

Abstract
Disorders of iron metabolism are largely attributed to an excessive or insufficient expression of hepcidin, the master regulator of systemic iron homeostasis. Here, we investigated whether drugs targeting genetic regulators of hepcidin can affect iron homeostasis. We focused our efforts on drugs approved for clinical use to enable repositioning strategies and/or to reveal iron-related side effects of widely prescribed therapeutics. To identify hepcidin-modulating therapeutics, we re-evaluated data generated by a genome-wide RNAi screen for hepcidin regulators. We identified ‘druggable’ screening hits and validated those by applying RNAi of potential drug targets and small-molecule testing in a hepatocytic cell line, in primary murine and human hepatocytes and in mice. We initially identified spironolactone, diclofenac, imatinib and Suberoylanilide hydroxamic acid (SAHA) as hepcidin modulating drugs in cellular assays. Among these, imatinib and spironolactone further suppressed liver hepcidin expression in mice. Our results demonstrate that a commonly used anti-hypertensive drug, spironolactone, which is prescribed for the treatment of heart failure, acne and female hirsutism, as well as imatinib, a first-line, lifelong therapeutic option for some frequent cancer types suppress hepcidin expression in cultured cells and in mice. We expect these results to be of relevance for patient management, which needs to be addressed in prospective clinical studies.
Introduction
Hepcidin is a liver-expressed regulatory hormone that adjusts iron fluxes to body iron requirements.1 Conditions of high iron load or inflammation activate hepatic hepcidin production, thereby reducing plasma iron levels. Iron deficiency or augmented erythropoietic activity inhibit hepcidin expression to increase systemic iron availability. Disorders of iron metabolism belong to the most common diseases worldwide and are to a large extent attributed to excessive or insufficient levels of hepcidin. The frequent iron overload disorder hereditary hemochromatosis (HH) is caused by inappropriately low hepcidin expression.1 Likewise, hepcidin deficiency leads to iron accumulation in several subtypes of hereditary anemias (e.g. thalassemias), termed iron-loading anemias, that are hallmarked by ineffective yet augmented hematopoiesis.2 Of clinical importance, progressive tissue iron deposition, especially in the heart, represents the major cause of mortality in these disorders.3 Furthermore, alterations of iron levels or its misdistribution exacerbates pathologies of common acquired diseases such as chronic liver disease,4 diabetes,5 cardiovascular disease or neurodegenerative disorders.6 Conversely, the anemia of inflammation (AI), caused by iron retention within body iron reservoirs and hypoferremia, develops due to hepcidin induction by immune activation.7 The progression of the AI increases morbidity rates, often prolongs time of hospitalization and is associated with a poorer prognosis in a variety of disease conditions.8
Current therapies for iron-related pathologies target symptoms of the disease rather than the underlying molecular mechanisms. Standard treatment of HH involves repetitive phlebotomies, which is not suitable for other patients with iron overload (e.g. for those suffering from concomitant anemias) and may be additionally compromised by suboptimal compliance.9 Treatment of choice for iron-loading anemias involves iron chelation, which, although effective, causes significant side effects.10 If combating the underlying pathology is not feasible, the current therapeutic options for the AI include blood transfusions, erythropoiesis stimulating agents (ESA) and parenteral iron administration.8 However, blood transfusions were shown to be associated with multiorgan failure in critically ill patients.11 ESA therapy for cancer-associated anemia increased incidence of tumor progression, cardiovascular pathologies and death,12 whereas the long-term safety of intravenous iron supplementations is still unknown. Because available therapies show limitations and adverse effects, there is substantial interest in identifying novel approaches that modify hepcidin levels or impact on its molecular function. Several novel strategies are currently under development, which include minihepcidins (hepcidin agonists), RNAi therapeutics targeting hepcidin regulators, anti-hepcidin drugs such as spiegelmers, anticalins or monoclonal antibodies, and non-coagulating heparins.13 Despite successful application of these agents in animal models, their clinical use may be challenging.13 Moreover, since these new therapeutics have not yet been approved for patients’ therapy, their long-term safety and efficacy are unknown.
The list of drugs licensed for therapy in patients consists of approximately 1900 small molecules, which are expected to impact on numerous molecular processes and for which a substantial amount of clinical data is available (http://www.fda.gov/). The spectrum of signals that control hepcidin expression extends beyond the originally identified bone morphogenetic proteins or inflammatory cues,1 and involves growth factors,14 erythroferrone,15 sex hormones,16 nutrient stimuli17 or retinoids.18 This demonstrates that multiple signaling events converge at the hepcidin promoter, which may imply that several drugs approved for clinical practice may modulate hepcidin transcription. Such a possibility opens avenues for the re-purposing of known therapeutics for the management of iron-related diseases,19 an approach which may offer an attractive alternative to both current and novel iron-focused therapies. Furthermore, the existence of a broad range of pathways that modulate hepcidin levels implies that disturbances of iron homeostasis may represent an overlooked side effect of licensed drugs. In this study, we reanalyzed data generated by a genome-wide RNAi screen for hepcidin regulators17 to identify putative drug targets and applied extensive small-molecule testing to identify drugs and drug targets that modulate hepcidin expression. We identified two widely prescribed drugs administered long term in patients, imatinib and spironolactone, as suppressors of hepcidin expression in primary hepatocytes and in mice. These findings may be of clinical relevance in that iron-related side effects of these therapeutics may be identified in further clinical studies.
Methods
Cell culture and drug treatment
The human hepatocarcinoma cell line Huh7 was obtained from ATCC (Wesel, Germany). Human liver tissue was obtained from macroscopically healthy human liver tissue that remained from resected human liver of patients with primary or secondary liver tumors or benign local liver diseases. Informed consent of the patients for the use of tissue for research purposes was obtained according to the ethical guidelines of the Charite University Hospital Berlin. Protocols for isolation of human and murine primary hepatocytes (PHs) as well as culture conditions for all cell models used in the study are described in the Online Supplementary Appendix.
