

Abstract
Although the primary origin of sickle cell disease is a hemoglobin disorder, many types of cells contribute considerably to the pathophysiology of the disease. The adhesion of neutrophils to activated endothelium is critical in the pathophysiology of sickle cell disease and targeting neutrophils and their interactions with endothelium represents an important opportunity for the development of new therapeutics. We focused on endothelin-1, a mediator involved in neutrophil activation and recruitment in tissues, and investigated the involvement of the endothelin receptors in the interaction of neutrophils with endothelial cells. We used fluorescence intravital microscopy analyses of the microcirculation in sickle mice and quantitative microfluidic fluorescence microscopy of human blood. Both experiments on the mouse model and patients indicate that blocking endothelin receptors, particularly ETB receptor, strongly influences neutrophil recruitment under inflammatory conditions in sickle cell disease. We show that human neutrophils have functional ETB receptors with calcium signaling capability, leading to increased adhesion to the endothelium through effects on both endothelial cells and neutrophils. Intact ETB function was found to be required for tumor necrosis factor α-dependent upregulation of CD11b on neutrophils. Furthermore, we confirmed that human neutrophils synthesize endothelin-1, which may be involved in autocrine and paracrine pathophysiological actions. Thus, the endothelin-ETB axis should be considered as a cytokine-like potent pro-inflammatory pathway in sickle cell disease. Blockade of endothelin receptors, including ETB, may provide major benefits for preventing or treating vaso-occlusive crises in sickle cell patients.
Introduction
Sickle cell disease (SCD) is a genetic hemoglobinopathy resulting from a unique mutation in the β-globin gene. SCD is characterized by hemolytic anemia, painful vaso-occlusive crises (VOC) and progressive organ failure. Although red blood cell dysfunction is the major contributor to disease development and progression, other types of cells, which are not affected by the genetic mutation (endothelial cells, leukocytes, platelets1,2), are also key actors in the pathophysiology of SCD. Several studies have highlighted the important role of polymorphonuclear neutrophils (neutrophils), both during an acute VOC3 and in the associated long-term morbidity and mortality.4 Interestingly, a high, steady-state, peripheral white cell count is a risk factor for both significant morbidity – stroke, pulmonary complications, nephropathy – and early SCD-related death.4–8
The central role of neutrophils in the pathophysiology of SCD has recently been explored.3,9 In vitro studies have shown that, compared to neutrophils from healthy controls, neutrophils from SCD patients have an increased expression of adhesion molecules,10–12 rendering them more susceptible to inflammatory stimuli.13 A relationship between clinical manifestations of SCD and the expression of adhesion molecules on neutrophils has also been reported.2,14 It is likely that activated neutrophils engage in a complex process of abnormal interactions between activated endothelial cells, platelets and circulating red blood cells contributing to decreased blood flow and to endothelial injury. This further accentuates erythrocyte sickling, neutrophil recruitment and tissue ischemia.9 Targeting the mechanisms of neutrophil-endothelial cell interactions would, therefore, represent a novel and potentially important therapeutic opportunity in SCD.
Endothelin-1 (ET-1) is the most potent endogenous vasoconstrictor.15 It is released by activated endothelial16 and non-endothelial cells17 in response to hypoxia and reduced nitric oxide bioavailability in several animal models.18 The effects of ET-1 are mediated via two receptors, the ETA and ETB receptors.15 We previously found that mixed ETA/B receptor antagonism has profound effects on organ injury and mortality in a mouse model of SCD.19 In addition to inhibition of tonic ET-1-dependent vasoconstriction during experimental VOC, we also observed an unexpected but powerful inhibition of neutrophil recruitment in the lungs and kidneys although we could not link this effect to a direct action of ET-1 receptors on neutrophil-endothelial interactions. We, therefore, hypothesized that activation of ET receptors might promote a pathogenic pro-inflammatory role for neutrophils in SCD.
In the present study, we combined intravital videomicroscopy of the microcirculation in a murine model of SCD with quantitative microfluidic fluorescence microscopy of human blood to investigate the involvement of ET receptors in the interaction of neutrophils with endothelial cells.
Methods
Animal model
Animals were used in accordance with the National Institutes of Health Guide for the care and use of laboratory animals (NIH publication n. 85-23) and the study protocol was approved by the French ministry of agriculture. SAD1 (SAD) Hbβ single/single hemizygous mice were used in this study. This strain harbors a recombinant hβ-globin gene construct expressing human hemoglobin SAD (A2β2SAD), which contains two mutations [Antilles (β23I) and D-Punjab (β121Q)] in addition to the βS6V mutation.19,20 This strain is bred on the C57BL/6J genetic background (with more than 30 backcrosses).
Intravital videomicroscopy: experimental protocol
The complete protocol is described in the Online Supplementary Methods and illustrated in Online Supplementary Figure S1. ETA antagonist (BQ123, A.G. Scientific®, San Diego, CA, USA or ETB antagonist (BQ788, Tocris Bioscience®, Bristol, UK) were injected 10 min before (10 mg/kg) and 3 h 15 min after (5 mg/kg) intrascrotal injection of 0.5 μg tumor necrosis factor (TNFα, R&D Systems, Minneapolis, MN, USA) and neutrophils were monitored by injection of labeled phycoerythrin-conjugated anti-Ly6G antibody (BD Biosciences Pharmingen, Sparks, MD, USA).
The neutrophil rolling flux fraction, adhesion density, adhesion efficiency and transmigration were measured from 2.5 h to 5 h after the TNFα injection at 30-min intervals.
Patients and healthy volunteers
The study was approved by the Ethical Evaluation Committee for Biomedical Research Projects (CEERB) of Robert Debré Hospital, France (n. 2014/155). All patients were included during a scheduled medical consultation, at steady state. Blood samples were collected from 2- to 18-year old patients with SCD (SS genotype), and from young healthy adult donors (AA genotype) from the local blood bank (Etablissement Français du Sang). Patients in a chronic transfusion therapy program, those having received blood transfusion in the 3 months preceding enrollment and those treated with hydroxycarbamide (hydroxyurea) were excluded. Exceptions were made for the quantitative polymerase chain reaction analyses, as mentioned below.
Human neutrophil preparation
Human neutrophils were isolated (98% purity) from fresh whole blood within 4 h after blood sampling with EDTA as anticoagulant, using a MACSxpress Neutrophil Isolation Kit followed by MACSxpress Erythrocyte Depletion Kit (Miltenyi Biotec, Paris, France). Details on the flow adhesion assays are provided in the Online Supplementary Methods.
Real-time quantitative polymerase chain reaction
ET-1, ETA and ETB mRNA levels were determined by real-time quantitative polymerase chain reaction as described previously.21 Beta-2 microglobulin mRNA was used as a housekeeping gene The relative expression of ET-1, ETA and ETB mRNA in neutrophils was calculated following the ΔΔCt method,22 with the values obtained in HMEC-1 as a reference.
