

Abstract
Malaria infections cause several systemic and severe single- or multi-organ pathologies, killing hundreds of thousands of people annually. Considering the existing widespread resistance of malaria parasites to anti-parasitic drugs and their high propensity to develop drug resistance, alternative strategies are required to manage malaria infections. Because malaria is a host immune response–driven disease, one approach is based on gaining a detailed understanding of the molecular and cellular processes that modulate malaria-induced innate and adaptive immune responses. Here, using a mouse cerebral malaria model and small-molecule inhibitors, we demonstrate that inhibiting MEK1/2, the upstream kinases of ERK1/2 signaling, alters multifactorial components of the innate and adaptive immune responses, controls parasitemia, and blocks pathogenesis. Specifically, MEK1/2 inhibitor treatment up-regulated B1 cell expansion, IgM production, phagocytic receptor expression, and phagocytic activity, enhancing parasite clearance by macrophages and neutrophils. Further, the MEK1/2 inhibitor treatment down-regulated pathogenic pro-inflammatory and helper T cell 1 (Th1) responses and up-regulated beneficial anti-inflammatory cytokine responses and Th2 responses. These inhibitor effects resulted in reduced granzyme B expression by T cells, chemokine and intracellular cell adhesion molecule 1 (ICAM-1) expression in the brain, and chemokine receptor expression by both myeloid and T cells. These bimodal effects of the MEK1/2 inhibitor treatment on immune responses contributed to decreased parasite biomass, organ inflammation, and immune cell recruitment, preventing tissue damage and death. In summary, we have identified several previously unrecognized immune regulatory processes through which a MEK1/2 inhibitor approach controls malaria parasitemia and mitigates pathogenic effects on host organs.
Keywords: inflammation, malaria, mitogen-activated protein kinase (MAPK), pathogenesis, phagocytosis
Introduction
Approximately 50% of the global population is at risk of contracting malaria, and ∼0.5 million people die annually (1, 2). Several Plasmodium parasite species cause malaria, but Plasmodium falciparum and, to a much lesser extent, Plasmodium vivax cause severe and fatal malaria (1, 2). Mass vaccination is the best strategy to prevent malaria. However, producing an effective vaccine remains challenging (3, 4). The use of anti-parasitic drugs, such as quinine derivatives, is becoming increasingly problematic because parasites have developed widespread resistance (5), and substantial resistance has also emerged to artemisinins, which are currently used (6). Malaria parasites have a high propensity to develop drug resistance; therefore, alternative strategies are required to treat malaria. Considering that severe malaria complications are immune-mediated, modulators of immune responses are attractive alternatives to prevent severe malaria pathogenesis and concurrently to control infection. Immunomodulators may also help to increase the efficacy of a vaccine. This approach may circumvent the problem of parasites developing drug resistance, but more information about the regulation of immune-mediated pathology is required before such approaches can be pursued.
The initial clinical manifestations of malaria, such as periodic fever, chills, headache, and malaise, are the outcome of elevated levels of pro-inflammatory mediators produced in response to the blood stage parasites (7, 8). In the case of P. falciparum, the most virulent among human malaria parasites, the infected red blood cells (IRBCs)3 are sequestered in microvascular capillaries of organs, including the brain, lungs, intestine, and liver, via binding to host endothelial cell receptors (9–11), establishing an inflammatory environment locally. As a consequence, immune cells infiltrate into organs to which parasites are sequestered, intensifying the pro-inflammatory environment and causing endothelial cell activation and up-regulation of adherence receptors, such as intercellular cell adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (12). Furthermore, infection-induced pro-inflammatory cytokines promote exaggerated Th1 responses, leading to enhanced cytotoxicity of lymphocytes. These processes are amplified in a cooperative feed-forward manner, contributing to chemotaxis-dependent infiltration of cytotoxic lymphocytes to the brain and other vital organs. Thus, when IRBCs adhere in organs, the eventual consequence is endothelial barrier damage and severe pathologies characterized by cerebral malaria (CM), pulmonary edema, intestinal bleeding, liver dysfunction, renal failure, splenic rupture, and intravascular hemolysis and fatality (8, 13–16). Because balanced production of both pro-/anti-inflammatory cytokines and Th1/Th2 responses results in positive clinical outcomes, modulation of inflammatory responses by blocking specific targets of signaling pathways involved in immune responses is likely to be effective in preventing malaria pathogenesis.
Pro-inflammatory cytokines produced by innate immune cells in response to malaria parasites are primarily triggered by TLR9 through the activation of ERK, p38, and JNK MAPK signaling pathways (17, 18). We previously showed that these pathways differentially regulate pro-inflammatory responses to malaria (19). MAPK pathways play crucial roles in immune responses to various pathogenic infections, including malaria, and MAPK-regulated processes trigger disease pathogenesis (20–22). Therefore, understanding how the MAPK signaling pathways modulate malaria-induced immune responses is likely to be useful in identifying specific targets to develop strategies to prevent severe malaria pathogenesis. However, very little is known about how MAPK signaling events regulate immune responses involved in malaria immunity and pathogenesis. In this study, by using P. berghei ANKA (PbA)-infected C57BL/6 mice, an established model of experimental cerebral malaria (ECM) (23, 24), we studied the immunomodulatory effects of inhibitors of MEK1/2, the kinases immediately upstream of ERK1/2 in the signaling cascade, on immune responses to malaria and ECM pathogenesis. Several interesting findings emerged. MEK1/2 inhibitor treatment resulted in B1 cell expansion, IgM production, phagocytic receptor expression, and phagocytosis of parasites by macrophages (Mφs) and neutrophils (PMNs) and thus contributed to a marked increase in parasite clearance. MEK1/2 inhibitor treatment also down-regulated pro-inflammatory responses by innate immune and T cells, reduced infiltration of immune cells into the brain, and prevented pathogenesis of ECM. Thus, our results provide significant new information on the effect of MEK1/2 inhibitor treatment in immune responses that contribute to malaria pathogenesis.
Results
Inhibitors of MEK1/2 prevent severe malaria pathogenesis
To determine the immunomodulatory role of MEK1/2 in severe malaria pathogenesis, we targeted these kinases by using small-molecule inhibitors and analyzed parasite growth kinetics, clinical episodes, and survival of the host in the mouse CM model. Although the present study used the ECM model, the knowledge gained may be generally applicable to other forms of severe malaria illnesses. The MEK1/2 were targeted with PD98059 (PD), a specific inhibitor. To ensure that the observed effects were not due to a direct anti-parasitic effect of PD, we tested whether PD has an inherent parasite growth–inhibitory property. Cultured P. falciparum treated with even 200 μm PD grew normally (Fig. 1A). PbA parasites treated ex vivo with 24 μg/ml or 48 μg/ml PD were as healthy as those of untreated control parasites, whereas parasites treated with 400 ng/ml chloroquine died as indicated by shrunken and disrupted mass (Fig. 1B). The maximum PD concentration in the blood of mice administered with 3-mg/kg dose is ∼38 μg/ml based on ∼2 ml of blood of an adult mouse weighing ∼25 g (Johns Hopkins University Animal Care and Use Procedure. The Mouse). Actually, the effective concentration of PD would be lower than 38 μg/ml because of in vivo absorption and clearance dynamics, so PD has no direct anti-parasitic effect on PbA at concentrations well above that used in infected mice (see below). Thus, PD was suitable to study the modulatory effects of MEK1/2 inhibition on parasitemia control and host immune responses and survival. In preliminary experiments, administering 2–5 mg of PD/kg body weight by injection intraperitoneally (i.p.) daily starting at 6 h postinfection (pi) showed that 3 mg/kg body weight is optimal to prevent PbA infection-induced ECM pathogenesis. Hence, we used this condition in all experiments performed here. At 1 day pi, measurable levels of ERK1/2, p38, and JNK activation were not observed (not shown) due to the fact that significant parasitemia was not established at this time. At 3 and 5 days pi, the time points at which significant infection was established, the phosphorylation of ERK1/2 was substantially reduced in splenocytes from PD-treated (PbA-PD) versus control (vehicle-treated, PbA) mice (Fig. 1, C and D). At 3 and 5 days pi, PD did not inhibit the activation of p38 and JNK, as indicated by the comparable levels of phospho-p38 and phospho-JNK in the splenocytes of vehicle-treated and PD-treated mice (Fig. 1, C and D). Consistent with the observed significant inhibition of ERK1/2 activation, the expression of c-Jun was significantly decreased in the splenocytes of PD-treated mice at both 3 and 5 days pi (Fig. 1, C and D). The expression of c-Fos was also significantly decreased in PD-treated mice at 5 days pi. However, at 3 days pi, c-Fos expression was very low in both untreated and PD-treated infected mice, and no measurable change was evident (Fig. 1, C and D). Together, the above results suggested that PD inhibits mainly the activation of ERK1/2 but not that of p38 and JNK signaling in malaria-infected mice.