If not specified, Huh7 cells and primary hepatocytes were treated with drugs in complete, serum-containing medium. The list of drugs used in the study is summarized in Online Supplementary Table S1.
Transfection of siRNAs and luciferase reporter constructs
As reported previously,17 we reverse transfected Huh7 cells with 10 pmol siRNA (pooled or individual duplexes; Dharmacon, Online Supplementary Table S2) in a 96-well format. For RNAi of weakly-expressed candidate genes we applied reverse transfection in 24-well plates with 13×104 cells in each well, using 50 pmol of siRNA and 1.5 μl Dharmafect1 (Dharmacon). Cells were cultured for 72 hours prior to harvesting of total RNA.
Luciferase reporter constructs that contain the full-length 2,762 bp hepcidin promoter (WT_2.7kb) or its mutant derivatives (STAT_BS_2.7kb and BMP_RE1_BMP_RE2_2.7kb) were described previously.20 Twenty-four hours after seeding Huh7 cells, these were transfected with reporter plasmids using Lipofectamine 2000 Transfection Reagent (2 μl/well; 200 ng of hepcidin promoter constructs, 20 ng of CMV-Renilla control plasmid). Twenty-four hours later cells were treated with drugs at the indicated concentrations and incubated for an additional 24 hours. Luciferase activity was measured as described before.21
Preparation of total RNA, reverse transcription, and quantitative real-time PCR analysis
Isolation of total RNA and the protocols for reverse transcription (RT) and real-time qPCR were described previously.20 Total RNA extraction from mouse livers was performed using Trizol (Invitrogen). Total RNA extraction for the secondary RNAi assays in 96-well plates was performed using the QuickExtract™ RNA Extraction Kit (Epicentre Biotechnologies, Madison, WI, USA) according to the manufacturer’s instructions.17 RNA extraction from all other cell-based experiments was performed using RNeasy® kit (Qiagen). Sequences of the qPCR primers are shown in Online Supplementary Table S3. Expression levels of human GAPDH (glyceraldehyde-3-phosphate-dehydrogenase) or murine PpIb or Rpl19 were used as normalization controls.
Mice
Experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of the EMBL Heidelberg. Male wild-type C57BL/6 mice were maintained with free access to food and water. At 11 weeks of age mice were administrated with the following drugs dissolved in drinking water for two weeks: imatinib (75 mg/kg/day), spironolactone (24 mg/kg/day), Suberoylanilide hydroxamic acid (SAHA; 50 mg/kg/day) and diclofenac (1 mg/kg/day). As described in detail in the Online Supplementary Appendix, the doses applied were chosen based on literature and U.S. Food and Drug Administration (FDA) approval documents to provoke desired biological responses without causing significant side effects. The drug doses used in mice correspond closely to the doses applied in human patients, based on the body surface area (BSA) dose escalation method.22,23 Since SAHA, spironolactone and diclofenac were difficult to dissolve in aqueous solutions, we increased their solubility in water by complexing with 2-hydroxypropyl-β-cyclodextrin (HOPβ-CD) (Online Supplementary Appendix for more details). Two different doses of HOPβ-CD were applied: 9 g/L for SAHA and diclofenac, and 1.1 mg/L for imatinib and spironolactone. As controls, mice administrated with an equivalent dose of HOPβ-CD dose were studied. Plasma levels of hepcidin peptide were measured using the ELISA kit from DRG Diagnostics according to the manufacturer’s protocol. Plasma iron levels were measured using SFBC and UIBC Iron Kits (Biolabor). Tissue non-heme iron content was measured using the bathophenanthroline method and calculated against dry weight tissue.24
Statistical analysis
Statistical analyses were performed using Prism v.6 (GraphPad). Data from cell-based experiments are shown as a fold change ± 95% confidence intervals (CI) compared to control conditions. Data from mouse experiments are shown as mean ± SEM. One sample t-test or two-tailed Student’s t-test were used for estimation of statistical significance, respectively.
Results
Identification of drug targets among putative hepcidin regulators
We recently reported the outcome of an unbiased genome-wide RNAi screen that generated a comprehensive list of putative hepatic hepcidin regulators.17 We now further analyzed the hit list of this screen by utilizing ‘Manually Annotated Targets and Drugs Online Resource’ (MATADOR; http://matador.embl.de/), a resource of drug and drug-target interactions.25 Among 2159 putative hepcidin regulators, our bioinformatic analysis identified approximately 160 screening hits that are potentially sensitive to pharmacological manipulation. Importantly, the initial selection process was unbiased in terms of gene function, specific pathways or expression profiles. To further enrich for clinically relevant gene-drug pairs we applied literature mining to manually exclude biologically ‘unspecific’ hits (e.g. those involved in general drug metabolism or those that were only distally related to real drug targets). From the remaining 56 direct and specific drug targets, we excluded 19 genes which could not be reliably detected at transcript level in Huh7 cells, a cellular model used in the RNAi screen (data not shown).17
Secondary validation assays confirm six drug targets as hepcidin regulators
Thirty-seven putative ‘druggable’ hepcidin modifiers were selected for RNAi-based validation in a 2-step manner. In a first step, we validated each drug target gene for its ability to modulate endogenous hepcidin mRNA levels upon siRNA-mediated knockdown with a pool of four single siRNAs. Thirteen genes, including 4 potential hepcidin suppressors and 9 potential activators were confirmed [Figure 1A; data for HDAC3 included also in Pasricha et al., (submitted manuscript)]. In a second step we individually applied four single siRNAs per gene and considered a gene as validated if at least two out of four siRNAs significantly altered hepcidin mRNA expression (Figure 1B). By applying these criteria, we identified histone deacetylase 3 (HDAC3) and retinoic acid receptor beta (RARB) as hepcidin suppressors. These results are consistent with previous findings that HDAC inhibitors26 and retinoic acid18 exert suppressive effects on hepcidin expression. Our RNAi validation strategy further identified 4 additional, functionally diverse genes as hepcidin activators: membrane progestin receptor gamma (MPRG), NAD(P)H quinone dehydrogenase 2 (NQO2), prostaglandin I2 (prostacyclin) synthase (PTGIS) and phospholipase C beta 3 (PLCB3). MPRG acts in intestinal endocrine cells to regulate glucose homeostasis,27 NQO2 oxidoreductase catalyzes metabolic detoxification of quinones28 and regulates levels of the tumor suppressor p53,29 PTGIS synthesizes prostacyclin,30 a potent vasodilator and inhibitor of platelet aggregation, and PLCB3 generates the second messenger molecules diacylglycerol and inositol 1,4,5-trisphosphate involved in signal transduction.31 Beside the fact that PTGIS uses iron as a co-factor, roles of the identified genes in iron metabolism have not been reported.
