistance to treatment would therefore occur as a purely stochastic consequence of clonal divergence. Genetic lesions that confer resistance to treatment might not be beneficial to the treatment-naïve clone, which may proliferate slowly or stagnate while other, more efficient, clones thrive and lead to initial lymphoma manifestations. Despite the initially small contribution to mass and volume, once treatment is applied and the dominant clone is eradicated, the former resistant subpopulation would survive and increase its growth potential by acquiring additional DNA lesions, and thus gradually evolve to tumor relapse. Potential implications for treatment
Figure 1.

Schematic representation of two patterns of genetic evolution in clonally-related relapses of aggressive B-cell lymphomas. Different colors represent genetically distinct clones that possess private genetic alterations due to independent genome evolution. (A) In the early-divergent/branching scenario, the divergence occurs early in tumor development. The majority of subpopulations stagnate but one clone eventually acquires the effective combination of drivers, expands and gives rise to a heterogeneous primary tumor. The dominant population is exterminated by the treatment, however, an intrinsically resistant subclone exists and gradually gives rise to a relapse. (B) In the late-divergent/linear evolution scenario, the tumor initially possesses a strong driver combination. Therefore, the neoplastic cells grow fast and unrestricted, giving rise to a rather homogeneous primary tumor. Such a tumor is almost exterminated by the treatment but an acquired resistance emerges. The resistant subclone already has drivers of effective growth and rapidly replenishes the tumor mass giving rise to a more rapid relapse.
If the above model is feasible, and late divergent and early divergent relapses do indeed reflect acquired and pre-existing (intrinsic) resistance, correct identification of relapse patterns may have important implications for treatment, especially in the era of targeted treatment.78 It has been shown that intrinsic and acquired resistance occur principally via distinct cellular mechanisms determined by the specific types of selective pressures applied to developing tumor populations.79 Acquired resistance primarily occurs due to mutations in the targeted molecule. In such a scenario, the initial response to treatment is usually observed until the accumulation of resistance-conferring mutations. The effects of such mutations can be variable and include: (i) increased target expression (through inactivation of negative regulation loops or gene amplifications); (ii) compromised binding of drug to target (specific mutations at drug binding sites); and (iii) changes modulating target activity (mutations in target functional domains).8 In contrast, intrinsic resistance to targeted treatment is characterized by alterations that are initially present upstream or downstream of the target signaling pathway. Target inhibition is, therefore, ineffective since the tumor has a pre-developed mechanism of circumventing the targets’ function by delivering important tumorigenic signals via alternative pathways. In such a scenario, the resistant cell population (possibly a minor clone in the context of the whole tumor, as discussed earlier) is unaffected by the therapy.
Both intrinsic and acquired resistance can contribute to relapses. For example, one study showed that two types of mutations are responsible for acquired resistance to the Bruton tyrosine kinase (BTK) inhibitor ibrutinib in chronic lymphocytic leukemia: (i) mutations in BTK itself that change ibrutinib-mediated BTK inactivation from irreversible to reversible, and (ii) gain-of-function mutations in phospholipase gamma 2 (PLCγ2), which is a downstream phosphorylation target of BTK.80 BTK mutations are likely to represent acquired resistance, while PLCγ2 mutations most probably are related to pre-existing resistance in small subpopulations. This hypothesis is strongly supported by the detection of PLCγ2 mutation in diagnostic chronic lymphocytic leukemia samples at very low frequencies.80 It is known that intrinsic resistance to ibrutinib in DLBCL occurs due to a (pre-existing) L265P mutation in MYD88 and activating mutations in CARD11, both of which result in constitutive activation of nuclear factor-κB (NF-κB) signaling.81 PIM1 mutations were also suggested to contribute to intrinsic resistance to ibrutinib in activated B-cell DLBCL.82 Mechanisms of acquired resistance to ibrutinib in DLBCL are still unknown.
In conclusion, information about early and late divergent patterns of genetic evolution at relapse can provide valuable insights into the potential type of treatment resistance in the recurrence, which can then aid in designing more efficaciously targeted treatment approaches.
