

Class I phosphoinositide-3-kinases (PI3Ks) convert phosphoinositol-4,5-bisphosphate to phosphoinositol-3,4,5-trisphosphate, a lipid second messenger produced mainly at plasma membranes where it recruits and activates pleckstrin homology domain-containing proteins, including the well known protein kinase AKT. Class IA PI3Ks are heterodimers composed of a catalytic subunit (p110α, p110β or p110δ) and a regulatory subunit (p85α, p85β or p85γ, p50α and p55α). The catalytic subunits consist of an adaptor-binding domain (ABD), a Ras-binding domain (RBD), a protein kinase C homology-2 (C2) domain, a helical domain and a kinase domain. All of the class IA regulatory subunits contain two Src homology 2 domains, nSH2 and cSH2, separated by a coiled-coil domain known as inter-SH2 domain (iSH2).1 The p110δ catalytic subunit encoded by the PIK3CD gene is predominantly expressed in leukocytes. So far, six heterozygous germline gain-of-function mutations affecting either C2, helical or kinase domains of p110δ have been described to be responsible for the autosomal dominant “Activated PI3K-δ Syndrome 1” (APDS1): N334K, C416R, E525K, E525A, E1021K and E1025G2–6 (Figure 1A, B). A clinically and biologically similar disease “Activated PI3K-δ Syndrome 2” (APDS2) is caused by mutations in the PIK3R1 gene encoding the regulatory subunit p85α, p55α and p50α: splice site mutations responsible for amino acid 434–475 deletion.7,8 Both diseases - although heterogeneous - share a common phenotype characterized mainly by recurrent respiratory tract infections since childhood, bronchiectasis, lymphoproliferative disorder, and predisposition to development of B-cell lymphoma as main clinical complications.9 Biologically, APDS patients present with hypogammaglobulinemia and B-cell lymphopenia with an increased percentage of transitional B cells and decreased naive T-cell counts, especially T CD4+ cells. Here, we report three unrelated patients with novel heterozygous mutations in PIK3CD (E81K and G124D) located in the ABD and the ABD-RBD linker region of p110δ as cause for APDS1. These two gain-of-function mutations thus affect domains not previously shown to be important in the increased p110δ activity characteristic of this syndrome.
Figure 1.
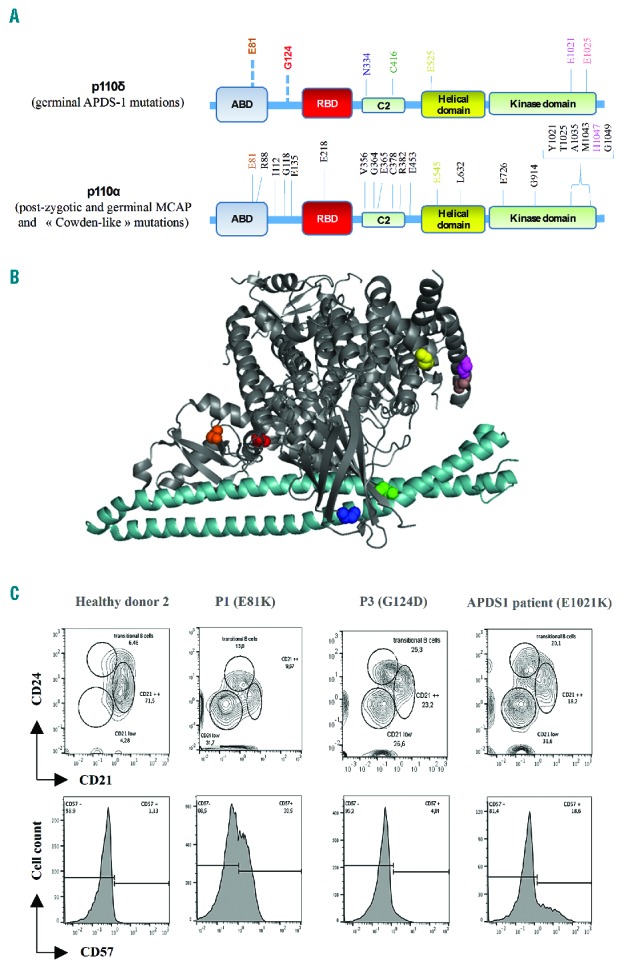
Localization of gain-of-function mutations in p110δ and phenotypic profile of lymphocytes. (A) Schematic representation of p110δ and p110α proteins, APDS1, MCAP and Cowden-like syndrome mutations are indicated, respectively.2–5,14 (B) Crystal structure of p110δ and the p85α iSH2 domain (light blue); PDB ID: 5DXU,15 the amino acids E81 (orange), G124 (red), N334 (blue), C416 (green), E525 (yellow), E1021 (magenta), and E1025 (salmon) in p110 δ are highlighted. (C) FACS plots of B cells (up) gated on CD19+ and T cells (down) gated on CD3+CD8+. Staining was performed on total blood from P1 and P3, a healthy donor and a known APDS1 patient.
