

Abstract
Mitochondria are multifaceted and indispensable organelles required for cell performance. Accordingly, dysfunction to mitochondria can result in cellular decline and possibly the onset of disease. Cells use a variety of means to recover mitochondria and restore homeostasis, including the activation of retrograde pathways such as the mitochondrial unfolded protein response (UPRmt). In this Minireview, we will discuss how cells adapt to mitochondrial stress through UPRmt regulation. Furthermore, we will explore the current repertoire of biological functions that are associated with this essential stress-response pathway.
Keywords: cell metabolism, cell signaling, cellular regulation, mitochondria, stress response, Mitochondrial dysfunction, mitochondrial quality control, mitochondrial stress signaling, mitochondrial unfolded protein response
Introduction
Mitochondria are double membrane organelles commonly associated with the production of cellular energy via oxidative phosphorylation (OXPHOS).2 Mitochondria are also required for the metabolism of nucleotides, amino acids, and lipids and have an essential function in regulating apoptosis. Maintaining mitochondrial integrity is therefore a key aspect in ensuring cellular and organismal viability. Consequently, a decline in mitochondrial function is frequently associated with the development of numerous diseases (1).
Mitochondria are dependent on a diverse compilation of proteins to carry out their vital functions. However, the mitochondrial proteome is faced with various challenges, most notably the partitioning of protein encoding genes between the mitochondrial and nuclear genomes. Remarkably, the human mitochondrial genome only encodes ∼1% of the total mitochondrial proteome with the remaining proteins being encoded by nuclear genes (2). Sophisticated mechanisms have evolved to efficiently transfer nuclear-encoded mitochondrial proteins to their proper organelle destination following translation on cytosolic ribosomes. To accomplish this complex task, proteins are sorted to mitochondria via targeting sequences that form characteristic amphipathic helices composed of positively charged residues. Such mitochondrial targeting sequences (MTS) are recognized and translocated via the mitochondrial Tom–Tim complex to their respective sub-organelle compartment (3). Exquisite coordination of expression between the mitochondrial and nuclear genomes exists during mitochondrial biogenesis, as failure in genome coordination can disrupt the precise stoichiometry of these OXPHOS complexes resulting in orphan subunit accumulation and proteotoxicity (4). Further contributing to mitochondrial proteotoxicity is the possible damage to the mitochondrial genome from reactive oxygen species (ROS) produced by OXPHOS machinery as well as the ill effects of various environmental toxins. Mechanisms must therefore exist to ensure the protection of the mitochondrial proteome.
Quality control of the mitochondrial proteome includes the functions of mitochondrial chaperones that assist in proper protein folding and proteases that promote clearance of misfolded proteins (5). Each sub-compartment of mitochondria houses its own quality control machinery to ensure protein homeostasis and organelle function. One way this is achieved is through retrograde signaling whereby stressed mitochondria signal to the nucleus to transcriptionally regulate a set of genes that assist in restoring mitochondrial activity. This Minireview will discuss the regulation and function of mitochondrial retrograde signaling with a focus on the mitochondrial unfolded protein response (UPRmt).
Mitochondrial UPR regulation
Caenorhabditis elegans
The UPRmt is a mitochondrial stress signaling pathway discovered in mammalian cells (6), but much of what is known with regard to its regulation was discovered in C. elegans (Fig. 1A). Conditions that increase mitochondrial proteotoxicity such as mitochondrial DNA depletion, impaired mitochondrial protein quality control machinery, and OXPHOS perturbation elicit the UPRmt (7, 8). A number of UPRmt regulators have been discovered using genetic approaches, including the mitochondrial matrix protease ClpP, the ubiquitin-like protein UBL-5, the transcription factor DVE-1, the mitochondrial ABC transporter HAF-1, and the bZIP transcription factor ATFS-1 (9, 10). The current paradigm suggests that ClpP proteolytically degrades improperly folded mitochondrial proteins with a subsequent release of the resulting peptides from mitochondria via HAF-1 leading to UPRmt activation (9, 10). How mitochondrial peptide extrusion affects the activity of the UPRmt is unclear, but it is clear that HAF-1 is not essential for UPRmt activation and instead serves as a modulator that impacts the nuclear accumulation of the bZIP transcription factor ATFS-1, a pivotal regulator of the UPRmt (7, 10).
Figure 1.
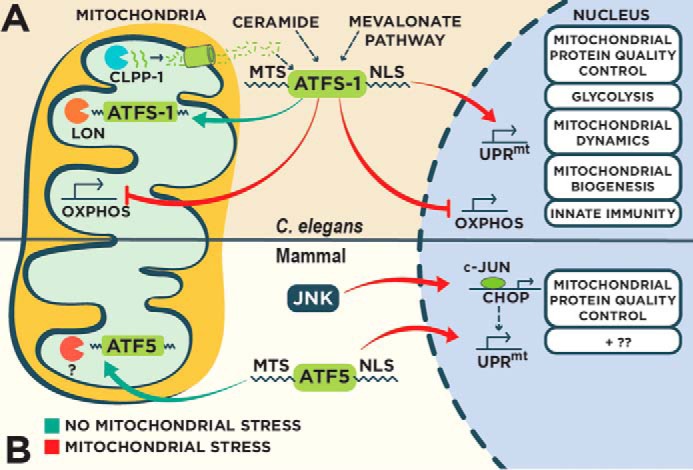
Regulation of the UPRmt in C. elegans and mammals. A, in C. elegans, the UPRmt is principally regulated by the bZIP transcription factor ATFS-1 that contains both mitochondrial and nuclear sorting sequences. In the absence of mitochondrial stress, ATFS-1 localizes to mitochondria and is degraded by the protease Lon. Perturbation to mitochondrial function reduces mitochondrial import efficiency causing an accumulation of cytosolic ATFS-1 and subsequent nuclear import. ATFS-1 regulates a diverse transcriptional program to recover mitochondrial function, including the attenuation of OXPHOS gene expression in both the nucleus and mitochondria. Multiple regulators of the UPRmt have been identified, including the protease ClpP and the ABC transporter HAF-1 that control UPRmt activity through an unidentified mechanism involving peptide efflux. B, mammalian UPRmt is regulated by the transcription factors CHOP and ATF5 that transcriptionally regulate genes to restore mitochondrial homeostasis. CHOP is transcriptionally activated by transcription factor c-Jun during mitochondrial stress. ATF5 contains mitochondrial and nuclear localization sequences similar to ATFS-1 that regulate UPRmt activity based on the efficiency of mitochondrial import.