Figure 1.

The effect of MEK1/2 inhibition on malaria parasite growth and malaria-induced activation of MAPK signaling. A, effect of PD on P. falciparum growth as assessed by a SYBR Green assay. Shown are the data from a representative of four independent experiments, each performed in triplicate. Chloroquine, which can completely inhibit the parasite growth, was used as a control. B, the effect of PD on PbA growth was assessed by culturing blood cells from infected mice in the presence of PD. Untreated and chloroquine-treated parasites were used as controls. Shown are photographs under light microscopy of Giemsa-stained smears of cells. C and D, PbA-infected mice were injected i.p. with 3 mg of PD/kg body weight or vehicle daily starting at 6 h pi. Separately, PbA-infected mice were treated orally with either vehicle or 5 mg of trametinib/kg body weight daily starting at 6 h postinfection. At the indicated time points, spleens of mice (n = 2/group) were analyzed for MAPK activation and for c-Jun and c-Fos expression by Western blotting (C). The protein bands on blots were quantified by densitometry measurements and plotted (D). Shown are the results of a representative of two independent experiments. UI, uninfected mice; PbA, vehicle-treated infected mice (control); PbA-PD, PD-treated infected mice; PbA-T, trametinib-treated infected mice. Error bars, S.D.
To confirm the observed effect of PD on MAPK signaling in malaria-infected mice, we tested trametinib (GSK1120212), a Food and Drug Administration-approved anti-cancer drug that has been reported to specifically target MEK1/2 (28). In a preliminary survival study, we treated PbA-infected mice with 0.5, 1, 2, 4, or 5 mg of trametinib/kg body weight. 0.5-, 1-, and 2-mg/kg doses showed partial protection; only 33–50% of mice survived for 10–12 days. However, with 4- and 5-mg/kg doses, 80–90% of mice survived for >20 days; of these two doses, 5 mg/kg showed somewhat better protection. Therefore, subsequently, detailed studies were performed using the 5-mg/kg dose. As in the case of PD, at both 3 and 5 days pi, trametinib efficiently inhibited ERK1/2 phosphorylation and the expression of c-Jun in splenocytes of infected mice (Fig. 1, C and D). Trametinib efficiently inhibited the expression of c-Fos as well at 5 days pi. However, in contrast to PD, at 3 and 5 days pi, trametinib inhibited moderate to substantial levels the phosphorylation of p38 but not that of JNK. Thus, trametinib, in addition to efficiently inhibiting the activation of ERK and the expression of c-Fos and c-Jun, significantly inhibited the phosphorylation of p38.
To determine the immunoregulatory effect of MEK1/2 inhibitor treatment on malaria pathogenesis, we assessed the progression of ECM by treating PbA-infected mice with PD or vehicle. By 7 days pi, all control mice developed severe CM, exhibiting ataxia, limb paralysis, seizures, coma, and lack of sensory and motor functions that are the characteristic neurological symptoms of CM (25–27) (Fig. 2A). All control mice died between 7 and 10 days pi (Fig. 2B). In contrast, the majority (∼85%) of PD-treated mice showed negligible CM symptoms and survived for 2–3 weeks even after the treatment was stopped. The remaining ∼15% of the treated mice died despite exhibiting mild clinical symptoms. Interestingly, parasitemia was significantly reduced during PD treatment, continued to decline until 12 days pi, and remained low until 16 days pi, although treatment was stopped at 7 days pi (Fig. 2C). Because PD has no direct anti-parasitic effect, the data suggested that immune-mediated mechanisms control parasitemia. Nine days after PD treatment was stopped, parasitemia increased exponentially. Despite the parasitemia increase, the PD-treated mice did not experience CM symptoms, likely because of significantly reduced cytotoxic activity of T cells and decreased infiltration of T cells (see Fig. 11), which are the key factors that contribute to CM in this malaria model (15). Eventually, the PD-treated mice died due to marked consumption of red blood cells.
Figure 2.

MEK1/2 inhibitor treatment controls malaria parasitemia and prevents ECM pathogenesis. PbA-infected mice (n = 15/group) were treated i.p. with 3 mg of PD/kg body weight or vehicle daily starting at 6 h pi. In separate experiments, PbA-infected mice (n = 10 mice/group) were treated orally with either vehicle or 5 mg of trametinib/kg body weight daily starting at 6 h pi. CM symptoms, parasitemia, and survival rates were assessed. Plots of CM clinical scores (A and D), survival rates (B and E), and parasitemia (C and F) are shown. The arrow in each panel indicates the time point (7 days pi) at which inhibitor treatment was stopped. PbA, vehicle-treated infected mice; PbA-PD, PD-treated infected mice; PbA-Trametinib, trametinib-treated infected mice. Mice survival data (B and E) were analyzed by log–rank (Mantel–Cox) test, and parasitemia results (C and F) were analyzed by unpaired two-tailed t test. Error bars, S.D. **, p < 0.01; ***, p < 0.001.
Figure 11.

MEK1/2 inhibitor treatment reduces chemotaxis-dependent brain infiltration of immune cells in malaria-infected mice. PbA-infected mice were treated i.p. with 3 mg of PD/kg body weight or vehicle daily starting at 6 h pi. A, cell numbers in the brain of PbA-infected mice at 6 days pi. The levels of PMNs and Mφs, CCR2+ Mφs, CXCR2+ PMNs, and CXCR3+ CD4+ and CD8+ T cells infiltrated to the brain are shown. B, CCL5, CXCL1, and CXCL10 chemokine levels in brain homogenates analyzed by ELISA. C, images of the sections of brains from uninfected mice (UI), PbA-infected mice (PbA), and PbA-infected and PD-treated mice (PbA-PD) at 7 days pi stained for ICAM-1. D, ICAM-1+ blood vessels in 20 microscopic fields of brain sections were counted and data plotted. E and F, brain cells were stained with antibodies against T cell markers, CD11a (LFA-1), and granzyme B. The stained cells were analyzed by flow cytometry. The gating of brain-infiltrated cells and the gating on cells expressing chemokine receptors, CD11a, and granzyme B are shown in supplemental Fig. S5. E, infiltrated CD4+ and CD8+ T cell numbers per brain. F, percentage of granzyme+ CD8+ T cells and the granzyme+ CD8+ T cell number per brain. The data are representative of three or four independent experiments; n = 3 mice/group in each experiment. Statistical analysis was performed by either one-way ANOVA with Newman–Keuls correction for multiple comparisons (D) or unpaired two-tailed t test (A, B, E, and F). Error bars, S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To confirm the observed immunomodulatory effects of PD on parasitemia and pathogenesis, we examined the effects of trametinib on ECM. Trametinib treatment also decreased neurological symptoms and fatality and reduced parasitemia (Fig. 2, D–F). Together, the data demonstrated that the MEK1/2 inhibitor treatment positively contributes to host immune responses that control parasitemia and block pathogenesis.