Figure 1.

Two-step validation RNAi assays identify six novel hepcidin regulators. (A) Thirteen screening hits were validated by the analysis of endogenous hepcidin mRNA levels upon knockdown with pools of 4 individual siRNAs in Huh7 cells. RNAi of SMAD7, SMAD4 and STAT3 served as a control. (B) Six regulators of hepcidin mRNA expression were validated by at least two independent siRNAs. Shown are mRNA levels of hepcidin and target genes after RNAi. Results are presented as a fold change (± 95% CI) compared to samples transfected with scrambled siRNA. The mean of at least three independent experiments is shown. Significant changes are indicated by asterisks (*P<0.05, **P<0.005, ***P<0.001).
Small-molecule testing identifies four approved therapeutics as hepcidin modulators in Huh7 cells
To identify hepcidin-modifying drugs, we next retrieved those compounds from the MATADOR database that target the 13 genes validated by application of siRNA pools (Table 1 and Figure 1A). We did not further consider retinoic acid in our experiments as it was previously shown to affect hepcidin mRNA levels in Huh7 cells.18 Furthermore, administration of diazepam, the GABA receptor agonist, was not possible due to legal reasons. As shown in Table 1, most of the selected drugs are routinely used in clinical practice and are prescribed for a wide range of diseases.
Table 1.
Drugs targeting hepcidin regulators identified by an RNAi screen.

We next tested these drugs in a time- and dose-dependent manner. To establish the optimal incubation time, Huh7 cells were exposed to the selected small molecules for various time intervals, including 4, 8, 16 and 24 hours (data not shown). Subsequently, different drug doses were tested. In these experiments we identified five therapeutics as modulators of hepcidin mRNA levels. We show that diclofenac, an analgesic classified as a non-steroidal anti-inflammatory agent (NSAID) and the histone deacetylase inhibitor (HDACi) SAHA (Vorinostat) induce hepcidin mRNA expression in a dose-dependent manner (Figure 2A). In addition, the anti-hypertensive drug spironolactone and the pan-tyrosine kinase inhibitor imatinib, which is broadly applied in cancer therapies, suppress hepcidin mRNA expression in hepatoma Huh7 cells (Table 1 and Figure 2A). Of note, the potassium-sparing diuretic amiloride induced hepcidin expression in Huh7 cells (data not shown), but was excluded from our analysis due to its toxicity in primary hepatocyte cultures.
Figure 2.

Drugs identified as hepcidin modulators in Huh7 cells and primary hepatocytes. (A,C,D) Cells were exposed to increasing concentrations of individual drugs for the indicated time points. (B) Effects of 24-hour drug treatments (10 μM imatinib, 50 μM spironolacton) on hepcidin promoter activity in cells transfected with wild-type (WT_2.7) and mutant hepcidin promoter constructs (ST: STAT-BS mutant, B1/B2: double mutant of BMP-RE1 and 2). Results are presented as a fold change (± 95% CI) of hepcidin mRNA levels (A,C,D) or hepcidin promoter activity (Firefly/Renilla luciferase signal) (B) compared to vehicle-treated control cells. The mean of at least three independent experiments is shown. Significant changes are indicated by asterisks: (*P<0.05, **P<0.005, ***P<0.001).
We next wondered whether the drugs that modulate hepcidin expression act by involving well-established signaling pathways, the inflammatory JAK-STAT or the BMP pathway.1 To test this we utilized luciferase reporter constructs driven by the hepcidin full-length promoter and its mutant derivatives.20,32 We did not detect significant responses of the hepcidin promoter to SAHA, diclofenac and amiloride treatment (data not shown). By contrast, promoter activity was reduced following treatment with spironolactone and imatinib. The drug responses were preserved upon mutation of the STAT-binding site in the hepcidin promoter, but were attenuated in promoter constructs lacking functional BMP-responsive elements (REs) 1 and 2. These data suggest that intact BMP signaling at the level of the hepcidin promoter contributes to the modulation of hepcidin transcription by these hepcidin-suppressing drugs.
Diclofenac, imatinib, SAHA and spironolactone affect hepcidin levels in primary hepatocyte cultures
We next exposed murine and human primary hepatocytes (PHs) to the selected drugs (Figure 2B and C). Diclofenac, imatinib and SAHA altered hepcidin mRNA levels consistently in murine and human hepatocytes, whereas spironolactone was only active in human PHs, where it potently suppressed hepcidin mRNA expression. Taken together, these data demonstrate that the response of hepcidin to the tested therapeutics is not restricted to hepatoma cells and is preserved in primary hepatocytes.