Flow cytometry assessment of adhesion molecules and intracellular Ca2+ measurements in neutrophils
Three hours after treatment with TNFα with or without ET receptor antagonists, mice were anesthetized; blood was collected on EDTA by intracardiac puncture and immediately processed for flow cytometry staining for viability and CD11a, CD11b and CD62L surface expression on Ly5G+ neutrophils (see Online Supplementary Methods).
Isolated human neutrophils were fluorescently labeled with fluo-4-acetoxymethyl ester (Fluo-4-AM; Invitrogen). Neutrophils were incubated with BQ123 or BQ788 (both from Sigma®, Saint-Quentin Fallavier, France) at 10 μmol/L for 15 min and analyzed immediately by flow cytometry (FACSCanto II, BD Biosciences, Sparks, MD, USA). After 2 min of signal acquisition, the analysis was interrupted for less than 10 s to add 100 nM of ET-1 (Sigma®) to the tube before the analysis was resumed.
Endothelin-1 immunoassay
Isolated neutrophils were incubated at 37°C in RPMI medium supplemented with TNFα (10 ng/mL) for 2 h. Supernatants were frozen at −80°C until assay. The concentration of ET-1 was determined using a Quantikine® enzyme-linked immunosorbent assay (R&D systems.
Flow adhesion assays
The experimental protocol is described in the Online Supplementary Methods. Briefly, human neutrophils were stained in whole blood with anti-CD16 antibody together with BQ123 (1 μM) or BQ788 (1 μM). The time between drawing blood and starting the anti-CD16 staining was always less than 3 h and reproducible. If more than 3 h elapsed, samples were discarded. Whole blood samples were perfused through biochip channels containing HMEC-1 monolayers or coated recombinant proteins (P-selectin, VCAM-1 and ICAM-1).
Statistics
Results are presented as means plus or minus the standard error of the mean (SEM), Data were analyzed using GraphPad Prism 6 software by ANOVA with the Tukey test, Wilcoxon rank test and Mann-Whitney test. P values less than 0.05 were considered statistically significant.
Results
Acute ETA and ETB receptor antagonism does not alter leukocyte blood count or microvascular hemodynamics in wild-type and SAD mice
In keeping with clinical SCD, SAD mice (n=5) showed an increase in circulating leukocytes with a neutrophil predominance compared to wild-type (WT) littermate mice, both at baseline and after TNFα administration (Online Supplementary Table S1). Treatment of TNFα-challenged animals with either BQ123 or BQ788 did not affect any of the peripheral cell counts. As changes in microvascular wall shear stress may influence neutrophil adhesion, we determined the effects of acute ETA and ETB receptor blockade on wall shear rate in cremasteric venules. There were no significant differences in venular wall shear rates between groups treated with TNFα + vehicle, TNFα + BQ123 and TNFα + BQ788 (Online Supplementary Table S2).
ETA and ETB receptors control neutrophil rolling in inflamed venules
In WT mice, neither BQ123 nor BQ788 affected neutrophil rolling flux throughout the time course of the experiment. As in WT mice, ETA antagonism with BQ123 had no effect on neutrophil rolling flux in SAD mice. In sharp contrast, ETB blockade with BQ788 profoundly inhibited neutrophil rolling flux in SAD mice, compared to that in the vehicle-treated SAD group, throughout the experiment (Figure 1A).
Figure 1.

The rolling, adhesion and emigration of neutrophils in inflamed cremasteric venules are attenuated in tumor necrosis factor-alpha challenged wild-type and SAD mice treated with the selective endothelin receptor inhibitors BQ123 and BQ788. (A, B) Rolling neutrophil flux fraction in TNFα inflamed venules of WT and SAD mice. The number of rolling neutrophils per minute (flux) was counted and the neutrophil rolling flux fraction was determined by dividing the rolling neutrophil flux by the total neutrophil flux (all neutrophils passing through the venule). (C, D) Neutrophil adhesion density, defined as the number of adherent neutrophils per square millimeter, in TNFα inflamed venules after BQ123 and BQ788 treatment in WT and SAD mice. (E, F) Neutrophil adhesion efficiency, defined as the number of adhered neutrophils per square millimeter normalized by the number of circulating neutrophils passing through the venule. (G, H) Neutrophil emigration, defined as the number of emigrated neutrophils per square millimeter of extravascular space in inflamed venules after BQ123 and BQ788 treatment in WT and SAD mice. Emigrated neutrophils were visualized and quantified by optical sectioning and two-dimensional maximum intensity projection. Fifteen venules (5 mice) were analyzed in each group [12 venules and 3 mice for control with phosphate-buffered saline (PBS)]. Data are presented as mean ± SEM. Two-way ANOVA with the Tukey multiple comparison test. *P<0.05 compared with WT-TNFα; **P<0.01 compared with WT-TNFα; ***P<0.001 compared with WT-TNFα; ****P<0.0001 compared with WT-TNFα. †P<0.05 compared with SAD-TNFα. ††P<0.01 compared with SAD-TNFα. †††P<0.001 compared with SAD-TNFα. ††††P<0.0001 compared with SAD-TNFα.
ETA and ETB receptor antagonism decreases neutrophil adhesion in inflamed venules from sickle mice
Overall, in WT mice, neutrophil adhesion density was not influenced by BQ123 or BQ788 treatment. Over the time course of the study, adhesion decreased in all three groups of WT mice from about 670 adherent neutrophils/mm2 at 2 h 30 min after the inflammatory challenge to reach about 250 adherent neutrophils/mm2 at 5 h. In SAD mice, the inflammatory challenge with TNFα induced a marked increase in neutrophil adhesion compared to that in the WT counterparts (1000 versus 586 adherent neutrophils/mm2, P<0.05). The adhesion remained elevated and stable for 90 min and then decreased slightly. At the end of the experiment, adhesion density in SAD mice challenged with TNFα was still increased compared to that in WT challenged mice (731 versus 271 adherent neutrophils/mm2, P<0.05). In SAD mice, both ETA and ETB receptor blockade markedly inhibited adhesion of neutrophils throughout the time course of the study with the degree of adhesion being similar to that observed in WT controls (Figure 1B).
To define the neutrophil adhesion better, we calculated the efficiency of neutrophil adhesion as the ratio between adhering neutrophils and those that are estimated to be available to adhere (Figure 1C). We found that adhesion efficiency was, overall, unaffected by BQ123 and BQ788 in WT mice. In contrast, adhesion efficiency was markedly and significantly decreased in SAD mice treated with BQ788 compared to that in the TNFα and vehicle-treated SAD group throughout the time course of the experiment; no significant differences were observed in SAD mice treated with BQ123. Taken together, these data show a differential effect of ETA versus ETB receptor antagonism on neutrophil adhesion (Figure 1C–F).
