

Abstract
Phosphoinositide 3-kinases (PI3Ks) are ubiquitous lipid kinases that activate signaling cascades controlling cell survival, proliferation, protein synthesis, and vesicle trafficking. PI3Ks have dual kinase specificity: a lipid kinase activity that phosphorylates the 3′-hydroxyl of phosphoinositides and a protein-kinase activity that includes autophosphorylation. Despite the wealth of biochemical and structural information on PI3Kα, little is known about the identity and roles of individual active-site residues in catalysis. To close this gap, we explored the roles of residues of the catalytic domain and the regulatory subunit of human PI3Kα in lipid and protein phosphorylation. Using site-directed mutagenesis, kinetic assays, and quantitative mass spectrometry, we precisely mapped key residues involved in substrate recognition and catalysis by PI3Kα. Our results revealed that Lys-776, located in the P-loop of PI3Kα, is essential for the recognition of lipid and ATP substrates and also plays an important role in PI3Kα autophosphorylation. Replacement of the residues His-936 and His-917 in the activation and catalytic loops, respectively, with alanine dramatically changed PI3Kα kinetics. Although H936A inactivated the lipid kinase activity without affecting autophosphorylation, H917A abolished both the lipid and protein kinase activities of PI3Kα. On the basis of these kinetic and structural analyses, we propose possible mechanistic roles of these critical residues in PI3Kα catalysis.
Keywords: ATP, phosphatidylinositide 3-kinase (PI 3-kinase), phosphoinositide, protein kinase, serine/threonine protein kinase, lipid kinase, p110a, p110ap85a
Introduction
Phosphoinositide 3-kinases (PI3Ks), critical for initiating the AKT/mTOR signaling pathway, phosphorylate phosphoinositides at position 3 of the inositol ring to produce 3-phosphoinositides, which play key roles in regulating cell survival, proliferation, motility, protein synthesis, and vesicle trafficking (1). Dysregulation of the PI3Kpathway has been implicated in various diseases such as cancer, diabetes, arthritis, inflammation, and respiratory illnesses (2–5). PI3Ks are grouped into 3 classes (Class I-A&B, II, and III) based on their substrate specificity and mode of activation (6). PI3Kα, a key player in cancer pathogenesis, belongs to Class IA and is frequently mutated in breast, colon, brain, head and neck, gastric, and endometrial cancers (7–14). It is a heterodimer consisting of a catalytic subunit, p110α, and a regulatory subunit, p85α (15, 16). The p110α subunit is composed of five domains: an adaptor-binding domain (ABD)3 that binds to p85α, a Ras-binding domain, a C2 domain that binds to cell membranes, a helical domain of unknown function, and a catalytic/kinase domain consisting of an N-terminal lobe (N-lobe; residues 697–851) and a C-terminal lobe (C-lobe; residues 852–1068) homologous to those in protein kinases (Fig. 1A). The p85α subunit also contains five domains: an Src homology 3 (SH3) domain, a GTPase-activating protein (GAP-like or BH) domain, and two Src homology 2, N-terminal SH2 (nSH2) and C-terminal SH2 (cSH2) (SH2 and cSH2) domains separated by an inter-SH2 domain (iSH2) that binds to p110α (Fig. 1A).
Figure 1.

Structural basis of site-directed mutations in PI3Kα. A, domain organization of the p110α and p85α subunits of PI3Kα. RBD, Ras-binding domain; GAP, GTPase-activating protein. Mutated residues are indicated with asterisks. B, structure of the WT p110α/nip85α (Miller et al.; Ref. 31) in complex with the diC4-PIP2 lipid substrate (PDB ID 4OVV) and ATP modeled from the p110γ+ATP structure (PDB ID 1E8X). A second lipid-binding site (PIP2′) is shown. The kinase domain (with the activation, catalytic, and P-loops), the iSH2 domain and the ABD are colored in purple, red, and yellow, respectively. The amino acid residues mutated in this study (six in the p110α kinase domain and two in the p85α iSH2 domain) are illustrated as sticks. C, expanded view of the lipid substrate-binding site in the same orientation as B.
PIK3CA, the gene that encodes the catalytic subunit of PI3Kα, is somatically mutated in diverse tumor types. The majority of mutations in solid tumors (>80%) are in the conserved regions of the helical and kinase domains (7), most commonly E542K and E545R in the helical domain and H1047R in the kinase domain (17). These mutations result in PI3Kα enzymes with increased phosphoinositide kinase activity (18–21), leading to up-regulation of downstream signaling events that modulate the function of numerous substrates involved in the regulation of cell survival, cell cycle progression, and cellular growth (18).
In addition to its lipid kinase activity, PI3Kα displays a protein kinase activity that results in the autophosphorylation of the p85α subunit (22, 23). The effect of this autophosphorylation is controversial; although in some studies it is reported that autophosphorylation of Ser-608 of p85α inhibits the PI3Kα lipid kinase activity (23–25), at least one study reported that autophosphorylation did not affect the lipid kinase activity (26). Differences in the preparation of the protein may account for the conflicting results; the latter work used recombinant protein, whereas the former used immunoprecipitated enzyme. In contrast to the vast knowledge of the biological significance of PI3Kα lipid kinase activity in cell signaling, the physiological relevance of the protein kinase activity of PI3Kα is still not well characterized (27, 28).
Structural studies on PI3Ks have revealed many features of the kinase domain including the presence of the phosphate-binding loop/P-loop and of the catalytic and activation loops described in the well-characterized protein kinases (29, 30). In the previously published structure of PI3Kα with diC4-PIP2 (PDB ID 4OVV), we determined that the PIP2 substrate binds in a groove formed by the P-loop (p110α residues 772–778), the activation loop of the kinase domain (p110α residues 935–958), and the iSH2 coiled-coil (31) (Fig. 1B). Modeling of bound ATP shows that the 3′ hydroxyl group of diC4-PIP2 is oriented toward the γ-phosphate of the ATP (Fig. 1B). Furthermore, the short-chain lipid substrate is in a position accessible to the membrane, which would allow the C-18 and C-20 hydrophobic tails of the physiological PIP2 substrate to be buried into the membrane. Being negatively charged, its placement in the structure makes it well-suited to interact with the basic residues on the surface of the iSH2 domain and those of the loops of the C2 and kinase domains as originally proposed in a model of the lipid membrane interaction with the p110α/niSH2 heterodimer (30). Despite the favorable orientation of the PIP2 substrate, the structure of this complex represents an inactive conformation of PI3Kα, as the reactive 3-OH of the PIP2 is too far (∼6 Å) from the γ-phosphate of the ATP for a productive phosphoryl transfer. An additional characteristic suggesting that this is an inactive enzyme conformation is a salt bridge between Lys-948 of the activation loop and Glu-342 of the nSH2 domain, which locks the kinase domain and prevents the nSH2 domain from binding to effectors (31). However, unlike in most of the previous p110 structures, in the PI3Kα–PIP2 complex structure the activation loop is ordered; it is nested near the iSH2 and nSH2 domains of p85α and positioned in a way that allows communication between p85α and the kinase domain of p110α.