Genetic events related to (clonally-related) recurrences
Aggressive B-cell lymphoma relapses occur, at least in part, as a consequence of genetic evolution of the primary tumor. It is, therefore, conceivable that investigation of the exact changes that occur during this evolution will result in identification of genetic drivers of the process, prognostic as well as predictive markers and, it is to be hoped, “actionable mutations” that could be targeted to improve currently available treatments. Two primary experimental approaches are utilized for such identification. In one approach, matched pairs of primary and relapse tumors from the same patient are genetically profiled and compared, assuming that those events, which are relapse-specific or increase from low to high allelic frequency are related to recurrence. This approach has several advantages in that it allows: (i) studying only genetic alterations that occur specifically at relapse; (ii) identification of patient-specific shared somatic mutations that represent early drivers in tumorigenesis; (iii) exclusion of patient-specific single nucleotide polymorphisms that are not annotated in databases; and (iv) investigation of the genetic evolution of the tumors. However, paired samples with sufficient and good quality material are scarce because relapses are not routinely biopsied during the diagnostic process. Consequently, there are only a few of such studies and the overall number of cases analyzed is limited.10,11,21,71 In the alternative approach, unpaired relapsed/refractory tumors are analyzed and frequencies of detected genetic alterations are compared to the respective frequencies in independent primary tumor collectives. This approach allows analysis of larger cohorts, but it has a significant drawback in that it is nearly impossible to accurately differentiate between mutations present at the primary stage and those that are relapse-specific. This problem can be illustrated by the ambiguous status of the MYD88 L265P mutation in DLBCL. In our previous study it was detected as a heterozygous early driver lesion present in both primary and relapsed tumors of the same patients.21 In contrast, an earlier DLBCL genome sequencing study reported that classic lymphomagenic events, such as mutations of MYD88, CD79A/B and EZH2, can also be subclonally present within primary DLBCL.76 The MYD88 mutation can, therefore, occur both as an early and a late event in DLBCL development, but this discrimination is highly problematic in cases in which only relapsed tumor samples are examined. The above mentioned shortcomings and the genuine heterogeneity of DLBCL potentially explain the limited overlap between five dedicated genomic studies on DLBCL relapses,10,11,21,71,83 and such studies in other types of aggressive lymphomas are even more limited (Table 2).69,70,72
Table 2.
Findings of the dedicated studies on aggressive B-cell lymphoma relapses.
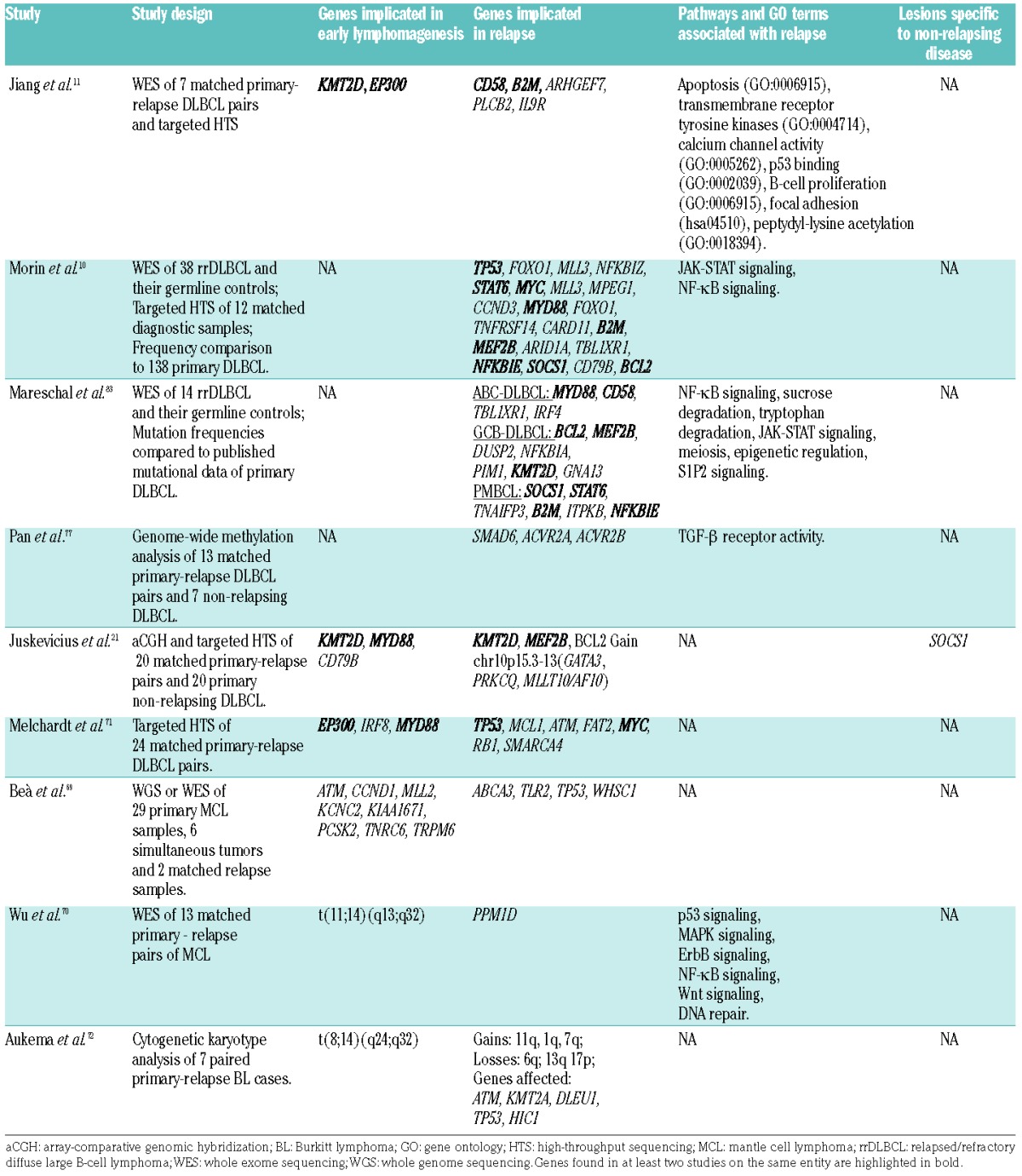
In general, all studies of paired samples evidenced ongoing genetic evolution and acquisition of a variable number of additional mutations unique to the relapsed tumors or increased in frequency compared to the primary tumors through the process of clonal selection. A high diversity of alterations was observed, reflecting the many ways through which tumors adapt to the changed microenvironment after treatment. Pathway and gene ontology term enrichment analysis of the mutated genes at DLBCL relapse implicated activation of NF-κB, JAK-STAT, trans-membrane receptor tyrosine kinase signaling and biological processes such as apoptosis, calcium channel activity, epigenetic regulation, cell cycle regulation and immunosurveillance (Table 2).