ATFS-1 harbors two sorting signals able to target it to both mitochondria and the nucleus (7, 11). Under physiological conditions, the MTS directs the localization of ATFS-1 to mitochondria where it is degraded by the protease Lon (7). However, mitochondrial import efficiency is reduced during conditions that perturb mitochondrial function (7, 12–14), causing ATFS-1 to accumulate in the cytosol and subsequently be imported into the nucleus via its nuclear localization sequence. ATFS-1 regulates a diverse transcriptional response to recover mitochondrial function, including the induction of mitochondrial proteases and chaperones, xenobiotic and ROS-detoxifying genes, and metabolic regulators (7). The concept of mitochondrial import efficiency regulating the UPRmt is reminiscent of Pink1 kinase regulation in the detection and removal of defective mitochondria by mitophagy (15). During mitophagy, damaged mitochondria are marked for removal by Pink1 leading to recruitment of the autophagic machinery (15). Pink1 is imported into unstressed mitochondria and degraded (16). However, mitochondrial dysfunction reduces the import efficiency of Pink1 allowing it to accumulate on the outer mitochondrial membrane where it initiates a cascade leading to mitochondrial clearance (17). Thus mitochondrial import efficiency may be used as a general indicator of perturbed mitochondrial function to activate mitochondrial recovery programs such as the UPRmt and mitophagy.
Additional regulators of the UPRmt have been identified that potentially shed light into the inputs of this protective program. Using a genetic approach, two metabolic pathways were identified as important regulators of the UPRmt (18). First, knockdown of components in the sphingolipid biosynthesis pathway, including the enzymes serine palmitoyltransferase and ceramide synthase, results in UPRmt attenuation that can be rescued with ceramide supplementation. Interestingly, ceramide was found to accumulate at mitochondria prior to UPRmt activation (18). Also, inhibition of the sphingolipid biosynthesis pathway could not block UPRmt activation from a gain-of-function ATFS-1 that is constitutively nuclear irrespective of mitochondrial stress (18, 19). Therefore, ceramide accumulation at mitochondria may be an early step in the activation of the UPRmt. In the same genetic screen, knockdown of 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) synthase, a component of the mevalonate pathway, also blocked UPRmt activation in the presence of mitochondrial stress (18). The mevalonate pathway is necessary for the synthesis of isoprenoids such as cholesterol. Interestingly, constitutive nuclear accumulation of ATFS-1 rescues the ill effects of statins, a class of cholesterol-lowering drugs that inhibit the mevalonate pathway (19) likely by increasing ubiquinone biosynthesis that is impaired by the statins. Intriguingly, statins block UPRmt activation during mitochondrial stress suggesting that the side effects experienced while taking this drug may be due to an inability to sense and respond to conditions that perturb mitochondrial function (18). In addition to the synthesis of cholesterol, the mevalonate pathway is also important for the production of prenylated lipids such as farnesyl pyrophosphate that can be blocked with statin treatment (20). Protein prenylation involves the addition of the farnesyl moiety to client proteins (21), a reaction that is particularly crucial for the activity of small GTPases (22). Importantly, gain-of-function ATFS-1 animals are also resistant to the toxicity exerted by gliotoxin (19), which blocks the addition of farnesyl to small GTPases through the inhibition of farnesyl transferase. This suggests that impaired prenylation may contribute to the negative effects of statins. Perhaps GTPase prenylation is required for UPRmt activation that can be mitigated through constitutive activation of ATFS-1. In support of this possibility, the small GTPase Rheb is required for UPRmt activity (9).
Recent observations suggest that chromatin remodeling is also critically required for UPRmt activity. In the first study, the histone lysine demethylases JMJD-1.2 and JMJD-3.1 were discovered in a genetic screen for positive UPRmt regulators (23). Both show specificity for histone H3K27me2/me3 and are required and sufficient for the UPRmt. Consistent with mediating a mitochondrial protective transcriptional response, genes differentially expressed from JMJD-1.2 and JMJD-3.1 overexpression show significant overlap with gene expression profiles of mitochondrially stressed animals with perturbed OXPHOS (23). Analogous findings were also observed with the mammalian homologs of JMJD-1.2 (Phf8) and JMJD-3.1 (Jmjd3), suggesting a conserved mechanism of UPRmt regulation (23). In the second study, the H3K9 methyltransferase MET-2 and the novel protein LIN-65 were found to positively regulate the UPRmt (24). LIN-65 is initially cytosolic but localizes to the nucleus in the presence of mitochondrial stress in a MET-2-dependent manner (24), suggesting they are functionally related. Indeed, both MET-2 and LIN-65 are required for the methylation of histone H3K9 that is needed for accumulation of the UPRmt transcriptional regulator DVE-1 into discrete nuclear puncta, specifically to chromatin regions thought to represent sites of active gene expression (9, 24). These findings add an extra layer of complexity to the regulation of the UPRmt, suggesting that epigenetic modifications are a necessary step for the activation of this mitochondrial recovery program.