Previous biochemical studies on PI3Ks have reported that Lys-802 in p110α (Lys-833 in p110γ) is involved in phosphate transfer reaction (29, 32). It was also shown that the activation loop of p110α is a determinant of lipid substrate specificity and not protein kinase activity and that Lys-942 and Lys-949 within the activation loop of p110α (Lys-973 and Ar-980, respectively, in the activation loop of p110γ) are required for lipid substrate recognition (33). Furthermore, in the conserved DRHXXN motif of the catalytic loop, Asp-731 and Asn-736 of Vps34p (Class III PI3K) and Arg-916 of p110α are essential for the kinase activity of PI3Ks (23, 34).
Despite the wealth of information on the PI3Kα active site, the identity and role of many of the individual residues in lipid and protein phosphorylation remain largely unknown. To answer these fundamental questions, we used the structure of the PI3Kα–PIP2 complex as a template and selected residues that lie within 9 Å of the catalytic PIP2-binding site for analyzing the effect of substitutions on these residues. Six p110α mutants, H917A-p110α/p85α, H936A-p110α/p85α, K776E-p110α/p85α, R777E-p110α/p85α, E722A-p110α/p85α, and K724E-p110α/p85α, and two p85α mutants, p110α/R461E-p85α and p110α/R465E-p85α, were expressed and studied (Fig. 1, A and B). Kinetic characterization, quantitative mass spectrometry, and analysis of the structure of PI3Kα–PIP2 complex provided insights into the binding sites for lipid, ATP, and protein substrates and the catalytic mechanisms involved in lipid and protein phosphorylation by PI3Kα.
Results
The site-directed mutations investigated do not affect the oligomeric state of PI3Kα
All proteins (wild-type (WT) PI3Kα (p110α/p85α)) and mutants (H917A-p110α/p85α, H936A-p110α/p85α, K776E-p110α/p85α, R777E-p110α/p85α, E722A-p110α/p85α, K724E-p110α/p85α, p110α/R461E-p85α, p110α/R465E-p85α) were expressed in Sf9 insect cells and purified to homogeneity by nickel affinity chromatography followed by anion exchange chromatography. The resulting proteins were ∼85–95% pure (Fig. 2A), and the yield was ∼2–3 mg per 4 liters of Sf9 cell culture. To confirm that the mutations do not disrupt the quaternary structure and structural integrity of PI3Kα, we performed gel-filtration chromatography and compared the chromatograms of the WT enzyme with those of the mutants. The mutants eluted as a single, symmetrical peak with the same retention volume as the WT enzyme, indicating that the single-point mutations do not disrupt PI3Kα folding or oligomerization (Fig. 2B).
Figure 2.

Lys-776, His-917, and His-936 on p110α are critical regulators of PI3Kα lipid kinase activity. A, SDS-PAGE analysis of PI3Kα constructs purified by anion-exchange chromatography. WT and eight mutants are shown. B, gel-exclusion chromatograms of the wild-type enzyme (shown in blue) and one of the mutants-H917A-p110α/p85α (shown in orange) as an example to illustrate the preservation of the oligomeric state of the mutants. Molecular weight standards (Bio-Rad) are shown in green, and peaks A, B, C, D, and E represent 678 kDa, 158 kDa, 44 kDa, 17 kDa, and 1.35 kDa, respectively. mAU, milliabsorbance units. C and D, comparison of lipid kinase activity of WT and mutants. Michaelis-Menten plots are shown with ATP (C) and PIP2 (D). Activity (V) is expressed in pmol/min. Error bars represent S.D.
Lys-776, His-936, and His-917 are crucial for the lipid kinase activity of PI3Kα
The kinetics of the lipid kinase activity of WT PI3Kα and the mutants were studied using a fluorescence polarization assay. The Km of WT PI3Kα was 1.77 ± 0.03 μm for PIP2 and 2.0 ± 0.5 μm for ATP with a Vmax of 1.78 ± 0.06 pmol/min (Table 1). Of the eight PI3Kα mutants, three showed a dramatic change in their kinetic parameters when compared with those of the WT enzyme (Table 1, Fig. 2, C and D). K776E-p110α/p85α showed a very high Km for both ATP and PIP2 substrates (Table 1), suggesting a role of Lys-776 in the recognition of both substrates. H917A-p110α/p85α and H936A-p110α/p85α of the catalytic and activation loops, respectively, showed a ∼50–100× reduction in their maximal activity compared with WT PI3Kα (Table 1), indicating that these residues play a role in catalysis. In the structure of the PI3Kα complex with PIP2, these two histidine residues do not make a direct interaction with either of the substrates and are too far to act as catalytic bases, but conformational changes during the catalytic cycle may bring these residues closer to a catalytically competent position as has been shown for protein kinases (35, 36).
Table 1.
Comparative kinetic analysis of the lipid kinase activity of WT PI3Kα and the site-directed mutants
The Michaelis-Menten constant Km and maximum velocity Vmax for the production of PIP3 were estimated from non-linear regression by fitting the reaction velocities to Michaelis-Menten equation. Km and Vmax values are shown as the mean ± S.E. The data are representative of three independent experiments each performed in duplicate.
| Enzyme |
Km |
Vmax | |
|---|---|---|---|
| PIP2 | ATP | ||
| μm | pmol/min | ||
| WT (p110α/p85α) | 1.80 ± 0.03 | 2.00 ± 0.50 | 1.70 ± 0.06 |
| H917A-p110α/p85α | 0.40 ± 0.00 | 0.10 ± 0.04 | 0.02 ± 0.00 |
| H936A-p110α/p85α | 0.20 ± 0.00 | 0.40 ± 0.10 | 0.03 ± 0.00 |
| K776E-p110α/p85α | 55.2 ± 3.24 | 170.4 ± 34.2 | 1.10 ± 0.10 |
| E722A-p110α/p85α | 1.28 ± 0.31 | 4.00 ± 2.40 | 1.66 ± 0.06 |
| K724E-p110α/p85α | 4.24 ± 0.90 | 6.43 ± 0.97 | 1.40 ± 0.20 |
| R777E-p110α/p85α | 5.38 ± 0.27 | 4.32 ± 0.02 | 1.76 ± 0.16 |
| p110α/R461E-p85α | 1.12 ± 0.03 | 3.29 ± 0.18 | 1.64 ± 0.05 |
| p110α/R465E-p85α | 1.72 ± 0.14 | 2.26 ± 0.08 | 1.76 ± 0.10 |
The kinetic parameters of the other PI3Kα mutants (E722A-p110α/p85α, K724E-p110α/p85α, R777E-p110α/p85α, p110α/R461E-p85α, and p110α/R465E-p85α) did not change significantly from those of the WT PI3Kα. Glu-722 of p110α, located on the N-lobe, is close to a second PIP2-binding site identified in the PI3Kα–PIP2 complex structure (31). Although kinetic analysis shows that it is not involved in the recognition of the catalytic PIP2, this assay, performed with soluble PIP2, does not rule out that it may interact with the second lipid-binding site, as seen in the crystal structure. If, as suggested, this second site aids in anchoring PI3Kα to the plasma membrane, this mutation may have an effect on the in vivo activity. Lys-724 of p110α, also present on the N-lobe, is closer than Glu-722 to the PIP2 in the active site of the enzyme, and being positively charged, it could have a role in stabilizing the negative charge of PIP2; however, the kinetic analysis presented here suggests that it does not. Arg-777 in the P-loop of p110α is not involved in substrate recognition, in contrast to its neighboring residue, Lys-776. The lack of conservation of this arginine in the P-loop of protein kinases suggests that it does not play a role in the recognition of ATP. On the other hand, the lack of effect on the catalytic activity of the p85α mutants (p110α/R461E-p85α and p110α/R465E-p85α) argues against a role of these arginine residues in compensating the negative charge of the phosphates at positions 4 and 5 on the inositol ring of PIP2 but does not rule out the possibility of their participation in hydrogen bonding to PIP2.