On the background of profound variability, a small number of recurrent alterations were detected. Jiang et al. suggested that recurrences might be driven by mutations in immunosurveillance genes. Exome-sequencing revealed relapse-specific inactivating alterations in either CD58 or B2M, or both, in five of seven investigated pairs.11 Another study found that B2M and CD58 mutations were enriched in relapses of primary mediastinal B-cell lymphomas (PMBCL) and activated B-cell DLBCL, respectively.83 β2-microglobulin (encoded by B2M) is necessary for the assembly and function of the major histocompatibility complex class I, which is involved in antigen presentation to cytotoxic T cells. CD58 is required for antigen adhesion and activation of NK and T cells.84 However, inactivating mutations in one or both of these genes were also frequently detected in primary DLBCL.85
Alterations in JAK-STAT pathway components were also implicated in DLBCL relapse. Morin et al. found that 36% of relapsed germinal center B-cell-like DLBCL and 38% of transformed follicular lymphomas bore STAT6 mutations of the DNA binding domain specifically affecting the D419 residue.10 Furthermore, this mutational status correlated with the overexpression of phosphorylated STAT6 levels in affected tumors, indicating that mutated STAT6 activates the JAK/STAT signaling cascade. Other dedicated studies, however, did not confirm the increased prevalence of this mutation at relapse, and clonal STAT6 D419 mutations were reportedly not rare in primary lymphomas, including DLBCL.86–88 In addition to STAT6, inactivating mutations of SOCS1, a negative regulator of the JAK/STAT pathway, were frequently detected, predominantly in PMBCL relapses.83 However, it is not clear how relapse-specific these mutations are, since no paired tumors were investigated in the study and SOCS1 mutations are frequently detectable in primary tumors (PMBCL and DLBCL).88 Another study found evidence of clonal selection of SOCS1 mutations at relapse, providing further confirmation that JAK/STAT signaling is important for recurrences.10 Importantly, SOCS1 is frequently mutated in primary DLBCL tumors88 and it was suggested that SOCS1 mutational status has a prognostic value: Schif et al. found that patients with primary tumors with truncating SOCS1 mutations had excellent overall survival, whereas those with non-foreshortening point mutations and minor consequences for the protein had poor overall survival.89 We found that SOCS1 mutations occurred only in non-relapsing primary DLBCL (5/20 cases), were absent in relapsing primary DLBCL (0/20),21 and were associated with excellent progression-free survival in uniformly treated (R-CHOP-14) cohort of patients with primary DLBCL.90
NF-κB pathway activation is well known in the pathogenesis of aggressive B-cell lymphomas, especially activated B-cell DLBCL. Two independent studies found identical 4 bp deletions of the NFKBIE gene that encodes an inhibitor of NF-κB signaling, IκBε, in three relapsing DLBCL and two PMBCL relapses.10,83 The same mutation was found by the authors in two additional cases of untreated DLBCL and was previously reported to occur in chronic lymphocytic leukemia, classical Hodgkin lymphoma and PMBCL.91–93 Additional NF-κB pathway-related genes shown to be enriched in relapses include NFKBIA and NFKBIZ, encoding IκBα and IκBζ, both of which have previously been implicated in the molecular pathogenesis of classical Hodgkin lymphoma and DLBCL, respectively.94,95
Inactivating aberrations (loss-of-function mutations and gene deletions) of TP53 and CDKN2A were also enriched in DLBCL relapses and are thought to be associated with resistance to chemotherapy.83 Defects in these two genes as well as gain-of-function mutations of NOTCH1 and MYC are most frequently implicated in Richter transformation, as excellently reviewed elsewhere.96
It has recently been demonstrated that, in addition to genetic mechanisms, changes of the epigenetic landscape can also play a role in DLBCL relapse.77,97 Pan et al. found that, compared to the primary tumors, relapses have significantly decreased intra-tumoral methylation heterogeneity.77 Furthermore, they showed that greater intra-tumoral methylation heterogeneity of primary tumors correlated with increased probability of relapse. Enrichment analysis of genes located in the differentially methylated regions between primary tumors and relapses suggested several deregulated pathways, such as anti-apoptotic and tumor necrosis factor activity. In particular, genes associated with transforming growth factor-β receptor activity were overrepresented among the hypomethylated genes at relapse. Methylation-dependent transforming growth factor-β regulation has previously been implicated in DLBCL chemoresistance; Clozel et al. showed that SMAD1, a regulator of the transforming growth factor-β pathway, is more frequently hypermethylated in treatment-resistant DLBCL, and that demethylating agents administered prior to chemotherapy sensitize DLBCL and result in increased cytotoxic activity.97