ETA and ETB receptor antagonism decreases neutrophil transmigration in inflamed venules from sickle mice
We counted neutrophils that had migrated into the extravascular space adjacent to the observed venules by optical sectioning (Online Supplementary Figure S1, Figure 1G,H). TNFα-induced emigration of neutrophils was significant in both groups of mice compared to the emigration in animals treated with vehicle (phosphate-buffered saline) but distinct kinetics were observed in SAD and WT animals. While neutrophil emigration reached a plateau at 3 h 30 min in the WT cremasteric microcirculation and subsequently resolved to baseline values, sickle SAD mice experienced a more sustained and more intense increase in emigration which was double that measured in WT after 4 h and lasted until the end of the experiments (Figure 1 G,H). In WT mice, TNFα-induced emigration was significantly decreased by BQ123 and BQ788 administration but only at early time points (∼2.3-fold decrease at 2 h 30 min, P<0.01 for BQ123, ∼3.5-fold decrease at 2 h 30 min and ∼2.1-fold decrease at 3 h for BQ788, P<0.001 versus vehicle-treated mice). In SAD mice that displayed more intense and prolonged emigration of neutrophils both ETA and ETB blockade significantly prevented trans-endothelial migration of neutrophils to tissues, especially at late points [BQ123: ∼2.3-fold decrease at 2 h 30 min (P<0.01 versus TNFα only-treated mice); BQ788: ∼3.5-fold decrease at 2 h 30 min (P<0.001 versus TNFα-treated mice) and ∼2.1-fold decrease at 3 h (P<0.01 versus TNFα-treated mice)] (Figure 1H). Representative images indicating less neutrophil recruitment in the presence of ETA and ETB, particularly in SAD mice, are shown in Figure 2.
Figure 2.

Kinetics of neutrophil recruitment. Representative images of cremasteric venules for each group witthout stimulation (administered phosphate - buffered saline, PBS) or after local TNFα stimulation associated or not with specific blocking of ETA or ETB receptors. White arrows indicate emigrated neutrophils. Scale bars: 10 μm.
All these experiments suggest that blocking ET receptors influences neutrophil recruitment in mice, especially in the context of sustained and intense neutrophil recruitment specific to experimental SCD.
ETA and ETB receptor antagonism reduces tumor necrosis factor α-induced high CD11b/Mac1 expression on neutrophils and does not alter cell viability
CD11b, also known as Mac-1α or integrin αM chain, is part of the CD11b/CD18 heterodimer, also known as the C3 complement receptor. Increased adhesion has been linked to engagement of CD11b membrane expression on neutrophils from sickle mice.23 We, therefore, measured CD11b expression on neutrophils from SAD and WT mice without or after TNFα administration. In the latter case, we evaluated the effect of BQ123 and BQ788 antagonists 3 h after injection of TNFα or vehicle (phosphate-buffered saline), when ET receptor antagonists displayed inhibitory effects on neutrophil adhesion in intravital imaging experiments.
TNFα challenge induced a very significant upregulation of CD11b expression in neutrophils from both WT and SAD animals within 3 h (Online Supplementary Figure S2). This effect was markedly increased in neutrophils from SAD mice compared to that in WT mice (+40 %, P<0.05). Both ET receptor antagonists limited CD11b-associated mean fluorescence surface intensity and the proportion of neutrophils with high CD11b expression in neutrophils from TNFα-challenged SAD animals, whereas neither antagonist had any effect on CD11b expression in normal mice (Online Supplementary Figure S2). This suggests that involvement of the ET system in pro-inflammatory CD11b upregulation and neutrophil adhesion is specific to the SCD condition. No effect of either TNFα or the ET receptor antagonists was observed on the surface expression of CD11a and CD62L on neutrophils (Online Supplementary Figure S2) and no difference in cell viability (apoptosis or necrosis affected less than 3% of blood neutrophils) was observed between the different groups (data not shown).
We next investigated the expression and the role of ET receptors on human neutrophils from SCD patients and healthy controls.
ETA and ETB mRNA expression in neutrophils from healthy volunteers and patients with sickle cell disease
As very few studies report on the expression of ETB receptors in neutrophils from normal individuals24–26 and none in SCD patients, we first investigated the expression of the EDNRA and EDNRB genes encoding ETA and ETB, respectively, in endothelial cells (HMEC-1) and in human neutrophils from SCD patients (SS) and healthy individuals (AA). ETA and ETB mRNA are expressed both in endothelial cells and in human neutrophils and at similar levels in neutrophils isolated from healthy controls and SCD patients (Figure 3).
Figure 3.
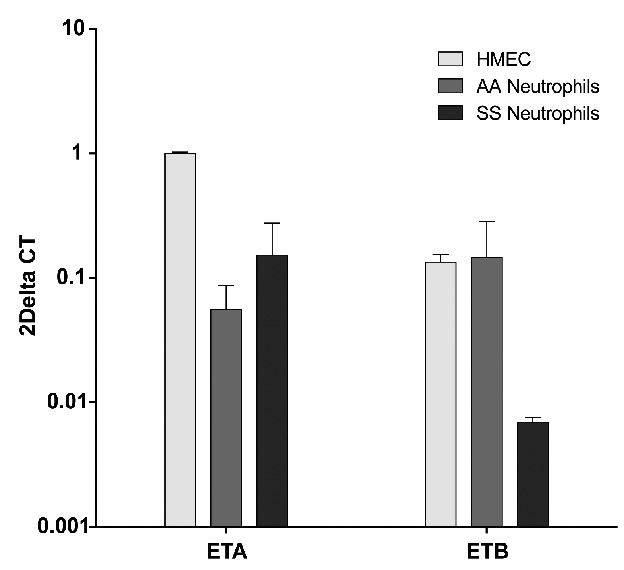
Endothelial cells and neutrophils from both healthy controls (AA), and SCD patients (SS) express ETA and ETB transcripts. Results of real time quantitative reverse transcriptase polymerase chain reaction are expressed as a comparison with ETA/B mRNA transcripts in endothelial cells (HMEC-1) with the ΔΔCT method. The reference gene used was the β2 microglobulin gene. Results are means (± SEM) of eight independent samples (4 controls, 4 patients) in duplicate.
To complete these results and confirm the presence of functional ET receptors on human neutrophils, we investigated the potential effect of neutrophil stimulation on the ET-1-mediated calcium response.
Endothelin-1 elicits an ETB-mediated calcium response in neutrophils from healthy controls and individuals with sickle cell disease
Although ETA and ETB receptors have been shown to be expressed on many cell types, their presence on the surface of neutrophils and their signaling pathways are poorly understood. We investigated the activation of a calcium-dependent signaling pathway in response to stimulation with ET-1 (Figure 4A,B). In the presence of ET-1 in neutrophils from both SCD individuals and healthy volunteers, we found a rapid increase in intra-cytoplasmic calcium concentration, which progressively decreased to steady state in less than 100 s. The same experiment was performed with neutrophils pre-incubated for 15 min with BQ123 or BQ788. Neither antagonist altered baseline values (Figure 4C). ETA antagonism did not prevent the ET-1-elicited calcium response (Figure 4A,C) whereas ETB receptor antagonism provoked a significant reduction in the calcium response after the addition of ET-1 both in AA and SS neutrophils (Figure 4A,C). These results indicate the presence of a functional ETB receptor on the surface of neutrophils.
Figure 4.