Bisubstrate kinetic analysis of PI3Kα indicates an ordered sequential mechanism
The kinetic mechanism of the wild-type enzyme was elucidated with initial velocity studies analyzed using double-reciprocal plots. Lineweaver-Burk analysis of velocities with varying concentrations of ATP at different fixed concentrations of PIP2 displayed convergence of a set of lines in the second quadrant (Fig. 3A). A similar analysis of rate data with varying concentrations of PIP2 at different fixed concentrations of ATP yielded a set of lines intersecting on the 1/v axis (Fig. 3B). This pattern of intersecting lines on the vertical axis is indicative of an ordered sequential mechanism involving the formation of a ternary complex (37). The dissociation constants, Ka for ATP and Kb for PIP2, were determined from the global fit of the data by nonlinear regression analysis to the ordered bisubstrate kinetic model. The dissociation constants Ka and Kb for the WT PI3Kα were 10.45 ± 0.25 μm and 2.49 ± 0.21 μm, respectively. The values of Ka and Kb for the K776E-p110α/p85α mutant were 146.23 ± 2.62 μm and 11.0 ± 0.01 μm, respectively. The high dissociation constants for K776E-p110α/p85α mutant compared with the wild-type enzyme are in agreement with the low affinity of this mutant for both the substrates, as observed in the lipid kinase assays described above.
Figure 3.

Ternary complex formation is required for PI3Kα catalysis. A, Lineweaver-Burk plot of velocity versus ATP concentration while varying ATP concentrations (5–15 μm) at different fixed concentrations of PIP2 (5, 7.5, 10, and 15 μm). B, Lineweaver-Burk plot of velocity versus PIP2 concentration (5–15 μm) at different fixed concentrations of ATP (5, 7.5, 10, and 15 μm). The data were fitted to bisubstrate sequential models using SigmaPlot 13. Data shown are the mean of duplicate determinations from a single experiment and are representative of three such experiments.
Lys-776 and His-917 are critical for autophosphorylation
In addition to PIP2 phosphorylation, the catalytic subunit of PI3Kα, p110α, phosphorylates Ser-608 of its regulatory subunit, p85α. This phosphorylation has been proposed to have a regulatory effect on the activity of PI3K (23). To assess autophosphorylation, we used radioactive γ-32P-labeled ATP in the PI3Kα kinase reaction. We separated the proteins by SDS-PAGE and visualized the radioactive bands by autoradiography. The reaction carried out with WT PI3Kα displayed a strong signal for p85α, clearly indicating its autophosphorylation (Fig. 4A). On the other hand, the PI3Kα mutants H917A-p110α/p85α and K776E-p110α/p85α lack protein kinase activity (Fig. 4A). These results are consistent with our lipid kinase assays that showed H917A-p110α/p85α was inactive for lipid kinase activity and K776E-p110α/p85α showed a very low-binding affinity for ATP. The other mutants (H936A-p110α/p85α, R777E-p110α/p85α, E722A-p110α/p85α, and K724E-p110α/p85α) displayed a signal for p85α phosphorylation comparable with that of the WT, suggesting that these residues do not play a role in the substrate-binding and catalytic mechanism of protein kinase activity of PI3Kα. Interestingly, in the presence of lipid substrate PIP2, the autophosphorylation activity of the wild-type enzyme was almost negligible compared with the reaction in the absence of PIP2 (Fig. 4B). The high PIP2 concentration (50 μm) may compete with the protein substrate for the catalytic site of the enzyme, suggesting that the PIP2 substrate site and the protein substrate site overlap at least partially.
Figure 4.

Lys-776 and His-917 are required for autophosphorylation on p85α. A, SDS-PAGE analysis and autoradiography for WT and mutant PI3Kα proteins. Autophosphorylation of p85α using radiolabeled ATP as substrate in the absence of PIP2. The negative control is unphosphorylated p85α in the absence of p110α. The positive control is PTEN-phosphorylated by CK2 kinase. B, autophosphorylation of PI3Kα with and without PIP2. C, relative quantification of Ser-608 phosphorylation in p85α for WT PI3Kα and mutants by tandem mass spectrometry (MS/MS) of TMT-labeled peptides. D, tandem mass spectrum of TMT labeled peptides showing phosphorylation of Ser-608 (marked by arrows on y15) and reporter ions region (boxed in blue).
Cellular phosphorylation of WT PI3Kα and mutants
To assess the extent of phosphorylation of Ser-608 of the p85α subunit of PI3Kα in cells, we purified the enzymes from infected Sf9 insect cells and mapped the phosphorylated sites in the recombinant WT PI3Kα enzyme and mutants by phospho-amino acid analysis using tandem mass tag (TMT) mass spectrometry. For this, we differentially labeled WT PI3Kα and mutants with isobaric tandem mass tags that release specific reporter ions upon fragmentation. Reporter ion intensities obtained by MS/MS were used to quantify the phosphorylated peptides in WT PI3Kα and mutants. We compared the ratios of phosphorylated to unphosphorylated Ser-608 for the WT and mutants. Three mutants (H936A-p110α/p85α, K776E-p110α/p85α, and H917A-p110α/p85α) showed a remarkable decrease in phosphorylation of Ser-608 compared with the WT enzyme (Fig. 4C). The phosphorylation levels of Ser-608 in K776E-p110α/p85α and H917A-p110α/p85α in insect cells are in agreement with our in vitro lipid and protein kinase assays which show that these mutants have highly impaired enzymatic activity.
Furthermore, MS/MS analysis of WT PI3Kα showed that the p85α subunit is phosphorylated at Ser-43, Ser-154, Ser-208, and Thr-603 in addition to Ser-608. In the available PI3Kα structure, the positions of these residues are not within the reach of the catalytic site, suggesting that these phosphorylations most likely are due to the catalytic action of other endogenous protein kinases present during recombinant PI3Kα expression in insect cells.
