

Abstract
We here describe a leukemogenic role of the homeobox gene UNCX, activated by epigenetic modifications in acute myeloid leukemia (AML). We found the ectopic activation of UNCX in a leukemia patient harboring a t(7;10)(p22;p14) translocation, in 22 of 61 of additional cases [a total of 23 positive patients out of 62 (37.1%)], and in 6 of 75 (8%) of AML cell lines. UNCX is embedded within a low-methylation region (canyon) and encodes for a transcription factor involved in somitogenesis and neurogenesis, with specific expression in the eye, brain, and kidney. UNCX expression turned out to be associated, and significantly correlated, with DNA methylation increase at its canyon borders based on data in our patients and in archived data of patients from The Cancer Genome Atlas. UNCX-positive and -negative patients displayed significant differences in their gene expression profiles. An enrichment of genes involved in cell proliferation and differentiation, such as MAP2K1 and CCNA1, was revealed. Similar results were obtained in UNCX-transduced CD34+ cells, associated with low proliferation and differentiation arrest. Accordingly, we showed that UNCX expression characterizes leukemia cells at their early stage of differentiation, mainly M2 and M3 subtypes carrying wild-type NPM1. We also observed that UNCX expression significantly associates with an increased frequency of acute promyelocytic leukemia with PML-RARA and AML with t(8;21)(q22;q22.1); RUNX1-RUNX1T1 classes, according to the World Health Organization disease classification. In summary, our findings suggest a novel leukemogenic role of UNCX, associated with epigenetic modifications and with impaired cell proliferation and differentiation in AML.
Introduction
Homeobox (HB) genes encode transcription factors involved in key cellular processes such as body patterning, embryonic organogenesis, and cell-fate decisions.1,2 A growing body of literature has highlighted the crucial role of HB genes in normal hematopoiesis and leukemogenesis.3–6 Among HB genes, clustered HOX genes can promote the proliferation and inhibit the differentiation of hematopoietic progenitor cells and cause acute myeloid leukemia (AML)7 and acute lymphoid leukemia.8 Furthermore, several non-clustered HB genes, such as those belonging to the NKL subclass2 or to the Parahox (CDX)9 HB gene family, are critically involved in normal hematopoiesis and in leukemogenesis through their deregulation or ectopic expression. Notably, recent studies have highlighted a correlation between HB gene overexpression and mutations in epigenetic regulators.8,10 Alterations of DNA methylation are now widely considered a hallmark of cancer,11 although the precise leukemogenic mechanisms involving HB genes have still not been completely elucidated. Recently, Jeong et al. identified extended genomic regions exhibiting a low methylation level (≤10%), named “canyons”, spanning conserved domains that frequently harbor transcription factor genes (mainly HB genes) in murine hematopoietic stem cells (HSCs).10 Since several deregulated HB genes in AML fall inside canyons, this mechanism was proposed as a novel model for gene expression alteration in leukemogenesis.10
The starting point of this study was the identification of an M5 AML patient harboring a t(7;10)(p22;p14) translocation as the sole cytogenetic abnormality. This rearrangement was associated with the ectopic expression of the HB gene UNCX (NM_001080461, 7p22.3), likely as a result of a position effect.
UNCX has tissue-specific expression in the eye, brain, and kidney, and it encodes a transcription factor involved in somitogenesis12,13 and neurogenesis.14 The murine Uncx gene was shown to map within a large canyon (23 kb) entirely covered by the repressive H3K27me3 histone mark in HSCs.10
Notably, UNCX expression has never been associated with cancer. We thus investigated the ectopic expression of UNCX in an independent and extensive AML cohort and performed genomic and functional studies to investigate its contribution to leukemogenesis.
Methods
Patients, cell lines, and normal tissues
We studied 62 AML patients (Table 1), including Case 1 with the t(7;10)(p22;p14) translocation, 75 AML and 14 additional cancer cell lines, and 6 normal tissues (Online Supplementary Table S1A and B). The use of samples was approved by the Policlinico S. Orsola-Malpighi Ethics Committee (ref. n. 253/2013/O/Tess of 29/10/2013).
Table 1.
Clinical, cytogenetic features, UNCX expression levels and DNMT3A, FLT3, and NPM1 mutational status of the 62 acute myeloid leukemia patients included in the study.
Assessment of UNCX expression levels in AML
UNCX expression was evaluated by RT-qPCR15,16 using a TaqMan UNCX Gene expression assay (Applied Biosystems, Milan, Italy). The TBP Endogenous Control (Applied Biosystems) was used as reference and Case 1 at onset (1-Dx) as calibrator. We classified patients on a median value of UNCX expression level (2−ΔΔCt=0.01300) as UNCX+ and UNCX−.
Methylation analysis of the UNCX canyon
DNA methylation ratios (MRs) of the UNCX canyon were determined through gene-specific amplification using in vitro transcription coupled with mass spectrometry (MassARRAY platform, Sequenom, San Diego, CA, USA).17 Statistical significance was obtained by comparing MRs of UNCX+ and UNCX− cases by using Student’s t-test; the Satterthwaite correction was applied when unequal variance was present. To clearly visualize the methylation differences, the MR values were submitted to angular transformation.
Methylation and correlation analysis of the UNCX canyon in AML samples from The Cancer Genome Atlas (TCGA)
We selected a total of 111 AML samples from the GDC Data Portal (https://gdc.cancer.gov), with both methylation and expression profile data. These subjects were divided according to the median value of UNCX expression (FPKM=0.0259) in UNCX-TCGA-positive (n=55) and UNCX-TCGA-negative (n=56). We compared the DNA methylation profile of UNCX as well as the whole genome (considering a minimum difference of 2-folds between groups) by the Mann-Whitney test. Spearman correlation was calculated between methylation and expression values within both sample sets. Correlation values were deemed significant at P<0.05.