Identification of drug target - drug relationships
The combination of RNAi and small-molecule testing identified 5 genes and 4 drugs that operate as hepcidin modulators (Table 2). Application of SAHA (Vorinostat), a broad-range HDAC inhibitor (HDACi), induced hepcidin mRNA expression in all three hepatocytic cell models (Figure 3A–C), supporting previous data that the HDACi trichostatin A induces hepcidin in hepatoma cells.26 Increased hepcidin mRNA levels in response to SAHA treatment of Huh7 cells correlated well with the hepcidin response to RNAi of HDAC3 (Figure 1), one target of SAHA. Further analysis showed that HDAC3 may be critical for hepcidin regulation, because Panobinostat (LBH-589) and Trapoxin A, two pan-HDACi targeting HDAC3 within their inhibitory spectrum, induced hepcidin levels (Figure 3A). Furthermore, hepcidin mRNA levels remained unaltered upon treatment with those compounds that do not target HDAC3, including the HDAC8-specific inhibitor Compound 2 and Entinostat (MS-275), an inhibitor targeting preferentially HDAC1 (Figure 3A). Surprisingly, murine and human PHs exposed to the same panel of HDAC inhibitors showed a different pattern of hepcidin alteration (Figure 3B and C). These data suggest that contributions of specific HDACs to hepcidin suppression may differ between hepatoma cells and primary cell models, and that HDACs other than HDAC3 may be involved in modulating hepcidin levels. This conclusion is supported by the finding that RNAi to Hdac3 in murine primary hepatocytes failed to alter hepcidin mRNA levels (data not shown). Independent of the specific HDAC involved in modulating hepcidin mRNA expression, these data, together with previous findings26 suggest that inhibition of histone deacetylation could represent an important intervention point for modulating hepcidin expression.
Table 2.
Summary of drug targets and drugs drugs identified as hepcidin modulators in hepatocytic cellular models.

Figure 3.
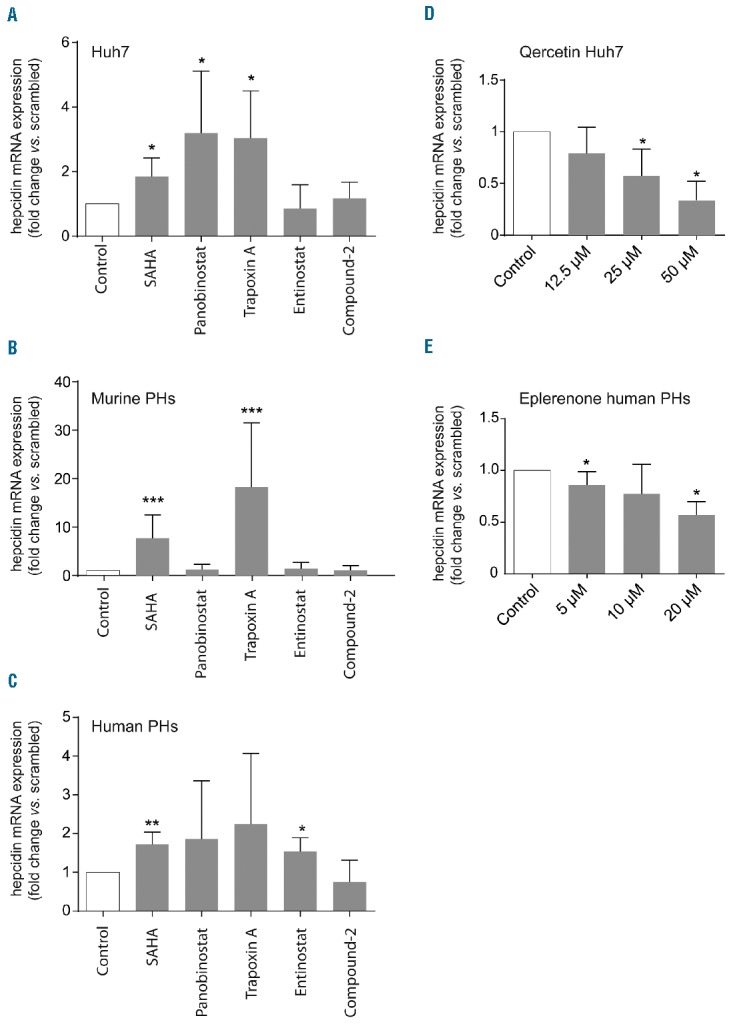
Refinement of drug target-drug relationships. (A–C) The indicated cell models were exposed to a panel of histone deacetylase (HDAC) inhibitors, SAHA (1 μM), Panobinostat (0.1 μM), Trapoxin A (0.08 μM), Entinostat (1 μM) and Compound-2 (250 nM) for 8 hours (h), in complete medium (A), or in serum-free medium (after a 24-h serum starvation) (B and C). (D) Huh7 cells were treated with increasing concentrations of quercetin for 24 h. (E) Human primary hepatocytes (PH) were exposed to increasing doses of eplerenone for 24 h. All results are presented as a fold change (± 95%CI) of hepcidin mRNA levels. The mean of at least three independent experiments is shown. Significant changes are indicated by asterisks: *P<0.05, **P<0.005, ***P<0.001.
Imatinib was selected from the MATADOR database, because it targets two tyrosine kinases (PDGFRB and TEC) identified in the RNAi screen as candidate hepcidin regulators (Table 1 and Figure 1A), a finding that we could not validate by further RNAi-based analysis (data not shown). Detailed literature mining however suggested NQO2 as a non-kinase target of imatinib.33 NQO2 was likewise identified as a hepcidin activator in our study. Interestingly, quercetin, a potent NQO2 inhibitor,34 attenuates hepcidin mRNA expression (Figure 3D), suggesting that NQO2 and its inhibitor imatinib may control hepcidin expression. It is of note that according to the experimental data NQO2 is insensitive to dicumarol,34 a compound linked to NQO2 in the MATADOR database (Table 1) due to its similarity to the known dicumarol target NQO1. Hence, the lack of dicumarol effect on hepcidin expression (Table 2) is consistent with previous knowledge.