Discussion
The structure of the complex of PI3Kα (harboring p110α and nip85α) with PIP2 most likely reflects a nonproductive binding mode and does not provide clear candidates for residues that participate in catalysis. To close this gap in knowledge, we cast a wide net to identify possible key residues and selected 8 residues within 9 Å of the PIP2-binding site for further studies. The rationale behind selecting residues at distances as large as 9 Å from the substrates was based on the assumption that closing of the active site upon ATP binding, as described in protein kinases, would bring the substrates, ATP and PIP2, closer to each other and would also position the catalytic residues to facilitate transfer of the phosphoryl group (38). Furthermore, the phosphate tail of the ATP is flexible and any small conformational change in the active site could bring the γ-phosphate of the ATP much closer to the PIP2.and form an active enzyme complex for catalysis. Using mutational and kinetic analysis, we identified three p110α residues that are essential for catalytic activity: Lys-776, His-917, and His-936.
We determined that Lys-776 in the P-loop of p110α is involved in the recognition of ATP as well as PIP2. This residue is also crucial for autophosphorylation of the p85α subunit. Despite the low sequence conservation of the P-loop, its three-dimensional structure is conserved in most mononucleotide-binding proteins, including protein kinase families. In most of the ATP- and GTP- binding proteins, a conserved lysine residue in the P-loop is implicated in binding to the γ-phosphate of NTPs (39, 40). Furthermore, in the crystal structure of PI3Kα in complex with PIP2 and with ATP modeled, the P-loop (residues 772–778) shows a conformation similar to that found in other P-loop-containing proteins, with Lys-776 oriented toward the β and γ phosphates of ATP (Fig. 5, A and B). The side chain of this lysine faces the 1-phospho group of PIP2 and may form a water-mediated hydrogen bond (31). This arrangement points toward the role of this lysine in phosphoinositide interaction (Fig. 5, A and B) even though it might not be able discriminate between different phosphoinositides. These observations are in agreement with the large effect of the K776E mutation on the Km of the enzyme for both the substrates. The charge reversal mutation K776E was selected to investigate whether this lysine is required to compensate the negative charge of ATP and/or PIP2, as hypothesized by Pirola et al. (33). The K776E mutation would amplify the effect of any charge-dependent interaction, making it easier to detect. Because both the substrates are highly negatively charged, a charge reversal in the active site might result in a loss of activity regardless of the function of this residue. However, our results from a similar charge reversal mutation on the neighboring residue, R777E, did not show any change in the enzyme activity and substrate affinity. This suggests that the results from the K776E mutation are indeed reflective of a role in substrate-binding rather than a nonspecific effect. Structurally, Lys-776 is not close enough to either of the substrates to interact with them; however, conformational changes in the N- and C-lobes and in the flexible P-loop region upon ATP-binding could bring this lysine closer to the two substrates.
Figure 5.
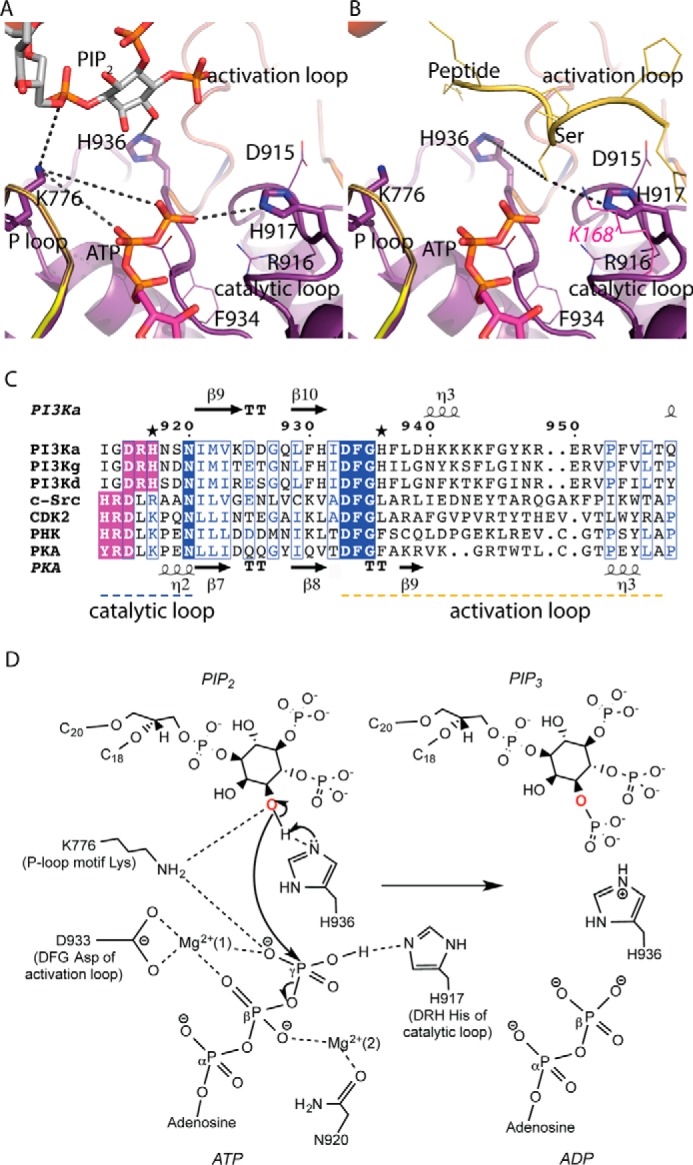
Structural insights into the catalytic mechanism of PI3Kα. Shown is the structure of the kinase domain of p110α harboring the P-loop and catalytic and activation loops with the lipid substrate (A) and the peptide substrate modeled from a cyclin-dependent protein kinase (CDK2) structure (B) (PDB ID 1QMZ). Lys-168 of PKA, shown in magenta, aligns with His-917 of p110α. C, sequence alignments of the catalytic loop (residues 912–920) and activation loop (residues 935–958) segments of class IA isoforms of PI3K (α, γ, δ) with different protein kinases (c-Src, tyrosine kinase; PHK, phosphorylase kinase; CDK2, cyclin-dependent kinase 2; PKA, protein kinase A). The conserved DFG motif of the activation loop and the XXDRH/HRDXK motif of the catalytic loop are highlighted in blue and purple, respectively. Mutated histidine residues His-917 and His-936 are marked with stars. D, molecular contacts between the substrates, conserved active-site residues, and the Mg2+ ions are shown. Asp-933 of the conserved DFG motif in the activation loop binds to the Mg2+ ion to orient the γ-phosphate of ATP for transfer. Asn-920 interacts with a second Mg2+ ion, which chelates the β-phosphate of ATP. Lys-776 of the conserved P-loop may support interactions with the γ-phosphate of ATP as well as 3′-OH of PIP2. His-936 of the activation loop is proposed to act as the catalytic base. His-917 of the conserved XXDRH motif in the catalytic loop, interacts with the γ-phosphate of ATP, and helps in the phosphoryl transfer step.
