

Relapse of T-cell acute lymphoblastic leukemia (T-ALL) has a dismal prognosis, with only 20% of afflicted children surviving.1 Children with relapsed T-ALL are commonly treated within high-risk arms of second-line treatment protocols that include mandatory hematopoietic stem cell transplantation (HSCT) if a second complete remission can be induced.2 Second-line treatment, however, fails in 80% of relapsed T-ALL patients.1 Novel risk markers are needed to identify patients who are unlikely to respond to conventional second-line treatment, and to support their reallocation into alternative treatment protocols, such as phase I/II trials including targeted drugs, as early as possible. Mutations in NOTCH1/FBXW7, NRAS/KRAS and PTEN and, for instance, the early T-cell precursor immunophenotype have previously been associated with outcome in pediatric and/or adult primary T-ALL,3–7 but their clinical value in children with relapsed T-ALL has not yet been investigated. TP53 mutations and deletions have been shown to occur more frequently in relapsed compared to primary ALL, and are adversely associated with survival following second-line therapy.8,9 We retrospectively analyzed genetic alterations as well as immunophenotype in T-ALL relapses from children enrolled in the German multicenter ALL-REZ BFM (Acute Lymphoblastic Leukemia Relapse Berlin-Frankfurt-Münster) 95/96 and 2002 trials with the aim of defining markers for risk stratification in treatment decisions. We show that NOTCH1 mutation, TP53 alteration and myeloid antigen expression predict outcome heterogeneity in relapsed pediatric T-ALL and, therefore, can aid in molecular risk assessment.
We assessed TP53, NRAS/KRAS, PTEN and NOTCH1/FBXW7 mutations by Sanger sequencing of key exons in 81 and 74 samples, respectively; TP53 copy number in 81 samples and immunophenotype in 74 samples of relapsed pediatric T-ALL (Figure 1). Experimental details are described in the Online Supplementary Methods. All patients (n=104) had their first T-ALL relapses with bone marrow involvement (patients with extramedullary relapse were excluded). The genetics and immunophenotype cohorts overlapped by 51 patients [48 patients for NOTCH1/FBXW7; (Figure 1)] and were representative subsets of the total cohort recruited by the trials (Online Supplementary Table S1).
Figure 1.

Genetic alterations and immunophenotypes in children with first relapse of T-ALL. The heatmap shows genetic alterations and immunophenotypes (rows) detected in 104 samples of children with first relapse of T-ALL (columns). Absolute and relative frequencies of genetic alterations and immunophenotypes are depicted on the right-hand side. Bars below the chart indicate patient cohorts of the present study. ETP: early T-cell precursor; MyAg: myeloid antigen.