Next, to assess the brain pathology, we analyzed PD-treated and control PbA-infected mice. Common features of CM pathology in both humans and mice include adherence of IRBCs in the brain, infiltration of immune cells, edema, blood coagulation, micro-hemorrhages, disruption of blood–brain barrier (BBB), and vascular leakage in the brain (15, 25–27). We examined the function of the BBB by injecting Evans blue, a dye that binds to serum albumin. Extraction of the perfused brains revealed substantially lower amounts of the dye in the brains of PD-treated mice than in control mice, indicating that PD treatment markedly prevented vascular leakage (Fig. 3, A and B). Consistent with these results, substantially enlarged and disrupted blood vessels and massive coagulation of RBCs were prominent in the H&E-stained brain sections of control mice (Fig. 3, C2, D2, and E–G). Additionally, sequestered IRBCs (black arrows), infiltrated lymphocytes (arrowheads), and condensed neuronal cells (white arrows) were seen in the brains of control mice (Fig. 3, D2, E, and F). These abnormalities were rarely present in uninfected (Fig. 3, C1 and D1) or PD-treated, infected mice (Fig. 3, C3 and D3). Treatment with trametinib also similarly prevented vascular leakage (Fig. 3, H and I), blood coagulation, endothelia damage, and infiltration of lymphocytes (Fig. 3, J–N). Overall, the above results indicated that MEK1/2 inhibitor treatment significantly enhances parasite clearance and impedes ECM pathogenesis.
Figure 3.

MEK1/2 inhibitor treatment blocks BBB damage and ECM pathology. PbA-infected mice were treated i.p. with 3 mg of PD/kg body weight or vehicle daily starting at 6 h pi. In separate experiments, PbA-infected mice were treated orally with either vehicle or 5 mg of trametinib/kg body weight daily starting at 6 h pi. A and H, photographs of the brains of two groups of mice in the PD treatment experiment (A, n = 7 mice/group) and one group of mice in the trametinib treatment experiment (H, n = 6 mice/group). B and I, amount of Evans blue leaked into the brain tissues of mice in the PD treatment experiment (B, n = 7 mice/group) and trametinib treatment experiment (I, n = 6 mice/group). C–F, images of H&E-stained brain sections of a representative group in the PD treatment experiment. C, ×10 images (C1–C3); D, ×40 images (D1–D3) corresponding to the middle portion of ×10 images (C1–C3); E, ×100 image of the boxed area in C2; F, ×100 image of the boxed area in D2. D2, E, and F, endothelial damage, sequestered parasites (black arrows), infiltrated lymphocytes (arrowheads), and condensed neuronal cells (white arrows) in the brains of PbA mice are indicated. These features were either negligible or absent in the brains of uninfected (D1) and PbA-PD (D3) mice. J–M, ×10 (J), ×40 (K), and ×100 (L and M) images of H&E-stained brain sections of a representative group in trametinib treatment experiment. Sequestered parasites (black arrows), infiltrated lymphocytes (arrowheads), and damaged BBB (white arrows) are shown in L and M. G and N, numbers of damaged blood vessels in 20 randomly selected fields in the H&E-stained tissue sections of the PD-treated (G) and trametinib-treated groups (N) were counted and plotted; n = 4 mice/group). Statistical analysis was by one-way ANOVA with Newman–Keuls correction for multiple comparisons. Error bars, S.D. (I and B) and S.E. (G and N). **, p < 0.01; ***, p < 0.001.
MEK1/2 inhibitor treatment enhances phagocytic clearance of parasites, controlling parasitemia
In malaria infection, the growth of parasites is controlled primarily by the phagocytic clearance of IRBCs (29), and large parasite biomass due to rapid growth of parasites is a risk factor for malaria pathogenesis (11, 30, 31). To determine the effect of MEK1/2 inhibitor treatment in controlling parasitemia and the underlying mechanisms, we assessed the phagocytosis of IRBCs by spleen and liver Mφs and PMNs by injecting CFSE-stained PbA IRBCs into PD-treated and control mice at 4 days pi. After 18 h, the phagocytosis of IRBCs by spleen PMNs, marginal zone Mφs (MM), marginal zone metallophilic Mφs (MMM), and red pulp Mφs (RPM) (32), gated as shown in Fig. 4A, was ∼2–17-fold higher in PD-treated mice than in control mice (Fig. 4B and supplemental Fig. S1). The phagocytosis of IRBCs by liver PMNs, inflammatory Mφs (Ly6ChiLy6G−), CD11bhiF4/80hi Mφs, and Kupffer cells (CD11bloF4/80hi) (Fig. 4C) was ∼2–18-fold higher in PD-treated mice than in control mice (Fig. 4D and supplemental Fig. S1), suggesting that enhanced phagocytic activity upon MEK1/2 inhibitor treatment contributes to the observed reduced parasitemia. Thus, we showed that immune responses modulated by MEK1/2 inhibitor treatment up-regulate the capacity of spleen and liver PMNs and various Mφ subsets to clear parasites by phagocytosis.
Figure 4.

Treatment with MEK1/2 inhibitor enhances phagocytic receptor expression and phagocytosis of parasites. PbA-infected mice were treated i.p. with 3 mg of PD/kg body weight or vehicle daily starting at 6 h pi. A–D, at 4 days pi, ∼2.5 × 107 CFSE-labeled PbA-IRBCs were administered. After 18 h, spleen and liver cells were stained with antibodies against the indicated marker proteins and analyzed by flow cytometry. SSC, side scatter; FSC, forward scatter. A and B, spleen PMNs and Mφ subsets were gated as shown in A, and the percentage of CFSE+ cells, gated as shown in supplemental Fig. S1, was plotted (B). C and D, liver PMNs, inflammatory Mφs, and Kupffer cells were gated as shown in C, and the percentages of CFSE+ PMNs, inflammatory Mφs, CD11bhiF4/80hi Mφs, and Kupffer cells, gated as shown in supplemental Fig. S1, were plotted (D). E and F, at 5 days pi, spleen (E) and liver (F) cells were analyzed by flow cytometry. The cells were gated as shown in A and C, and the percentages of phagocytic receptor+ PMNs and the indicated Mφ subsets, analyzed as shown in supplemental Fig. S2A (spleen cells) and supplemental Fig. S2B (liver cells), were plotted. Results are representative of three (A–D) or four (E and F) independent experiments. A–F, n = 3–4 mice/group. Statistical analysis was by unpaired two-tailed t test. Error bars, S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Scavenger receptors, especially CD36, Fc receptors, and complement receptors (CRs), play important roles in IRBC phagocytosis (29, 33, 34). Consistent with increased phagocytosis, at 5 days pi, the frequencies of spleen PMNs, expressing FcγII/IIIR and CD36, and several Mφ subsets, expressing various phagocytic receptors, were significantly higher in PD-treated mice than in control mice (Fig. 4E and supplemental Fig. S2A). Specifically, the Mφ subsets that expressed increased levels of phagocytic receptors in PD-treated mice include MMM and RPM expressing CD36, MM and RPM expressing MARCO (another scavenger receptor), MM and MMM expressing Fcα/μR, and MMM expressing CR1/CR2. Similarly, the frequencies of CD36-, Fcα/μR-, FcγII/IIIR-, and CR1/CR2-expressing liver PMNs, Fcα/μR- and CR1/CR2-expressing inflammatory Mφs, CR1/CR2-expressing Kupffer cells, and Fcα/μR-, FcγII/IIIR-, and CR1/CR2-expressing CD11bhiF4/80hi Mφs were substantially higher in PD-treated mice than in control mice (Fig. 4F and supplemental Fig. S2B). Thus, these results indicated that MEK1/2 inhibitor treatment up-regulates the expression of phagocytic receptors by spleen and liver PMNs and various Mφ subsets in response to malaria infection, increasing the phagocytic clearance of parasites.