Our data show that His-936, located in the activation loop of PI3Kα, is critical for lipid kinase activity but not required for protein phosphorylation. In the p110α sequence, His-936 is present immediately after the conserved DFG motif (Fig. 5C). All protein kinases known contain a conserved DFG sequence (Mg2+-orienting loop) in the activation segment of their catalytic domains, and this motif has a critical function in catalysis (41). His-936 is conserved in all class I PI3Ks but not in protein kinases (Fig. 5C), suggesting that this histidine is required for lipid phosphorylation and not for the protein kinase activity. Moreover, His-936 is in the vicinity of the 3′ hydroxyl on the inositol ring of PIP2, but is relatively far from the serine hydroxyl group on the peptide substrate modeled in our p110α/nip85α structure (Fig. 5B). On the basis of this structural observation, we propose that His-936 may act as a catalytic base to deprotonate the 3′ hydroxyl of the PIP2 substrate and promote the nucleophilic attack on the γ-phosphate of ATP. Although this histidine is not close enough to the 3′-OH of PIP2 to deprotonate it, it is possible, as observed in protein kinases, that a conformational change involving the N- and C-lobes could bring His-936, present on the flexible activation loop, closer to PIP2. In addition, physiological activation of PI3Kα, triggered by the binding of phosphorylated proteins to the nSH2 domain of the regulatory subunit, could release the network of hydrogen bonds between the activation loop and the nSH2 domain (31, 42) and allow the activation loop bearing the catalytic His-936 to adopt an active conformation in closer proximity to the lipid substrate.
His-917, located on the catalytic loop, is required for the lipid as well as protein kinase activities of PI3Kα as shown by the lack of activity of the H917A-p110α/p85α mutant in this study. His-917 is part of the conserved XXDRH motif in the catalytic loop of PI3Ks. In contrast, the catalytic loop of protein kinases contains a conserved HRDXK motif at the corresponding position in the sequence (Fig. 5C). Sequence as well as structural alignment of PI3Kα with a prototypical protein kinase PKA (cAMP-dependent protein kinase) shows that the lysine (Lys-168) in the HRDXK motif of PKA is at the position of the PI3K XXDRH histidine (Fig. 5, B and C). The lysine of the HRDXK motif is conserved in all Ser/Thr protein kinases and is replaced by an arginine in tyrosine protein kinases (43, 44). In light of these facts, we propose that His-917 of PI3Kα, due to its proximity to ATP, may interact electrostatically with the γ-phosphate of ATP and mediate phosphoryl transfer as reported for the corresponding Lys-168 of PKA (45, 46).
The HRDXK histidine of protein kinases has been described as one of the residues of the R-spine (R= regulatory). R-spine is a highly conserved spatial motif consisting of four non-consecutive residues that connects the two lobes of kinases and is found only in the active protein kinases (36). Disruption of R-spine affects the enzyme activity of protein kinases, suggesting a regulatory function (47). However, structural alignment of PI3Kα with protein kinases does not show a similar spatial arrangement of residues characteristic of the R-spine of protein kinases. This suggests that for PI3Ks, the residues of the R-spine should be redefined. Furthermore, recent studies in Aurora kinases reveal the involvement of the HRDXK histidine in preserving the active conformation of the DFG motif through a hydrophobic interaction between this histidine and the DFG phenylalanine (48). This arrangement defines another important role for the HRDXK histidine in protein kinases. However, in the PI3Kα structure, the arginine of the conserved XXDRH motif seems to play a role in the interaction with the DFG motif (Fig. 5A).
The lipid kinase mechanism of PI3Kα that emerges from the kinetic and structural analyses involves a nucleophilic attack by the 3′-OH of the inositol on the γ-phosphate of ATP (Fig. 5D). His-936, being closer to the inositol ring of the lipid substrate, is the catalytic base required for the deprotonation of the 3′ hydroxyl. Two Mg2+ ions and Lys-776 of the P-loop compensate the charge of the bound ATP (Fig. 5D). Furthermore, the proximity of Lys-776 to the 3′-OH of the PIP2 substrate aids the reaction by lowering the hydroxyl pKa. His-917 of the catalytic loop is in a position to make an electrostatic interaction with the γ-phosphate of ATP and help in phosphotransfer (Fig. 5D). The proposed roles of Lys-776, His-936, and His-917 in this mechanism provide a rationale for the observed effects of their mutations in lipid and protein kinase activities of PI3Kα. Besides elucidating substrate specificity and catalytic mechanism, our studies highlight the kinetic mechanism of PI3Kα. We performed classic bisubstrate steady-state kinetic analysis of PI3Kα that suggests a ternary complex mechanism involving direct phosphoryl transfer from ATP to PIP2.
Taken together, our studies report the first detailed kinetic analysis of PI3Kα active-site residues that resulted in the identification of three residues critical for PI3Kα catalysis and allowed us to propose a catalytic mechanism. In addition, we showed similarities and differences between the lipid and protein kinase specificities of PI3Kα. Our data suggest that the lipid and protein kinase activities are mediated in part by different structural motifs in the kinase domain of PI3Kα. Moreover, we analyzed for the first time the bisubstrate kinetic mechanism of the PI3Kα lipid kinase activity. These findings constitute a significant advance in the characterization of dual kinase specificity of PI3Kα, a key enzyme in the AKT/mTOR signaling pathway that regulates diverse cellular processes like survival, proliferation, and motility as well as protein synthesis and vesicle trafficking. The identification of PI3Kα residues involved in substrate recognition and catalysis will have a significant impact in many fields and disciplines ranging from the regulation of glucose homeostasis to the design of cancer chemotherapeutic agents.
Experimental procedures
Cloning and expression of PI3Kα
WT human PI3Kα subunits p110α and p85α were cloned into pFastBac HT-B (encoding an N-terminal His tag) and pFastBac1 vectors (Invitrogen), respectively. These recombinant constructs of p110α and p85α were transformed into competent DH10Bac Escherichia coli cells for transposition into bacmid DNA. The resulting p110α and p85α recombinant bacmids were used to transfect two separate stocks of Sf9 insect cells, and recombinant baculoviruses for each were isolated. These baculoviral stocks were amplified twice to generate high-titer stocks (107-108 infectious units per ml (IFU/ml)), which we used to infect insect cells for large-scale expression of PI3Kα. The baculoviral titers were determined by BacPAK Baculovirus Rapid Titer kit (Clontech). p110α mutants H917A-p110α/p85α, H936A-p110α/p85α, K776E-p110α/p85α, R777E-p110α/p85α, E722A-p110α/p85α, K724E-p110α/p85α and p85α mutants p110α/R461E-p85α and p110α/R465E-p85α were produced by site-directed mutagenesis. WT and mutant proteins were expressed in the presence of a PI3K inhibitor J32 (42). Sf9 insect cells were grown as suspension culture in Sf 900 III serum-free media (Invitrogen) supplemented with 1% penicillin-streptomycin at 27 °C. PI3Kα was expressed by co-infecting Sf9 cells at a density of 4 × 106 cells/ml with p110α and p85α baculoviruses at a multiplicity of infection ratio of 3:2. Cells were harvested 48 h after infection.