DNMT3A mutational analysis
A full description of the analytical methods used is provided in the Online Supplementary Methods.
Gene-expression profiling and pathway analyses
Exon array was performed in two subgroups of our patient cohort [UNCX+ (ns. 1-Dx, 9, and 16) and UNCX− (ns. 13, 41, and 49)]. Similarly, 173 AML TCGA samples were divided according to the median value of UNCX expression in UNCX-TCGA-positive (n=47) and UNCX-TCGA-negative (n=126) cases. Pathway analysis was conducted on differentially expressed genes through QIAGEN’s Ingenuity Pathway Analysis software (IPA, QIAGEN, Redwood City, CA, USA).
Retrovirus-mediated UNCX expression in CB CD34+ cells
Ectopic UNCX expression was achieved by retrovirus-mediated transduction of human cord blood (CB) CD34+ cells.18 Proliferation and differentiation rates were determined by colony forming cell (CFC) assays at 14 days after seeding. Flow cytometry analysis provided quantitative information regarding the maturation stage of infected cells.18 Cell morphology was assessed by May-Grunwald-Giemsa staining.
Correlation between UNCX expression and clinical/molecular features in TCGA patients
A total of 161 out of 173 TCGA AML samples were analyzed for potential associations between UNCX and clinical/molecular features. UNCX-TCGA-positive and -negative patients (Online Supplementary Table S2) were compared using the χ2 test or Fisher’s exact test. Continuous variables and medians of distributions were analyzed by Mann-Whitney U test or Kruskal-Wallis test for multiple comparisons.
Results
A novel t(7;10)(p22;p14) translocation in AML is associated with ectopic expression of the HB gene UNCX
FISH results obtained in Case 1-Dx (Table 1) identified a breakpoint region on 7p22.3 within the fosmid clone G248P85449H7 (WI2-1959P13) that contained UNCX as the only target gene (Online Supplementary Figure S1A and C). A breakpoint region on 10p14 was mapped to the G248P8034H6 (WI2-3164O12) fosmid clone within the coding sequence of the housekeeping gene UPF2, which is involved in nonsense-mediated decay of mRNAs19 and normally expressed in the bone marrow (BM) (http://biog-ps.org) (Online Supplementary Figure S1B and C). As a consequence of this rearrangement, UNCX was juxtaposed to the 3′ end of UPF2 in the derivative chromosome 7 [der(7)], as shown by FISH (Online Supplementary Figure S1), and ectopically expressed in Case 1-Dx, as detected by RT-qPCR (Table 1 and Figure 1). In addition, Case 1-Dx presented mutations in FLT3-TDK (Table 1) and WT1 (data not shown), which were not confirmed in Case 1 at complete remission (1-Rem), and wild-type NPM1 (Table 1).
Figure 1.
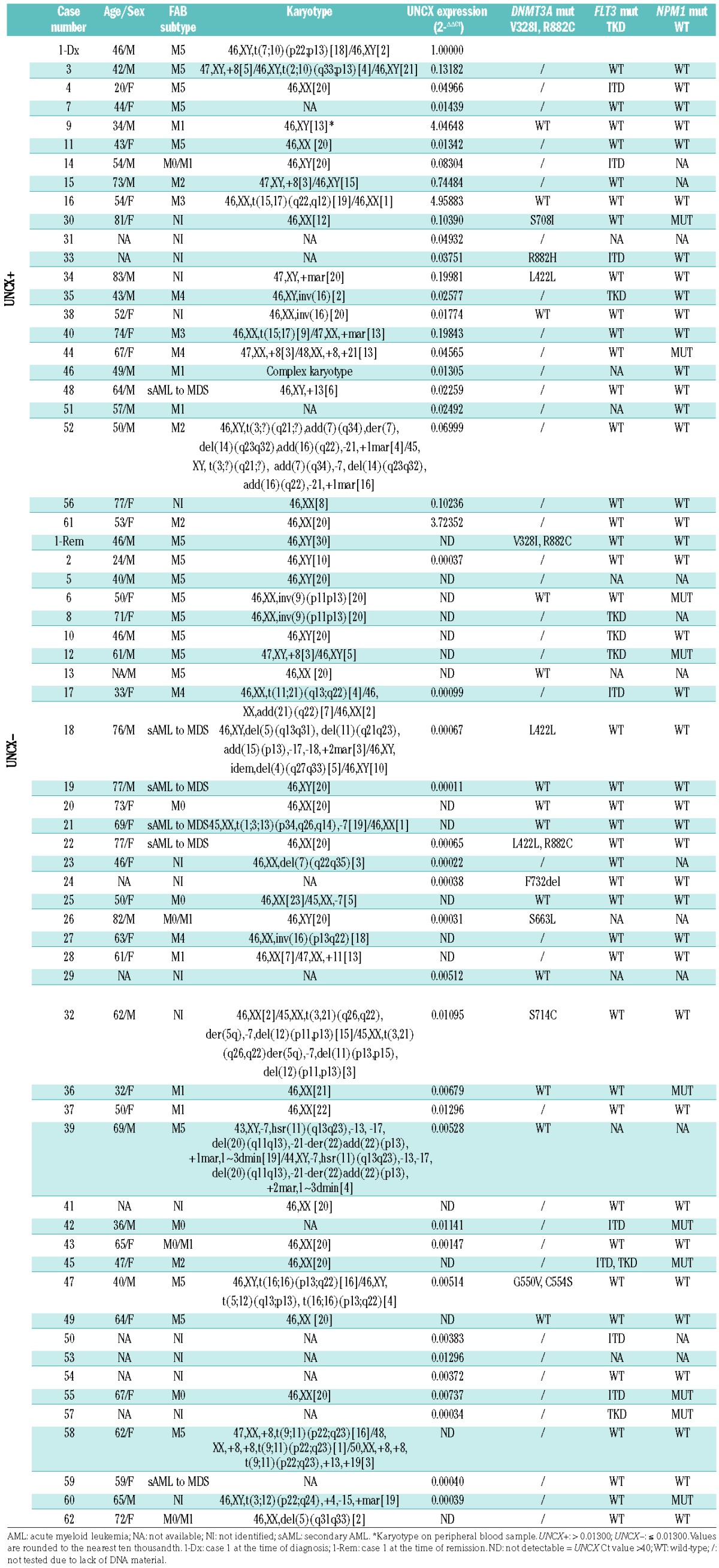
Expression levels of UNCX in acute myeloid leukemia (AML) patients and cell lines. RT-qPCR results showing UNCX expression in AML patients (A) and AML cell lines (B) in comparison to Case 1-Dx. Only positive samples exhibiting an expression level ≥0.10 are reported. The experiments were performed once and each sample was analyzed in triplicate.