Overall, recurrently altered genes and pathways in relapses overlap significantly with those implicated in the primary pathogenesis of aggressive B-cell lymphomas. It is, therefore, unclear whether they truly confer relapse-specific phenomena such as treatment resistance or merely reflect means of tumor clones to gain fitness. With regard to treatment resistance, it is thought to occur via two main mechanisms: (i) decreased drug accumulation or increased drug inactivation or (ii) biological pathways that evade cell death from applied agents; the majority of genetic alterations and pathways described in this review point towards the latter mechanism in DLBCL.
To sum up, it is evident that considerable progress has been made in the last 5 years toward understanding relapses of aggressive B-cell lymphomas. Important insights have been gained from numerous studies examining different aspects of recurrences, from patterns of tumor evolution to recurrent genetic and epigenetic alterations. However, the substantial inter- and intra-tumoral heterogeneity, small study groups and varied study designs mean that we are just beginning to uncover the underlying mechanisms. Since there is an obvious lack of sufficient numbers of suitable specimens to conduct large-scale comprehensive studies on human samples, it could be worthwhile to pursue alternative strategies. One potential opportunity would be to conduct comprehensive studies of selected single cases at multiple, currently accessible levels (i.e. genome, epigenome, transcriptome, proteome, metabolome) in an attempt to develop a state-of-the-art individual tumor/tumor-pair molecular profile at great depth to identify actionable alterations. Another complementary strategy could be based on modeling DLBCL relapses in currently available or newly established animal models,98,99 in which potential relapse-causing events might be identified, functionally tested and subsequently screened by focused assays on human sample cohorts.
Supplementary Material
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/7/1139
References
- 1.Okosun J, Cwynarski K. Non-Hodgkin Lymphoma: High Grade. In: Postgraduate Haematology. Oxford, UK: John Wiley & Sons, Ltd; 2015. p. 631–650. [Google Scholar]
- 2.Coiffier B, Thieblemont C, Van Den Neste E, et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d’Etudes des Lymphomes de l’Adulte. Blood. 2010;116(12):2040–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Den Neste E, Schmitz N, Mounier N, et al. Outcome of patients with relapsed diffuse large B-cell lymphoma who fail second-line salvage regimens in the International CORAL study. Bone Marrow Transplant. 2016;51(1):51–57. [DOI] [PubMed] [Google Scholar]
- 4.Philip T, Guglielmi C, Hagenbeek A, et al. Autologous bone marrow transplantation as compared with salvage chemotherapy in relapses of chemotherapy-sensitive non-Hodgkin’s lymphoma. N Engl J Med. 1995;333(23):1540–1545. [DOI] [PubMed] [Google Scholar]
- 5.Friedberg JW. Relapsed/refractory diffuse large B-cell lymphoma. Hematology. 2011;2011(1):498–505. [DOI] [PubMed] [Google Scholar]
- 6.Gisselbrecht C, Glass B, Mounier N, et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol. 2010;28(27):4184–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jain S, Shah N, Gregory S. Relapsed diffuse large B-cell lymphoma–10 years later. Clin Adv Hematol Oncol. 2011;9(9):704–708. [PubMed] [Google Scholar]
- 8.Asi K. Dominant mechanisms of primary resistance differ from dominant mechanisms of secondary resistance to targeted therapies. Crit Rev Oncol Hematol. 2016;97:178–196. [DOI] [PubMed] [Google Scholar]
- 9.Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med. 2009;15(9):1010–1012. [DOI] [PubMed] [Google Scholar]
- 10.Morin RD, Assouline S, Alcaide M, et al. Genetic landscapes of relapsed and refractory diffuse large B-cell lymphomas. Clin Cancer Res. 2016;22(9):2290–2300. [DOI] [PubMed] [Google Scholar]