Guided by information obtained from the MATADOR database, the analgesic drug diclofenac was selected to target PTGIS, a gene encoding the prostacyclin synthase. As shown in Table 2, the results obtained from the analyses of this drug-drug target pair are inconsistent. However, diclofenac may not directly target PTGIS, but, as other NSAIDs, may predominantly inhibit cyclooxygenases (COX-1 and COX-2).35 Cyclooxygenases are the rate-limiting enzymes in the synthesis of prostaglandin H2, that is then further converted to the vasoactive prostacyclin by PTGIS.35 Importantly, treatment of cells with Aspirin, another member of the NSAID family known to inhibit COX-1 and to a lesser extent COX-2,35 failed to control hepcidin mRNA levels (data not shown). Taken together, these findings suggest that diclofenac controls hepcidin expression via a mechanism independent of PTGIS and cyclooxygenases. Spironolactone was selected for analysis, because it targets and antagonizes OSBPL10, a member of the oxysterol-binding protein-related proteins (ORP) family. Despite suppressing effects of spironolactone on hepcidin expression, OSBPL10 was not validated by our RNAi assays. More detailed analysis of an additional drug-target database ‘DrugBank’ (www.drugbank.ca)36 revealed that ORPs are only distally related to bona fide targets of spironolactone, aldosterone and androgen receptors.36–38 To further corroborate the role of spironolactone in hepcidin regulation and to address its mode of action, we treated human PHs with eplerenone, a highly specific blocker of the aldosterone receptor.36 Interestingly, we found a dose-dependent reduction of hepcidin mRNA levels in response to eplerenone supplementation (Figure 3E), suggesting that aldosterone, but not androgen signaling may cross-talk to hepcidin transcriptional control in primary hepatocytes.
Imatinib and spironolactone modulate hepcidin levels in vivo
We next investigated whether the drugs that we identified in cellular assays (spironolactone, diclofenac, imatinib and SAHA) modulate hepcidin levels in mice, when administered in the drinking water. Drug doses were selected based on literature and FDA documentation (see Online Supplementary Appendix) in such a way that they would provoke the desired biological responses without causing significant side effects. Mice were treated for a period of two weeks, shown by previous studies to be sufficient to affect systemic iron parameters.13
Generally, the applied drugs were well tolerated and did not affect the weight of the mice (data not shown). Diclofenac caused a mild decrease of red blood cell numbers, hemoglobin levels and hematocrit (Table 3), whereas blood parameters remained unaltered following treatment with all other drugs. Hepatic hepcidin mRNA levels remained unchanged upon administration of SAHA and diclofenac (Figure 4A), while treatment with imatinib and spironolactone significantly reduced hepcidin mRNA levels (Figure 4A). Spironolactone treatment of mice further decreased plasma hepcidin levels (Figure 4B). Hepatotoxicity unlikely contributes to diminished hepcidin expression, because plasma levels of the liver damage marker alanine aminotransferase (ALT) remained unaffected (Figure 4C). Imatinib and spironolactone treatment further caused a decrease of unsaturated iron binding capacity (UIBC), and an increase of transferrin saturation, without a significant change in serum iron levels (Figure 4D). Two weeks of drug supplementation was not sufficient to alter tissue iron levels (Online Supplementary Figure S1). Taken together, our data show that imatinib and spironolactone reduce hepcidin levels in cellular assays and mice, and mildly affect plasma iron parameters, suggesting that altered body iron levels may arise as a complication of long-term drug application. This will have to be tested in future studies.
Table 3.
Blood parameters in mice treated with hepcidin-modifying drugs.

Figure 4.

Treatment with imatinib and spironolactone suppresses hepcidin in mice. Wild-type 11-week old mice received imatinib (75 mg/kg/day), spironolactone (24 mg/kg/day), suberoylanilide hydroxamic acid (SAHA) (50 mg/kg/day) and diclofenac (1 mg/kg/day) for two weeks with drinking water. (A) Relative liver mRNA expression of hepcidin and (B) plasma hepcidin levels in drug-treated mice compared to respective controls. (C) Plasma alanine aminotransferase (ALT) levels for the indicated treatment groups. (D) Plasma iron content (SFBC), unsaturated iron binding capacity (UIBC) and transferrin saturation in response to drug administration. All data are presented as a mean±SEM from the indicated number of mice per treatment group. t-test, *P<0.05.
Discussion
The knowledge about processes and signaling mechanism which modulate hepcidin expression is constantly growing. We hypothesized that licensed therapeutics affect some of these pathways and thus systemic iron levels. This assumption has two medically-relevant implications. First, it may open a possibility for drug re-positioning, a time- and cost-effective approach of finding new applications for compounds approved for clinical practice.19 Second, it may suggest iron-related side effects caused by FDA-approved drugs, which may not be detected by routine diagnostics.
We therefore reanalyzed data from our recently reported RNAi screen17 for hepcidin regulators to select genes that are ‘druggable’ hepcidin modulators. These genes were validated by RNAi and small-molecule testing was applied to identify the corresponding drugs as hepcidin modulators. This approach complements and extends previously reported drug screens in search for hepcidin regulators,39,40 in that it starts with a ‘pre-selected’ set of drugs based on a comprehensive RNAi approach, applies time-and dose-response experiments, and involves validations in primary cells and mice. With this approach we identified four compounds that modulate hepcidin mRNA levels in primary hepatocyte models (Figure 2). Two of these, imatinib and spironolactone, further affected hepcidin levels in mice, causing a mild increase of transferrin saturation, without causing hepatotoxicity (Figure 4). Whether these relatively modest alterations of iron parameters affect tissue iron content during long-term therapy in patients requires further investigation.
Assessment of the potential for drug re-positioning
The safety profile of imatinib excludes this compound from the possibility for drug re-positioning. Imatinib may cause peripheral edema, nausea, vomiting, rash, fatigue and abdominal pain.41,42 In addition, patients on imatinib may develop congestive heart failure due to severe left ventricular dysfunction.41
By comparison, spironolactone shows relatively minor adverse effects. It was approved in 1959 and initially was used as supplementary therapy for refractory hypertension.37 Although it is well-tolerated, antiandrogenic effects of spironolactone were reported, mainly due to cross-reactivity with the androgen receptor and alterations of peripheral metabolism of testosterone.38 These properties of spironolactone were linked to undesirable effects in males (e.g. gynecomatia), but in women were exploited to successfully treat acne and hirsutism.43,44 In the latter case spironolactone therapy commonly caused menstrual irregularities and increased diuresis. These side effects were transient and usually not considered as sufficient for treatment discontinuation. Taken together, the safety profile of spironolactone, supported by its potential to affect hepcidin levels in vivo, make it promising for re-purposing for diseases hallmarked by inappropriately high hepcidin synthesis. Whether spironolactone can modulate hepcidin expression under disease-associated conditions, such as inflammation, and whether prolonged spironolactone-mediated hepcidin suppression affects tissue iron content remain to be tested.