Remarkably, mitochondrial damage originating from one tissue can induce a UPRmt in distal tissues via cell non-autonomous signaling (Fig. 2) (25). For example, neuronal mitochondrial dysfunction can induce the UPRmt in the intestine through the production of secreted “mitokines” (25). Presumably, sensory neurons that are in contact with the external environment housing potentially damaging toxins would be ideal to relay mitochondrial stress signals to tissues, like the intestine, that are likely to be impacted by toxin exposure during feeding. Two recent advancements have been made into the identity of the mitokines that regulate the cell non-autonomous control of the UPRmt. In the first study, the bioamine neurotransmitter serotonin was found to be required for activation of the UPRmt in the intestine as a consequence of mitochondrial protein aggregate accumulation in neurons (26). In a parallel study, interneuron secretion of the neuropeptide FLP-2 was necessary and sufficient for UPRmt activation in the intestine (27). Flp-2 transcript levels are enhanced in the presence of neuronal mitochondrial stress, consistent with a role in the cell non-autonomous regulation of the UPRmt. Indeed, overexpression of FLP-2 specifically activates the UPRmt and not other stress responses (27). Together, these studies suggest that neuronal control of the UPRmt in distal tissues is likely complex involving the secretion of multiple mitokine signals. Going forward, it will be interesting to understand how these various mitokines are regulated in a coordinated manner in response to varying forms of mitochondrial dysfunction as well as their role in regulating whole-animal metabolism.
Figure 2.
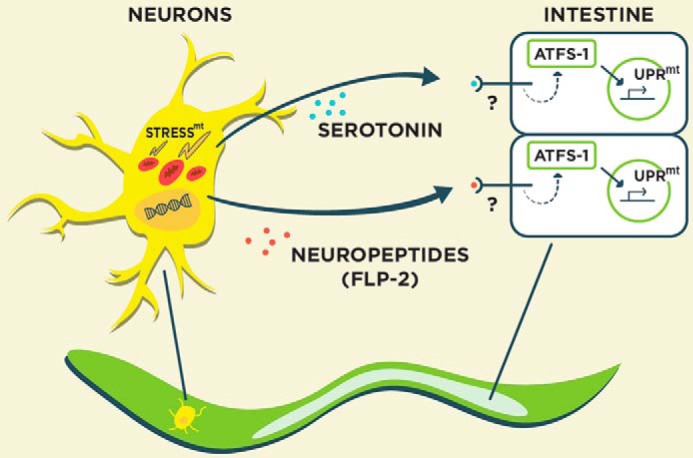
Cell non-autonomous regulation of the UPRmt. Mitochondrial dysfunction in neurons activates the UPRmt in distal tissues such as the intestine through diffusible “mitokine” signals, including the neurotransmitter serotonin and the neuropeptide FLP-2. stressmt, mitochondrial stress.
Mammals
The UPRmt was first discovered in mammalian cultured cells expressing a misfolded mitochondrial protein that resulted in increased expression of mitochondrial protein quality control genes in the nucleus (Fig. 1B) (6). This transcriptional response to mitochondrial dysfunction is thought to require the C/EBP transcription factor CHOP as the corresponding consensus binding site is present in multiple genes that are induced during the UPRmt (28, 29). Consistently, CHOP is transcriptionally induced during the UPRmt presumably by the transcription factor c-Jun (6, 28).
Inspired by the discovery of ATFS-1, Fiorese et al. (30) identified the bZIP transcription factor ATF5 as an additional regulator of the UPRmt response. Mitochondrial import efficiency appears to regulate ATF5 in a manner akin to ATFS-1 because ATF5 also contains a presequence that directs its localization to mitochondria in the absence of mitochondrial stress (30). Similar to ATFS-1, ATF5 transcriptionally regulates mitochondrial protective gene expression that is necessary for cell growth during mitochondrial stress (30). Interestingly, ATF5 can induce a UPRmt in C. elegans in the absence of ATFS-1 suggesting that it is a conserved mammalian homolog (30). However, there are still questions left unanswered with regard to ATF5 and the mammalian UPRmt. First, is ATF5 proteolytically degraded in healthy mitochondria by LONP1 similar to ATFS-1? LONP1 expression is transcriptionally induced during the UPRmt by ATF5 suggesting a functional relationship (30). Second, does ATF5 accumulation in the nucleus during the UPRmt require ClpP-derived peptide efflux via the mammalian homolog of HAF-1 (TAP)? Finally, how are ATF5 and CHOP coordinated during the UPRmt? Interestingly, the CHOP consensus sequence is present in some genes that are also regulated by ATF5 during the UPRmt (28, 30) suggesting some degree of overlap in target gene regulation. Further work is therefore needed to better understand the specific mechanisms of ATF5 regulation during the UPRmt.
The mitochondrial intermembrane space (IMS) contains its own suite of protein quality machinery and potentially distinct stress response. Perturbation to the IMS protein folding environment elicits a protective response through the phosphorylation of the estrogen receptor α by Akt (31). In a recent study, proteotoxicity to the mitochondrial matrix and IMS activated the deacetylase SirT3 to promote mitochondrial recovery through the activation of anti-oxidant machinery and the stimulation of mitophagy (32). Presumably, the SirT3 response during mitochondrial stress is mediated by the transcription factor FOXOA3 that accumulates in the nucleus following SirT3-mediated deacetylation (32).