Purification of WT PI3Kα and mutants
Cell pellets harvested from a 4-liter culture of Sf9 cells co-infected with p110α and p85α recombinant baculoviruses were resuspended in 250 ml of resuspension buffer (50 mm sodium phosphate, pH 8.0, 400 mm NaCl, 10 mm imidazole, 1% Triton X-100, 5% glycerol, 1 mm sodium orthovanadate, 5 tablets of Complete Protease Inhibitor Mixture (Roche Applied Science)) and lysed by sonication. The soluble supernatant was loaded onto nickel-nitrilotriacetic acid beads (Qiagen). The complex of p110α and p85α was eluted with 250 mm imidazole. The nickel-nitrilotriacetic acid elutes containing both the subunits were pooled and desalted using HiPrep 26/10 desalting column (GE Healthcare). The protein was further purified by anion-exchange chromatography using a Mono Q column (GE Healthcare). PI3Kα was eluted using a linear gradient of 0–500 mm NaCl. The anion-exchange fractions containing pure PI3Kα were pooled, and the protein concentration was determined by the Bradford assay (Bio-Rad). All PI3Kα mutants used in the study (six p110α mutants and two p85α mutants) were purified by the same protocol. A Superose 6 (10/300 GL) gel-filtration column was used to determine and compare the oligomeric state of WT PI3Kα and mutants. The native molecular weight of WT PI3Kα was determined from a standard curve for this column.
Fluorescence polarization activity assay for PI3Kα
To characterize the lipid kinase activity of WT PI3Kα and its mutants, we used a fluorescence polarization assay (Echelon Biosciences). 10 μl of PI3-kinase reactions were carried out in low-binding flat-bottom 384-well plates in 1X KBZ buffer (Echelon Biosciences), 5 mm DTT, 200 μm ATP, 40 μm diC8-PIP2 substrate, and 50 ng of PI3Kα. The Vmax and Km for ATP were determined by varying the concentration of ATP from 0.78 to 200 μm at a constant 40 μm concentration of PIP2. Kinetics with PIP2 substrate was determined by varying the concentration of PIP2 from 0.78–50 μm at a constant 200 μm concentration of ATP. The reaction mixtures were incubated at 30 °C for 45 min and quenched by the addition of 10 μl of phosphatidylinositol 3,4,5-trisphosphate detector protein. Detection of PIP3 was done according to the manufacturer's recommendation. The polarization signal was measured with a Tecan Infinite M-1000 plate reader. Data were analyzed by the GraphPad Prism Software.
Bisubstrate kinetics
Initial velocity studies of PI3Kα were performed using the fluorescence polarization assay described above to determine both the bisubstrate kinetic mechanism of WT PI3Kα and the dissociation constants of the reaction. Double-reciprocal plots were plotted for velocities versus substrate concentrations by varying ATP concentration from 5 to 15 μm at different fixed non-saturating concentrations of PIP2 (5, 7.5, 10, and 15 μm). The data were fitted to bisubstrate sequential equations using SigmaPlot 13, and the dissociation constants Ka for ATP and Kb for PIP2 were calculated by nonlinear regression analysis. Equations for ordered (Equation 1) and random bi-bi mechanisms (Equation 2) are listed below, respectively (49).
| (Eq. 1) |
| (Eq. 2) |
Radioactive PI3Kα protein kinase assay
We used a radioactive-based protein kinase assay to analyze the autophosphorylation of the p85α subunit of WT PI3Kα and mutants. The reaction mixture contained 20 mm HEPES, pH 7.5, 10 mm MgCl2, 0.25 mm DTT, 1 μg of PI3Kα, and 1 μl of γ-32P-labeled ATP (3000 Ci/mmol, 10 mCi/ml, PerkinElmer Life Sciences) at a final concentration of 10 μm ATP in a total of 50 μl. To assess the autophosphorylation in the presence and absence of lipid substrate, 50 μm PIP2 was added to the reaction mixture. The incubation was done at 30 °C for 30 min. Samples were separated by 8–16% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and phosphorylated products were visualized by autoradiography.
Phosphorylation analysis and quantification by mass spectrometry
WT PI3Kα and six p110α mutants were identified for phosphorylation sites by TMT mass spectrometry. For this, samples were prepared by denaturation, reduction, alkylation, and tryptic digestion. Next, samples were differentially labeled with isobaric mass tags (TMT 10-plex, ThermoScientific) according to the manufacturer's specifications. All the TMT-labeled peptides were mixed and fractionated by basic reverse phase chromatography. Each fraction was analyzed by tandem mass spectrometry (MS/MS), and data analysis was performed. Peptide and fragment ion masses were extracted from the raw mass spectra in Proteome Discoverer (PD) software (v1.4, ThermoScientific) and searched using Mascot (v2.5.1) against a custom database containing the wild-type proteins and their respective mutants. The reporter ions from all peptides were quantile-normalized to minimize any technical variation in sample preparation and TMT labeling. For quantification, the intensities of seven reporter ions obtained by MS/MS were used to compare the relative amounts of phosphorylated to unphosphorylated peptides in all seven samples, and the results were reported as ratios of phosphorylated to unphosphorylated peptides for WT PI3Kα and its mutants.
Author contributions
S. M., M. S. M., and S. B. G. conceived and designed the experiments. S. M., M. S. M., R. O., and R. N. C. performed the research, S. M., M. S. M., L. M. A., and S. B. G. analyzed the data. S. M., L. M. A., and S. B. G. wrote the paper.
Acknowledgments
We thank the Cole laboratory for the gift of CK2 kinase, Dr. Valerie O'Shea for help with the radioactive experiments, and Dr. B. Vogelstein and Dr. K. Kinzler for helpful discussions. Part of this research was undertaken in the Eukaryotic Tissue Culture Facility, which is supported by The Johns Hopkins University. We thank Dr. Li, manager of the facility, for help and suggestions on insect cell protein expression. We acknowledge the use and service of the Johns Hopkins University School of Medicine Mass Spectrometry and Proteomics Core supported by the Sidney Kimmel Comprehensive Cancer Center (NCI Grant 2P30 CA006973).
This work was supported by National Institutes of Health Grants CA062924 and CA043460, Department of Defense Congressionally Directed Medical Research Program (CDMRP) BC151831, and the Alexander and Margaret Stewart Trust. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ABD
- adaptor binding domain
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- PIP3
- phosphatidylinositol 3,4,5-trisphosphate
- TMT
- tandem mass tag
- SH
- Src homology
- nSH2
- N-terminal SH2
- cSH2
- C-terminal SH2
- iSH2
- inter-SH2 domain.