The patients were born at term and presented with recurrent upper and lower respiratory tract infections since childhood. For patient 1 (P1), a 13-year-old boy, no relevant family history was reported. He presented with hypogammaglobulinemia with decreased IgG and IgA but normal IgM serum levels (Table 1). Current complications include bronchiectasis, a lymphoproliferative syndrome with splenomegaly and hepatic fibrosis responsible for portal hypertension associated with gastrointestinal bleedings. P2, a 10-year-old boy, presented with recurrent otitis media and sinusitis since his first year of life. He had an adenoidectomy at 3 years of age. He presented with high IgM but normal IgG and IgA serum levels (Table 1). The serum levels of IgG2 (0.23; N: 0.56) and IgG4 (<0.002; N: 0.018) subclasses were low. P3, a 9-year-old girl, presented with growth retardation since 6 months of age (currently −3 SD of height and −2.5 SD of weight). Lymphadenopathy and splenomegaly were noted at the age of 8 years, and adenoidectomy and tonsillectomy were performed. She had decreased IgA but normal IgG and IgM serum levels. Her mother had a similar clinical phenotype and died from Hodgkin lymphoma. Unfortunately, no genetic analysis of the mother could be performed since no DNA was available. All three patients received IgG replacement therapy. A CD4+ T-cell lymphopenia affecting especially the naïve compartment was noticed in all three cases. In P1, an elevated number of CD8+ CD57+ senescent T cells was found (Figure 1C). In addition, the three patients developed a progressive B-cell lymphopenia with decreased counts of memory B cells and increased percentage of transitional B cells (Table 1 and Figure 1C). Overall, the clinical and biological phenotype of the three patients was compatible with APDS. Thus, mutations known to cause APDS1 or APDS2 were searched and excluded by Sanger sequencing. Subsequently, to identify the genetic cause of the disease, whole exome sequencing was performed in P1. A heterozygous G to A mutation at position 9775698 (GRCh37; NM_005026.3) on chromosome 1, c.241 G>A in the PIK3CD gene was detected. Neither our in-house variant database nor the Exome Sequencing Project and Exome Aggregation Consortium databases contained a record of a nucleotide variation at this position. In the Catalogue of Somatic Mutation in Cancer database, the nucleotide variation was reported as somatic mutation in a lymphoid neoplasm and in a lung carcinoma. The variation encodes an amino acid substitution from glutamic acid to lysine at position 81 (E81K) located in the ABD at the N-terminal part of p110δ (Figure 1A). Sanger sequencing confirmed the mutation in the patient, which was not present in either parents. The de novo mutation was predicted to be highly damaging and deleterious by in silico prediction algorithms PolyPhen (0.99) and SIFT (0.01), respectively.
Table 1.
Immunological features.