Endothelin-1 elicits an ETB-mediated calcium response in neutrophils from healthy controls and individuals with sickle cell disease. (A) Representative images of the intracellular calcium measurements in isolated neutrophils: rapid increase of calcium concentration after addition of ET-1, similar effect on neutrophils previously incubated with BQ123, significant reduction of the ET-1-elicited calcium response in neutrophils previously incubated with BQ788. (B) Gating strategy for the comparison of the fluorescence intensity before and after addition of ET-1. (C) Comparison between the percentages of the Fluo4 AM-associated highly fluorescent sub-population (population “P7”) from seven independent samples (neutrophils from 4 healthy controls and 3 SCD patients) in the different conditions: (C) Percentages of the Fluo4 AM-associated highly fluorescent neutrophil sub-population (population “P7”) from AA individuals (green squares) and from SS patients (red circles) previously incubated with phosphate-buffered saline (unlabeled), BQ123, or BQ788. (D) Percentages of the Fluo4 AM-associated highly fluorescent neutrophil sub-population measured within the first 30 s after addition of ET-1 (period P3), after phosphate-buffered saline, BQ123 or BQ788. Paired comparisons were made. *P<0.05 vs. ET-1 + PBS.
We next investigated the potential secretion of ET-1 by human neutrophils.
Endothelin-1 secretion by neutrophils from healthy individuals and patients with sickle cell disease
We investigated the expression of preproET-1 mRNA in neutrophils from healthy controls and SCD patients, and compared this to expression levels in the endothelial HMEC-1 cell line. As shown in Online Supplementary Figure S3, preproET-1 mRNA is significantly expressed in neutrophils but at a much lower level than in endothelial cells.
To confirm the ability of neutrophils to secrete ET-1, we measured the concentration of ET-1 in the supernatant of isolated human neutrophils activated or not by TNFα (Figure 5). We found detectable concentrations of ET-1 in the supernatant of isolated neutrophils at baseline (data not shown) and after stimulation with TNFα. The level of extracellular ET-1 in the presence of TNFα was higher in supernatants of neutrophils from SS individuals than those from AA healthy controls (0.27 ± 0.15 pg/mL versus 1.64 ± 0.45 pg/mL respectively; P=0.033) and from SS patients treated with hydoxycarbamide (0.20 ± 0.08 pg/mL; P=0.04) (Figure 5).
Figure 5.
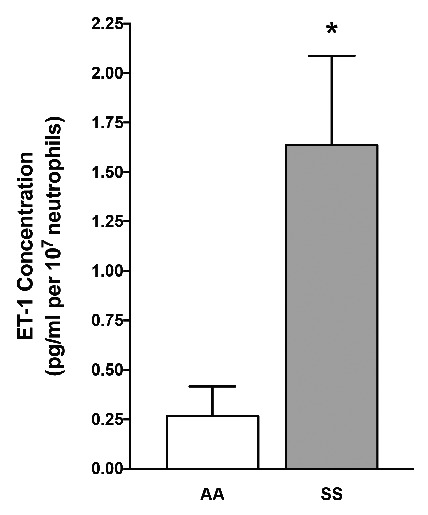
Endothelin-1 secretion by neutrophils from healthy controls and sickle cell disease patients at steady state. ET-1 concentration in supernatants of human neutrophils. Results represent means of 22 independent samples [9 healthy controls (AA), 9 SCD patients without hydroxycarbamide therapy (SS)]. Concentration is normalized for 107 neutrophils per condition. *P<0.05 vs. AA.
These data highlight the ability of neutrophils to secrete ET-1, which may then activate endothelial cells and neutrophils themselves, with an autocrine and paracrine mechanism. Thus, we next investigated the effects of ET receptor antagonists on human neutrophil adhesion to endothelial cells.
ETB receptor antagonism prevents neutrophil adhesion to endothelial cells in controls and patients with sickle cell disease
To mimic in vivo circulation accurately, we infused whole blood onto a layer of activated endothelial cells under conditions of laminar flow. Adhesion of neutrophils to TNFα-primed endothelium was similar in controls and SCD patients (Figure 6A). As shown in Figure 6B,C, ETA receptor antagonism with BQ123 did not affect this. Conversely, adhesion of BQ788-treated neutrophils to endothelial cells was decreased by 42% (± 26%) compared to the adhesion of untreated neutrophils (P=0.0008) (Figure 6B,C). These data indicate an unexpected stimulatory role for the ETB receptor in human neutrophil adhesion to endothelial cells, paralleling that observed in vivo in sickle mice.
Figure 6.

ETB but nor ETA blockade prevents adhesion of neutrophils from healthy individuals and patients with sickle cell disease on activated endothelial cells and recombinant adhesion proteins. (A) Adhesion of neutrophils to endothelial cells (HMEC-1) over a 30 min period in a microfluidic system at a shear stress of 1 dyn/cm2, expressed as median area of neutrophil-associated fluorescence per field comparing six healthy controls (AA) to 12 SCD patients (SS). (B) Representative photomicrographs of anti-CD16 Alexa 488 (in green)-labeled neutrophils that have gradually adhered to the endothelial cells in microfluidic channels. Three channels were infused with the same batch of whole blood. Conditions were: NT: untreated blood; BQ123: blood preincubated with 1 μM BQ123; BQ788: blood previously treated with 1 μM BQ788. (C) Relative change in neutrophil adhesion on activated HMEC-1 after 30 min of infusion of whole blood from seven healthy AA controls (green circles) and 14 SS SCD patients (red triangles) previously incubated with 1 μM BQ123 or 1 μM BQ788 compared with non-treated blood. (D) Relative change in neutrophil adhesion 30 min after infusion of whole blood from five healthy donors (green circles) and five SCD patients (red triangles) on activated HMEC previously incubated with BQ123 or BQ788 compared with untreated HMEC. (E) Neutrophil adhesion on recombinant P-selectin, VCAM-1 and ICAM-1 proteins 30 min after infusion of whole blood from four healthy donors (green circles) and four SCD subjects (red triangles) previously incubated with BQ123 or BQ788 compared with non-treated blood. *P<0,05, ***P<0,001 with paired, non-parametric t-test (Wilcoxon test) vs. untreated blood or HMEC.
ETB receptor antagonism prevents neutrophil adhesion to endothelial cells through effects on both endothelial cells and neutrophils
During the adhesion experiments described above, ET receptor antagonists were added to whole blood, then infused directly onto endothelial cells. We were not, therefore, able to exclude any potential effect of the pharmacological antagonists on endothelial cells. To discriminate between the effects of the antagonists on endothelial and neutrophil ET receptors, we performed the same experiments by incubating only endothelial cells with the two selective antagonists. We found a 28% (±33%) significant decrease in the adhesion of neutrophils following treatment of endothelial cells with BQ788 (but not with BQ123) compared to the untreated condition (P=0.02) (Figure 6D).