Patients with relapsed T-ALL harbored activating NOTCH1 mutations in 52.7% (39/74), and inactivating FBXW7 mutations in 25.7% (19/74) of cases (Figure 1; Online Supplementary Table S2). NRAS, KRAS and PTEN mutations were present in 9.9% (8/81), 1.2% (1/81) and 9.9% (8/81) of patients with relapsed T-ALL, respectively (Figure 1; Online Supplementary Table S2). TP53 alterations were detected in 7.4% (6/81) of cases of relapsed T-ALL, which included mutations in 6.2% (5/81; 3 cases also harbored deletion of the other TP53 allele) and deletions in 3.7% (4/81; 1 case harbored only a TP53 deletion) (Figure 1; Online Supplementary Table S2). In the two relapses with TP53 mutation only, the mutation appeared homozygous, and single nucleotide polymorphism (SNP) array analysis confirmed the presence of uniparental disomy on chromosome 17p (Online Supplementary Table S2). Thus, five of six patients with TP53 alterations showed complete loss of the wild-type allele. The frequency of TP53 alterations at relapse is similar to our previous observation from a subset of this patient cohort (n=47),8 but it is significantly lower than that described by Diccianni et al. three decades ago (24%).9 However, Diccianni et al. observed a different spectrum of TP53 mutations, with the majority being located in exon 5 versus exon 7 in our study and with 50% of mutations being in a heterozygous state versus 0% in our study. This may relate to methodological differences (Diccianni et al. additionally analyzed exon 4), but also to differences in the ethnic background of patients and in the selective pressure of front-line treatment. Immunophenotype classification by maturation stage identified a cortical T-ALL subtype in 46.0% (34/74), an immature pre-/pro-T-ALL subtype in 32.4% (24/74), and a mature stage of T-ALL lymphoblasts in 21.6% (16/74) of relapsed T-ALL patients (Figure 1; Online Supplementary Table S3). An early T-cell precursor immunophenotype was detected in 10.8% (8/74) and expression of myeloid antigens CD13 and/or CD33 and/or CD65 in ≥20% of lymphoblasts in 36.5% (27/74) of T-ALL relapse samples (Figure 1; Online Supplementary Table S3). As expected, we found significant overlaps between the more immature immunological subtypes, i.e., pre-/pro-T-ALL, early T-cell precursor and myeloid antigen positive T-ALL (P values ≤0.001), whereas myeloid antigen expression was rare in cortical T-ALL relapses in our study (P<0.001; Online Supplementary Figure S1). Relapses of the pre-/pro-T-ALL subtype were characterized by an increased frequency of NRAS/KRAS mutations (P=0.039) and TP53 alterations (by trend, P=0.082) compared to non-pre-/pro-T-ALL relapses (Online Supplementary Figure S1). NOTCH1 mutations were absent from the early T-cell precursor subgroup (P=0.022), but enriched in relapses of the cortical T-ALL subtype (by trend, P=0.082; Online Supplementary Figure S1). Comparing frequencies of gene mutations and immunophenotypes in relapsed T-ALL patients in our study to frequencies described in primary pediatric T-ALL,3,10–14 we found that FBXW7 mutations were increased from 14% at primary T-ALL to 26% at relapse (P=0.014; Online Supplementary Figure S2A). The frequency of cortical T-ALL was decreased (P=0.001; Online Supplementary Figure S2B), whereas those of the pre-/pro-T-ALL subtype and of myeloid antigen positive T-ALL were significantly increased in patients with relapsed T-ALL (P=0.031 and P=0.026, respectively; Online Supplementary Figure S2B).