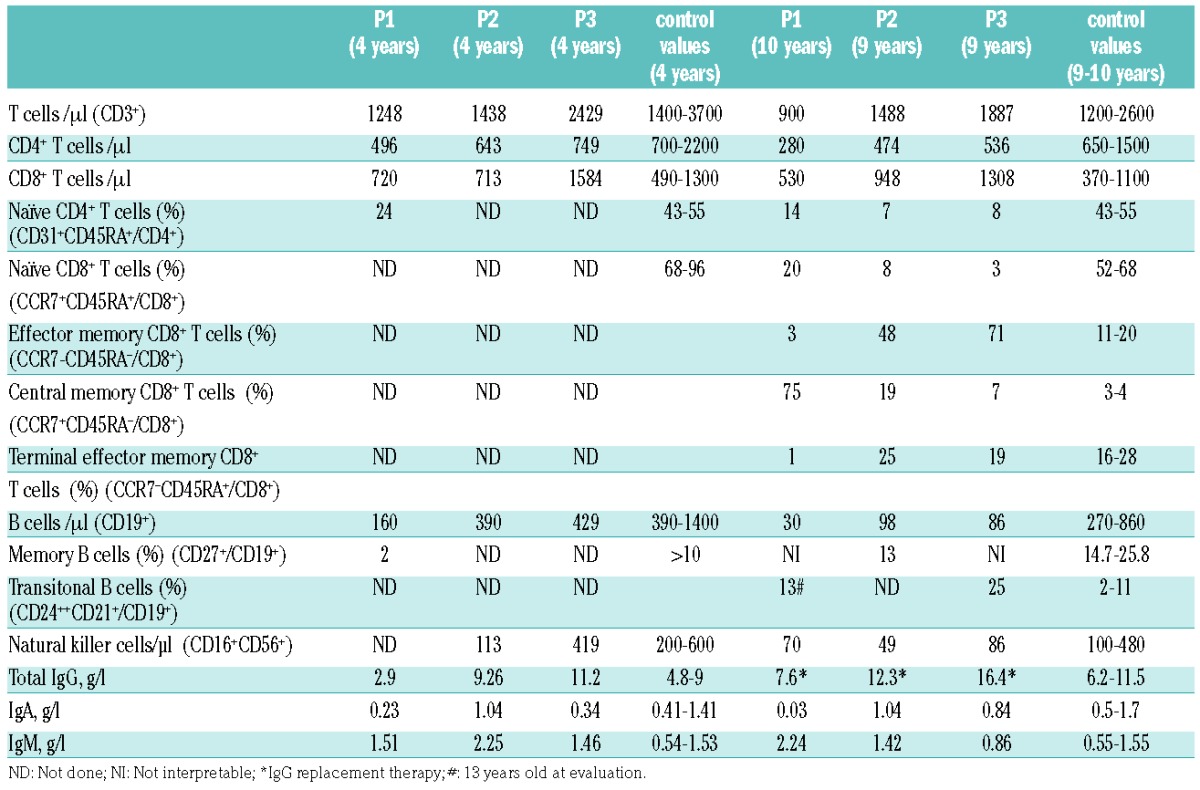
For P2 and P3, a targeted Next-Generation sequencing of a primary immunodeficiencies gene panel including PIK3CD and PIK3R1 was performed. A heterozygous G to A mutation at position 9775907 (GRCh37; NM_005026.3) in the PIK3CD gene was detected. The mutation was not found in our in-house variant database, and was predicted to be highly damaging in PolyPhen (1.00). The mutation leads to an amino acid substitution from glycine to aspartic acid at position 124 (G124D) located in the linker region between the ABD and the RBD at the N-terminal part of p110δ (Figure 1A).
To investigate whether the E81K and G124D mutations lead to a gain-of-function of p110δ activity, phosphorylation levels of AKT, a downstream protein of the PI3Kδ-signalling pathway, were determined in patients’ T-cell blasts. Higher levels of phosphorylated AKT at Ser473 were observed both basally and on stimulation with OKT3 in T-cell blasts derived from P1 an P2 versus healthy control T-cell blasts (Figure 2A). Treatment with a p110δ specific inhibitor (IC87114) abrogated those differences, indicating that PI3Kδ-signalling was responsible for the high level of AKT phosphorylation at Ser473. In addition, phosphorylation of S6, another downstream protein of the PI3K/AKT signalling pathway, was evaluated ex vivo in total blood lymphocytes. B-lymphocytes from P1 and P3 consistently presented with elevated phosphorylation at Ser235/236 of S6 compared to control samples. In contrast, phosphorylation at Ser235/236 of S6 in T lymphocytes was variable as only elevated in P1 (Figure 2B). Together, our functional analysis demonstrated that the de novo E81K mutation located in ABD and the G124D mutation located in the ABD-RBD linker region of p110δ are gain-of-function mutations. The difference in phosphorylation at Ser235/236 of S6 level ex vivo in patient T lymphocytes, together with the difference in the frequency of CD8+CD57+ T cells, further reflect the variability described in APDS patient cohorts,2,10,11 possibly explained by infection histories, and environmental and genetic factors.