References
- 1. Fruman D. A., Meyers R. E., and Cantley L. C. (1998) Phosphoinositide kinases. Annu. Rev. Biochem. 67, 481–507 [DOI] [PubMed] [Google Scholar]
- 2. Chang H. W., Aoki M., Fruman D., Auger K. R., Bellacosa A., Tsichlis P. N., Cantley L. C., Roberts T. M., and Vogt P. K. (1997) Transformation of chicken cells by the gene encoding the catalytic subunit of PI 3-kinase. Science 276, 1848–1850 [DOI] [PubMed] [Google Scholar]
- 3. Cho H., Mu J., Kim J. K., Thorvaldsen J. L., Chu Q., Crenshaw E. B. 3rd, Kaestner K. H., Bartolomei M. S., Shulman G. I., and Birnbaum M. J. (2001) Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKBβ). Science 292, 1728–1731 [DOI] [PubMed] [Google Scholar]
- 4. Maira S. M., Finan P., and Garcia-Echeverria C. (2010) From the bench to the bed side: PI3K pathway inhibitors in clinical development. Curr. Top. Microbiol. Immunol. 347, 209–239 [DOI] [PubMed] [Google Scholar]
- 5. Koorella C., Nair J. R., Murray M. E., Carlson L. M., Watkins S. K., and Lee K. P. (2014) Novel regulation of CD80/CD86-induced phosphatidylinositol 3-kinase signaling by NOTCH1 protein in interleukin-6 and indoleamine 2,3-dioxygenase production by dendritic cells. J. Biol. Chem. 289, 7747–7762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Domin J., and Waterfield M. D. (1997) Using structure to define the function of phosphoinositide 3-kinase family members. FEBS Lett. 410, 91–95 [DOI] [PubMed] [Google Scholar]
- 7. Samuels Y., Wang Z., Bardelli A., Silliman N., Ptak J., Szabo S., Yan H., Gazdar A., Powell S. M., Riggins G. J., Willson J. K., Markowitz S., Kinzler K. W., Vogelstein B., and Velculescu V. E. (2004) High frequency of mutations of the PIK3CA gene in human cancers. Science 304, 554. [DOI] [PubMed] [Google Scholar]
- 8. Broderick D. K., Di C., Parrett T. J., Samuels Y. R., Cummins J. M., McLendon R. E., Fults D. W., Velculescu V. E., Bigner D. D., and Yan H. (2004) Mutations of PIK3CA in anaplastic oligodendrogliomas, high-grade astrocytomas, and medulloblastomas. Cancer Res. 64, 5048–5050 [DOI] [PubMed] [Google Scholar]
- 9. Campbell I. G., Russell S. E., Choong D. Y., Montgomery K. G., Ciavarella M. L., Hooi C. S., Cristiano B. E., Pearson R. B., and Phillips W. A. (2004) Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 64, 7678–7681 [DOI] [PubMed] [Google Scholar]
- 10. Levine D. A., Bogomolniy F., Yee C. J., Lash A., Barakat R. R., Borgen P. I., and Boyd J. (2005) Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin Cancer Res. 11, 2875–2878 [DOI] [PubMed] [Google Scholar]
- 11. Bachman K. E., Argani P., Samuels Y., Silliman N., Ptak J., Szabo S., Konishi H., Karakas B., Blair B. G., Lin C., Peters B. A., Velculescu V. E., and Park B. H. (2004) The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol. Ther. 3, 772–775 [DOI] [PubMed] [Google Scholar]
- 12. Pedrero J. M., Carracedo D. G., Pinto C. M., Zapatero A. H., Rodrigo J. P., Nieto C. S., and Gonzalez M. V. (2005) Frequent genetic and biochemical alterations of the PI 3-K/AKT/PTEN pathway in head and neck squamous cell carcinoma. Int. J. Cancer 114, 242–248 [DOI] [PubMed] [Google Scholar]
- 13. Miyaki M., Iijima T., Yamaguchi T., Takahashi K., Matsumoto H., Yasutome M., Funata N., and Mori T. (2007) Mutations of the PIK3CA gene in hereditary colorectal cancers. Int. J. Cancer 121, 1627–1630 [DOI] [PubMed] [Google Scholar]
- 14. Velho S., Oliveira C., Ferreira A., Ferreira A. C., Suriano G., Schwartz S. Jr., Duval A., Carneiro F., Machado J. C., Hamelin R., and Seruca R. (2005) The prevalence of PIK3CA mutations in gastric and colon cancer. Eur. J. Cancer 41, 1649–1654 [DOI] [PubMed] [Google Scholar]
- 15. Escobedo J. A., Navankasattusas S., Kavanaugh W. M., Milfay D., Fried V. A., and Williams L. T. (1991) cDNA cloning of a novel 85-kDa protein that has SH2 domains and regulates binding of PI3-kinase to the PDGF β-receptor. Cell 65, 75–82 [DOI] [PubMed] [Google Scholar]
- 16. Hiles I. D., Otsu M., Volinia S., Fry M. J., Gout I., Dhand R., Panayotou G., Ruiz-Larrea F., Thompson A., and Totty N. F. (1992) Phosphatidylinositol 3-kinase: structure and expression of the 110-kDa catalytic subunit. Cell 70, 419–429 [DOI] [PubMed] [Google Scholar]
- 17. Samuels Y., and Velculescu V. E. (2004) Oncogenic mutations of PIK3CA in human cancers. Cell Cycle 3, 1221–1224 [DOI] [PubMed] [Google Scholar]
- 18. Kang S., Bader A. G., and Vogt P. K. (2005) Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc. Natl. Acad. Sci. U.S.A. 102, 802–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ikenoue T., Kanai F., Hikiba Y., Obata T., Tanaka Y., Imamura J., Ohta M., Jazag A., Guleng B., Tateishi K., Asaoka Y., Matsumura M., Kawabe T., and Omata M. (2005) Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res. 65, 4562–4567 [DOI] [PubMed] [Google Scholar]
- 20. Zhao L., and Vogt P. K. (2008) Helical domain and kinase domain mutations in p110α of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc. Natl. Acad. Sci. U.S.A. 105, 2652–2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Carson J. D., Van Aller G., Lehr R., Sinnamon R. H., Kirkpatrick R. B., Auger K. R., Dhanak D., Copeland R. A., Gontarek R. R., Tummino P. J., and Luo L. (2008) Effects of oncogenic p110α subunit mutations on the lipid kinase activity of phosphoinositide 3-kinase. Biochem. J. 409, 519–524 [DOI] [PubMed] [Google Scholar]