Comparison of the UPRmt and UPRER
Much like mitochondria, the endoplasmic reticulum (ER) is prone to the accumulation of misfolded proteins and similarly responds to increases in proteotoxicity through the transcriptional up-regulation of ER chaperones and proteases to restore protein homeostasis via the UPRER (33). Although conceptually similar to the UPRmt, the UPRER possesses distinct signaling mechanisms that are involved in its regulation. There are three main regulatory pathways that govern the UPRER: the serine/threonine kinase/endonuclease IRE1; the bZIP transcription factor ATF6; and the kinase PERK. All three proteins are bound in an inactive state to the ER chaperone BiP and are released in the presence of ER stress. Active IRE1 results in preferential mRNA splicing of the bZIP transcription factor XBP1 resulting in its activation and subsequent transcriptional up-regulation of proteostasis genes. ATF6 also positively regulates the expression of proteostasis genes following its activation by proteolytic cleavage.
In addition to transcriptionally regulating the expression of ER protective genes, the UPRER helps restore ER homeostasis by attenuating protein translation through PERK-dependent phosphorylation of the translation initiation factor eIF2α (34). PERK is among four kinases that constitute the integrated stress response (ISR) that diminishes protein translation in response to various cellular stresses (35). Interestingly, the ISR kinase GCN2 is activated by increased ROS during mitochondrial stress and helps restore mitochondrial homeostasis by lowering cytosolic protein translation via phosphorylation of eIF2α, thus reducing the load of incoming unfolded mitochondrial proteins (36). Although general protein translation is attenuated during the ISR, mRNAs containing upstream open reading frames (uORFs) are favorably translated. Intriguingly, ATF5 is among the uORF containing mRNAs that are preferentially translated following eIF2α phosphorylation during ER stress (37). Whether mitochondrial stress results in a similar preferential translation of ATF5 is still unknown.
It is noteworthy that the UPRmt and UPRER share some common regulatory proteins, suggesting a possible intersection in the regulatory inputs of both stress responses. For example, CHOP is also activated during ER stress downstream of the bZIP transcription factor ATF4 (38). In this context CHOP assumes a pro-apoptotic role by transcriptionally regulating ATF5, but whether a similar mode of regulation occurs during UPRmt activation is not known (39). In contrast to the pro-apoptotic role of CHOP and ATF5 during ER stress, these transcription factors adopt a pro-survival function during mitochondrial stress through the regulation of mitochondrial protective gene expression (6, 30). Possibly, CHOP and ATF5 undergo stress-specific post-translational modifications or heterodimerization that dictate their particular function.
Physiological roles of the UPRmt
Protection against pathogen infection
Mitochondria are desirable targets for bacterial pathogens during infection. For example, various pathogens manipulate mitochondrion-dependent cell death pathways to promote their own survival (40). Mitochondria also play an important role in regulating the innate immune response (41) that can be compromised during infection by certain pathogens (42). As such, bacterial pathogens have evolved various means to exploit mitochondrial function, including the precise targeting of protein virulence factors directly to mitochondria (43).
Host resistance to infection is dependent on innate and adaptive immune responses that limit the colonization of pathogenic microbes. In contrast, host tolerance during infection is defined as the ability to withstand the damage caused by harmful pathogens and possibly the self-damage inflicted by the host resulting from the inflammatory response (44). The UPRmt supports host tolerance by regulating mitochondrial recovery while additionally promoting host resistance through the transcriptional regulation of various innate immunity genes such as secreted anti-microbial peptides and lysozymes (Fig. 3A) (45). Various microbes such as the opportunistic pathogen Pseudomonas aeruginosa can activate the UPRmt through the production of mitochondrial toxins (18, 45). Consistent with a role in protecting the host from infection, ATFS-1 is required and sufficient for survival during P. aeruginosa infection (45, 46). Interestingly, the mitochondrial chaperone HSP-60 that is transcriptionally up-regulated during the UPRmt by ATFS-1 (7, 10, 11) promotes host resistance during infection by enhancing the PMK-1/p38 MAPK innate immunity pathway through direct binding and stabilization of SEK-1/MAPK kinase 3 (46).
Figure 3.
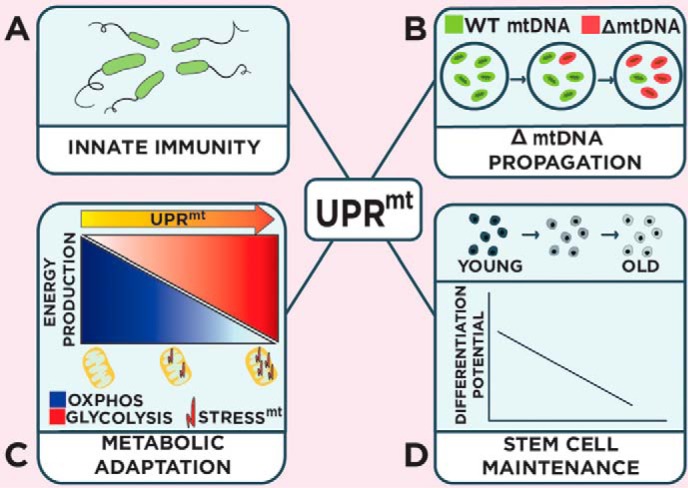
Biological roles of the UPRmt. The UPRmt supports a variety of organismal functions, including the regulation of innate immunity during pathogen infection (A); the control of deleterious mitochondrial genome numbers (B); mediating metabolic adaptations during mitochondrial stress (C); and promoting stem cell maintenance (D). stressmt, mitochondrial stress.