Antibody-mediated control of parasitemia through opsonization of IRBCs and merozoites and subsequent phagocytosis represents an important component of protective immunity to malaria (35). At early stages of infection, IgM is the predominant antibody produced in response to pathogenic infections, including malaria. IgM is known to control the growth of various pathogens, including malaria parasites, and provide resistance against infections (36, 37). In addition to promoting phagocytosis, IgM can also reduce inflammatory responses by promoting the clearance of dead Mφs and PMNs and producing IL-10 (38–41). Further, IgM may play a role in clearance of phagocytic cells that die after ingesting IRBCs and uninvaded merozoites, thus removing stimulus for inflammation. At 5 days pi, the levels of total IgM and parasite-reactive IgM were ∼2- and 5-fold higher, respectively, in PD-treated mice than those in control mice (Fig. 5A). Significantly increased parasite-specific IgM was evident even at 3-day pi in PD-treated mice. Total IgM (natural plus parasite-specific IgM) was also increased at 3 days pi in PD-treated mice, but this increase was modest, because of high levels of preexisting natural IgM. These results demonstrated that MEK1/2 inhibitor treatment augments IgM production during early stages of infection.
Figure 5.

Treatment of malaria-infected mice with PD leads to increased B1 cell expansion, IgM production, and phagocytosis of parasites. A–D, PbA-infected mice were treated i.p. with 3 mg of PD/kg body weight or vehicle daily starting at 6 h pi. A, serum IgM levels (n = 4 mice/group) at 3 days and 5 days pi assessed by ELISA. B and C, flow cytometry analysis of splenic B1a and B1b B cells at 3 days and 5 days pi (n = 3 mice/group). Shown are the gating strategy and FMO control for gating CD23− cells (B) and B1a and B1b cell frequency (top) and cell numbers per spleen (bottom) in control and PD-treated mice relative to uninfected mice (C). D, the phagocytosis of IRBCs pretreated with 1:10-diluted sera from control or PD-treated mice at 5 days pi by spleen and liver PMNs and MM, MMM, and RPM from PD-treated mice at 5 days pi. Experiments were performed in triplicate. Data are a representative of four independent experiments; n = 3 mice/group in each experiment. A, C, and D, statistical analysis was by one-way ANOVA with Newman–Keuls correction for multiple comparisons. Error bars, S.D. E, the phagocytosis of CFSE-labeled IRBCs pretreated with 1:10-diluted sera from PD-treated mice at 5 days pi by liver PMNs and CD11bhiF4/80hi Mφs from PD-treated mice at 5 days pi treated with either anti-Fcα/μR antibody or anti-CR1/CR2 antibody. PMNs and CD11bhiF4/80hi Mφs from PD-treated mice not treated with antibodies were analyzed as controls. Experiments were performed in triplicate. Data represent the mean of two independent experiments, and statistical analysis was by two-way ANOVA. Error bars, S.E. Blocking of Fcα/μR and CR1/CR2 with antibodies significantly inhibited phagocytic uptake of IRBCs. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
IgM is mainly produced by B1 B cells, which are rapidly mobilized upon sensing of pathogens; B1a and B1b subpopulations produce natural IgM and antigen-specific IgM, respectively (41, 42). Consistent with the increased IgM production upon PD treatment, at 5 days pi, the frequency and numbers of spleen B1a cells (CD5+CD19hiCD23−) and B1b cells (CD5−CD19hiCD23−) were significantly increased in PD-treated mice compared with control mice (Fig. 5, B and C). Even at 3 days pi, B1a and B1b cell numbers were increased (Fig. 5C).
To assess the role of IgM in clearance of parasites, we examined the phagocytosis of IRBCs treated with serum from the PD-treated infected mice compared with IRBCs treated with serum from control mice. The frequencies of spleen MM, MMM, and RPM Mφ subsets from PD-treated mice that internalized IRBCs were significantly higher when IRBCs were treated with sera from PD-treated mice (Fig. 5D). The frequencies of liver PMNs and CD11bhiF4/80hi Mφs from PD-treated mice were also significantly higher when IRBCs were treated with sera from PD-treated mice. Given that the expression of Fcα/μR and CR1/CR2 by phagocytes was markedly up-regulated in MEK1/2 inhibitor-treated mice (see Fig. 4, E and F), we tested the effect of blocking these receptors. The frequencies of liver PMNs and CD11bhiF4/80hi Mφs that phagocytosed IRBCs treated with sera from PD-treated mice were substantially reduced when phagocytic cells were pretreated with anti-Fcα/μR or anti-CR1/CR2 antibody (Fig. 5E). In PbA-infected mice, the early antibody response consists mainly of IgM, significant IgG response usually occurs after 12 days pi, and immune complexes during the first week of infection consist mainly of IgM (43). Fcα/μR and CR1/CR2 mediate the uptake of IgM- and complement-fixed antibody-bound IRBCs (44, 45). Thus, the inhibitory effects of anti-Fcα/μR and anti-CR1/CR2 antibodies suggest that IgM– and IgM-immune complex–mediated phagocytosis play significant roles in parasite clearance. Together, the above results strongly suggested that rapid B1 cell expansion, up-regulation of total and parasite-specific IgM, and elevated Fc receptor and CR1/CR2 expression by phagocytic cells contribute to the observed increased phagocytosis of IRBCs in MEK1/2 inhibitor–treated mice. Thus, MEK1/2 inhibitor treatment positively regulates the phagocytic clearance of malaria parasites by PMNs and Mφs.
Treatment with MEK1/2 inhibitor differentially modulates pro-/anti-inflammatory responses to malaria
Blood stage malaria infection induces elevated production of pro-inflammatory cytokines, typically TNF-α and IFN-γ, during the early stages of infection, promoting robust Th1 responses and contributing to malaria pathogenesis (8, 13, 15). Ideally, as the infection progresses, pro-inflammatory cytokine responses would be opposed by the production of anti-inflammatory cytokines and consequent Th2 polarization, resulting in balanced pro- and anti-inflammatory responses that reduce pathogenesis (46–48). To determine the effect of MEK1/2 inhibitors on malaria parasite-induced cytokine production, we analyzed serum TNF-α, IFN-γ, and IL-10 after PbA infection and PD treatment. Compared with control mice, serum TNF-α and IFN-γ levels were markedly decreased in PD-treated mice at both 4 and 6 days pi. However, compared with control mice, the serum level of IL-10 in PD-treated mice was higher at 4 days pi and lower at 6 days pi (Fig. 6A), suggesting that the kinetics of IL-10 response is faster in PD-treated mice than in untreated control mice.
Figure 6.

MEK1/2 inhibitor treatment differentially modulates malaria-induced pro- and anti-inflammatory responses by DCs in vivo. A–D, PbA-infected mice were treated i.p. with 3 mg of PD/kg body weight or vehicle daily starting at 6 h pi. A, serum cytokine levels at 4 and 6 days pi in PbA-infected mice assessed by ELISA. B–D, spleen cells from the infected mice were analyzed by flow cytometry for cytokine production by DCs at 3 and 5 days pi (C) and costimulatory molecule expression (D) by DCs at 4 days pi. The gating strategy for the CD11chiMHCIIhiCD8α+ and CD11chiMHCIIhiCD8α− DC subsets is shown in B. The gating strategy for cytokine+ spleen DCs is shown in supplemental Fig. S3. Isotype-specific antibodies were used as control. A–D, the results are a representative of four independent experiments; n = 3 mice/group in each experiment. Statistical analysis was by one-way ANOVA with Newman–Keuls correction for multiple comparisons. Error bars, S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
DCs directly respond early to malaria infection and produce pro-inflammatory cytokines, which promote a strong Th1 development (49, 50). Consistent with the observed decreased levels of serum pro-inflammatory cytokines (see Fig. 6A), PD-treated mice at both 3 and 5 days pi showed significantly lower frequency of TNF-α-producing CD8α+ and CD8α− DCs (Fig. 6 (B and C) and supplemental Fig. S3). Also, by 5 days pi, PD-treated mice had a significantly higher proportion of IL-10-producing CD8α+ and CD8α− DCs than control mice (Fig. 6C). Further, in agreement with the reduced pro-inflammatory cytokine production and increased IL-10, both CD8α+ and CD8α− DCs in PD-treated mice were significantly less mature, as indicated by the decreased surface expression of costimulatory molecules, CD40, CD80, and CD86 (Fig. 6D).