Iron-related side effects caused by FDA-approved drugs
Growing evidence suggests that a dysregulated systemic iron balance represents an important modifying factor for several common diseases, e.g. chronic liver disease, diabetes or atherosclerosis.4–6 Furthermore, epidemiological studies indicate that an increased body iron status may increase cancer risk.45 In this context it is of clinical relevance to identify iron-related side effects of frequently prescribed therapeutics. Imatinib represents a lifelong first-line therapeutic option in chronic myeloid leukemia, gastrointestinal stoma tumors and several other rare disorders (e.g. those hallmarked by platelet-derived growth factor aberrations).46 Here we report that imatinib treatment decreased hepcidin mRNA levels in vivo (Figure 4A). Whether this will cause imbalanced systemic iron levels under long-term treatment needs to be investigated. Interestingly, liver iron overload was reported in a patient treated with imatinib.47 It remained unclear whether in this single case a hepatotoxic response contributes to hepcidin suppression. Our data in mice however suggest that imatinib affects hepcidin transcription independently of liver damage (Figure 4A and C).
It will be interesting to systematically analyze iron-related parameters, such as serum ferritin, transferrin saturation and hepcidin levels in patients treated with imatinib. In cases in which iron overload is observed in these patients it may contribute to progression of primary tumors due to pro-oxidative and nutritional roles of iron in cancer cells.45 Furthermore, iron overload in response to long-term imatinib treatment may cause heart iron deposition contributing to the reported cardiotoxicity of this drug.41 In such a case, a diet with limited iron content, phlebotomies47 or iron chelation therapies may be indicated to alleviate the severity of congestive heart failure observed in some patients on imatinib.
Edema, a condition hallmarked by water and sodium retention in the body, is commonly observed in clinical practice.48 Congestive heart failure is one of the most life-threatening causes of edema. Spironolactone was shown to combat hyperaldosteronism and subsequent edema and decreased morbidity and mortality rates in patients with severe heart failure.49 As described above, hepcidin deficiency upon long-term spironolactone therapy may cause iron overload also in the heart, which could aggravate heart disease and thus should be diagnosed and prevented. Another important indication for spironolactone is ascites, an accumulation of fluid in the abdominal cavity. Interestingly, ascites is tightly linked to liver cirrhosis,48 a condition in which hepcidin deficiency and increased iron levels were reported.50 Further suppression of hepcidin levels by spironolactone treatment (Figure 4A) may cause an even more pronounced dysregulation of iron homeostasis in cirrhotic patients, which may outweigh its beneficial effects on edema and thus provide a rationale for the application of alternative agents.
Mechanistic insights
Our data further generate insight into the potential mode of action of imatinib and spironolactone. For them to modulate hepcidin expression, these drugs require preserved BMP signaling mediated by the BMP responsive elements located in the hepcidin promoter (Figure 2B).17 Imatinib displays a much broader range of inhibitory activity than originally described.33 Our data demonstrate that in the context of hepcidin regulation, imatinib may suppress its non-kinase target oxidoreductase NQO2,33 which was identified as a hepcidin activator in our study (Figure 1B). Consistently, quercetin, another agent that suppresses NQO2, also reduces hepcidin mRNA levels (Figure 3D). How NQO2 regulates hepcidin remains to be established. Previous studies suggested that it controls the protein levels of p53,29 a tumor suppressor that was reported as a hepcidin activator.51 Likewise, NQO2 may regulate the stability of other hepcidin activators. It is of note that we observe decreased hepcidin mRNA expression in response to quercetin which contrasts previously reported results showing hepcidin activation upon quercetin treatment.52 This discrepancy may arise from different cell lines used, drug doses applied or time periods of drug supplementation studied.
Our results consistently reveal that two aldosterone receptor antagonists, spironolactone and eplerenone, suppress hepcidin in primary human hepatocytes (Figures 2D and 3E). This effect is unlikely to be explained by a cross-reactivity of this drug with testosterone signaling, which is known to suppress hepcidin.16 Thus, our data suggest that aldosterone-mediated signaling in hepatocytes controls hepcidin expression, a finding that is consistent with a growing spectrum of extra-renal roles of aldosterone.53
Diclofenac was identified as hepcidin modulator in primary hepatocytes (Figure 2), but failed to affect hepcidin expression in mice (Figure 4A). Side effects of diclofenac therapy include hepatotoxicity, gastrointestinal ulcers and bleeding.54 According to reports accompanying the FDA approval for diclofenac, the dose used in this study was expected to cause mild anemia, but no other adverse effects. Consistently, we observe a mild reduction of hemoglobin levels, hematocrit and red blood cell counts in mice treated with diclofenac (Table 3). However, a modulation of iron homeostasis is unlikely to contribute to the mild anemic phenotype, which may rather arise from minor intestinal injury.
Our work further demonstrates that SAHA and other HDAC inhibitors (HDACi) induce hepcidin expression in cultured cells and primary hepatocytes (Figure 2) but not in mice (Figure 4A). It is of note that the HDAC inhibitors tested modulate hepcidin expression to different degrees dependent on the in vitro model studied (Figure 3). Our data obtained in Huh7 cells suggest that HDAC3 may predominantly control hepcidin promoter accessibility (Figures 3A and 1B). However, further studies in murine and human PH suggested the involvement of other HDACs. Previous reports already identified HDACi as hepcidin modulators. Miura et al. showed that trichostatin A induces hepcidin in hepatoma cells, prevents hepatitis virus C-mediated hepcidin suppression and increases binding of the core transcriptional activators, STAT3 and CEBPα, to the hepcidin promoter.26 Furthermore, SAHA was independently identified as hepcidin-inducing agent by a drug screen for hepcidin regulators.39 Although in our hands SAHA did not alter hepcidin levels in vivo, it remains to be established whether different dosing or administration routes, or the employment of alternative HDACi modulates systemic iron parameters. According to the DrugBank database, HDACi are used predominantly in cancer therapy. Their hepcidin-inducing potential may thus be beneficial to limit iron availability and prevent disease progression.45 Nevertheless, such effects of HDACi may likely exacerbate cancer-related anemia, frequently observed in several malignancies.55
Taken together, our study investigated a large panel of approved drugs, and identified imatinib and spironolactone as modulators of hepcidin levels in primary hepatocytes and in mice. These data suggest that prolonged administration of these therapeutics may affect body iron balance, and possibly the course of an underlying disease. Such knowledge is of high relevance for patient care. It merits the study of potential iron-related side effects of these therapeutics in prospective clinical studies.