UNCX is ectopically expressed in a subset of AML patients and cell lines
To verify whether UNCX is expressed in AML independently of the t(7;10) translocation, UNCX transcript level was assessed by RT-qPCR in 61 additional AML cases. UNCX expression was detected in 37.1% (23 of 62) of our AML patient cohort (Table 1 and Figure 1A) and 8% (6 of 75) of the AML cell lines (Figure 1B and Online Supplementary Table S1A) at variable levels, regardless of the French-American-British (FAB) subtype. Interestingly, UNCX expression was not detectable in Case 1-Rem (Table 1). None of the other investigated cases (ns. 9, 16, and 61) or cell lines harbored the t(7;10)(p22;p14) balanced translocation, as confirmed by FISH, or mutations in NPM1, FLT3 (Table 1), and WT1 (data not shown).
UNCX expression was also detected in MEG-01 [chronic myeloid leukemia (CML)] and in brain cells. In normal cells, UNCX was not expressed in total BM, peripheral blood (PB), CB or BM CD34+ stem-progenitor cells (Online Supplementary Table S1B), and in hematopoietic lineages at different stage of maturation, according to Blueprint Epigenome data portal (http://blueprint-data.bsc.es/release_2016-08/#!/) (Online Supplementary Table S1C).
In addition, we identified two alternative transcript iso-forms for UNCX [UNCX-alternative 1 (UNCX-a1), GenBank KM587719 and UNCX-alternative 2 (UNCX-a2), GenBank KM587718], generated through the retention of distinct portions of intron II in both AML patients and cell lines (Online Supplementary Figure S2 and further details in the Online Supplementary Results). Moreover, to evaluate UNCX expression at protein level, we tested two different polyclonal antibodies specific for the N- and the C-terminus of the UNCX protein (ab105966, Abcam, Cambridge, UK, and AV47546, Sigma, respectively) by Western blot (WB). However, the lack of UNCX antibodies specificity (due to the detection of multiple non-specific bands) prevented the identification of the protein in HEK293T and Jurkat cell lines, indicated by the companies as WB positive controls, as well as in our AML cell lines. Notably, none of these antibodies has been referenced in any publication so far (further details in the Online Supplementary Methods).
UNCX ectopic expression is significantly associated with DNA methylation increase at UNCX canyon borders but is not correlated with DNMT3A mutations
To assess whether epigenetic changes were responsible for the ectopic expression of UNCX in AML patients, we analyzed DNA methylation. UNCX exon 1 and also a 2.5-kb region upstream of UNCX were significantly hypomethylated in all analyzed samples, regardless of UNCX expression level (Online Supplementary Figure S3A), as expected for genes within methylation canyons.10
Interestingly, we detected a significant increase of the MRs at UNCX 5′ and 3′ canyon borders in UNCX+ versus UNCX− samples. Specifically, three (Figure 2A, Online Supplementary Table S3A and Online Supplementary Figure S3B–D) and seven amplicons (Figure 2B, Online Supplementary Table S3B and Online Supplementary Figure S3E–M) at the 5′ and 3′ canyon borders, respectively, exhibited a significant increase in the average MR of CpGs.
Figure 2.

DNA methylation levels at UNCX canyon in our acute myeloid leukemia (AML) patient cohort and TCGA samples. Mean values of DNA methylation ratios (MRs) of MassARRAY amplicons obtained for each of the 20 amplicons tested at both 5′ (A) and 3′ (B) UNCX canyon borders; P-values are reported above each amplicon showing a significant increase in the average MR in UNCX+ versus UNCX− patients. (C) Box and Whisker plot showing DNA methylation β values at the UNCX canyon in UNCX-TCGA-positive (n=55, median 0.3471, interquartile range 0.2898–0.3981) and UNCX-TCGA-negative (n=56, median 0.2771, interquartile range 0.2273–0.3348) patients. Significant differences were identified using the Mann-Whitney U-test (P<0.0001).