In vivo, DCs interact with other immune cells, especially NK and T cells, resulting in DC-NK cell cross-talk and DC-T cell cross-talk, influencing immune responses. Thus, to determine whether PD directly targets DCs, we analyzed IRBC-induced cell activation and cytokine responses by bone marrow cell-derived FLT3 ligand-differentiated DCs (FL-DCs). The activation level of DCs treated with PD was substantially lower than that of untreated DCs, as indicated by the lower surface expression of CD40, CD80, and CD86 in the former (Fig. 7A). Further, PD-treated DCs produced significantly lower levels of IL-12 than untreated DCs (Fig. 7B). These results indicated that PD treatment directly modulates immune responses of DCs.
Figure 7.
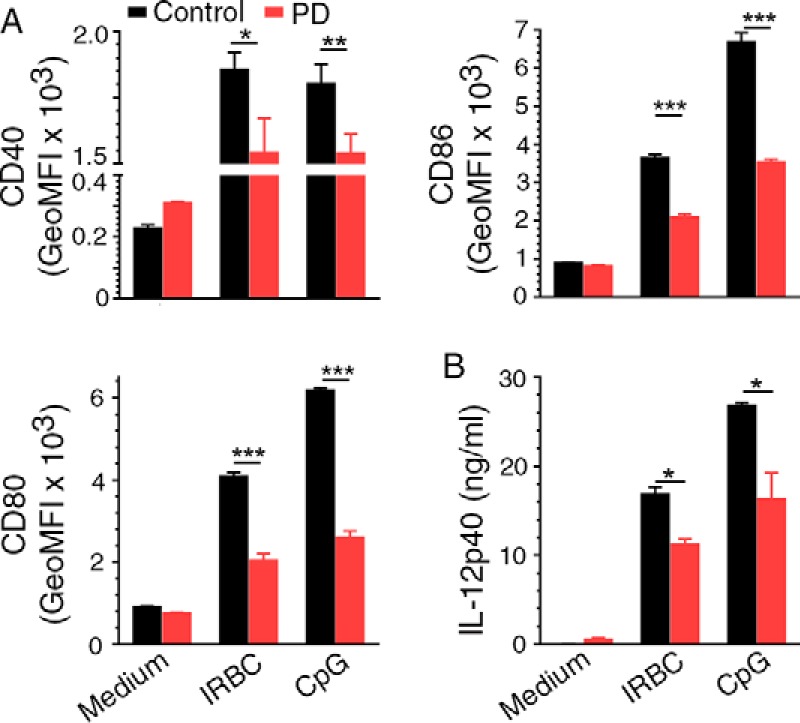
PD treatment modulates malaria parasite-induced immune responses by DCs. A and B, FL-DCs either untreated or treated with 20 μm PD were incubated with 3D7 IRBCs in triplicate, and cell activation was measured by flow cytometry (A) and cytokine production was measured by ELISA (B). FL-DCs stimulated with CpG ODN were analyzed as controls. Statistical analysis was by unpaired two-tailed t test. The data are representative two separate experiments. Error bars, S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
IFN-γ, a strong Th1 promoting cytokine, plays a key role in ECM and other forms of severe pathogenesis, and NK cells are a major source of IFN-γ at early stages of malaria infection (15, 51). NK cells from PD-treated mice produced substantially decreased levels of IFN-γ and increased levels of IL-10 as indicated, respectively, by the decreased frequency of IFN-γ-producing and increased frequency of IL-10-producing NK cells (Fig. 8, A and B). Further, NK cells from uninfected mice, treated in vitro with PD, produced significantly lower IFN-γ in response to IL-12/IL-18 stimulation than untreated NK cells (Fig. 8C), indicating that MEK1/2 inhibition directly modulates immune responses of NK cells. Because DC cytokine production shapes NK cell responses, the above results suggested that DC cytokine responses modulated upon MEK1/2 inhibitor treatment cooperatively contributed to decreased pro-inflammatory and increased anti-inflammatory responses by NK cells. This conclusion is supported by the results of DC and NK cocultures. Upon stimulation with the TLR9 ligand CpG ODN 1826, cocultures of NK cells from uninfected mice and DCs from PD-treated mice produced significantly lower levels of IFN-γ and higher levels of IL-10 than cocultures of NK cells from uninfected mice and DCs from control mice (Fig. 8D). Furthermore, cocultures of FL-DCs and NK cells from uninfected mice in which either cell type was treated or both were treated with PD also produced significantly decreased IFN-γ in response to P. falciparum IRBCs (Fig. 8E). Treatment of both DCs and NK cells with PD resulted in an almost complete loss of the IFN-γ response and markedly increased production of IL-10. These data demonstrated that PD not only can act directly upon NK cells to reduce IFN-γ production but also can act on DCs to decrease IFN-γ and increase IL-10 production by NK cells. Thus, the inhibition of MEK1/2 results in reduced production of pro-inflammatory cytokines and increased production of anti-inflammatory cytokines by DCs and NK cells. Together, the above results showed that the MEK/12 inhibitor treatment not only directly down-regulates pro-inflammatory cytokine responses and up-regulates anti-inflammatory cytokine responses to malaria by DCs and NK cells, but also modulates cooperative immune responses of DC and NK cell cross-talk.
Figure 8.

MEK1/2 inhibitor treatment differentially regulates malaria-induced pro- and anti-inflammatory responses by NK cells. A and B, PbA-infected mice were treated i.p. with 3 mg of PD/kg body weight or vehicle daily starting at 6 h pi. Spleen NK cells from the infected mice at 4 days pi were analyzed by flow cytometry for cytokine production. Shown are the gating of CD49b+CD3ϵ− NK cells (A) and quantification of IFN-γ+ and IL-10+ cells (B). The results are representative of four independent experiments; n = 3 mice/group in each experiment. C, PD-treated and -untreated liver NK cells from uninfected mice were stimulated with IL-12 (1 ng/ml) and IL-18 (50 ng/ml), and IFN-γ production was measured by ELISA. D, cocultures of FACS-sorted spleen DCs from PD-treated and untreated infected mice at 3 days and 5 days pi and liver NK cells from uninfected mice stimulated with 2 μg/ml CpG ODN. Cytokines in the culture supernatants were analyzed ELISA. E, cocultures of FL-DCs and liver NK cells stimulated with P. falciparum IRBCs. Prior to coculturing, DCs alone, NK cells alone, or both DCs and NK cells were pretreated with PD for 1 h; cells not treated with PD were used as controls. Cytokines in culture supernatants were analyzed by ELISA. D and E, the results are representative of four independent experiments. Statistical analysis was by either one-way ANOVA with Newman–Keuls correction for multiple comparisons (B, D, and F) or unpaired two-tailed t test (C). Error bars, S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
MEK1/2 inhibitor treatment reduces infiltration of immune cells and T cell effector function in ECM
In ECM pathogenesis, granzyme B-expressing cytotoxic CD8+ T cells that migrate to the inflamed brain are the major effector cells in the disruption of BBB (15, 52). In addition, CD4+ T cells enhance the infiltration of CD8+ T cells by producing IFN-γ to induce chemokine production (15, 51). IFN-γ-producing CD4+ T and CD8+ T cell numbers were substantially decreased in the spleens of PD-treated mice at both 4 and 6 days pi (Fig. 9 (A–C) and supplemental Fig. S4). Also, PD treatment significantly increased IL-10+ T cell numbers as early as 4 days pi. At 6 days pi, PD-treated mice showed an increased number of CD4+ T cells producing IL-4, a Th2 cytokine. Consistent with a decreased Th1 response and an increased Th2 response, as indicated by cytokine responses, the numbers of splenic CD4+ T and CD8+ T cells that express T-bet, a transcription factor that promotes Th1 development, as well as numbers of cytotoxic CD8+ T cells, were substantially lower in PD-treated mice (Fig. 9, B and C). This correlated with an observed decrease in the number of granzyme B-expressing CD8+ T cells in PD-treated mice (Fig. 9C). Thus, MEK1/2 inhibitor treatment suppresses malaria-induced Th1 responses and promotes Th2 responses, accompanied by a marked decrease in the production of pro-inflammatory cytokines and granzyme B-expressing CD8+ T cells.