Supplementary Material
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/7/1173
Funding
MUM acknowledges funding from the Deutsche Forschungsgemeinschaft (SFB1036) and the DietmarHopp Stiftung. KM-S acknowledges support from the University of Heidelberg (Walter Erb-Stiftung; D.10052215.03) and from the National Science Centre, Poland (POLONEZ fellowship UMO-2015/19/P/NZ2/03278; this project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 665778). GD and DS acknowledge support from the German Federal Ministry of Education and Research (BMBF) project Virtual Liver (0315741).
References
- 1.Hentze MW, Muckenthaler MU, Galy B, Camaschella C. Two to tango: regulation of Mammalian iron metabolism. Cell. 2010;142(1):24–38. [DOI] [PubMed] [Google Scholar]
- 2.Papanikolaou G, Tzilianos M, Christakis JI, et al. Hepcidin in iron overload disorders. Blood. 2005;105(10):4103–4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kremastinos DT, Farmakis D. Iron overload cardiomyopathy in clinical practice. Circulation. 2011;124(20):2253–2263. [DOI] [PubMed] [Google Scholar]
- 4.Deugnier Y, Turlin B. Pathology of hepatic iron overload. World J Gastroenterol. 2007;13(35):4755–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rajpathak SN, Crandall JP, Wylie-Rosett J, Kabat GC, Rohan TE, Hu FB. The role of iron in type 2 diabetes in humans. Biochim Biophys Acta. 2009;1790(7):671–681. [DOI] [PubMed] [Google Scholar]
- 6.Altamura S, Muckenthaler MU. Iron toxicity in diseases of aging: Alzheimer’s disease, Parkinson’s disease and atherosclerosis. J Alzheimers Dis. 2009;16(4):879–895. [DOI] [PubMed] [Google Scholar]
- 7.Nemeth E, Rivera S, Gabayan V, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113(9):1271–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiss G, Goodnough LT. Anemia of chronic disease. N Engl J Med. 2005; 352(10):1011–1023. [DOI] [PubMed] [Google Scholar]
- 9.Pietrangelo A. Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology. 2010;139(2):393–408. [DOI] [PubMed] [Google Scholar]
- 10.Al-Khabori M, Bhandari S, Al-Huneini M, Al-Farsi K, Panjwani V, Daar S. Side effects of Deferasirox Iron Chelation in Patients with Beta Thalassemia Major or Intermedia. Oman Med J. 2013;28(2):121–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vincent JL, Baron JF, Reinhart K, et al. Anemia and blood transfusion in critically ill patients. JAMA. 2002;288(12):1499–1507. [DOI] [PubMed] [Google Scholar]
- 12.Bohlius J, Schmidlin K, Brillant C, et al. Recombinant human erythropoiesis-stimulating agents and mortality in patients with cancer: a meta-analysis of randomised trials. Lancet. 2009;373(9674):1532–1542. [DOI] [PubMed] [Google Scholar]
- 13.Ruchala P, Nemeth E. The pathophysiology and pharmacology of hepcidin. Trends Pharmacol Sci. 2014;35(3):155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goodnough JB, Ramos E, Nemeth E, Ganz T. Inhibition of hepcidin transcription by growth factors. Hepatology. 2012; 56(1):291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. 2014; 46(7):678–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Latour C, Kautz L, Besson-Fournier C, et al. Testosterone perturbs systemic iron balance through activation of EGFR signaling in the liver and repression of hepcidin. Hepatology. 2013;59(2):683–694. [DOI] [PubMed] [Google Scholar]
- 17.Mleczko-Sanecka K, Roche F, da Silva AR, et al. Unbiased RNAi screen for hepcidin regulators links hepcidin suppression to proliferative Ras/RAF and nutrient-dependent mTOR signaling. Blood. 2014; 123(10):1574–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsuchiya H, Akechi Y, Ikeda R, et al. Suppressive effects of retinoids on iron-induced oxidative stress in the liver. Gastroenterology. 2009;136(1):341–350. [DOI] [PubMed] [Google Scholar]
- 19.Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov. 2004; 3(8):673–683. [DOI] [PubMed] [Google Scholar]
- 20.Casanovas G, Mleczko-Sanecka K, Altamura S, Hentze MW, Muckenthaler MU. Bone morphogenetic protein (BMP)-responsive elements located in the proximal and distal hepcidin promoter are critical for its response to HJV/BMP/SMAD. J Mol Med. 2009;87(5):471–480. [DOI] [PubMed] [Google Scholar]
- 21.Mleczko-Sanecka K, Casanovas G, Ragab A, et al. SMAD7 controls iron metabolism as a potent inhibitor of hepcidin expression. Blood. 2010;115(13):2657–2665. [DOI] [PubMed] [Google Scholar]
- 22.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. Faseb J. 2008;22(3):659–661. [DOI] [PubMed] [Google Scholar]
- 23.Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7(2):27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Torrance JD, Bothwell TH. A simple technique for measuring storage iron concentrations in formalinised liver samples. S Afr J Med Sci. 1968;33(1):9–11. [PubMed] [Google Scholar]
- 25.Gunther S, Kuhn M, Dunkel M, et al. SuperTarget and Matador: resources for exploring drug-target relationships. Nucleic Acids Res. 2008;36:D919–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miura K, Taura K, Kodama Y, Schnabl B, Brenner DA. Hepatitis C virus-induced oxidative stress suppresses hepcidin expression through increased histone deacetylase activity. Hepatology. 2008; 48(5):1420–1429. [DOI] [PubMed] [Google Scholar]