To corroborate this evidence, DNA methylation at UNCX canyon was found to be significantly higher (P<0.0001) in AML samples from TCGA expressing UNCX (UNCX-TCGA-positive) than in the UNCX-TCGA-negative cohort (Figure 2C). Intriguingly, there was a significant correlation between this DNA methylation increase and UNCX expression levels (rs=0.649, P<0.0001) (Online Supplementary Table S3C) in the UNCX-TCGA-positive set. Moreover, a genome-wide methylation analysis yielded 270 genomic regions containing 289 differentially methylated genes, differing from UNCX-TCGA-positive and UNCX-TCGA-negative samples by at least 2-fold (Online Supplementary Table S3D).
We then evaluated whether mutations in DNMT3A could explain the altered methylation status of our UNCX+ cases. We detected approximately the same rate of DNMT3A mutation in UNCX+ and UNCX− patients from our internal cohort, with heterozygous lesions at position R882 and at other amino acid residues [3 of 8 (37.5%) in UNCX+, 6 of 19 (31.6%) in UNCX−] (Table 1). Notably, Case 1 showed the same mutations in both 1-Dx and 1-Rem samples (Online Supplementary Table S3E), as expected for pre-leukemic lesions.20 In the TCGA cohort, we observed a significant association between mutations in DNMT3A and lack of UNCX expression (33.3% of mutant cases among UNCX-TCGA-negative vs. 9.1% among UNCX-TCGA-positive, P=0.0022 considering all patients’ subtypes; 33.6% of mutant cases among UNCX-TCGA-negative vs. 13.3% among UNCX-TCGA-positive, P=0.041 considering non-M3 AML). By analyzing the mutational status of genes involved in DNA methylation (DNMT1, 3A, 3B, IDH1/2, TET1/2, WT1), we observed a significant association with UNCX-TCGA-negative cases (58.9% of mutated cases among UNCX-TCGA-negative, vs. 25% among UNCX-TCGA-positive AML considering all cases, P=0.0002; 59.3% of mutated cases among UNCX-TCGA-negative vs. 36.7% among UNCX-TCGA-positive AML, P=0.0384 considering non-M3 AML).
Ectopic expression of UNCX in AML patients induces deregulation of genes involved in cell proliferation and differentiation
To characterize the transcriptional program associated with UNCX expression in AML, we performed gene expression profiling of UNCX+ (ns. 1, 9, and 16) and UNCX− (ns. 13, 41, and 49) cases. A total of 596 genes were differentially expressed (414 up-regulated and 182 down-regulated in UNCX+ cases; Array Express accession n. E-MTAB-4098) (Online Supplementary Table S4A and Online Supplementary Figure S4A). TCGA AML samples were divided into 47 (27.2%) UNCX-TCGA-positive and 126 (72.8%) UNCX-TCGA-negative patients, according to the UNCX median expression value, which was zero, since more than half (i.e. 126) the individuals did not express UNCX at all. To check whether the partitioning strategy applied to both data sets matched, we considered the set of UNCX-TCGA-negative patients and sought for a gene fingerprint that better transcriptionally discriminated them from a subset of 12 “extremely positive” patients, namely expressing UNCX over the 75th percentile. A fingerprint of 199 on 20,530 genes was found by means of the sPLS-DA. This set of genes was fed to a PCA that, when applied on the samples of our internal data set, differentiated perfectly UNCX+ from UNCX− individuals (Online Supplementary Figure S4B and C).
Analyses performed using Ingenuity Pathway Analysis software (IPA) (Online Supplementary Table S4B–D) identified a number of altered pathways, biological functions or diseases, and networks of genes involved in cell proliferation [mainly activation of mitogen-activated protein kinases (MAPKs), transforming growth factor-β (TGF-β), and phosphatidylinositol 3-kinase (PI3K) signaling pathways], cell-cycle regulation, hematopoiesis and hematologic disease, and cell death and survival. These results supported our hypothesis that UNCX could play an important role in differentiation (Figure 3A).
Figure 3.

Association analysis of TCGA acute myeloid leukemia (AML) cohort. (A) Treemap showing the activation state of the enriched processes for commonly differentially expressed genes between exon array and TCGA data; green: reduced activity, orange: enhanced activity. Higher significance levels for a process are reflected by a larger enclosing rectangle. (B and D) UNCX transcript levels were obtained by TCGA RNASeq data. (B) Mean value and standard deviation across FAB types are shown. UNCX expression was significantly higher in M3 cases compared with the other FAB subtypes (P<0.0001, except for M6/7). (C) Graph showing the distribution of UNCX-TCGA-positive and UNCX-TCGA-negative AML cases across FAB types (excluding M3). Percentages are reported in the graphs. (D) Mean value and standard deviation across World Health Organization (WHO) classes (here reported with abbreviations) are shown (AML with BCR-ABL1 and AML with t(9;11)(p21.3;q23.3); MLLT3-KMT2A are reported together due to low number of cases. UNCX expression was higher in acute promyelocytic leukemia (APL) with PLM-RARA and AML with t(8;21)(q22;q22.1); RUNX1-RUNX1T1.
Among the set of deregulated genes obtained from the analyses of both our cohort of samples and TCGA data, we tested the differential expression of 3 genes with an established role in leukemia (upregulation of CCNA1 and PIK3CB and downregulation of MAP2K1) by RT-qPCR in all patients under study. We observed a statistically significant upregulation of CCNA1 and downregulation of MAP2K1 only in the 4 patients (ns. 1-Dx, 9, 16, and 61) showing the highest UNCX expression levels (UNCX≥1) (Online Supplementary Figure S4D), and not in the overall cohort of UNCX+ patients (Online Supplementary Figure S4E).