- 11.Jiang Y, Redmond D, Nie K, et al. Deep-sequencing reveals clonal evolution patterns and mutation events associated with relapse in B-cell lymphomas. Genome Biol. 2014;15(8):432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kreso A, O’Brien CA, van Galen P, et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science. 2013;339(6119):543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele JWV. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Fourth Edition. IARC Press; Lyon: 2008. [Google Scholar]
- 14.Alizadeh AA, Aranda V, Bardelli A, et al. Toward understanding and exploiting tumor heterogeneity. Nat Med. 2015;21(8): 846–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alt FW, Oltz EM, Young F, et al. VDJ recombination. Immunol Today. 1992;13 (8):306–314. [DOI] [PubMed] [Google Scholar]
- 16.Gazzola A, Mannu C, Rossi M, et al. The evolution of clonality testing in the diagnosis and monitoring of hematological malignancies. Ther Adv Hematol. 2014;5(2): 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coiffier B. Treatment of aggressive non-Hodgkin’s lymphoma. Semin Oncol. 1999;26(5 Suppl 14):12–20. [PubMed] [Google Scholar]
- 18.Shioyama Y, Nakamura K, Kunitake N, et al. Relapsed non-Hodgkin’s lymphoma: detection and treatment. Radiat Med. 2000;18(6):369–375. [PubMed] [Google Scholar]
- 19.Cappelaere P. Secondary non-Hodgkin’s lymphomas. Bull Cancer. 1998;85(3): 217–231. [PubMed] [Google Scholar]
- 20.Geurts-Giele WR, Wolvers-Tettero IL, Dinjens WN, Lam KH, Langerak AW. Successive B-cell lymphomas mostly reflect recurrences rather than unrelated primary lymphomas. Am J Clin Pathol. 2013;140(1): 114–126. [DOI] [PubMed] [Google Scholar]
- 21.Juskevicius D, Lorber T, Gsponer J, et al. Distinct genetic evolution patterns of relapsing diffuse large B-cell lymphoma revealed by genome-wide copy number aberration and targeted sequencing analysis. Leukemia. 2016;30(12):2385–2395. [DOI] [PubMed] [Google Scholar]
- 22.de Jong D, Glas AM, Boerrigter L, et al. Very late relapse in diffuse large B-cell lymphoma represents clonally related disease and is marked by germinal center cell features. Blood. 2003;102(1):324–327. [DOI] [PubMed] [Google Scholar]
- 23.Lee SE, Kang SY, Yoo HY, et al. Clonal relationships in recurrent B-cell lymphomas. Oncotarget. 2016;7(11):12359–12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mao Z, Quintanilla-Martinez L, Raffeld M, et al. IgVH mutational status and clonality analysis of Richter’s transformation: diffuse large B-cell lymphoma and Hodgkin lymphoma in association with B-cell chronic lymphocytic leukemia (B-CLL) represent 2 different pathways of disease evolution. Am J Surg Pathol. 2007;31(10):1605–1614. [DOI] [PubMed] [Google Scholar]
- 25.Rossi D, Spina V, Deambrogi C, et al. The genetics of Richter syndrome reveals disease heterogeneity and predicts survival after transformation. Blood. 2011;117(12): 3391–3401. [DOI] [PubMed] [Google Scholar]
- 26.Obermann EC, Mueller N, Rufle A, et al. Clonal relationship of classical Hodgkin lymphoma and its recurrences. Clin Cancer Res. 2011;17(16):5268–5274. [DOI] [PubMed] [Google Scholar]
- 27.Pasqualucci L, Khiabanian H, Fangazio M, et al. Genetics of follicular lymphoma transformation. Cell Rep. 2014;6(1):130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okosun J, Bödör C, Wang J, et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet. 2014;46(2):176–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matolcsy A, Casali P, Warnke RA, Knowles DM. Morphologic transformation of follicular lymphoma is associated with somatic mutation of the translocated Bcl-2 gene. Blood. 1996;88(10):3937–3944. [PubMed] [Google Scholar]
- 30.SEER Stat Fact Sheets: Non-Hodgkin Lymphoma [Internet]. 2016. [cited 2017 Jan 2]. Available from: https://seer.cancer.gov/statfacts/html/nhl.html
- 31.Aricò M, Mussolin L, Carraro E, et al. Non-Hodgkin lymphoma in children with an associated inherited condition: a retrospective analysis of the Associazione Italiana Ematologia Oncologia Pediatrica (AIEOP). Pediatr Blood Cancer. 2015;62(10): 1782–1789. [DOI] [PubMed] [Google Scholar]
- 32.Wang SS, Slager SL, Brennan P, et al. Family history of hematopoietic malignancies and risk of non-Hodgkin lymphoma (NHL): a pooled analysis of 10 211 cases and 11 905 controls from the International Lymphoma Epidemiology Consortium (InterLymph). Blood. 2007;109(8):3479–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vannata B, Arcaini L, Zucca E. Hepatitis C virus-associated B-cell non-Hodgkin’s lymphomas: what do we know? Ther Adv Hematol. 2016;7(2):94–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bagg A, Dunphy CH. Immunosuppressive and immunomodulatory therapy-associated lymphoproliferative disorders. Semin Diagn Pathol. 2013;30(2):102–112. [DOI] [PubMed] [Google Scholar]