- 27.Flock GB, Cao X, Maziarz M, Drucker DJ. Activation of enteroendocrine membrane progesterone receptors promotes incretin secretion and improves glucose tolerance in mice. Diabetes. 2013;62(1):283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vella F, Ferry G, Delagrange P, Boutin JA. NRH:quinone reductase 2: an enzyme of surprises and mysteries. Biochem Pharmacol. 2005;71(1–2):1–12. [DOI] [PubMed] [Google Scholar]
- 29.Khutornenko AA, Roudko VV, Chernyak BV, Vartapetian AB, Chumakov PM, Evstafieva AG. Pyrimidine biosynthesis links mitochondrial respiration to the p53 pathway. Proc Natl Acad Sci USA. 2010; 107(29):12828–12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stitham J, Midgett C, Martin KA, Hwa J. Prostacyclin: an inflammatory paradox. Front Pharmacol. 2011;2:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kadamur G, Ross EM. Mammalian phospholipase C. Annu Rev Physiol. 2013; 75:127–154. [DOI] [PubMed] [Google Scholar]
- 32.Verga Falzacappa MV, Vujic Spasic M, Kessler R, Stolte J, Hentze MW, Muckenthaler MU. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood. 2007;109(1):353–358. [DOI] [PubMed] [Google Scholar]
- 33.Bantscheff M, Eberhard D, Abraham Y, et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat Biotechnol. 2007;25(9):1035–1044. [DOI] [PubMed] [Google Scholar]
- 34.Wu K, Knox R, Sun XZ, et al. Catalytic properties of NAD(P)H:quinone oxidoreductase-2 (NQO2), a dihydronicotinamide riboside dependent oxidoreductase. Arch Biochem Biophys. 1997;347(2):221–228. [DOI] [PubMed] [Google Scholar]
- 35.Fosslien E. Cardiovascular complications of non-steroidal anti-inflammatory drugs. Ann Clin Lab Sci. 2005;35(4):347–385. [PubMed] [Google Scholar]
- 36.Wishart DS, Knox C, Guo AC, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ouzan J, Perault C, Lincoff AM, Carre E, Mertes M. The role of spironolactone in the treatment of patients with refractory hypertension. Am J Hypertens. 2002;15(4 Pt 1):333–339. [DOI] [PubMed] [Google Scholar]
- 38.Corvol P, Michaud A, Menard J, Freifeld M, Mahoudeau J. Antiandrogenic effect of spirolactones: mechanism of action. Endocrinology. 1975;97(1):52–58. [DOI] [PubMed] [Google Scholar]
- 39.Gaun V, Patchen B, Volovetz J, et al. A chemical screen identifies small molecules that regulate hepcidin expression. Blood Cells Mol Dis. 2014;53(4):231–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patchen B, Koppe T, Cheng A, Seo YA, Wessling-Resnick M, Fraenkel PG. Dietary supplementation with ipriflavone decreases hepatic iron stores in wild type mice. Blood Cells Mol Dis. 2016;60:36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kerkela R, Grazette L, Yacobi R, et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med. 2006; 12(8):908–916. [DOI] [PubMed] [Google Scholar]
- 42.van Oosterom AT, Judson I, Verweij J, et al. Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I study. Lancet. 2001; 358(9291):1421–1423. [DOI] [PubMed] [Google Scholar]
- 43.Cumming DC, Yang JC, Rebar RW, Yen SS. Treatment of hirsutism with spironolactone. JAMA. 1982;247(9):1295–1298. [PubMed] [Google Scholar]
- 44.Shaw JC, White LE. Long-term safety of spironolactone in acne: results of an 8-year followup study. J Cutan Med Surg. 2002;6(6):541–545. [DOI] [PubMed] [Google Scholar]
- 45.Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer. 2013;13(5):342–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jones RL, Judson IR. The development and application of imatinib. Expert Opin Drug Saf. 2005;4(2):183–191. [DOI] [PubMed] [Google Scholar]
- 47.Maiti B, Setrakian S, Daw HA. Hepatic iron overload, a possible consequence of treatment with imatinib mesylate: a case report. Cases J. 2009;2:7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O’Brien JG, Chennubhotla SA, Chennubhotla RV. Treatment of edema. Am Fam Physician. 2005;71(11):2111–2117. [PubMed] [Google Scholar]
- 49.Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341(10):709–717. [DOI] [PubMed] [Google Scholar]
- 50.Tan TC, Crawford DH, Franklin ME, et al. The serum hepcidin:ferritin ratio is a potential biomarker for cirrhosis. Liver Int. 2012; 32(9):1391–1399. [DOI] [PubMed] [Google Scholar]
- 51.Weizer-Stern O, Adamsky K, Margalit O, et al. Hepcidin, a key regulator of iron metabolism, is transcriptionally activated by p53. Br J Haematol. 2007;138(2):253–262. [DOI] [PubMed] [Google Scholar]
- 52.Bayele HK, Balesaria S, Srai SK. Phytoestrogens modulate hepcidin expression by Nrf2: Implications for dietary control of iron absorption. Free Radic Biol Med. 2015;89:1192–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baudrand R, Gupta N, Garza AE, et al. Caveolin 1 Modulates Aldosterone-Mediated Pathways of Glucose and Lipid Homeostasis. J Am Heart Assoc. 2016;5(10):e003845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Banks AT, Zimmerman HJ, Ishak KG, Harter JG. Diclofenac-associated hepatotoxicity: analysis of 180 cases reported to the Food and Drug Administration as adverse reactions. Hepatology. 1995;22(3):820–827. [PubMed] [Google Scholar]
- 55.Bohlius J, Weingart O, Trelle S, Engert A. Cancer-related anemia and recombinant human erythropoietin-an updated overview. Nat Clin Pract Oncol. 2006; 3(3):152–164. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.