To study the specific effects of ETA and ETB antagonists on leukocytes, we used a different protocol since it was not possible to rinse the whole blood and totally exclude contact between the antagonists and the endothelial cells. We therefore infused whole blood directly onto channels coated with purified adhesion proteins (P-selectin, VCAM-1 and ICAM-1). The results illustrated in Figure 6E show that neutrophil adhesion after incubation with BQ123 was not different to that in the untreated condition. In contrast, there was a 16% (±18%) decrease in the adhesion of neutrophils on recombinant adhesion molecules after incubation with BQ788 compared to the untreated sample (P=0.03) (Figure 6E). We could also rule out that ET-1 altered human and mouse neutrophil viability as neither exogenous ET-1 nor ETA or ETB antagonists altered the proportion of apoptotic and necrotic neutrophils in vitro and in vivo, respectively (data not shown).
Overall these results indicate that activation of the ETB receptor promotes neutrophil adhesion through action on two compartments: endothelial cells and neutrophils.
Discussion
Polymorphonuclear neutrophils play a major role in the pathogenesis of endothelial injury characteristic of conditions such as ischemia-reperfusion and SCD.9 Sequestration of neutrophils into the microvasculature is the essential initiating step in neutrophil recruitment in inflammation and this exacerbates vaso-occlusion in SCD.2 Neutrophils from SCD patients and mice are activated, and show increased adhesion.2,11 The identification of specific mediators of neutrophil recruitment in SCD is important given that the broad spectrum and non-specific inhibition of neutrophil-endothelium interactions may lead to an increased risk of bacterial infections and trigger VOC and death.
Following studies indicating that the ET system is activated in SCD individuals,27–32 our group previously showed that ET receptor antagonism not only blunts experimental VOC-associated vasoconstriction but also substantially inhibits acute VOC-induced increases of the numbers of peripheral blood total leukocytes (and specifically neutrophils), neutrophil infiltration of the bronchoalveolar space, renal and pulmonary myeloperoxidase activity and organ damage.19 Furthermore, early studies suggested that ET-1 acts on neutrophils by increasing intracellular free calcium mobilization,33 N-formyl-L-methionyl- L-leucyl-L-phenylalanine-mediated superoxide anion production,34 or aggregation.35
We, therefore, hypothesized that the ET system may play a direct role in neutrophil-endothelial interactions in SCD. We found that ET receptors are involved in several steps of neutrophil microvascular recruitment in SCD mice. Whereas rolling adhesion in sickle SAD mice involves the ETB receptor alone, firm adhesion and post-adhesive dynamic behavior with transmigration involve both the ETA and ETB receptors. We also demonstrated an unexpected stimulatory role for the ETB receptor in adhesion of human neutrophils to endothelial cells under laminar flow conditions. Our findings suggest a differential role for ET receptors between mice and humans given that we found no pro-adhesive role for the ETA receptor in human neutrophil adhesion, in contrast to the situation in sickle mice.
Another feature was the more intense and prolonged neutrophil adhesion to endothelial cells in vivo in sickle mice compared to control mice, whereas no differences were found in human neutrophil adhesion in vitro. Our study with human samples may have lacked statistical power, given the greater variability in neutrophil adhesion compared to that measured in mice. This might be due to species specificities and greater genetic heterogeneity in humans than in mice, a factor that should be taken into account when translating animal studies into the clinical setting. One limitation to the in vitro experiments was the lack of significant matching of AA with SS individuals providing blood, due to ethical constraints. Differences in models caused by in vitro manipulation of human blood with phenotypic heterogeneity of cultured endothelial cells should also be considered. Moreover, chronic in vivo exposure of the endothelium to cytokines, products of hemolysis and blood cell micro-vesicles may alter endothelial phenotype and trigger expression of different patterns of adhesion molecules.36 Nevertheless, both murine and human models provide consistent evidence for a powerful anti-adhesive role for ETB receptor antagonism.
Our work also challenges the common opinion that adhesion of neutrophils to endothelial cells is always increased in SCD subjects. In fact, Fadlon et al., using a different approach (counting the radioactivity associated with adherent extensively centrifuged, washed and radio-labeled isolated neutrophils bound to layers of human umbilical vein endothelial cells in a static condition), could not show any difference in adhesion of neutrophils from 25 control subjects and 25 patients out of crisis to untreated and TNF-treated endothelial cells.12 Thus, our measurements of in-flow adhesion of neutrophils in whole blood from six healthy control subjects and 12 patients are consistent with the seminal results of Fadlon et al. In contrast, peripheral blood neutrophils from the majority of SCD patients in crisis were more adhesive to cultured endothelial monolayers than neutrophils from patients out of crisis or from healthy control subjects.162 Likewise, TNFα−injected SCD mice exhibited significant increases in neutrophil adhesion compared to AS mice, as measured by multi-channel fluorescence intravital microscopy analyses.2,37 In retrospect it is interesting to note that despite the absence of anemia in young animals, the SAD mouse model still presents remarkable neutrophil features in common with other SCD mouse models such as the βS “BERK” and SS knock-in sickle strains, with high counts of circulating leukocytes and neutrophils at steady state and during experimental VOC, thus mimicking what is observed in humans,19 and exquisite sensitivity to TNFα-induced leukocyte adhesion.2,37 One of the advantages of the SAD strain is its robustness with a fully controlled congenic C57Bl6/J background and easy breeding thus enabling experiments without the use of fetal liver or bone marrow transplant. It has also proven its relevance as a model of vaso-occlusion20,38–40 and of typical chronic degenerative organ injury.20,38–41 Thus, the SAD mouse model displays a characteristic inflammatory microvascular disease. At least at the level of neutrophil adhesion to the endothelium, our study indicates that this model shares common pathways relevant to the human condition.
Contrasting with the findings of previous studies, which used pharmacological blockade or genetic deficiency models42–45 to unravel physiological functions of the ETB receptor, our current data suggest that neutrophil endothelial recruitment is primarily dependent upon an unblocked ETB receptor. However, previous studies used both non-SCD animal models and human subjects at steady state, in which the primary role of endothelial ETB is to contribute to ET-1 clearance42 and stimulation of nitric oxide production which is essential for termination of ET-1 signaling.45,46 Our novel findings demonstrate an important contribution of the ETB receptor to vascular inflammation in SCD.
Here, we provide the first series of systematic investigations aimed at unravelling the role of ET receptors in the different phases of the neutrophil-endothelial interaction. They complement earlier work showing that endothelins are chemoattractants for neutrophils47 in vitro. The first in vivo studies used perfusion of exogenous ET-1; this led to a time- and dose-dependent sequential entrapment of platelets and neutrophils in the pulmonary circulation.48 Similarly, the multi-step recruitment of rabbit peritoneal neutrophils was stimulated by ET-1 and inhibited by a specific antagonist of the ETA receptor.49 These findings were recently confirmed and expanded, as treatment with a mixed ETA/B receptor antagonist, bosentan, and selective ETA and ETB receptor antagonists (BQ-123 and BQ-788, respectively) inhibited ET-1 and ovalbumin-induced neutrophil migration to the peritoneal cavity, suggesting that ET-1, acting through both ETA and ETB, is an important mediator of neutrophil recruitment in adaptive inflammation.50 Further studies will be needed to explore these chemotactic actions of ET-1.