- 35.Ponce RA, Gelzleichter T, Haggerty HG, et al. Immunomodulation and lymphoma in humans. J Immunotoxicol. 2014;11(1):1–12. [DOI] [PubMed] [Google Scholar]
- 36.Kramer S, Hikel SM, Adams K, Hinds D, Moon K. Current status of the epidemiologic evidence linking polychlorinated biphenyls and non-Hodgkin lymphoma, and the role of immune dysregulation. Environ Health Perspect. 2012;120(8): 1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alicandro G, Rota M, Boffetta P, La Vecchia C. Occupational exposure to polycyclic aromatic hydrocarbons and lymphatic and hematopoietic neoplasms: a systematic review and meta-analysis of cohort studies. Arch Toxicol. 2016;90(11):2643–2656. [DOI] [PubMed] [Google Scholar]
- 38.Merhi M, Raynal H, Cahuzac E, et al. Occupational exposure to pesticides and risk of hematopoietic cancers: meta-analysis of case-control studies. Cancer Causes Control. 2007;18(10):1209–1226. [DOI] [PubMed] [Google Scholar]
- 39.Filipovich AH, Mathur A, Kamat D, Shapiro RS. Primary immunodeficiencies: genetic risk factors for lymphoma. Cancer Res. 1992;52(19 Suppl):5465s–5467s. [PubMed] [Google Scholar]
- 40.Brimo F, Michel RP, Khetani K, Auger M. Primary effusion lymphoma: a series of 4 cases and review of the literature with emphasis on cytomorphologic and immunocytochemical differential diagnosis. Cancer. 2007;111(4):224–233. [DOI] [PubMed] [Google Scholar]
- 41.Campana S, Corradini P, Astolfi M, et al. Analysis of the immunoglobulin heavy-chain gene rearrangement providing molecular evidence of second lymphoma in a patient in apparent relapse after autotrans-plantation. Bone Marrow Transplant. 1997;20(4):341–343. [DOI] [PubMed] [Google Scholar]
- 42.Lossos A, Ashhab Y, Sverdlin E, et al. Late-delayed cerebral involvement in systemic non-hodgkin lymphoma: a second primary tumor or a tardy recurrence? Cancer. 2004;101:1843–1849. [DOI] [PubMed] [Google Scholar]
- 43.Menter T, Juskevicius D, Alikian M, et al. Mutational landscape of B-cell post-transplant lymphoproliferative disorders. Br J Haematol. [Epub ahead of print] 2017. [DOI] [PubMed] [Google Scholar]
- 44.Forbes SA, Beare D, Gunasekaran P, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43(Database issue):D805–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skibola CF, Curry JD, Nieters A. Genetic susceptibility to lymphoma. Haematologica. 2007;92(7):960–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126(1):9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kikushige Y, Ishikawa F, Miyamoto T, et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell. 2011;20(2):246–259. [DOI] [PubMed] [Google Scholar]
- 48.Xiao W, Chen WW, Sorbara L, et al. Hodgkin lymphoma variant of Richter transformation: morphology, Epstein-Barr virus status, clonality, and survival analysis-with comparison to Hodgkin-like lesion. Hum Pathol. 2016;55:108–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Dongen JJ, Langerak AW, Bruggemann M, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2003;17(12):2257–2317. [DOI] [PubMed] [Google Scholar]
- 50.Meier VS, Rufle A, Gudat F. Simultaneous evaluation of T- and B-cell clonality, t(11;14) and t(14;18), in a single reaction by a four-color multiplex polymerase chain reaction assay and automated high-resolution fragment analysis: a method for the rapid molecular diagnosis of lymphoproliferative disorders applicable to fresh frozen and formalin-fixed, paraffin-embedded tissues, blood, and bone marrow aspirates. Am J Pathol. 2001;159(6):2031–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Langerak AW, Groenen PJTA, Brüggemann M, et al. EuroClonality/BIOMED-2 guidelines for interpretation and reporting of Ig/TCR clonality testing in suspected lymphoproliferations. Leukemia. 2012;26(10): 2159–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Langerak AW, van Dongen JJM. Multiple clonal Ig/TCR products: implications for interpretation of clonality findings. J Hematop. 2012;5(1–2):35–43. [Google Scholar]
- 53.Boyd SD, Marshall EL, Merker JD, et al. Measurement and clinical monitoring of human lymphocyte clonality by massively parallel VDJ pyrosequencing. Sci Transl Med. 2009;1(12):12ra23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robins H. Detecting and monitoring lymphoma with high-throughput sequencing. Oncotarget. 2011;2(4):287–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.He J, Wu J, Jiao Y, et al. IgH gene rearrangements as plasma biomarkers in non-Hodgkin’s lymphoma patients. Oncotarget. 2011;2(3):178–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Georgiou G, Ippolito GC, Beausang J, et al. The promise and challenge of high-throughput sequencing of the antibody repertoire. Nat Biotechnol. 2014;32(2):158–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Appenzeller S, Gilissen C, Rijntjes J, et al. Immunoglobulin rearrangement analysis from multiple lesions in the same patient using next-generation sequencing. Histopathology. 2015;67(6):843–858. [DOI] [PubMed] [Google Scholar]