The coupling of host tolerance and resistance to one stress-response pathway is an efficient means of protecting the host during infection considering that mitochondria are targeted by a number of pathogens (18, 43, 45). Furthermore, the UPRmt may be regarded as an additional sensor of infection from the damage that is inflicted by harmful microbes targeting mitochondrial function. It is unclear whether all metazoans couple the UPRmt to the regulation of innate immunity during infection. However, the homolog of ATFS-1 in the whiteleg shrimp induces the expression of antimicrobial peptides and is required for survival during pathogen infection suggesting that UPRmt-mediated regulation of host resistance may be conserved (47).
Accumulation of deleterious mitochondrial genomes
Deletions in mitochondrial DNA (mtDNA) occur as an organism ages and contributes to various diseases (48). However, each cell houses hundreds of mitochondrial genomes as a mixture of wild-type and mutant forms, a phenomenon known as heteroplasmy (49), and accordingly the number of deleterious mitochondrial genomes only needs to exceed a certain threshold to be pathogenic (typically comprising more than 60% of the total number of mitochondrial genomes) (49). The mechanism that regulates the number of deleterious mitochondrial genomes has remained elusive until the discoveries made by two recent independent studies (50, 51). Here, a C. elegans heteroplasmic strain harboring mtDNA with a 3.1-kb deletion (ΔmtDNA (52)) activated the UPRmt as a result of reduced OXPHOS activity (50, 51). Surprisingly, loss of the UPRmt through ATFS-1 inhibition reduced the number of ΔmtDNA copies, whereas constitutive activation of ATFS-1 promoted the accumulation of ΔmtDNA, further reducing OXPHOS efficiency and causing impaired development (50, 51).
How does ATFS-1 promote the accumulation of deleterious ΔmtDNA? Potentially, the UPRmt limits mitophagy by promoting mitochondrial health, but ATFS-1 appears to preferentially promote ΔmtDNA accumulation independent from mitophagy (50, 51). ATFS-1 does promote the expression of genes involved in mitochondrial biogenesis, including regulators of mtDNA replication such as mtDNA polymerase and TFAM (51) as well as transcriptionally regulating genes involved in mitochondrial dynamics (7, 51). Reducing regulators of mtDNA replication and mitochondrial dynamics attenuate the increased ΔmtDNA number that is observed in constitutively active ATFS-1 animals suggesting that both are involved in the ATFS-1-dependent propagation of ΔmtDNA (51). Together, these studies reveal an unexpected toxic consequence of the UPRmt mitochondrial recovery program that expands ΔmtDNA through the stimulation of mitochondrial biogenesis (Fig. 3B).
Metabolic adaptations
Paradoxically, in addition to nuclear accumulation, ATFS-1 also accumulates in mitochondria during stress despite reduced mitochondrial import efficiency (7, 11). Multiple isoforms of ATFS-1 are present in mitochondria during the UPRmt, but the exact identity of these isoforms is still unknown. However, two splice variants of ATFS-1 exist with a difference of just 16 amino acids (11). Although both the short and long ATFS-1 isoforms contain the MTS, it is not known which isoform accumulates in mitochondria during stress. Within mitochondria ATFS-1 binds the D-loop of mtDNA, an A-T-rich region residing in the control region adjacent to transcriptional regulatory elements (11). Here, ATFS-1 limits the abundance of OXPHOS transcripts encoded by the mitochondrial genome. ATFS-1 regulates mitochondrial OXPHOS transcript numbers potentially through transcriptional repression via its association with the D-loop region; however, its effects on transcript stability have yet to be studied. Interestingly, ATFS-1 also negatively regulates nuclear-encoded OXPHOS gene transcript abundance through direct or indirect binding of their gene promoters (11). Thus, ATFS-1 appears to transcriptionally attenuate or limit OXPHOS biogenesis until the protein folding capacity is restored as a means of promoting mitochondrial recovery. In addition to repressing the abundance of OXPHOS transcripts, ATFS-1 also transcriptionally induces the expression of genes involved in glycolysis (7, 11). By positively regulating glycolysis gene expression, ATFS-1 presumably ensures adequate ATP levels in lieu of reduced OXPHOS activity in an effort to promote mitochondrial repair and cell survival (Fig. 3C).
Hematopoietic stem cell maintenance
Quiescent stem cells have relatively little mitochondrial activity that increases during differentiation (53). In the study by Mohrin et al. (54), the histone deacetylase SIRT7 was found to reduce mitochondrial translation by complexing with the central metabolic regulator of mitochondrial gene expression NRF1 and repressing transcription of mitochondrial ribosomes and translation factors. Loss of SIRT7 consequently results in enhanced mitochondrial translation and biogenesis leading to the activation of the UPRmt. Interestingly, inhibiting SIRT7 in hematopoietic stem cells (HSC) enhances mitochondrial biogenesis and activates the UPRmt, thus reducing quiescence and the ability to differentiate. Consistent with a role in maintaining stem cell quiescence, SIRT7 levels are elevated during the quiescent stage and low in aged stem cell populations (54). Consequently, increasing SIRT7 levels in aged stem cells decrease mitochondrial proteotoxicity and improve their differentiation potential. Although it is still unclear whether specific loss of UPRmt regulators impacts HSC maintenance, this study suggests the importance of mitochondrial protein homeostasis in stem cell vitality (Fig. 3D).