It is intriguing that ET receptor blockers had a significantly more potent effect on neutrophil adhesion in sickle SAD mice than in their WT counterparts. This may be due to a potentially stronger chemotactic ET-1 concentration gradient emanating from the pathological endothelium, creating higher ET-1 concentrations in the SCD circulation. We hypothesize that SCD-specific vascular inflammation may prime neutrophils and cause increased neutrophil adhesion and trans-endothelial migration of neutrophils to tissues through increased local ET-1 availability from endothelial cells, neutrophils and, potentially, other, yet to be identified blood cells. Another complementary hypothesis could be that neutrophils from SCD individuals may differ in their ability to degrade local endothelial-derived and autocrine-derived ET-1, as reported for human neutrophils in vitro.51 Our findings may also foster further studies to investigate the role of ET-3, a selective ETB agonist that may be induced in SCD.52
Increased potency of ET receptor blockers on neutrophil adhesion in SCD conditions may also be related to distinct patterns of ET receptor expression, although we did not observe this, at least in human neutrophils. Excess of ligand usually promotes desensitization of G-protein-coupled receptors such as ET receptors, a counter-regulatory mechanism that may be defective in SCD neutrophils or endothelial cells. We did not perform any comparative dose-response studies to evaluate neutrophil sensitivity to ET-1 in SCD and control subjects. Defective desensitization or SCD-specific bias in post-receptor signaling could cause increased ET receptor-dependent cell adhesion. This would be consistent with the more intense and prolonged adhesion of neutrophils to post-capillary venules induced by TNFα in sickle mice compared to that in their WT congenic controls. Thus, the SAD mouse model exhibits exquisite sensitivity to TNFα, reminiscent of TNF-induced leukocyte stasis in the cremasteric venules, which was fatal in a high percentage of mice transplanted with bone marrow from the severe βS “BERK” mouse model.2,37 The reason for this magnified response to TNFα is still unclear. Such induced blood stasis may trigger VOC in the context of infections, as classically reported in septic patients.
The current studies set the stage for investigation of the ET system in neutrophil recruitment in other conditions beyond SCD. Indeed, early studies suggested a particular role for neutrophil recruitment in models of ischemia-reperfusion injury53,54 a condition that is particularly relevant to SCD-associated VOC. Conflicting reports regarding the respective roles of the ETA and ETB receptors suggest that pathophysiological context matters. Overall, both receptor subtypes have been shown to be involved in undefined steps of neutrophil recruitment55,56 and our findings further demonstrate the potent effect of ET receptor antagonists on neutrophil adhesion and transmigration in SCD with a particular emphasis on the role of the ETB receptor.
Our results with human neutrophils and cultured endothelial cells in microfluidic flow chambers indicate that the ETB receptor promotes neutrophil adhesion through activation at both the neutrophil and the endothelial cell surface.
In endothelial cells, the exact mechanisms whereby recruitment of neutrophils is dependent on an unblocked ETB receptor remain to be clarified. As discussed above, more efficient coupling of the ETB receptor to adhesion molecules may be at play.
On the other hand, ET-1 was shown to promote neutrophil aggregation (homotypic adhesion)35,57 as well as β2-integrin-dependent attachment of neutrophils to cultured human and bovine vascular endothelial cells (heterotypic cell adhesion). We provide here the first direct evidence of functional ETB receptors coupled to calcium signaling in neutrophils from control and sickle cell subjects. Together with our results showing that selective ETB blockade alleviated adhesion of human neutrophils to adhesion molecules, this suggests that ETB signaling may trigger calcium-induced activation of integrins. Interestingly, increased adhesion has been linked to the higher membrane expression of CD11b/CD18 on neutrophils from SCD patients than on those from controls.13 Furthermore, blocking CD11b/Mac-1 using M1/70 antibody was sufficient to diminish the erythrocyte–leukocyte interaction significantly, preventing VOC and prolonging survival in SCD mice challenged by TNFα and surgical trauma.23 These studies highlighted the role of CD11b in the pathophysiology of acute VOC in SCD. It is worth noting that expression of CD18 and CD11b on the surface of neutrophils was also increased by ET-1 in vitro53,58,59 although it was not investigated which ET receptor was involved. In line with these results, administration of TNFα induced very significant upregulation of CD11b expression in neutrophils from both WT and SAD animals but this effect was markedly asymmetric with a further 50% increase in the sickle cell condition. Surprisingly, ET receptor antagonists limited CD11b-associated mean fluorescence surface intensity and the proportion of neutrophils with high CD11b expression in neutrophils from TNFα−challenged SAD animals. Moreover, ETA and ETB antagonists had no effect on CD11b expression in normal mice. This suggests that involvement of the ET system in pro-inflammatory CD11b upregulation and neutrophil adhesion is specific to the SCD condition. Moreover, the expression of the integrin is probably not the only regulatory mechanism involved, given that rapid alterations in β2 integrin conformation, and consequently ligand affinity,60 could also play a very important role in neutrophil adhesion.
Surprisingly, and contrary to previous reports, we found no difference in CD62L expression in neutrophils from sickle mice compared to normal ones.9,11 An explanation for this phenomenon might be that, in vivo, strongly activated neutrophils may have left the circulation and are therefore underrepresented in the blood samples used for flow cytometry analysis.
In summary, our current findings suggest that human neutrophils display functional ETB receptors with calcium signaling capability, leading to accentuated adhesion to the endothelium. Using our high-speed imaging platform, we observed that the ETB response promoted rolling and firm adhesion of neutrophils, resulting in significant subsequent migration of these cells through venular walls to tissue, specifically in sickle cell mice. Furthermore, we confirmed that human neutrophils synthesize ET-1, which may be involved in autocrine and paracrine pathophysiological actions, and may contribute to the higher plasma levels of ET-1 reported in SCD individuals. Thus, the ET-1-ETB axis should be considered as a cytokine-like potent pro-inflammatory pathway in SCD. If ET receptor antagonists are proven to be safe and effective in the prevention or treatment of acute VOC in the clinical setting, they should include anti-ETB potency. Such antagonists may provide major clinical benefits, improve quality of life, and prolong survival of SCD patients.