Figure 9.

Treatment with MEK1/2 inhibitor suppresses malaria-induced Th1 development and CD8+ T cell cytotoxicity. PbA-infected mice were treated i.p. with 3 mg of PD/kg body weight or vehicle daily starting at 6 h pi. Spleen cells from the PbA-infected mice at 4 days and 6 days pi were stained with antibodies against surface markers and the indicated intracellular proteins and analyzed by flow cytometery. A–C, shown are the gating strategy for CD4+ and CD8+ T cells (A) and IFNγ+, IL-10+, IL-4+, T-bet+, and granzyme B+ CD4+ and/or CD8+ T cell numbers per spleen (B and C). The cytokine+, T-bet+, and granzyme B+ cells were gated as shown supplemental Fig. S4, A and B. The data are representative of three or four independent experiments; n = 3 or 4 mice/group in each experiment. Statistical analysis was by one-way ANOVA with Newman–Keuls correction for multiple comparisons. Error bars, S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
DCs are known to activate and induce proliferation of T cells and shape T cells for antigen-specific immune responses. To determine the effect of MEK1/2 inhibitor in directly regulating T cell responses, we analyzed the effect of PD on cell activation, proliferation, and cytokine responses. Treatment of T cells from uninfected mice with PD and activation with anti-CD3ϵ/anti-CD28 antibodies resulted in reduced frequencies of CD69-expressing CD4+ T and CD8+ T cells (Fig. 10, A and B); CD69 is an early T cell activation marker. Further, treatment with PD and activation with the antibodies resulted in decreased proliferation of CD4+ T and CD8+ T cells compared with those of untreated cells, as indicated by the CFSE intensity (Fig. 10C). Furthermore, both CD4+ T and CD8+ T cells showed reduced expression of IFN-γ and TNF-α (Fig. 10, D and E). In addition, CD4+ T cells exhibited increased expression of IL-4 and IL-10 (Fig. 10, F and G). These results demonstrated that MEK1/2 inhibitor treatment down-regulates T cell activation and proliferation and altered cytokine expression in response to immune stimulation. Given the down-regulated pro-inflammatory responses and up-regulated anti-inflammatory responses by DCs and that DCs shape T cell responses (53, 54), the above data suggest that MEK1/2 inhibitor treatment results in the regulation of immune responses by DCs and T cells and their cross-talk.
Figure 10.

The effect of PD on anti-CD3ϵ/anti-CD28 stimulation-induced T cell responses. A–D, spleen T cells from uninfected mice were either untreated or treated with PD and then stimulated with 5 μg/ml of anti-CD3ϵ coated on plates and 2 μg/ml anti-CD28 antibodies in triplicate, and cells were analyzed by flow cytometry for activation, proliferation, and cytokine responses. A, gating of CD4+ and CD8+ T cells. B, percentage of CD69-expressing CD4 and CD8 T cells. C, percentage of T cells having decreased CFSE intensity. D and E, percentage of IFN-γ- and TNF-α-expressing T cells, respectively. F and G, percentage of IL-4- and IL-10-producing CD4+ T cells, respectively. The data are representative of three (D–G), four (C), and five (B) separate experiments. Statistical analysis was by unpaired two-tailed t test. **, p < 0.01; ***, p < 0.001.
Infiltration of monocytes/Mφs, NK cells, and T cells contributes to the disruption of BBB and ECM pathogenesis (55, 56). The numbers of total immune cells, PMNs, inflammatory Mφs, and CD4+ T and CD8+ T cells were all markedly lower in the brains of PD-treated mice compared with control mice (Fig. 11A and supplemental Fig. S5). This reduction was reflected in a PD-induced decrease in the number of PMNs expressing CXCR2, the receptor for the PMN chemoattractant CXCL1; Mφs expressing CCR2, the receptor for the Mφ chemoattractant CCL5; and CD4+ T and CD8+ T cells expressing CXCR3, the receptor for the IFN-γ-induced T cell chemoattractant CXCL10 (Fig. 11A). Consistent with these results, the production of chemokines CCL5, CXCL1, and CXCL10 was significantly lower in the brains of PD-treated mice (Fig. 11B). Whereas the surface expression of chemokine receptor guides the chemotactic migration of T cells into the brain, cell adhesion molecules, predominantly ICAM-1 (a ligand for LFA-1), support the sequestration of T cells in the brain vasculature (57). Immunohistochemical analysis of brain sections showed higher levels of ICAM-1 in the vascular beds of control mice than that in PD-treated mice (Fig 11, C and D). Consistent with these results, numbers of CD11a (LFA-1)-expressing CD4+ T and CD8+ T cells were significantly lower in the brains of PD-treated mice (Fig. 11E and supplemental Fig. S5). Importantly, the number of granzyme B+ cytotoxic CD8+ T cells was markedly lower in the brains of PD-treated mice than in control mice (Fig. 11F). Together, these results showed that MEK1/2 inhibitor treatment prevents brain pathology by reducing infiltration of inflammatory myeloid cells and cytotoxic T cells.
Discussion
Pathogenesis of cerebral and other severe malaria involves the participation of a series of host immune responses that are initiated upon blood stage infection and subsequently amplified, ultimately causing organ damage and dysfunction. The initial trigger of pathogenesis is excessive and dysregulated production of pro-inflammatory cytokines by DCs upon pathogen receptor–mediated recognition of parasites (8, 13, 16). Subsequently, cytokine-mediated signaling and interaction of activated DCs with other cells of the innate immune systems, such as NK and γδ T cells, amplifies the pro-inflammatory response, leading to strong Th1 and cytotoxic CD8+ T cell effector responses. In parallel, the sequestration of parasites via the adherence of IRBCs results in the activation of endothelia and subsequent infiltration of activated myeloid cells and then Th1 cells, creating a strong inflammatory environment in organs. Consequently, granzyme B- and perforin-expressing cytotoxic CD8+ T cells infiltrate and cause BBB damage and subsequent organ dysfunction (15).
The malaria-induced pro-inflammatory responses by DCs are the downstream output of MAPK signaling pathways upon primarily Toll-like receptor-mediated recognition of parasites (17, 49, 58). The MAPK signaling pathways also regulate inflammatory responses of other cells of the immune system that interact with DCs or are activated by DC-produced cytokines. As such, the MAPK pathways play crucial roles in a multitude of immunological processes, many of which contribute to both development of malaria immunity and severe pathogenesis. Thus, knowledge of how the MAPK signaling pathways regulate malaria-induced immune responses is likely to be valuable in developing a vaccine and/or immunomodulators for malaria. However, very little is known about the manner in which the MAPK pathways regulate various malaria-induced immune responses and how these responses contribute to malaria pathogenesis.
Although in this study, the two small-molecule inhibitors, PD and trametinib, efficiently inhibited ERK1/2, and trametinib also significantly inhibited p38 signaling at 5 days pi. Because PD did not inhibit p38 signaling, it is likely that the observed effects on immune responses to malaria are primarily due to the inhibition of the ERK1/2 signaling pathway. However, it is possible that both inhibitors exert effects on some other kinases of MAPK pathway to a certain degree, especially upon prolonged treatment. Therefore, we refrain from attributing the results solely to the inhibition of the ERK1/2 signaling pathway, but instead we cautiously ascribe our observations to the effects of these inhibitors.