The future of the UPRmt
A decline in mitochondrial function likely impacts a wide range of cellular activities because of the essential nature of the organelle. Consequently, mitochondrial stress presumably activates parallel cytoprotective pathways. For instance, mitochondrial stress results in the activation of multiple signaling modules in addition to the UPRmt, including the hypoxia-response transcription factor HIF-1, the AMP-activated kinase pathway, and the transcription factors TAF-4 and CEH-23 (55–57). As well, increased mitochondrial ROS can activate an oxidative stress response regulated by the transcription factor SKN-1 (58). Intriguingly, ATFS-1 regulates the expression of skn-1 as part of the UPRmt transcriptional response (9). Finally, accumulation of mitochondrial precursor proteins in the cytosol as a result of impaired mitochondrial import elicits a separate stress-response pathway involving reduced protein translation and increased proteasome activity (12, 14). How the UPRmt integrates with these and other cellular stress pathways is therefore an area worthy of further investigation.
Also, as the UPRmt is an important regulator of mitochondrial recovery, it may be an attractive therapeutic target for those diseases that result from mitochondrial dysfunction. The therapeutic potential of the UPRmt is particularly promising in the field of cancer research. The relationship between mitochondrial dysfunction and cancer was first proposed based on the observation by Otto Warburg that cancer cells utilize glycolysis in the presence of oxygen presumably due to a defect in mitochondrial respiration and function (59). Although this remains true with certain cancers, it can by no means be generalized. Indeed, certain cancers display normal or even enhanced mitochondrial function (60) suggesting a considerable heterogeneity in cancer subtypes. It is clear though that some cancer cells express higher levels of mitochondrial chaperones indicative of an activated UPRmt (30, 61–63). The UPRmt in this context could help support tumor survival from the mitochondrial dysfunction that may arise from genetic mutations (64) or through the increase in mitochondrial biogenesis used to support tumor growth and proliferation (65). Interestingly, inhibiting mitochondrial chaperones or the UPRmt regulator ATF5 increases tumor cell death while having little to no effect on healthy cells (30, 63, 66), supporting the targeting of the UPRmt as an anti-cancer therapy. How the UPRmt might be pursued for other mitochondrially related pathologies such as neurological disease and diabetes is still relatively unexplored and is likely to be an exciting avenue of research in the field.
This work was supported by Cancer Prevention Research Institute of Texas Grant RR160053 and the University of Texas Arlington (to M. W. P.) and Grant R01AG047182 (to C. M. H.). The authors declare that they have no conflicts of interest with the contents of this article.
- OXPHOS
- oxidative phosphorylation
- ROS
- reactive oxygen species
- ISR
- integrated stress response
- MTS
- mitochondrial targeting sequence
- IMS
- intermembrane space
- ER
- endoplasmic reticulum
- uORF
- upstream open reading frame
- HSC
- hematopoietic stem cell.
References
- 1. Nunnari J., and Suomalainen A. (2012) Mitochondria: in sickness and in health. Cell 148, 1145–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Calvo S. E., and Mootha V. K. (2010) The mitochondrial proteome and human disease. Annu. Rev. Genomics Hum. Genet. 11, 25–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schmidt O., Pfanner N., and Meisinger C. (2010) Mitochondrial protein import: from proteomics to functional mechanisms. Nat. Rev. Mol. Cell Biol. 11, 655–667 [DOI] [PubMed] [Google Scholar]
- 4. Rugarli E. I., and Langer T. (2012) Mitochondrial quality control: a matter of life and death for neurons. EMBO J. 31, 1336–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baker B. M., and Haynes C. M. (2011) Mitochondrial protein quality control during biogenesis and aging. Trends Biochem. Sci. 36, 254–261 [DOI] [PubMed] [Google Scholar]
- 6. Zhao Q., Wang J., Levichkin I. V., Stasinopoulos S., Ryan M. T., and Hoogenraad N. J. (2002) A mitochondrial specific stress response in mammalian cells. EMBO J. 21, 4411–4419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nargund A. M., Pellegrino M. W., Fiorese C. J., Baker B. M., and Haynes C. M. (2012) Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337, 587–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yoneda T., Benedetti C., Urano F., Clark S. G., Harding H. P., and Ron D. (2004) Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J. Cell Sci. 117, 4055–4066 [DOI] [PubMed] [Google Scholar]
- 9. Haynes C. M., Petrova K., Benedetti C., Yang Y., and Ron D. (2007) ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 13, 467–480 [DOI] [PubMed] [Google Scholar]
- 10. Haynes C. M., Yang Y., Blais S. P., Neubert T. A., and Ron D. (2010) The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol. Cell 37, 529–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nargund A. M., Fiorese C. J., Pellegrino M. W., Deng P., and Haynes C. M. (2015) Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol. Cell 58, 123–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang X., and Chen X. J. (2015) A cytosolic network suppressing mitochondria-mediated proteostatic stress and cell death. Nature 524, 481–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wright G., Terada K., Yano M., Sergeev I., and Mori M. (2001) Oxidative stress inhibits the mitochondrial import of preproteins and leads to their degradation. Exp. Cell Res. 263, 107–117 [DOI] [PubMed] [Google Scholar]
- 14. Wrobel L., Topf U., Bragoszewski P., Wiese S., Sztolsztener M. E., Oeljeklaus S., Varabyova A., Lirski M., Chroscicki P., Mroczek S., Januszewicz E., Dziembowski A., Koblowska M., Warscheid B., and Chacinska A. (2015) Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature 524, 485–488 [DOI] [PubMed] [Google Scholar]