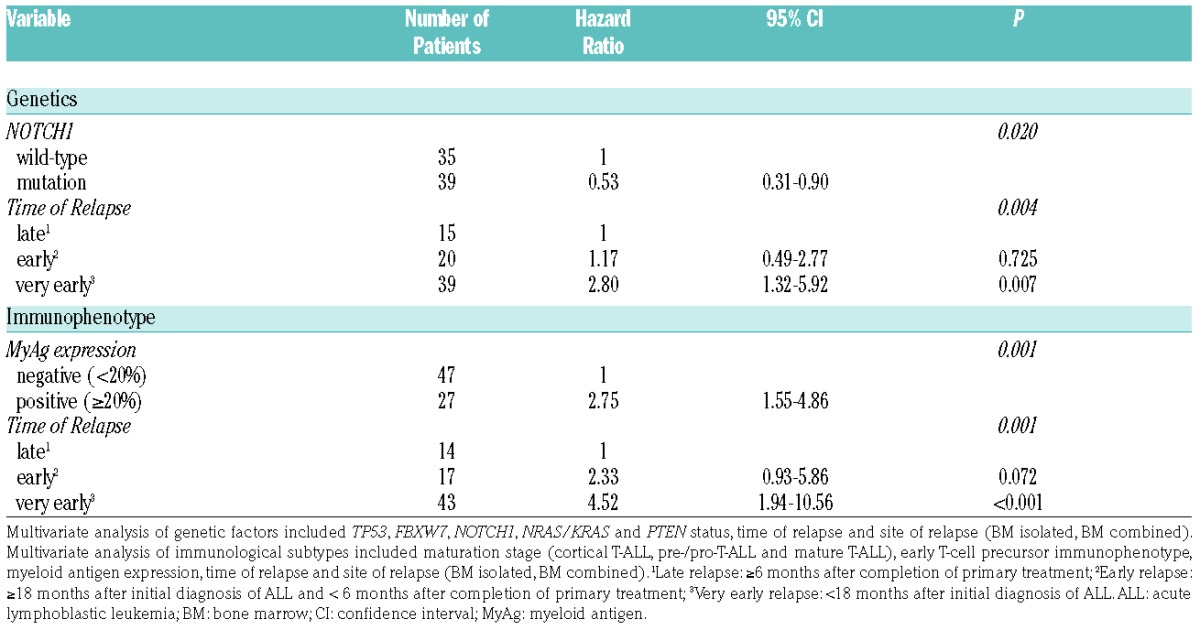
Survival analysis revealed that the probability of event-free survival (pEFS) and overall survival (pOS) of patients with relapsed T-ALL harboring NOTCH1 mutations was significantly better than that of patients with relapsed T-ALL harboring wild-type NOTCH1 (pEFS: P=0.034, 0.359±0.077 vs. 0.114±0.063, Figure 2A; pOS: P=0.026, 0.379±0.079 vs. 0.133±0.071, Online Supplementary Figure S3A). Accordingly, patients with relapsed T-ALL harboring NOTCH1 mutations achieved a second complete remission significantly more often (77% vs. 51%; P=0.022) and exhibited nonresponse to treatment less often (21% vs. 40%) than those patients with relapsed T-ALL harboring wild-type NOTCH1 (Online Supplementary Table S4). A favorable treatment response of patients with primary T-ALL harboring NOTCH1 mutations has been observed in most studies to date,3,4,15,16 although this translated into favorable outcome in only some trials.3,4 The favorable effect of NOTCH1 mutation on the outcome of relapsed patients in our cohort was further pronounced in the patients who reached a second complete remission and subsequently received HSCT (pEFS: 0.604±0.102 vs. 0.235±0.123, P=0.049; Online Supplementary Figure S3B). Although inactivating FBXW7 mutations act in the same pathway as activating NOTCH1 mutations, patients in our cohort with relapsed T-ALL harboring FBXW7 mutations responded no differently to treatment and had similar outcomes to relapsed patients lacking FBXW7 mutations (Online Supplementary Figure S3C). However, survival analysis revealed two distinct groups among FBXW7 mutation positive patients (Online Supplementary Figure S3D). Relapsed patients harboring both FBXW7 and NOTCH1 mutations had a 46% pEFS (0.462±0.138), whereas those relapsed patients harboring solely FBXW7 mutations had only a 17% pEFS (0.167± 0.152), and those relapsed patients harboring only NOTCH1 mutations were intermediately placed with a 31% pEFS (0.308± 0.091, P=0.12). In agreement with primary pediatric T-ALL,10,11 but in contrast to adult T-ALL,5,17 we did not identify a prognostic significance of NRAS/KRAS and PTEN mutations in our relapse cohort (Online Supplementary Figure S4). NRAS/KRAS and, in particular, PTEN mutations were rare in relapsed patients with NOTCH1 mutations (n=2 for NRAS/KRAS and n=1 for PTEN, P=0.095 for PTEN; Online Supplementary Figure S1). Therefore, this small number of patients prevented us from studying a prognostic impact of NRAS/KRAS and PTEN mutations within the favorable NOTCH1 mutant group that was described previously in primary adult T-ALL.5,17 However, it