- 58.Carlotti E, Wrench D, Rosignoli G, et al. High Throughput sequencing analysis of the immunoglobulin heavy chain gene from flow-sorted B cell sub-populations define the dynamics of follicular lymphoma clonal evolution. PLoS One. 2015;10(9):e0134833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Faham M, Zheng J, Moorhead M, et al. Deep-sequencing approach for minimal residual disease detection in acute lym-phoblastic leukemia. Blood. 2012;120(26): 5173–5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Turchaninova MA, Davydov A, Britanova OV, et al. High-quality full-length immunoglobulin profiling with unique molecular barcoding. Nat Protoc. 2016;11(9): 1599–1616. [DOI] [PubMed] [Google Scholar]
- 61.Khan TA, Friedensohn S, de Vries ARG, et al. Accurate and predictive antibody repertoire profiling by molecular amplification finger-printing. Sci Adv. 2016;2(3):e1501371–e1501371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Friedensohn S, Khan TA, Reddy ST. Advanced methodologies in high-throughput sequencing of immune repertoires. Trends Biotechnol. 2017;35(3): 203–214. [DOI] [PubMed] [Google Scholar]
- 63.Bolotin DA, Poslavsky S, Mitrophanov I, et al. MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods. 2015;12(5):380–381. [DOI] [PubMed] [Google Scholar]
- 64.Gupta NT, Vander Heiden JA, Uduman M, et al. Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics. 2015;31(20):3356–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barak M, Zuckerman NS, Edelman H, Unger R, Mehr R. IgTree: creating immunoglobulin variable region gene lineage trees. J Immunol Methods. 2008;338(1–2):67–74. [DOI] [PubMed] [Google Scholar]
- 66.Kuchenbecker L, Nienen M, Hecht J, et al. IMSEQ–a fast and error aware approach to immunogenetic sequence analysis. Bioinformatics. 2015;31(18):2963–2971. [DOI] [PubMed] [Google Scholar]
- 67.Chen YJ, Yeh SH, Chen JT, et al. Chromosomal changes and clonality relationship between primary and recurrent hepatocellular carcinoma. Gastroenterology. 2000;119(2):431–440. [DOI] [PubMed] [Google Scholar]
- 68.Aparicio S, Caldas C. The implications of clonal genome evolution for cancer medicine. N Engl J Med. 2013;368(9):842–851. [DOI] [PubMed] [Google Scholar]
- 69.Beà S, Valdés-Mas R, Navarro A, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci USA. 2013;110(45): 18250–18255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu C, de Miranda NF, Chen L, et al. Genetic heterogeneity in primary and relapsed mantle cell lymphomas: impact of recurrent CARD11 mutations. Oncotarget. 2016;7(25):38180–38190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Melchardt T, Hufnagl C, Weinstock DM, et al. Clonal evolution in relapsed and refractory diffuse large B-cell lymphoma is characterized by high dynamics of subclones. Oncotarget. 2016;7(32):51494–51502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aukema SM, Theil L, Rohde M, et al. Sequential karyotyping in Burkitt lymphoma reveals a linear clonal evolution with increase in karyotype complexity and a high frequency of recurrent secondary aberrations. Br J Haematol. 2015;170(6):814–825. [DOI] [PubMed] [Google Scholar]
- 73.Ruminy P, Jardin F, Picquenot J-M, et al. S mutation patterns suggest different progression pathways in follicular lymphoma: early direct or late from FL progenitor cells. Blood. 2008;112(5):1951–1959. [DOI] [PubMed] [Google Scholar]
- 74.Green MR, Gentles AJ, Nair RV, et al. Hierarchy in somatic mutations arising during genomic evolution and progression of follicular lymphoma. Blood. 2013;121(9): 1604–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Landau DA, Carter SL, Stojanov P, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152(4):714–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Morin RD, Mungall K, Pleasance E, et al. Mutational and structural analysis of diffuse large B-cell lymphoma using whole-genome sequencing. Blood. 2013;122(7): 1256–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pan H, Jiang Y, Boi M, et al. Epigenomic evolution in diffuse large B-cell lymphomas. Nat Commun. 2015;6:6921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Roschewski M, Staudt LM, Wilson WH. Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nat Rev Clin Oncol. 