This study has made several important novel observations on how immunological processes modulated by MEK1/2 inhibitor treatment prevent ECM pathogenesis. Importantly, the data presented here show that MEK1/2 inhibitor treatment positively regulates several malaria-induced immune responses that are involved in control of parasitemia, including B1 cell expansion, IgM production, expression of phagocytic receptors, and phagocytic activity of PMNs and Mφs. This is evident from our findings that MEK1/2 inhibitor treatment substantially reduced parasitemia by enhancing phagocytic clearance of parasites through increased IgM production and phagocytic receptor expression. In addition, MEK1/2 inhibitor treatment negatively regulated pro-inflammatory, Th1, and cytotoxic T cell responses. Moreover, the MEK1/2 inhibitor treatment negatively regulated chemotaxis-dependent infiltration of immune cells, including cytotoxic CD8+ T cells that are known to contribute to cerebral and also probably to other organ-specific malaria pathogenesis. Thus, collectively, our data support the conclusion that MEK1/2 inhibitors substantially reduce pro-inflammatory responses, infiltration of immune cells, and cytotoxic potential of CD8+ T cells, preventing organ damage and dysfunction and, eventually, fatality. MEK1/2 inhibition is also likely to protect against other forms of serve malaria pathology.
One of the key novel findings here is that MEK1/2 targeting positively regulates malaria infection–induced expansion of B1 cells and IgM production, as indicated by the finding that MEK1/2 inhibitor treatment causes significantly increased production of IgM through the expansion of B1a and B1b B cells. IgM is known to control dissemination of various infections, including malaria (36), and also plays an important role in the clearance of dead cells, limiting inflammatory and autoimmune responses. In malaria infection, in addition to IRBCs, there is accumulation of large numbers of uninvaded merozoites, erythrocyte debris, and hemozoin-containing digestive vacuoles (59). PMNs and Mφs that ingest IRBCs, merozoites, digestive vacuoles, and other parasite components undergo apoptotic death. All of these components need to be efficiently removed and degraded to prevent autoantibody production, pro-inflammatory responses, and their deleterious effects. IgM plays an important role in clearing debris, dead cells, immune complexes, and pathogens (36–42, 60). Consistent with this role of IgM, there were substantial levels of IgM-dependent phagocytosis of IRBCs by spleen and liver PMNs and Mφs. These results also agree with the reported finding that P. chabaudi-infected IgM−/− mice exhibit reduced survival and increased parasitemia compared with wild-type mice (36) and that IgM-polarized LPS-activated murine DCs acquire a regulatory phenotype that suppresses inflammatory responses (61). Thus, the rapid expansion of B1 cells, elevated levels of IgM, and increased IgM– and immune complex–dependent phagocytosis of IRBCs seen upon MEK1/2 inhibitor treatment indicate that MEK1/2-regulated immune responses to malaria significantly curtail the beneficial IgM-mediated immune responses required to avoid pathogenesis.
Another key novel finding of this study is that MEK1/2 inhibitors significantly increase the phagocytic capacity of PMN and Mφs and the clearance of parasites by up-regulating phagocytic receptor expression. Thus, our findings demonstrate that MEK1/2-mediated immune responses contribute to severe malaria pathogenesis, because inefficient clearance of parasites results in high parasite burdens, leading to severe illnesses with adverse clinical outcomes (33, 48). Malaria parasitemia is controlled mainly by phagocytic clearance of parasites, although other mechanisms contribute to a certain degree. In people who have acquired immunity to malaria, phagocytosis of IgG-opsonized parasites mediates efficient clearance. However, in non-immune people, at early stages of infection, parasite clearance is restricted to scavenger receptor–, complement receptor–, and IgM–mediated phagocytosis. Thus, increased expression of several phagocytic receptors, including scavenger as well as various Fc and complement receptors, and elevated production of IgM are essential for the efficient control of parasitemia when malaria–non-immune individuals are infected. Thus, our findings demonstrate that targeting MEK1/2 enhances parasite clearance through increased phagocytic activity, contributing to protection against pathogenesis.
One more important finding of this study is that MEK1/2 inhibitors efficiently reduce the harmful inflammatory responses and promote the beneficial anti-inflammatory responses, preventing pathogenesis. The pathogenic immune responses down-regulated upon MEK1/2 inhibitor treatment include reduced pro-inflammatory cytokine production by DCs, DC-induced IFN-γ production by NK and T cells, and development of cytotoxic CD8+ T cells. MEK1/2 inhibition also reduced the infiltration of immune cells, including cytotoxic CD8+ T cells, to the brain. TNF-α and IFN-γ play important roles in CM pathogenesis by up-regulating the expression of adhesion molecules such as ICAM-1 on the brain endothelia, facilitating the adherence of immune cells, particularly LFA-1+ T cells (8, 13). TNF-α and IFN-γ produced by myeloid and NK cells that have entered the brain are the initial inducers of chemokines such as CXCL1, CCL5, CXCL9, and CXCL10 by the brain endothelia (14, 51, 62). The chemokines play critical roles in CM pathogenesis by promoting infiltration of immune cells, which express chemokine receptors, such as CXCR2, CCR2, and CXCR3. CD4+ T cells that initially move to the brain produce IFN-γ, promoting further infiltration of CD4+ T and CD8+ T cells in a cooperative feed-forward manner. This amplifies CXCL9 and CXCL10 production and augments the infiltration of CXCR3+ pathogenic CD8+ T cells into the brain (15, 51). Accumulation of CD8+ T cells in the brain causes CM pathology. The expression of ICAM-1 in the vascular bed was significantly decreased upon PD treatment, as were the levels of infiltrated myeloid cells, chemokines, chemokine receptor-positive cells, and LFA-1+ T cells. Overall, these data support the notion that the inhibitors of MEK1/2 significantly down-regulate inflammatory immune responses that augment malaria pathogenesis and up-regulate anti-inflammatory responses.
The beneficial modulated immune responses observed in PD-treated infected mice are mainly the multifactorial output of MEK1/2-regulated immune responses of DC–NK and DC–T cell cross-talk. Activated DCs shape the innate and adaptive immune responses by inducing their function-specific responses in cells such as NK and T cells. Furthermore, cytokines, such as IL-12, IFN-γ, IL-10, and IL-4, also strongly modulate responses by DCs, NK cells, and T cells, resulting in a cytokine-mediated feedback loop, contributing to the responses by the immune cells. Given this and the observation that PD treatment directly modulates DC and NK and T cell responses, the data presented here indicate that MEK1/2 inhibitor treatment regulates malaria parasite-induced cytokine responses directly by targeting various cells of the innate and adaptive immune systems and indirectly by regulating the cytokine-induced secondary responses.
In summary, the results presented here show that inhibitors of MEK1/2 block severe malaria pathogenesis by differentially regulating the detrimental and beneficial immune responses. Specifically, MEK1/2 inhibitor treatment reduces (a) malaria parasite burden by promoting B1 cell expansion and IgM production, phagocytic receptor expression, and phagocytic activity of Mφs PMNs; (b) pro-inflammatory responses and expression of chemotactic mediators and endothelial cell adhesion molecules; and (c) infiltration of immune cells and cytotoxic effector functions of CD8+ T cells, which cause BBB damage and pathology. Thus, we conclude that MEK1/2-mediated signaling plays crucial roles in malaria-induced immune responses that promote malaria pathogenesis, and MEK1/2 are promising targets for developing treatment strategies for malaria. Additionally, considering that excessive inflammatory responses contribute to disease pathogenesis, our results have important implications for other pathogenic infections and inflammation-driven diseases.