Supplementary Material
Acknowledgments
The authors thank the “Région Ile-de-France” for funding for the intravital microscopy platform (SESAME 2007) and supporting Giulia Ghinatti through a CORDDIM fellowship. This study was supported by a grant from Labex GR-Ex. The Labex GR-Ex, reference ANR-11-LABX-0051, is funded by the “Investissements d’avenir” program of the French National Research Agency, reference ANR-11-IDEX-0005-02. We also thank the Fondation pour la Recherche Médicale – FRM (ING20121226435 to PL Tharaux) for supporting Dr. P. Nivoit and charitable funding from LVMH (2011/RDB/018, 2013/RDB/028). Finally, we thank Anna Chipont for excellent technical assistance and Elizabeth Huc and the ERI970 team for state-of-the-art animal care.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/7/1161
References
- 1.Kaul DK, Finnegan E, Barabino GA. Sickle red cell-endothelium interactions. Microcirculation. 2009;16(1):97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turhan A, Weiss LA, Mohandas N, Coller BS, Frenette PS. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc Natl Acad Sci USA. 2002;99(5):3047–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manwani D, Frenette PS. Vaso-occlusion in sickle cell disease: pathophysiology and novel targeted therapies. Blood. 2013;122(24):3892–3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639–1644. [DOI] [PubMed] [Google Scholar]
- 5.Anyaegbu CC, Okpala IE, Akren’Ova YA, Salimonu LS. Peripheral blood neutrophil count and candidacidal activity correlate with the clinical severity of sickle cell anaemia (SCA). Eur J Haematol. 1998;60(4):267–268. [DOI] [PubMed] [Google Scholar]
- 6.Castro O, Brambilla DJ, Thorington B, et al. The acute chest syndrome in sickle cell disease: incidence and risk factors. The Cooperative Study of Sickle Cell Disease. Blood. 1994;84(2):643–649. [PubMed] [Google Scholar]
- 7.Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91(1):288–294. [PubMed] [Google Scholar]
- 8.Wigfall DR, Ware RE, Burchinal MR, Kinney TR, Foreman JW. Prevalence and clinical correlates of glomerulopathy in children with sickle cell disease. J Pediatr. 2000;136(6):749–753. [PubMed] [Google Scholar]
- 9.Zhang D, Xu C, Manwani D, Frenette PS. Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood. 2016;127(7):801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okpala I, Daniel Y, Haynes R, Odoemene D, Goldman J. Relationship between the clinical manifestations of sickle cell disease and the expression of adhesion molecules on white blood cells. Eur J Haematol. 2002;69(3):135–144. [DOI] [PubMed] [Google Scholar]
- 11.Lard LR, Mul FP, de Haas M, Roos D, Duits AJ. Neutrophil activation in sickle cell disease. J Leukoc Biol. 1999;66(3):411–415. [DOI] [PubMed] [Google Scholar]
- 12.Fadlon E, Vordermeier S, Pearson TC, et al. Blood polymorphonuclear leukocytes from the majority of sickle cell patients in the crisis phase of the disease show enhanced adhesion to vascular endothelium and increased expression of CD64. Blood. 1998;91(1):266–274. [PubMed] [Google Scholar]
- 13.Lum AF, Wun T, Staunton D, Simon SI. Inflammatory potential of neutrophils detected in sickle cell disease. Am J Hematol. 2004;76(2):126–133. [DOI] [PubMed] [Google Scholar]
- 14.Benkerrou M, Delarche C, Brahimi L, et al. Hydroxyurea corrects the dysregulated L-selectin expression and increased H(2)O(2) production of polymorphonuclear neutrophils from patients with sickle cell anemia. Blood. 2002;99(7):2297–2303. [DOI] [PubMed] [Google Scholar]
- 15.Davenport AP, Hyndman KA, Dhaun N, et al. Endothelin. Pharmacol Rev. 2016;68(2):357–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phelan M, Perrine SP, Brauer M, Faller DV. Sickle erythrocytes, after sickling, regulate the expression of the endothelin-1 gene and protein in human endothelial cells in culture. J Clin Invest. 1995;96(2):1145–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Firth J, Ratcliffe J. Organ distribution of the three rat endothelin messengers RNAs and the effects of ischemia on renal gene expression. J Clin Invest. 1992;90(3):1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kedzierski RM, Yanagisawa M. Endothelin system: the double-edged sword in health and disease. Annu Rev Pharmacol Toxicol. 2001;41:851–876. [DOI] [PubMed] [Google Scholar]
- 19.Sabaa N, de Franceschi L, Bonnin P, et al. Endothelin receptor antagonism prevents hypoxia-induced mortality and morbidity in a mouse model of sickle-cell disease. J Clin Invest. 2008;118(5):1924–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trudel M, Saadane N, Garel MC, et al. Towards a transgenic mouse model of sickle cell disease: hemoglobin SAD. EMBO J. 1991;10(11):3157–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brun M, Bourdoulous S, Couraud PO, Elion J, Krishnamoorthy R, Lapoumeroulie C. Hydroxyurea downregulates endothelin-1 gene expression and upregulates ICAM-1 gene expression in cultured human endothelial cells. Pharmacogenomics J. 2003;3(4):215–226. [DOI] [PubMed] [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25(4):402–408. [DOI] [PubMed] [Google Scholar]
- 23.Hidalgo A, Chang J, Jang JE, Peired AJ, Chiang EY, Frenette PS. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat Med. 2009;15(4):384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elferink JG, de Koster BM. Stimulation and inhibition of neutrophil chemotaxis by endothelin-3. J Cardiovasc Pharmacol. 1995;26 (Suppl 3):S142–144. [PubMed] [Google Scholar]
- 25.Elferink JG, de Koster BM. The effect of endothelin-2 (ET-2) on migration and changes in cytosolic free calcium of neutrophils. Naunyn Schmiedebergs Arch Pharmacol. 1996;353(2):130–135. [DOI] [PubMed] [Google Scholar]
- 26.Elferink JG, de Koster BM. Modulation of human neutrophil chemotaxis by the endothelin-B receptor agonist sarafotoxin S6c. Chem Biol Interact. 1996;101(3):165–174. [DOI] [PubMed] [Google Scholar]
- 27.Graido-Gonzalez E, Doherty JC, Bergreen EW, Organ G, Telfer M, McMillen MA. Plasma endothelin-1, cytokine, and prostaglandin E2 levels in sickle cell disease and acute vaso-occlusive sickle crisis. Blood. 1998;92(7):2551–2555. [PubMed] [Google Scholar]
- 28.Werdehoff SG, Moore RB, Hoff CJ, Fillingim E, Hackman AM. Elevated plasma endothelin-1 levels in sickle cell anemia: relationships to oxygen saturation and left ventricular hypertrophy. Am J Hematol. 1998;58(3):195–199. [DOI] [PubMed] [Google Scholar]
- 29.Rybicki AC, Benjamin LJ. Increased levels of endothelin-1 in plasma of sickle cell anemia patients. Blood. 1998;92(7):2594–2596. [PubMed] [Google Scholar]
- 30.Tharaux PL, Hagege I, Placier S, et al. Urinary endothelin-1 as a marker of renal damage in sickle cell disease. Nephrol Dial Transplant. 2005;20(11):2408–2413. [DOI] [PubMed] [Google Scholar]