Similarly, HOXA10 expression was significantly increased only in patients with UNCX ≥1 (Online Supplementary Figure S4D). Notably, HOXA10 was not identified as a deregulated gene in the GEP analysis but was found to be up-regulated in LUNCXIΔN cells, as described below.
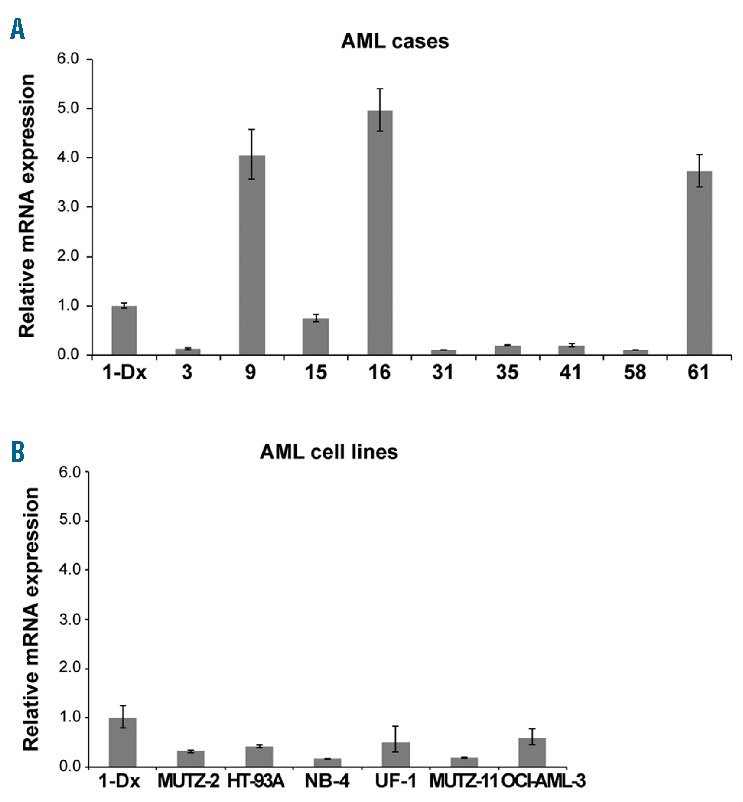
UNCX expression strongly affects the proliferation and differentiation of normal myeloid cells in vitro
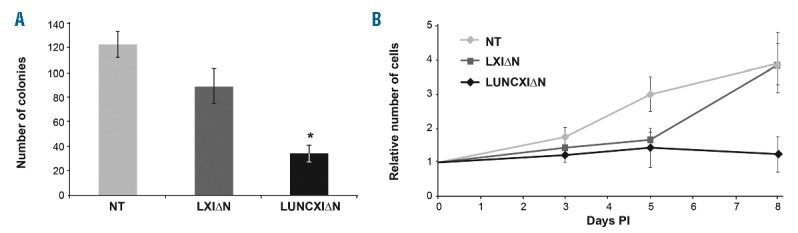
To clarify the biological function of UNCX in hematopoietic cells, we ectopically expressed UNCX in CD34+ CB cells (LUNCXIΔN cells). CFC assays showed a significantly reduced number of colonies in LUNCXIΔN cells compared with LXIΔN (cells transduced with an empty vector) or non-transduced (NT) cells (Figure 4A), reflecting an UNCX-mediated effect on cell proliferation. To further assess the role of UNCX in the proliferation of myeloid cells, LUNCXIΔN, LXIΔN, and NT cells were seeded at the same density and counted every two days after infection for eight days. The proliferation rate was already reduced at day 5 post-infection (PI) in LUNCXIΔN cells (Figure 4B). Flow cytometry analysis at days 7, 10, and 14 PI revealed that CD34 tended to increase in LUNCXIΔN cells (mean value 29.1%, 15.9%, and 7.4%, respectively) compared with control LXIΔN cells (mean value 20.9%, 3.4%, and 0.6%, respectively), and this increase became statistically significant at day 14 PI (Figure 5A). RT-qPCR analysis confirmed the differential expression of CD34 at the transcript level; CD34 mRNA levels were significantly increased starting from day 5 PI (Online Supplementary Figure S5) to day 14 PI (Figure 5B) in LUNCXIΔN cells compared with LXIΔN cells. We also evaluated the expression level of HOXA10, KLF4, and MAFB, which are master regulators of mono-macrophage differentiation.21–24 We observed increasing levels of HOXA10 and KLF4 transcripts from day 5 to 14 PI (Figure 5B and Online Supplementary Figure S5). In the same cells, MAFB expression was elevated at days 5 and 7, but decreased after day 10 PI in LUNCXIΔN cells compared with control cells (Figure 5B and Online Supplementary Figure S5).
Figure 4.
Colony forming cell (CFC) assay and cell-proliferation analysis of UNCX-transduced cells. Number of colonies obtained in CFC assays (A) and proliferation curves (B) of non-transduced (NT), LXI∆N, and LUNCXI∆N CD34+ cells. All experimental data were verified in three independent experiments in triplicate.
Figure 5.