2014;11(1):12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bozic I, Nowak MA. Timing and heterogeneity of mutations associated with drug resistance in metastatic cancers. Proc Natl Acad Sci USA. 2014;111(45):15964–15968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Woyach JA, Furman RR, Liu T-M, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Davis RE, Ngo VN, Lenz G, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463(7277):88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kuo H-P, Ezell SA, Hsieh S, et al. The role of PIM1 in the ibrutinib-resistant ABC subtype of diffuse large B-cell lymphoma. Am J Cancer Res. 2016;6(11):2489–2501. [PMC free article] [PubMed] [Google Scholar]
- 83.Mareschal S, Dubois S, Viailly P-J, et al. Whole exome sequencing of relapsed/refractory patients expands the repertoire of somatic mutations in diffuse large B-cell lymphoma. Genes Chromosomes Cancer. 2016;55(3):251–267. [DOI] [PubMed] [Google Scholar]
- 84.Altomonte M, Gloghini A, Bertola G, et al. Differential expression of cell adhesion molecules CD54/CD11a and CD58/CD2 by human melanoma cells and functional role in their interaction with cytotoxic cells. Cancer Res. 1993;53(14):3343–3348. [PubMed] [Google Scholar]
- 85.Challa-Malladi M, Lieu YK, Califano O, et al. Combined genetic inactivation of β2-microglobulin and CD58 reveals frequent escape from immune recognition in diffuse large B cell lymphoma. Cancer Cell. 2011;20(6):728–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ritz O, Guiter C, Castellano F, et al. Recurrent mutations of the STAT6 DNA binding domain in primary mediastinal B-cell lymphoma. Blood. 2009;114(6): 1236–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yildiz M, Li H, Bernard D, et al. Activating STAT6 mutations in follicular lymphoma. Blood. 2015;125(4):668–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dubois S, Viailly P-J, Mareschal S, et al. Next-generation sequencing in diffuse large B-cell lymphoma highlights molecular divergence and therapeutic opportunities: a LYSA study. Clin Cancer Res. 2016;22(12): 2919–2928. [DOI] [PubMed] [Google Scholar]
- 89.Schif B, Lennerz JK, Kohler CW, et al. SOCS1 mutation subtypes predict divergent outcomes in diffuse large B-cell lymphoma (DLBCL) patients. Oncotarget. 2013;4(1): 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Juskevicius D, Jucker D, Klingbiel D, et al. Mutations of CREBBP and SOCS1 are independent prognostic factors in diffuse large B cell lymphoma: mutational analysis of the SAKK 38/07 prospective clinical trial cohort. J Hematol Oncol. 2017;10(1):70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Emmerich F, Theurich S, Hummel M, et al. Inactivating I kappa B epsilon mutations in Hodgkin/Reed-Sternberg cells. J Pathol. 2003;201(3):413–420. [DOI] [PubMed] [Google Scholar]
- 92.Mansouri L, Sutton LA, Ljungstrom V, et al. Functional loss of IkappaBepsilon leads to NF-kappaB deregulation in aggressive chronic lymphocytic leukemia. J Exp Med. 2015;212(6):833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mansouri L, Noerenberg D, Young E, et al. Frequent NFKBIE deletions are associated with poor outcome in primary mediastinal B-cell lymphoma. Blood. 2016;128(23): 2666–2670. [DOI] [PubMed] [Google Scholar]
- 94.Lake A, Shield LA, Cordano P, et al. Mutations of NFKBIA, encoding IkappaB alpha, are a recurrent finding in classical Hodgkin lymphoma but are not a unifying feature of non-EBV-associated cases. Int J Cancer. 2009;125(6):1334–1342. [DOI] [PubMed] [Google Scholar]
- 95.Nogai H, Wenzel S-S, Hailfinger S, et al. IκB-ζ controls the constitutive NF-κB target gene network and survival of ABC DLBCL. Blood. 2013;122(13):2242–2250. [DOI] [PubMed] [Google Scholar]
- 96.Rossi D, Gaidano G. Richter syndrome: pathogenesis and management. Semin Oncol. 2016;43(2):311–319. [DOI] [PubMed] [Google Scholar]
- 97.Clozel T, Yang SN, Elstrom RL, et al. Mechanism-based epigenetic chemosensitization therapy of diffuse large B-cell lymphoma. Cancer Discov. 2013;3(9): 1002–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Calado DP, Zhang B, Srinivasan L, et al. Constitutive canonical NF-κB activation cooperates with disruption of BLIMP1 in the pathogenesis of activated B cell-like diffuse large cell lymphoma. Cancer Cell. 2010;18(6):580–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mandelbaum J, Bhagat G, Tang H, et al. BLIMP1 is a tumor suppressor gene frequently disrupted in activated B cell-like diffuse large B cell lymphoma. Cancer Cell. 2010;18(6):568–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.