- 22. Carpenter C. L., Auger K. R., Duckworth B. C., Hou W. M., Schaffhausen B., and Cantley L. C. (1993) A tightly associated serine/threonine protein kinase regulates phosphoinositide 3-kinase activity. Mol. Cell. Biol. 13, 1657–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dhand R., Hiles I., Panayotou G., Roche S., Fry M. J., Gout I., Totty N. F., Truong O., Vicendo P., and Yonezawa K. (1994) PI 3-kinase is a dual specificity enzyme: autoregulation by an intrinsic protein-serine kinase activity. EMBO J. 13, 522–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Buchanan C. M., Dickson J. M., Lee W. J., Guthridge M. A., Kendall J. D., and Shepherd P. R. (2013) Oncogenic mutations of p110α isoform of PI 3-kinase upregulate its protein kinase activity. PLoS ONE 8, e71337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Foukas L. C., Beeton C. A., Jensen J., Phillips W. A., and Shepherd P. R. (2004) Regulation of phosphoinositide 3-kinase by its intrinsic serine kinase activity in vivo. Mol. Cell. Biol. 24, 966–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Layton M. J., Saad M., Church N. L., Pearson R. B., Mitchell C. A., and Phillips W. A. (2012) Autophosphorylation of serine 608 in the p85 regulatory subunit of wild type or cancer-associated mutants of phosphoinositide 3-kinase does not affect its lipid kinase activity. BMC Biochem. 13, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thomas D., Powell J. A., Green B. D., Barry E. F., Ma Y., Woodcock J., Fitter S., Zannettino A. C., Pitson S. M., Hughes T. P., Lopez A. F., Shepherd P. R., Wei A. H., Ekert P. G., and Guthridge M. A. (2013) Protein kinase activity of phosphoinositide 3-kinase regulates cytokine-dependent cell survival. PLos Biol. 11, e1001515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hunter T. (1995) When is a lipid kinase not a lipid kinase? When it is a protein kinase? Cell 83, 1–4 [DOI] [PubMed] [Google Scholar]
- 29. Walker E. H., Perisic O., Ried C., Stephens L., and Williams R. L. (1999) Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature 402, 313–320 [DOI] [PubMed] [Google Scholar]
- 30. Huang C. H., Mandelker D., Schmidt-Kittler O., Samuels Y., Velculescu V. E., Kinzler K. W., Vogelstein B., Gabelli S. B., and Amzel L. M. (2007) The structure of a human p110α/p85α complex elucidates the effects of oncogenic PI3Kα mutations. Science 318, 1744–1748 [DOI] [PubMed] [Google Scholar]
- 31. Miller M. S., Schmidt-Kittler O., Bolduc D. M., Brower E. T., Chaves-Moreira D., Allaire M., Kinzler K. W., Jennings I. G., Thompson P. E., Cole P. A., Amzel L. M., Vogelstein B., and Gabelli S. B. (2014) Structural basis of nSH2 regulation and lipid binding in PI3Kα. Oncotarget 5, 5198–5208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wymann M. P., Bulgarelli-Leva G., Zvelebil M. J., Pirola L., Vanhaesebroeck B., Waterfield M. D., and Panayotou G. (1996) Wortmannin inactivates phosphoinositide 3-kinase by covalent modification of Lys-802, a residue involved in the phosphate transfer reaction. Mol. Cell. Biol. 16, 1722–1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pirola L., Zvelebil M. J., Bulgarelli-Leva G., Van Obberghen E., Waterfield M. D., and Wymann M. P. (2001) Activation loop sequences confer substrate specificity to phosphoinositide 3-kinase α (PI3Kα): functions of lipid kinase-deficient PI3Kα in signaling. J. Biol. Chem. 276, 21544–21554 [DOI] [PubMed] [Google Scholar]
- 34. Stack J. H., and Emr S. D. (1994) Vps34p required for yeast vacuolar protein sorting is a multiple specificity kinase that exhibits both protein kinase and phosphatidylinositol-specific PI 3-kinase activities. J. Biol. Chem. 269, 31552–31562 [PubMed] [Google Scholar]
- 35. Taylor S. S., Radzio-Andzelm E., Madhusudan, Cheng X., Ten Eyck L., and Narayana N. (1999) Catalytic subunit of cyclic AMP-dependent protein kinase: structure and dynamics of the active site cleft. Pharmacol. Ther. 82, 133–141 [DOI] [PubMed] [Google Scholar]
- 36. Taylor S. S., and Kornev A. P. (2011) Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem. Sci. 36, 65–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Segel I. H. (1975) Enzyme Kinetics, Behavior, and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems, pp. 320–329, John Wiley & Sons, Inc., New York [Google Scholar]
- 38. Huse M., and Kuriyan J. (2002) The conformational plasticity of protein kinases. Cell 109, 275–282 [DOI] [PubMed] [Google Scholar]
- 39. Saraste M., Sibbald P. R., and Wittinghofer A. (1990) The P-loop; a common motif in ATP- and GTP-binding proteins. Trends Biochem. Sci. 15, 430–434 [DOI] [PubMed] [Google Scholar]
- 40. Deyrup A. T., Krishnan S., Cockburn B. N., and Schwartz N. B. (1998) Deletion and site-directed mutagenesis of the ATP-binding motif (P-loop) in the bifunctional murine ATP-sulfurylase/adenosine 5′-phosphosulfate kinase enzyme. J. Biol. Chem. 273, 9450–9456 [DOI] [PubMed] [Google Scholar]
- 41. Schwartz P. A., and Murray B. W. (2011) Protein kinase biochemistry and drug discovery. Bioorg. Chem. 39, 192–210 [DOI] [PubMed] [Google Scholar]
- 42. Mandelker D., Gabelli S. B., Schmidt-Kittler O., Zhu J., Cheong I., Huang C. H., Kinzler K. W., Vogelstein B., and Amzel L. M. (2009) A frequent kinase domain mutation that changes the interaction between PI3Kα and the membrane. Proc. Natl. Acad. Sci. U.S.A. 106, 16996–17001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hanks S. K., and Quinn A. M. (1991) Protein kinase catalytic domain sequence database: identification of conserved features of primary structure and classification of family members. Methods Enzymol. 200, 38–62 [DOI] [PubMed] [Google Scholar]
- 44. Knighton D. R., Cadena D. L., Zheng J., Ten Eyck L. F., Taylor S. S., Sowadski J. M., and Gill G. N. (1993) Structural features that specify tyrosine kinase activity deduced from homology modeling of the epidermal growth factor receptor. Proc. Natl. Acad. Sci. U.S.A. 90, 5001–5005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zheng J., Knighton D. R., ten Eyck L. F., Karlsson R., Xuong N., Taylor S. S., and Sowadski J. M. (1993) Crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MgATP and peptide inhibitor. Biochemistry 32, 2154–2161 [DOI] [PubMed] [Google Scholar]
- 46. Madhusudan, Trafny E. A., Xuong N. H., Adams J. A., Ten Eyck L. F., Taylor S. S., and Sowadski J. M. (1994) cAMP-dependent protein kinase: crystallographic insights into substrate recognition and phosphotransfer. Protein Sci. 3, 176–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Strong T. C., Kaur G., and Thomas J. H. (2011) Mutations in the catalytic loop HRD motif alter the activity and function of Drosophila Src64. PLoS ONE 6, e28100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang L., Wang J. C., Hou L., Cao P. R., Wu L., Zhang Q. S., Yang H. Y., Zang Y., Ding J. P., and Li J. (2015) Functional role of histidine in the conserved His-X-Asp motif in the catalytic core of protein kinases. Sci. Rep. 5, 10115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. SigmaPlot (2015) SigmaPlot version 13. Systat Software, Inc., San Jose, CA [Google Scholar]