Delayed differentiation of CD34+ hematopoietic progenitor cells after retrovirus-mediated UNCX transfer. (A) Percentage values [including the calculated mean and the standard error of mean (SEM)] of CD34+ cells, measured in two independent UNCX transfer experiments (I and II), using flow cytometry at days 7, 10, and 14 post-infection (PI): non-transduced (NT) (blue line), LXI∆N (red line), and LUNCXI∆N (green line) CD34+ cells. *Two-tailed P≤0.05. (B) RT-qPCR results of the genes encoding specific surface markers and transcription factors implicated in hematopoiesis, including MAP2K1 and CCNA1; the assay was performed on RNA extracted from cells at day 14 PI. *Two-tailed P≤0.05. (C) Morphological analysis after May-Grunwald-Giemsa staining at day 14 PI in NT, LXI∆N, and LUNCXI∆N CD34+ cells; the myeloid immature forms, characterized by a basophilic cytoplasm and a despiralized nucleus, are indicated with a red arrow.
Notably, at day 14 PI, we observed a significant downregulation of MAP2K1 and upregulation of CCNA1 in LUNCXIΔN cells (confirming what was observed in AML patients; see above), which could indicate an effect of UNCX activation on cell proliferation and cell cycle control (Figure 5B). Moreover, morphological analysis with May-Grunwald-Giemsa staining demonstrated a persistence of immature myeloid cells at day 14 PI, which particularly affected the mono-macrophage lineage in the LUNCXIΔN sample (5% vs. 1% in the LXIΔN control sample) (Figure 5C). The persistence of undifferentiated cells of mono-macrophage lineage, even after several days PI, confirmed the flow cytometry and RT-qPCR results.
Ectopic expression of UNCX characterizes a specific subgroup of AML patients
The TCGA data showed that UNCX expression was associated with lower age (median age of UNCX-TCGA-positive patients 50 years; median age of UNCX-TCGA-negative patients 58.5 years; P=0.003) (Online Supplementary Table S2A), M3 FAB type (31.1% in UNCX-TCGA-positive patients; 0.9% in UNCX-TCGA-negative patients, with 93% of M3 cases expressing the gene; P<0.0001) (Figure 3B and Online Supplementary Table S2A), favorable prognosis according to karyotype-based classification (P=0.0001) (Online Supplementary Table S2A), and reduced fraction of bone marrow monocytes (median of UNCX-TCGA-positive patients 3.5%; median of UNCX-TCGA-negative patients 7.0%; P=0.0034) (Online Supplementary Table S2A). The same results were also confirmed by excluding M3 FAB cases from the analysis (Online Supplementary Table S2B), with a tendency towards increased UNCX expression in AML M2 cases compared to the other FAB types (P=0.0181). Accordingly, in the overall AML cohort M2 cases represented 45.1% of UNCX-TCGA-positive patients but only 20% of UNCX-TCGA-negative patients (P=0.0233) (Figure 3C and Online Supplementary Table S2B). The analysis of UNCX expression, according to the WHO disease classification, showed the highest UNCX levels in APL with PML-RARA and AML with t(8;21)(q22;q22.1); RUNX1-RUNX1T1 classes (P<0.0001) (Figure 3D and Online Supplementary Table S2A). On the other hand, we observed an increased frequency of AML with inv(16)(p13.1q22), AML with mutated RUNX1, and AML with mutated NPM1 among UNCX-TCGA-negative cases (Online Supplementary Table S2A). Indeed, UNCX expression showed an inverse correlation with NPM1c mutation, with 6.5% of UNCX-TCGA-positive and 32.1% of UNCX-TCGA-negative cases carrying the mutation (P=0.0029 excluding M3 cases). No association was observed with FLT3, RAS, and IDH1 mutational status (Online Supplementary Table S2C). The results suggest that UNCX expression characterized a specific subgroup of AML patients, according to their biological and molecular features.
Discussion
We report for the first time the ectopic expression of UNCX in both 23 AML patients of our cohort (37.1%) and 8 AML cell lines (8%). Notably, the involvement of this HB gene and the identity of its downstream target genes in cancer have not previously been reported.
Induced expression of UNCX in normal CD34+ CB cells in vitro markedly affected the proliferation and differentiation of the transduced cells. Indeed, UNCX expression resulted in both a reduction of the proliferation rate and an unprecedented persistence of CD34+ cells at days 10 (15.9%) and 14 (7.4%) PI. This result is noteworthy because in similar experiments,25 at day 10 PI, CD34+ HSCs were almost completely replaced by differentiated cells, and their contribution was reduced to approximately 7% of total cells. The observed reduction of CD34+ cell proliferation and their differentiation arrest at a very immature stage supported the hypothesis that UNCX exerts a potential leukemogenic effect over the myeloid lineage, which was corroborated by the transcriptional increase of HOXA10 and KLF4, along with the decrease of MAFB transcript levels. Notably, the expression of these genes, which are involved in mono-macrophage differentiation (HOXA10 and KLF4 in the early phase, MAFB in the late phase), was modulated along with UNCX activation in vitro. Upregulation of HOXA10 was also observed in a subgroup of UNCX+ patients of our cohort (UNCX expression level ≥1). This result is in line with the recent work of Yao et al. showing that Hoxa10 overexpression in murine bone marrow cells suppresses differentiation and induces expansion of a cell population derived from the common granulocyte/monocyte progenitor population and results in the AML phenotype.26 The identification of downstream target genes of UNCX will clarify its role in proliferation and differentiation.