- 15. Youle R. J., and Narendra D. P. (2011) Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thomas R. E., Andrews L. A., Burman J. L., Lin W. Y., and Pallanck L. J. (2014) PINK1-Parkin pathway activity is regulated by degradation of PINK1 in the mitochondrial matrix. PLoS Genet. 10, e1004279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Narendra D. P., Jin S. M., Tanaka A., Suen D. F., Gautier C. A., Shen J., Cookson M. R., and Youle R. J. (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8, e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu Y., Samuel B. S., Breen P. C., and Ruvkun G. (2014) Caenorhabditis elegans pathways that surveil and defend mitochondria. Nature 508, 406–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rauthan M., Ranji P., Aguilera Pradenas N., Pitot C., and Pilon M. (2013) The mitochondrial unfolded protein response activator ATFS-1 protects cells from inhibition of the mevalonate pathway. Proc. Natl. Acad. Sci. U.S.A. 110, 5981–5986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Möorck C., Olsen L., Kurth C., Persson A., Storm N. J., Svensson E., Jansson J. O., Hellqvist M., Enejder A., Faergeman N. J., and Pilon M. (2009) Statins inhibit protein lipidation and induce the unfolded protein response in the non-sterol producing nematode Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 106, 18285–18290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Konstantinopoulos P. A., Karamouzis M. V., and Papavassiliou A. G. (2007) Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat. Rev. Drug Discov. 6, 541–555 [DOI] [PubMed] [Google Scholar]
- 22. Itzen A., and Goody R. S. (2011) GTPases involved in vesicular trafficking: structures and mechanisms. Semin. Cell Dev. Biol. 22, 48–56 [DOI] [PubMed] [Google Scholar]
- 23. Merkwirth C., Jovaisaite V., Durieux J., Matilainen O., Jordan S. D., Quiros P. M., Steffen K. K., Williams E. G., Mouchiroud L., Tronnes S. U., Murillo V., Wolff S. C., Shaw R. J., Auwerx J., and Dillin A. (2016) Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell 165, 1209–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tian Y., Garcia G., Bian Q., Steffen K. K., Joe L., Wolff S., Meyer B. J., and Dillin A. (2016) Mitochondrial stress induces chromatin reorganization to promote longevity and UPR(mt). Cell 165, 1197–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Durieux J., Wolff S., and Dillin A. (2011) The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 79–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Berendzen K. M., Durieux J., Shao L. W., Tian Y., Kim H. E., Wolff S., Liu Y., and Dillin A. (2016) Neuroendocrine coordination of mitochondrial stress signaling and proteostasis. Cell 166, 1553–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shao L. W., Niu R., and Liu Y. (2016) Neuropeptide signals cell non-autonomous mitochondrial unfolded protein response. Cell Res. 26, 1182–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Horibe T., and Hoogenraad N. J. (2007) The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS ONE 2, e835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aldridge J. E., Horibe T., and Hoogenraad N. J. (2007) Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS ONE 2, e874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fiorese C. J., Schulz A. M., Lin Y. F., Rosin N., Pellegrino M. W., and Haynes C. M. (2016) The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr. Biol. 26, 2037–2043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Papa L., and Germain D. (2011) Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J. Cell Sci. 124, 1396–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Papa L., and Germain D. (2014) SirT3 regulates the mitochondrial unfolded protein response. Mol. Cell. Biol. 34, 699–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hetz C. (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102 [DOI] [PubMed] [Google Scholar]
- 34. Harding H. P., Zhang Y., Bertolotti A., Zeng H., and Ron D. (2000) Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 5, 897–904 [DOI] [PubMed] [Google Scholar]
- 35. Pakos-Zebrucka K., Koryga I., Mnich K., Ljujic M., Samali A., and Gorman A. M. (2016) The integrated stress response. EMBO Rep. 17, 1374–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baker B. M., Nargund A. M., Sun T., and Haynes C. M. (2012) Protective coupling of mitochondrial function and protein synthesis via the eIF2α kinase GCN-2. PLoS Genet. 8, e1002760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou D., Palam L. R., Jiang L., Narasimhan J., Staschke K. A., and Wek R. C. (2008) Phosphorylation of eIF2 directs ATF5 translational control in response to diverse stress conditions. J. Biol. Chem. 283, 7064–7073 [DOI] [PubMed] [Google Scholar]
- 38. Ma Y., Brewer J. W., Diehl J. A., and Hendershot L. M. (2002) Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J. Mol. Biol. 318, 1351–1365 [DOI] [PubMed] [Google Scholar]
- 39. Teske B. F., Fusakio M. E., Zhou D., Shan J., McClintick J. N., Kilberg M. S., and Wek R. C. (2013) CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol. Biol. Cell 24, 2477–2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ashida H., Mimuro H., Ogawa M., Kobayashi T., Sanada T., Kim M., and Sasakawa C. (2011) Cell death and infection: a double-edged sword for host and pathogen survival. J. Cell Biol. 195, 931–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. West A. P., Shadel G. S., and Ghosh S. (2011) Mitochondria in innate immune responses. Nat. Rev. Immunol. 11, 389–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Suzuki M., Danilchanka O., and Mekalanos J. J. (2014) Vibrio cholerae T3SS effector VopE modulates mitochondrial dynamics and innate immune signaling by targeting Miro GTPases. Cell Host Microbe 16, 581–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Escoll P., Mondino S., Rolando M., Buchrieser C. (2016) Targeting of host organelles by pathogenic bacteria: a sophisticated subversion strategy. Nat. Rev. Microbiol. 14, 5–19 [DOI] [PubMed] [Google Scholar]