is worth noting that NRAS/KRAS and PTEN mutations were absent from the most favorable NOTCH1/FBXW7 double mutant group of T-ALL relapse patients (Figure 1). TP53 mutation and/or deletion was associated with exceptionally poor outcome in relapsed T-ALL in this study, as all patients suffered a second event and died (pEFS: 0.00±0.00 vs. 0.261±0.052; P=0.051; Figure 2B). This confirms earlier findings that TP53 alterations predict poor outcome in pediatric relapsed ALL.8,9 However, TP53 alteration was not correlated with nonresponse to treatment in our cohort (Online Supplementary Table S4), as was observed in patients with relapsed B-cell precursor ALL.8 This is likely due to the overall higher percentage of nonresponse to treatment in patients with relapsed T-ALL.1 Survival analysis by immunophenotype revealed that relapsed patients with a cortical T-ALL subtype had improved pEFS (0.350±0.082) compared to patients with pre-/pro-T-ALL (0.167±0.08) and mature T-ALL immunophenotype (0.125±0.083, P=0.18; Online Supplementary Figure S5A). Although this result did not reach statistical significance, it is consistent with previous reports regarding primary T-ALL.18 The pEFS in relapsed patients with early T-cell precursor immunophenotype was reduced to zero (0.00±0.00 vs. 0.27±0.05; P=0.17; Figure 2C). Originally identified as a high-risk group,6 more recent studies have questioned the inferior outcome of patients with primary early T-cell precursor ALL treated on contemporary, intensive protocols.13 However, our results indicate a trend, namely that in a relapse situation, intensive treatment on ALL-REZ BFM protocols is insufficient to reach second continuous complete remissions in patients with this subtype. Although the prognostic significance of myeloid antigen expression has remained controversial in primary T-ALL,14 we found expression of each of the myeloid markers CD13, CD33 and CD65 in more than 20% of relapsed T-ALL cells to be associated with an inferior pEFS in our cohort (Online Supplementary Figure S5B-D). Collectively, the patients with expression of one or more of the myeloid markers CD13, CD33 and CD65 in their relapsed T-ALL cells had a significantly reduced pEFS compared to patients without myeloid antigen expression (0.037±0.072 vs. 0.359±0.070, P=0.008; Figure 2D). Multivariate Cox regression analysis of genetic factors and the clinical prognostic factors of time and site of relapse1 identified NOTCH1 mutation as an independent prognostic marker in addition to the time of relapse (P=0.02; Table 1). Notwithstanding the fact that TP53 alteration was a significant adverse factor in univariate analysis, we did not identify it as such in multivariate analysis, likely due to the small number of patients in this group. The value of myeloid antigen expression as a marker for poor outcome in children with relapsed T-ALL was confirmed by multivariate analysis, that identified it as an independent factor alongside time of relapse (P=0.001; Table 1).