In parallel, the analysis of the TCGA AML cohort indicates that UNCX expression characterizes blast cells at early phases of differentiation, preferentially the M2 and M3 FAB types, which generally contain wild-type NPM1. Accordingly, UNCX positivity and NPM1c mutation are mutually exclusive both in our patient cohort and in the TCGA cohort. Moreover, the comparison between TCGA RNA-Seq data and our exon array data suggests that UNCX expression is associated with a differentiation arrest in AML blasts, as confirmed by forcing the ectopic expression of UNCX in stem-progenitor cells, which induces an accumulation of immature cells of the mono-macrophage lineage. The different maturation stage of UNCX-TCGA-positive AML blasts and UNCX-transduced normal stem-progenitor cells may explain the differences in the transcriptional signatures associated with UNCX expression in the two settings. Our GEP data and pathway analyses clearly show that the 3 UNCX+ patients (UNCX expression level ≥1) displayed remarkable deregulation of genes involved in hemopoiesis, proliferation, and cell cycle, such as those implicated in MAPK, TGF-β, and PI3K signaling. All of these pathways play critical roles in proliferation, regulation of cell growth, differentiation, apoptosis, survival, and development in a wide range of cell types. Notably, although RT-qPCR analysis did not validate these data in the overall cohort of UNCX+ patients, we observed MAP2K1 downregulation and CCNA1 upregulation in LUNCXIΔN cells, which showed UNCX expression level ≥1. Therefore, we speculate that abnormal levels of UNCX expression might be associated with specific transcriptional signatures and signaling pathway activation through a gene-dosage effect. However, further investigation is required to confirm this hypothesis. In AML patients (in both our and the TCGA cohorts), UNCX activation was associated and significantly correlated with a substantial increase in DNA methylation at both the 5ʹ and 3ʹ canyon borders. This methylation change might drastically affect UNCX transcription and result in its ectopic expression. However, here we found that DNMT3A mutations (both R882 and additional point mutations, including a few mutations not yet reported in the COSMIC database), did not associate with UNCX positivity, in contrast to what has been reported in mice where Dnmt3a mutations were directly correlated with shifts in DNA methylation at gene canyon borders.10 Our results are in line with previous reports of somatic mutations in DNMT3A in approximately 20% of de novo AML cases and 36% of cytogenetically normal AML cases.27,28 This result clearly suggests that DNMT3A mutations in humans are not directly correlated with UNCX canyon shrinkage and gene activation. In addition, analysis of the TCGA cohort showed the mutational status of alternative epigenetic modifier genes, classified according to the literature,29–31 and we did not observe any significant association with UNCX expression. Hence, future analyses are mandatory to clarify the epigenetic mechanism behind the regulation of UNCX. Since the Uncx canyon was found to be enriched in the repressive histone mark H3K27me3,10 we speculate that the increase in methylation might also induce a change in the histone mark configuration at the shrunk canyon of UNCX, which would result in changes in chromatin condensation and in a remodeling of nuclear architecture. This hypothesis, together with that concerning a possible role of hydroxymethylation in UNCX activation, could not be verified here due to the lack of vital frozen cells for our patients. Notably, the future development of efficient anti-UNCX specific antibodies will help to understand the role of UNCX at the protein level in leukemogenesis. In fact, commercially available antibodies failed to identify UNCX in tumor cell lines, even in those with a high expression of the gene (Online Supplementary Table S1A and B). This finding could be explained by the documented poor expression of homeodomain-containing proteins, as reported in several studies32,33 and also for UNCX itself. Indeed, UNCX has been identified as a “missing protein” in the HEK293 cell line by means of bioinformatic predictions and high-throughput transcriptomic and proteomic technologies.34
In summary, our results suggest that ectopic activation of UNCX might represent an early epigenetic event in AML with a potential role in leukemogenesis because of its dramatic impact on the proliferation and differentiation of HSCs and progenitors. Our molecular and biological data indicate that UNCX expression may characterize a new subgroup of AML cases, which warrants further investigation. Moreover, although the molecular mechanisms underlying UNCX activation and its role in proliferation and differentiation remain to be clarified, this newly described alteration in AML, caused by an epigenetic modification affecting DNA methylation, introduces a previously undescribed scenario for the leukemogenic potential of epigenetic alterations.
Supplementary Material
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/7/1204
Funding
This work was supported by the AIRC (Associazione Italiana per la Ricerca sul Cancro; AIRC IG n. 15413 for CTS), European LeukemiaNet, AIL (Associazione Italiana contro le Leucemie-Linfomi e Mieloma), AIL Modena, Fondazione Del Monte di Bologna e Ravenna, FIRB 2006, Ateneo RFO grants, and Progetto Regione-Università 2010–12 (L. Bolondi).