Figure 2.
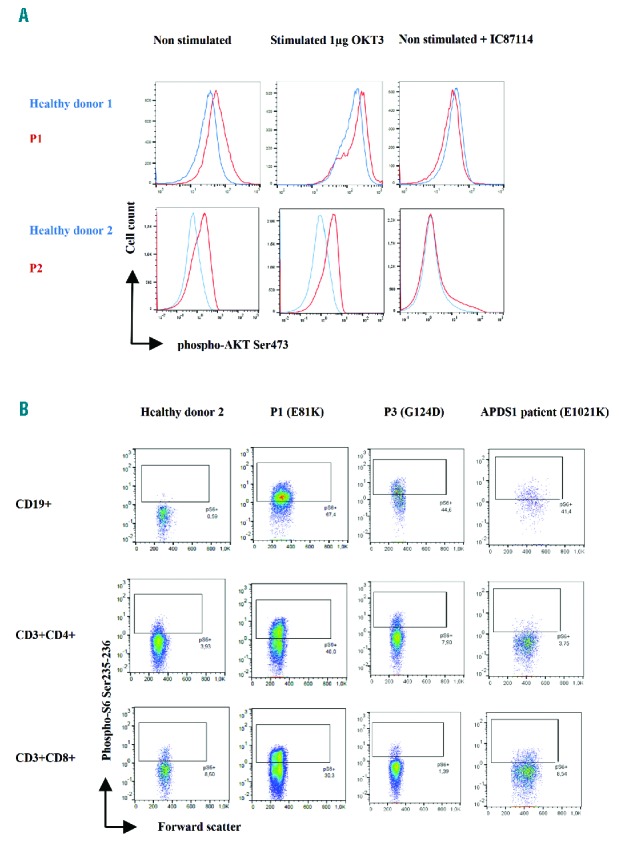
AKT and S6 phosphorylation study. (A) Intracellular staining of phospho-AKT Ser473 on T-cell blast (day 7 of IL2 culture) from patient and healthy donor, without and with stimulation with OKT3 or p110δ inhibitor IC87114 for 10 minutes. (B) Ex vivo phospho-S6 Ser235-236 intracellular staining performed on total blood from P1 and P3, a healthy donor and a known APDS1 patient.
All of the class IA p110 isoforms are inhibited by the presence of the iSH2 and the nSH2 domains of p85. This effect is mediated by contacts between the iSH2 and C2, and contacts of the nSH2 with the C2, helical and kinase domains.1 The published gain-of-function mutations of p110δ responsible for APDS1 map to these regulatory interfaces (Figure 1B). It has been reported that the iSH2 domain of the regulatory subunit interacts tightly with the ABD of the catalytic subunit.12 The E81K mutation located in ABD and the G214D mutation located in its linker domain thus likely activates p110δ by mimicking or enhancing specific conformational changes that are required for the PI3Kδ catalytic activity. The class IA catalytic subunits p110α (encoded by PIK3CA) and p110δ share a 72% amino acid homology.3 Intriguingly, gain-of-function mutations in PIK3CA at positions corresponding to mutations observed in PIK3CD causing APDS1 (including the E81K and G124D mutations) have been described in cancers, and some of them are responsible for the megalencephaly-capillary malformation-polymicrogyria syndrome (MCAP)13,14 (Figure 1A). This syndrome is characterized by overgrowth, brain and body asymmetry, cutaneous vascular malformations, digital anomalies, connective tissue dysplasia in joints, cortical brain malformations, and polymicrogyria. Thus, gain-of-function mutations in PIK3CD and PIK3CA responsible for APDS1 and MCAP, respectively, are likely to share similar activation mechanisms. Of note, mutations in the regulatory subunits encoded by PIK3R1 and PIK3R2 are responsible for APDS2 and MCAP, respectively.7,13
Precise genetic diagnosis of APDS is essential for patients to allow better prognostic measures and adequate treatment and follow up. Successful allogeneic hematopoietic stem cell transplantations (HSCT) as treatment for APDS were reported for severe cases.11 APDS patients presenting with signs of lymphoproliferation were started from their diagnosis on long-term rapamycin treatment, which appears efficient in most cases on clinical manifestations.3,11,10 Since the diagnosis of APDS1, P1 and P3 were treated with rapamycin and are under consideration for HSCT. Inhibitors specific for p110δ are currently in clinical trials (EudraCT Numbers: 2015-002900-10; 2015-005541-30; 2014-003876-22; 2015-004876-31) and could offer a new treatment option for APDS patients with possibly higher efficiency and less unwanted side effects.