- 44. Soares M. P., Teixeira L., and Moita L. F. (2017) Disease tolerance and immunity in host protection against infection. Nat. Rev. Immunol. 17, 83–96 [DOI] [PubMed] [Google Scholar]
- 45. Pellegrino M. W., Nargund A. M., Kirienko N. V., Gillis R., Fiorese C. J., and Haynes C. M. (2014) Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature 516, 414–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jeong D. E., Lee D., Hwang S. Y., Lee Y., Lee J. E., Seo M., Hwang W., Seo K., Hwang A. B., Artan M., Son H. G., Jo J. H., Baek H., Oh Y. M., Ryu Y., et al. (2017) Mitochondrial chaperone HSP-60 regulates anti-bacterial immunity via p38 MAP kinase signaling. EMBO J. 36, 1046–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen Y. G., Yue H. T., Zhang Z. Z., Yuan F. H., Bi H. T., Yuan K., Weng S. P., He J. G., and Chen Y. H. (2016) Identification and characterization of a mitochondrial unfolded protein response transcription factor ATFS-1 in Litopenaeus vannamei. Fish Shellfish Immunol. 54, 144–152 [DOI] [PubMed] [Google Scholar]
- 48. Greaves L. C., Nooteboom M., Elson J. L., Tuppen H. A., Taylor G. A., Commane D. M., Arasaradnam R. P., Khrapko K., Taylor R. W., Kirkwood T. B., Mathers J. C., Turnbull D. M., et al. (2014) Clonal expansion of early to mid-life mitochondrial DNA point mutations drives mitochondrial dysfunction during human ageing. PLoS Genet. 10, e1004620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stewart J. B., and Chinnery P. F. (2015) The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat. Rev. Genet. 16, 530–542 [DOI] [PubMed] [Google Scholar]
- 50. Gitschlag B. L., Kirby C. S., Samuels D. C., Gangula R. D., Mallal S. A., and Patel M. R. (2016) Homeostatic responses regulate selfish mitochondrial genome dynamics in C. elegans. Cell Metab. 24, 91–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lin Y. F., Schulz A. M., Pellegrino M. W., Lu Y., Shaham S., and Haynes C. M. (2016) Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 533, 416–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tsang W. Y., and Lemire B. D. (2002) Stable heteroplasmy but differential inheritance of a large mitochondrial DNA deletion in nematodes. Biochem. Cell Biol. 80, 645–654 [DOI] [PubMed] [Google Scholar]
- 53. Zhang J., Nuebel E., Daley G. Q., Koehler C. M., and Teitell M. A. (2012) Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell 11, 589–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mohrin M., Shin J., Liu Y., Brown K., Luo H., Xi Y., Haynes C. M., and Chen D. (2015) Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 347, 1374–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hwang A. B., Ryu E. A., Artan M., Chang H. W., Kabir M. H, Nam H. J., Lee D., Yang J. S., Kim S., Mair W. B., Lee C., Lee S. S., and Lee S. J. (2014) Feedback regulation via AMPK and HIF-1 mediates ROS-dependent longevity in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 111, E4458–E4467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Khan M. H., Ligon M., Hussey L. R., Hufnal B., Farber R. 2nd, Munkácsy E., Rodriguez A., Dillow A., Kahlig E., and Rea S. L. (2013) TAF-4 is required for the life extension of isp-1, clk-1, and tpk-1 Mit mutants. Aging 5, 741–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Walter L., Baruah A., Chang H. W., Pace H. M., and Lee S. S. (2011) The homeobox protein CEH-23 mediates prolonged longevity in response to impaired mitochondrial electron transport chain in C. elegans. PLoS Biol. 9, e1001084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schaar C. E., Dues D. J., Spielbauer K. K., Machiela E., Cooper J. F., Senchuk M., Hekimi S., and Van Raamsdonk J. M. (2015) Mitochondrial and cytoplasmic ROS have opposing effects on lifespan. PLoS Genet. 11, e1004972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liberti M. V., and Locasale J. W. (2016) The Warburg effect: how does it benefit cancer cells? Trends Biochem. Sci. 41, 211–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zu X. L., and Guppy M. (2004) Cancer metabolism: facts, fantasy, and fiction. Biochem. Biophys. Res. Commun. 313, 459–465 [DOI] [PubMed] [Google Scholar]
- 61. Cappello F., Bellafiore M., Palma A., Marciano V., Martorana G., Belfiore P., Martorana A., Farina F., Zummo G., and Bucchieri F. (2002) Expression of 60-kD heat shock protein increases during carcinogenesis in the uterine exocervix. Pathobiology 70, 83–88 [DOI] [PubMed] [Google Scholar]
- 62. Castle P. E., Ashfaq R., Ansari F., and Muller C. Y. (2005) Immunohistochemical evaluation of heat shock proteins in normal and preinvasive lesions of the cervix. Cancer Lett. 229, 245–252 [DOI] [PubMed] [Google Scholar]
- 63. Ghosh J. C., Dohi T., Kang B. H., and Altieri D. C. (2008) Hsp60 regulation of tumor cell apoptosis. J. Biol. Chem. 283, 5188–5194 [DOI] [PubMed] [Google Scholar]
- 64. Brandon M., Baldi P., and Wallace D. C. (2006) Mitochondrial mutations in cancer. Oncogene 25, 4647–4662 [DOI] [PubMed] [Google Scholar]
- 65. Lee C. H., Wu S. B., Hong C. H., Liao W. T., Wu C. Y., Chen G. S., Wei Y. H., and Yu H. S. (2011) Aberrant cell proliferation by enhanced mitochondrial biogenesis via mtTFA in arsenical skin cancers. Am. J. Pathol. 178, 2066–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Monaco S. E., Angelastro J. M., Szabolcs M., and Greene L. A. (2007) The transcription factor ATF5 is widely expressed in carcinomas, and interference with its function selectively kills neoplastic, but not nontransformed, breast cell lines. Int. J. Cancer 120, 1883–1890 [DOI] [PubMed] [Google Scholar]