References
- 1.Shah N, Sukumar S. The Hox genes and their roles in oncogenesis. Nat Rev Cancer. 2010;10(5):361–371. [DOI] [PubMed] [Google Scholar]
- 2.Homminga I, Pieters R, Meijerink JP. NKL homeobox genes in leukemia. Leukemia. 2012;26(4):572–581. [DOI] [PubMed] [Google Scholar]
- 3.Abramovich C, Pineault N, Ohta H, Humphries RK. Hox genes: from leukemia to hematopoietic stem cell expansion. Ann N Y Acad Sci. 2005;1044:109–116. [DOI] [PubMed] [Google Scholar]
- 4.Argiropoulos B, Humphries RK. Hox genes in hematopoiesis and leukemogenesis. Oncogene. 2007;26(47):6766–6776. [DOI] [PubMed] [Google Scholar]
- 5.Jiang Q, Liu WJ. [Relationship between the HOX gene family and the acute myeloid leukemia-review]. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2013;21(5):1340–1344. [DOI] [PubMed] [Google Scholar]
- 6.Alharbi RA, Pettengell R, Pandha HS, Morgan R. The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia. 2013;27(5):1000–1008. [DOI] [PubMed] [Google Scholar]
- 7.Eklund EA. The role of HOX genes in malignant myeloid disease. Curr Opin Hematol. 2007;14(2):85–89. [DOI] [PubMed] [Google Scholar]
- 8.Conway O’Brien E, Prideaux S, Chevassut T. The epigenetic landscape of acute myeloid leukemia. Adv Hematol. 2014;2014:103175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rawat VP, Humphries RK, Buske C. Beyond Hox: the role of ParaHox genes in normal and malignant hematopoiesis. Blood. 2012;120(3):519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeong M, Sun D, Luo M, et al. Large conserved domains of low DNA methylation maintained by Dnmt3a. Nat Genet. 2014;46(1):17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delpu Y, Cordelier P, Cho WC, Torrisani J. DNA methylation and cancer diagnosis. Int J Mol Sci. 2013;14(7):15029–15058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sewell W, Sparrow DB, Smith AJ, et al. Cyclical expression of the Notch/Wnt regulator Nrarp requires modulation by Dll3 in somitogenesis. Dev Biol. 2009;329(2):400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skuntz S, Mankoo B, Nguyen MT, et al. Lack of the mesodermal homeodomain protein MEOX1 disrupts sclerotome polarity and leads to a remodeling of the craniocervical joints of the axial skeleton. Dev Biol. 2009;332(2):383–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sammeta N, Hardin DL, McClintock TS. Uncx regulates proliferation of neural progenitor cells and neuronal survival in the olfactory epithelium. Mol Cell Neurosci. 2010;45(4):398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. [DOI] [PubMed] [Google Scholar]
- 16.Storlazzi CT, Lonoce A, Guastadisegni MC, et al. Gene amplification as double minutes or homogeneously staining regions in solid tumors: origin and structure. Genome Res. 2010;20(9):1198–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ehrich M, Bocker S, van den Boom D. Multiplexed discovery of sequence polymorphisms using base-specific cleavage and MALDI-TOF MS. Nucleic Acids Res. 2005;33(4):e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grande A, Montanari M, Tagliafico E, et al. Physiological levels of 1alpha, 25 dihydroxyvitamin D3 induce the monocytic com mitment of CD34+ hematopoietic progenitors. J Leukoc Biol. 2002;71(4):641–651. [PubMed] [Google Scholar]
- 19.Weischenfeldt J, Waage J, Tian G, et al. Mammalian tissues defective in nonsense-mediated mRNA decay display highly aberrant splicing patterns. Genome Biol. 2012;13(5):R35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shlush LI, Zandi S, Mitchell A, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506(7488):328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feinberg MW, Cao Z, Wara AK, Lebedeva MA, Senbanerjee S, Jain MK. Kruppel-like factor 4 is a mediator of proinflammatory signaling in macrophages. J Biol Chem. 2005;280(46):38247–38258. [DOI] [PubMed] [Google Scholar]
- 22.Gemelli C, Montanari M, Tenedini E, et al. Virally mediated MafB transduction induces the monocyte commitment of human CD34+ hematopoietic stem/progenitor cells. Cell Death Differ. 2006;13 (10):1686–1696. [DOI] [PubMed] [Google Scholar]
- 23.Gemelli C, Orlandi C, Zanocco Marani T, et al. The vitamin D3/Hox-A10 pathway supports MafB function during the mono-cyte differentiation of human CD34+ hemopoietic progenitors. J Immunol. 2008; 181(8):5660–5672. [DOI] [PubMed] [Google Scholar]
- 24.Gemelli C, Zanocco Marani T, Bicciato S, et al. MafB is a downstream target of the IL-10/STAT3 signaling pathway, involved in the regulation of macrophage de-activation. Biochim Biophys Acta. 2014;1843 (5):955–964. [DOI] [PubMed] [Google Scholar]
- 25.Salati S, Zini R, Bianchi E, et al. Role of CD34 antigen in myeloid differentiation of human hematopoietic progenitor cells. Stem Cells. 2008;26(4):950–959. [DOI] [PubMed] [Google Scholar]
- 26.Yao J, Fang LC, Yang ZL, et al. Mixed lineage leukaemia histone methylases 1 collaborate with ERalpha to regulate HOXA10 expression in AML. Biosci Rep. 2014; 34(6):e00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363(25):2424–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marcucci G, Metzeler KH, Schwind S, et al. Age-related prognostic impact of different types of DNMT3A mutations in adults with primary cytogenetically normal acute myeloid leukemia. J Clin Oncol. 2012; 30(7):742–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cancer Genome Atlas Research N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garg M, Nagata Y, Kanojia D, et al. Profiling of somatic mutations in acute myeloid leukemia with FLT3-ITD at diagnosis and relapse. Blood. 2015; 126(22): 2491–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rampal R, Alkalin A, Madzo J, et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 2014;9(5):1841–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Geng LN, Tyler AE, Tapscott SJ. Immunodetection of human double homeobox 4. Hybridoma. 2011;30(2):125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawagoe H, Humphries RK, Blair A, Sutherland HJ, Hogge DE. Expression of HOX genes, HOX cofactors, and MLL in phenotypically and functionally defined subpopulations of leukemic and normal human hematopoietic cells. Leukemia. 1999;13(5):687–698. [DOI] [PubMed] [Google Scholar]
- 34.Garin A, Odriozola L, Martinez-Val A, et al. Detection of missing proteins using the PRIDE database as a source of mass-spectrometry evidence. J Proteome Res. 2016; 15(11):4101–4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.