Figure 2.
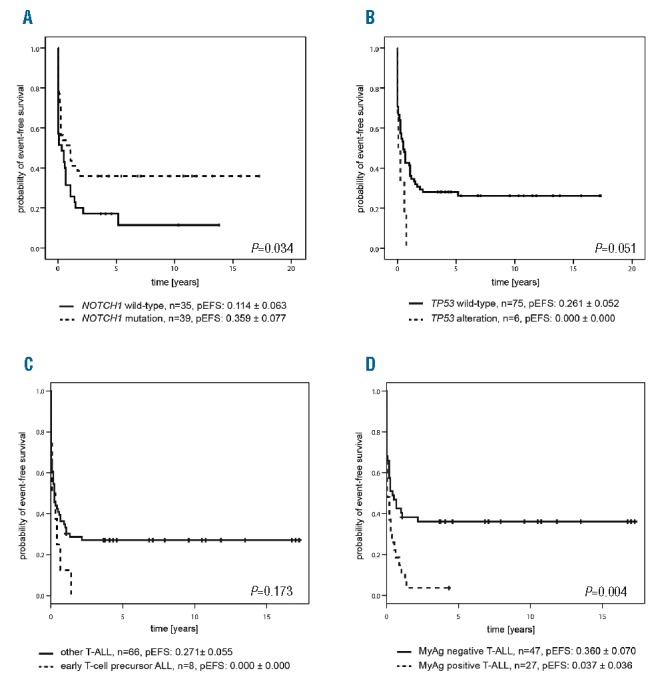
Survival of children with first relapse of T-ALL by genetic alterations and immunophenotype. (A) Kaplan-Meier analysis for the probability of EFS (pEFS) of patients with and without leukemic NOTCH1 mutation, P=0.034 by log-rank test. NOTCH1 mutation: n=39, censored n=14; NOTCH1 wild-type: n=35, censored n=5. (B) Kaplan-Meier analysis for the pEFS of patients with and without leukemic TP53 alteration (mutation and/or deletion), P=0.051 by log-rank test. TP53 alteration: n=6, censored n=0; TP53 wild-type: n=75, censored n=20. (C) Kaplan-Meier analysis for the pEFS of patients with and without early T-cell precursor immunophenotype, P=0.173 by log-rank test. Early T-cell precursor ALL: n=8, censored n=0; other T-ALL: n=66, censored n=18. (D) Kaplan-Meier analysis for the pEFS of patients with and without myeloid antigen expression (CD13 and/or CD33 and/or CD65), P=0.04 by log-rank test. MyAg positive T-ALL: n=27, censored n=1; MyAg negative T-ALL: n=47, censored n=17. ALL: acute lymphoblastic leukemia; EFS: event-free survival; MyAg: myeloid antigen; T-ALL: T-cell acute lymphoblastic leukemia.
Table 1.
Multivariate Cox regression analysis of patients with first relapse of T-ALL.
Herein we present a systematic analysis of the clinical value of genetic and immunophenotypic markers in children with relapsed T-ALL. It includes all patients with available T-ALL relapse samples from the pediatric trials ALL-REZ BFM 95/96 and 2002, thereby enabling correlative analysis in a comparatively large number of patients with this rare condition. We found that NOTCH1 mutations had a remarkable positive influence on outcome, with more than 60% pEFS in patients who reached a second complete remission and received HSCT. Since patients with relapsed T-ALL accompanied by bone marrow involvement have previously been thought to have a consistently poor outcome, this is a highly interesting result. By contrast, we found that TP53 alteration and myeloid antigen expression characterize patients with relapsed T-ALL with very poor outcome, which we confirmed by multivariate analysis for myeloid antigen expression. These markers can aid in identifying patients with relapsed T-ALL who are unlikely to benefit from standard second-line treatment, but who would be excellent candidates for enrollment in phase I/II trials, including targeted drugs or personalized treatment approaches. Our results reveal outcome heterogeneity in genetic and immunological relapsed T-ALL subgroups, and demonstrate proof of principle for the feasibility of molecular risk assessment in children with relapsed T-ALL.
Supplementary Material
Acknowledgements
The authors would like to thank K. Astrahantseff for critical reading of the manuscript.
Footnotes
Funding: this work was supported by grants of the German Childhood Cancer Foundation (DKS 2011.14 and DKS 2015.07 to RKS and CE, DKS 2013.12 to LK).