- 31.Chaar V, Tarer V, Etienne-Julan M, Diara JP, Elion J, Romana M. ET-1 and ecNOS gene polymorphisms and susceptibility to acute chest syndrome and painful vaso-occlusive crises in children with sickle cell anemia. Haematologica. 2006;91(9):1277–1278. [PubMed] [Google Scholar]
- 32.Lionnet F, Bachmeyer C, Stankovic K, Tharaux PL, Girot R, Aractingi S. Efficacy of the endothelin receptor blocker bosentan for refractory sickle cell leg ulcers. Br J Haematol. 2008;142(6):991–992. [DOI] [PubMed] [Google Scholar]
- 33.Lopez Farre A, Riesco A, Moliz M, et al. Inhibition by L-arginine of the endothelin-mediated increase in cytosolic calcium in human neutrophils. Biochem Biophys Res Commun. 1991;178(3):884–891. [DOI] [PubMed] [Google Scholar]
- 34.Ishida K, Takeshige K, Minakami S. Endothelin-1 enhances superoxide generation of human neutrophils stimulated by the chemotactic peptide N-formyl-methionyl-leucyl-phenylalanine. Biochem Biophys Res Commun. 1990;173(2):496–500. [DOI] [PubMed] [Google Scholar]
- 35.Gomez-Garre D, Guerra M, Gonzalez E, et al. Aggregation of human polymorphonuclear leukocytes by endothelin: role of platelet-activating factor. Eur J Pharmacol. 1992;224(2–3):167–172. [DOI] [PubMed] [Google Scholar]
- 36.Camus SM, De Moraes JA, Bonnin P, et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood. 2015;125(24):3805–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang D, Chen G, Manwani D, et al. Neutrophil ageing is regulated by the micro-biome. Nature. 2015;525(7570):528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trudel M, De Paepe ME, Chretien N, et al. Sickle cell disease of transgenic SAD mice. Blood. 1994;84(9):3189–3197. [PubMed] [Google Scholar]
- 39.De Franceschi L, Brugnara C, Rouyer-Fessard P, Jouault H, Beuzard Y. Formation of dense erythrocytes in SAD mice exposed to chronic hypoxia: evaluation of different therapeutic regimens and of a combination of oral clotrimazole and magnesium therapies. Blood. 1999;94(12):4307–4313. [PubMed] [Google Scholar]
- 40.de Franceschi L, Baron A, Scarpa A, et al. Inhaled nitric oxide protects transgenic SAD mice from sickle cell disease-specific lung injury induced by hypoxia/reoxygenation. Blood. 2003;102(3):1087–1096. [DOI] [PubMed] [Google Scholar]
- 41.De Paepe ME, Trudel M. The transgenic SAD mouse: a model of human sickle cell glomerulopathy. Kidney Int. 1994;46(5): 1337–1345. [DOI] [PubMed] [Google Scholar]
- 42.Fukuroda T, Fujikawa T, Ozaki S, Ishikawa K, Yano M, Nishikibe M. Clearance of circulating endothelin-1 by ETB receptors in rats. Biochem Biophys Res Commun. 1994;199(3):1461–1465. [DOI] [PubMed] [Google Scholar]
- 43.Ohuchi T, Kuwaki T, Ling GY, et al. Elevation of blood pressure by genetic and pharmacological disruption of the ETB receptor in mice. Am J Physiol. 1999;276(4 Pt 2):R1071–1077. [DOI] [PubMed] [Google Scholar]
- 44.Pollock JS, Pollock DM. Endothelin and NOS1/nitric oxide signaling and regulation of sodium homeostasis. Curr Opin Nephrol Hypertens. 2008;17(1):70–75. [DOI] [PubMed] [Google Scholar]
- 45.Goddard J, Eckhart C, Johnston NR, Cumming AD, Rankin AJ, Webb DJ. Endothelin A receptor antagonism and angiotensin-converting enzyme inhibition are synergistic via an endothelin B receptor-mediated and nitric oxide-dependent mechanism. J Am Soc Nephrol. 2004;15(10):2601–2610. [DOI] [PubMed] [Google Scholar]
- 46.Goligorsky MS, Tsukahara H, Magazine H, Andersen TT, Malik AB, Bahou WF. Termination of endothelin signaling: role of nitric oxide. J Cell Physiol. 1994;158(3):485–494. [DOI] [PubMed] [Google Scholar]
- 47.Wright CD, Cody WL, Dunbar JB, Jr, Doherty AM, Hingorani GP, Rapundalo ST. Characterization of endothelins as chemoattractants for human neutrophils. Life Sci. 1994;55(21):1633–1641. [DOI] [PubMed] [Google Scholar]
- 48.Helset E, Lindal S, Olsen R, Myklebust R, Jorgensen L. Endothelin-1 causes sequential trapping of platelets and neutrophils in pulmonary microcirculation in rats. Am J Physiol. 1996;271(4 Pt 1):L538–546. [DOI] [PubMed] [Google Scholar]
- 49.Elferink JG, de Koster BM. Endothelin-induced activation of neutrophil migration. Biochem Pharmacol. 1994;48(5):865–871. [DOI] [PubMed] [Google Scholar]
- 50.Zarpelon AC, Pinto LG, Cunha TM, et al. Endothelin-1 induces neutrophil recruitment in adaptive inflammation via TNFalpha and CXCL1/CXCR2 in mice. Can J Physiol Pharmacol. 2012;90(2):187–199. [DOI] [PubMed] [Google Scholar]
- 51.Sessa WC, Kaw S, Hecker M, Vane JR. The biosynthesis of endothelin-1 by human polymorphonuclear leukocytes. Biochem Biophys Res Commun. 1991;174(2):613–618. [DOI] [PubMed] [Google Scholar]
- 52.Makis AC, Hatzimichael EC, Kolios G, Bourantas KL. Circulating endothelin-3 levels in patients with sickle cell disease during hydroxyurea treatment. Haematologica. 2004;89(3):360–361. [PubMed] [Google Scholar]
- 53.Espinosa G, Lopez Farre A, Cernadas MR, et al. Role of endothelin in the pathophysiology of renal ischemia-reperfusion in normal rabbits. Kidney Int. 1996;50(3):776–782. [DOI] [PubMed] [Google Scholar]
- 54.Farmer DG, Kaldas F, Anselmo D, et al. Tezosentan, a novel endothelin receptor antagonist, markedly reduces rat hepatic ischemia and reperfusion injury in three different models. Liver Transplant. 2008;14(12): 1737–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khimenko PL, Moore TM, Taylor AE. Blocked ETA receptors prevent ischemia and reperfusion injury in rat lungs. J Appl Physiol. 1996;80(1):203–207. [DOI] [PubMed] [Google Scholar]
- 56.Ghandour S, Cetinel S, Kurtel H. Endothelin-3 induced mesenteric vasoconstriction and PMN infiltration in the rat small intestine: role of endothelin receptors. Regul Pept. 2004;119(1–2):125–131. [DOI] [PubMed] [Google Scholar]
- 57.Lopez-Farre A, Caramelo C, Esteban A, et al. Effects of aspirin on platelet-neutrophil interactions. Role of nitric oxide and endothelin-1. Circulation. 1995;91(7):2080–2088. [DOI] [PubMed] [Google Scholar]
- 58.Zouki C, Baron C, Fournier A, Filep JG. Endothelin-1 enhances neutrophil adhesion to human coronary artery endothelial cells: role of ET(A) receptors and platelet-activating factor. Br J Pharmacol. 1999;127(4):969–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fernandez-Patron C, Zouki C, Whittal R, Chan JS, Davidge ST, Filep JG. Matrix metalloproteinases regulate neutrophil-endothelial cell adhesion through generation of endothelin-1[1–32]. FASEB J. 2001;15(12): 2230–2240. [DOI] [PubMed] [Google Scholar]
- 60.Kim M, Carman CV, Springer TA. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science. 2003;301(5640):1720–1725. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.