Finally, our work describing two new PIK3CD gain-of-function mutations, E81K and G124D, demonstrated that mutations in the ABD and its linker region lead to APDS1. It highlights that mutations occurring in different parts of the gene can lead to the very same consequences, and should thus be screened in patients with a phenotype resembling APDS.
Supplementary Material
Acknowledgements
The authors would like to thank the clinical research team of the Imagine Institute for their support.
Footnotes
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Burke JE, Williams RL. Synergy in activating class I PI3Ks. Trends Biochem Sci. 2015;40(2):88–100. [DOI] [PubMed] [Google Scholar]
- 2.Angulo I, Vadas O, Garçon F, et al. Phosphoinositide 3-kinase δ gene mutation predisposes to respiratory infection and airway damage. Science. 2013;342(6160):866–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lucas CL, Kuehn HS, Zhao F, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat Immunol. 2014;15(1):88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crank MC, Grossman JK, Moir S, et al. Mutations in PIK3CD can cause hyper IgM syndrome (HIGM) associated with increased cancer susceptibility. J Clin Immunol. 2014;34(3):272–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsujita Y, Mitsui-Sekinaka K, Imai K, et al. Phosphatase and tensin homolog (PTEN) mutation can cause activated phosphatidylinositol 3-kinase δ syndrome-like immunodeficiency. J Allergy Clin Immunol. 2016;138(6):1672–1680.e10. [DOI] [PubMed] [Google Scholar]
- 6.Dulau Florea AE, Braylan RC, Schafernak KT, et al. Abnormal B-Cell Maturation in the Bone Marrow of Patients with Germline Mutations in PIK3CD. J Allergy Clin Immunol. 2016;139(3):1032–1035.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deau M, Heurtier L, Frange P, et al. A human immunodeficiency caused by mutations in the PIK3R1 gene. J Clin Immunol. 2014;124(9):3923–3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lucas CL, Zhang Y, Venida A, et al. Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J Exp Med. 2014;211(13):2537–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kracker S, Curtis J, Ibrahim MAA, et al. Occurrence of B-cell lymphomas in patients with activated phosphoinositide 3-kinase δ syndrome. J Allergy Clin Immunol. 2014;134(1):233–236.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elkaim E, Neven B, Bruneau J, et al. Clinical and immunologic phenotype associated with activated phosphoinositide 3-kinase δ syndrome 2: A cohort study. J Allergy Clin Immunol. 2016;138(1):210–218.e9. [DOI] [PubMed] [Google Scholar]
- 11.Coulter TI, Chandra A, Bacon CM, et al. Clinical spectrum and features of activated phosphoinositide 3-kinase δ syndrome: A large patient cohort study. J Allergy Clin Immunol. 2017;139(2):597–606.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dornan GL, Siempelkamp BD, Jenkins ML, et al. Conformational disruption of PI3Kδ regulation by immunodeficiency mutations in PIK3CD and PIK3R1. Proc Natl Acad Sci USA. 2017;114(8):1982–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rivière J-B, Mirzaa GM, O’Roak BJ, et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012; 44(8):934–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Donato N, Rump A, Mirzaa GM, et al. Identification and Characterization of a Novel Constitutional PIK3CA Mutation in a Child Lacking the Typical Segmental Overgrowth of “PIK3CA-Related Overgrowth Spectrum.” Hum Mutat. 2016;37(3):242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heffron TP, Heald RA, Ndubaku C, et al. The Rational Design of Selective Benzoxazepin Inhibitors of the α-Isoform of Phosphoinositide 3-Kinase Culminating in the Identification of (S)-2-((2-(1-Isopropyl-1H-1,2,4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl)oxy)propanamide. J Med Chem. 2016;59(3):985–1002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.