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Tallen G, Ratei R, Mann G, et al. Long-term outcome in children with relapsed acute lymphoblastic leukemia after time-point and site-of-relapse stratification and intensified short-course multidrug chemotherapy: results of trial ALL-REZ BFM 90. J Clin Oncol. 2010; 28(14):2339–2347. [DOI] [PubMed] [Google Scholar]
- 2.Locatelli F, Schrappe M, Bernardo ME, Rutella S. How I treat relapsed childhood acute lymphoblastic leukemia. Blood. 2012;120(14):2807–2816. [DOI] [PubMed] [Google Scholar]
- 3.Kox C, Zimmermann M, Stanulla M, et al. The favorable effect of activating NOTCH1 receptor mutations on long-term outcome in T-ALL patients treated on the ALL-BFM 2000 protocol can be separated from FBXW7 loss of function. Leukemia. 2010;24(12):2005–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jenkinson S, Koo K, Mansour MR, et al. Impact of NOTCH1/FBXW7 mutations on outcome in pediatric T-cell acute lymphoblastic leukemia patients treated on the MRC UKALL 2003 trial. Leukemia. 2013;27(1):41–47. [DOI] [PubMed] [Google Scholar]
- 5.Trinquand A, Tanguy-Schmidt A, Ben Abdelali R, et al. Toward a NOTCH1/FBXW7/RAS/PTEN-based oncogenetic risk classification of adult T-cell acute lymphoblastic leukemia: a Group for Research in Adult Acute Lymphoblastic Leukemia study. J Clin Oncol. 2013; 31(34):4333–4342. [DOI] [PubMed] [Google Scholar]
- 6.Coustan-Smith E, Mullighan CG, Onciu M, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009;10(2):147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jain N, Lamb AV, O’Brien S, et al. Early T-cell precursor acute lymphoblastic leukemia/lymphoma (ETP-ALL/LBL) in adolescents and adults: a high-risk subtype. Blood. 2016;127(15):1863–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hof J, Krentz S, van Schewick C, et al. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol. 2011;29(23):3185–3193. [DOI] [PubMed] [Google Scholar]
- 9.Diccianni MB, Yu J, Hsiao M, Mukherjee S, Shao LE, Yu AL. Clinical significance of p53 mutations in relapsed T-cell acute lymphoblastic leukemia. Blood. 1994;84(9):3105–3112. [PubMed] [Google Scholar]
- 10.Bandapalli OR, Zimmermann M, Kox C, et al. NOTCH1 activation clinically antagonizes the unfavorable effect of PTEN inactivation in BFM-treated children with precursor T-cell acute lymphoblastic leukemia. Haematologica. 2013;98(6):928–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jenkinson S, Kirkwood AA, Goulden N, Vora A, Linch DC, Gale RE. Impact of PTEN abnormalities on outcome in pediatric patients with T-cell acute lymphoblastic leukemia treated on the MRC UKALL2003 trial. Leukemia. 2016;30(1):39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wada M, Bartram CR, Nakamura H, et al. Analysis of p53 mutations in a large series of lymphoid hematologic malignancies of childhood. Blood. 1993;82(10):3163–3169. [PubMed] [Google Scholar]
- 13.Conter V, Valsecchi MG, Buldini B, et al. Early T-cell precursor acute lymphoblastic leukaemia in children treated in AIEOP centres with AIEOP-BFM protocols: a retrospective analysis. Lancet Haematol. 2016;3(2):e80–86. [DOI] [PubMed] [Google Scholar]
- 14.Ratei R, Schabath R, Karawajew L, et al. Lineage classification of childhood acute lymphoblastic leukemia according to the EGIL recommendations: results of the ALL-BFM 2000 trial. Klin Padiatr. 2013; 225 Suppl 1:S34–39. [DOI] [PubMed] [Google Scholar]
- 15.Zuurbier L, Homminga I, Calvert V, et al. NOTCH1 and/or FBXW7 mutations predict for initial good prednisone response but not for improved outcome in pediatric T-cell acute lymphoblastic leukemia patients treated on DCOG or COALL protocols. Leukemia. 2010;24(12):2014–2022. [DOI] [PubMed] [Google Scholar]
- 16.Clappier E, Collette S, Grardel N, et al. NOTCH1 and FBXW7 mutations have a favorable impact on early response to treatment, but not on outcome, in children with T-cell acute lymphoblastic leukemia (T-ALL) treated on EORTC trials 58881 and 58951. Leukemia. 2010; 24(12):2023–2031. [DOI] [PubMed] [Google Scholar]
- 17.Gianfelici V, Chiaretti S, Demeyer S, et al. RNA sequencing unravels the genetics of refractory/relapsed T-cell acute lymphoblastic leukemia. Prognostic and therapeutic implications. Haematologica. 2016;101(8):941–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pullen J, Shuster JJ, Link M, et al. Significance of commonly used prognostic factors differs for children with T cell acute lymphocytic leukemia (ALL), as compared to those with B-precursor ALL. A Pediatric Oncology Group (POG) study. Leukemia. 1999; 13(11):1696–1707. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.