Experimental procedures
Reagents
PD98059 and trametinib were purchased from LC Laboratories (Woburn, MA). ELISA kits for analysis of TNF-α, IFN-γ, IL-10, CCL5, CXCL1, and CXCL10 were from R&D Systems (Minneapolis, MN). DMEM and RPMI 1640 medium were from Mediatech (Manassas, VA). Penicillin/streptomycin solution and the CellTraceTM CFSE cell proliferation analysis kit were from Invitrogen. FBS was from Atlanta Biologicals (Lawrenceville, GA). Collagenase D was from Roche Applied Science (Mannheim, Germany). CpG oligodeoxynucleotide 1826 (referred to as CpG) was from Coley Pharmaceuticals (Kanata, Canada). Nonessential amino acids, sodium pyruvate, and 2-mercaptoethanol for cell culturing and Percoll were from Sigma-Aldrich. Anti-mouse CD11c (clone N418) antibody-, anti-human/mouse CD11b antibody-, and anti-mouse CD90.2-conjugated MicroBeads for the isolation of DCs, Mφs, and T cells, respectively, and NK cell isolation kit II were from Miltenyi Biotec (Auburn, CA). The mouse immunoglobulin isotype kit was from BD Biosciences. Phospho-specific anti-mouse ERK1/ERK2 (mouse monoclonal), p38 (rabbit polyclonal), JNK (rabbit monoclonal), and C-Jun (rabbit monoclonal) antibodies; rabbit polyclonal antibodies against ERK1/ERK2, p38, and JNK; and horseradish peroxidase-conjugated goat anti-mouse IgG and goat anti-rabbit IgG were from Cell Signaling Technology, Inc. (Danvers, MA). Rabbit polyclonal antibody against c-Fos was from Santa Cruz Biotechnology, Inc. (Dallas, TX).
Antibodies
The following anti-mouse antibody conjugates were used for cell staining: FITC- and APC-eFluor 780-anti-CD5 (clone 53-7.3), eFluor 450-anti-CD11a (M17/4), PE- and PerCP-Cy5.5-anti-CD11b (M1/70), PE- and APC-anti-CD11c (418N), APC- and Brilliant Violet (BV)421-anti-I-A/I-E (M5/114.15.2), APC-eFluor 780-anti-F4/80 (BM8), FITC-anti-CD16/32 (clone 93), PE-anti-CD36 (72-1), FITC- and BV421-anti-CD49b (DX5), PE-Cy5-anti-CD80 (16-10A1), PE-Cy5-anti-CD86 (GL-1), FITC-anti-granzyme B (NG2B), PerCP-Cy5.5-anti-T-bet (eBio4B10), eFluor 450- anti-TNF-α (MP6-XT22), and eFluor 450-anti-IFN-γ (XMG1.2) antibodies. These antibodies were from e-Biosciences (San Diego, CA). Functional grade anti-CD3ϵ (17A2), APC- and APC-Cy7-anti-CD3ϵ (145-2C11), Alexa Fluor 700-anti-CD4 (RM4-5), Alexa Fluor 700- and BV605-anti-CD8α (53-6.7), purified anti-CD16/32 (2.4G2), PE- and PE-CF594-anti-CD19 (1D3), functional grade, purified anti-CD21/35 (7G6), BV510-anti-CD21/35 (7G6), FITC-anti-CD23 (B3B4), functional grade anti-CD28 (37.51), APC-anti-CD40 (3/23), BV421-anti-CD86 (GL-1), APC-anti-Gr-1 (RB6-8C5), PE-CF594-anti-Ly6C (AL-21), Alexa Fluor 700-anti-Ly6G (1A8), PE-anti-NK1.1 (PK136), APC- and PerCP-Cy5.5-anti-TCRβ (H57–597), PerCP-Cy5.5-anti-IL-4 (11B11), PE-anti-IL-10 (JES5-16E3), APC- and FITC-anti-IFN-γ (XMG1.2), and FITC-anti-TNF-α (MP6-XT22) antibodies were from BD Biosciences. BV605-anti-CD169 (3D6.112), PE-Cy7-anti-CXCR3 (CXCR3–173), and functional grade, purified anti-Fcα/μR (TX61) and PE-anti-Fcα/μR (TX61) antibodies were from BioLegend (San Diego, CA). FITC-anti-CCR2 (475301), PE-anti-CXCR2 (242216), and APC-anti-MARCO (579511) antibodies were from R&D Systems (Minneapolis, MN).
Mice, malaria infection, and treatment with MEK1/2 inhibitors
10–12-week-old gender-matched C57BL/6 WT mice bred in-house, fed routinely with a regular-fat level (6.2%) diet but not high-fat (9.0%) diet, were used. Mice were infected with PbA strain by injection i.p. of blood containing 5 × 105 IRBCs from donor mice in 100 μl of saline. The parasites were obtained from Bei Resources-ATCC (Manassas, VA). For treatment with PD, a fresh 25-mg/ml stock solution in cell culture grade DMSO was prepared, aliquoted for daily use, and stored in the dark at −20 °C. The aliquots were diluted with saline and DMSO to obtain 30–40% final DMSO concentration, and 100 μl of solutions containing 3 mg of PD/kg body weight were administered i.p. Mice were treated daily starting at 6 h pi until 7 days pi. In the case of trametinib, a fresh 10-mg/ml stock solution in DMSO was prepared, aliquots were stored in the dark at −20 °C. The aliquots were diluted with saline containing sterile 0.5% hydroxymethyl cellulose and 0.2% Tween 80, and 100 μl of solution containing 5 mg of trametinib/kg body weight was administered orally daily starting at 6 h pi until 7 days pi. In both treatments, the vehicle-treated mice were used as controls.
The growth of parasites in mice was assessed on alternative days starting from days 2 pi by the light microscopic examination of Giemsa-stained thin blood smears. The control mice exhibited maximal CM symptoms on 7 and 8 days pi, and the majority died at 7–9 days pi. Mice were monitored twice daily for CM clinical symptoms: ataxia, limb paralysis, seizures, coma, and lack of sensory and motor functions. For each of these clinical symptoms, a score of 0, 1, or 2 was given, depending upon whether mice had no symptoms or exhibited a moderate or high level of symptoms, respectively. These scores were added to define the CM status; thus, mice exhibiting no symptoms received an overall score of 0, whereas those exhibiting maximal symptoms in all categories received a score of 10. The overall clinical score of each mouse was plotted.
Ethics statement
The animal care and experiments were performed in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The institutional animal care and use committee of the Pennsylvania State University College of Medicine approved the animal experimental protocols (approval 46661).
The human blood and plasma were purchased from the Blood Bank, Hershey Medical Center (Hershey, Pennsylvania). The institutional review board of the Pennsylvania State University College of Medicine (Hershey, Pennsylvania) approved the study (approval HY03-261).
Assessment of brain vascular leakage
When vehicle-treated mice exhibited peak ECM symptoms (7 days pi), 200 μl of 0.5% Evans blue in sterile saline was administered i.v. to PD-treated, vehicle-treated, and uninfected mice. Evans blue binds to albumin, which enters brain tissue if vascular damage has occurred and the BBB is disrupted (63). After 1 h, mice were euthanized and perfused with PBS. Brains were removed, weighed, and photographed, Evans blue-albumin that intercalated into the brains was extracted by incubating with 0.5 ml of N,N-dimethylformamide overnight at 55 °C. The extracts were centrifuged in a microcentrifuge at 4,000 rpm, and the absorption of the supernatants was measured at 610 nm. The amount of dye in the brain was quantified against a standard curve generated using known concentrations of Evans blue.
H&E staining and assessment of brain pathology
Brains of uninfected and PD- and vehicle-treated mice at 7 days pi were fixed with 4% paraformaldehyde and embedded in paraffin, and 4-μm sections on glass slides were deparaffinized and stained with H&E dye. The stained sections were assessed under light microscopy for pathological features, including sequestered parasites, IRBCs, infiltrated lymphocytes, blood coagulation, swollen and damaged blood vessels, and condensed neuronal cells.
Analysis of ICAM-1 expression
Deparaffinized, 4-μm-thick sections of formalin-fixed brains on glass slides, prepared as above, were treated with 3% hydrogen peroxidase solution to inhibit endoperoxidase activity. The sections were blocked with 5% nonfat milk in Tris-buffered saline, pH 7.6, treated successively with 1:25-diluted rat anti-mouse monoclonal ICAM-1 antibody (e-Biosciences, San Diego, CA), 1:500-diluted biotinylated goat anti-rat Ig (BD Biosciences), and 1:150-diluted HRP-conjugated streptavidin (KPL, Gaithersburg, MD). The bound antibodies were visualized by using 3,3′-diaminobenzidine color developing reagent. The sections were counterstained with Giemsa, mounted, and visualized under light microscopy. The numbers of ICAM-1-positive vessels in 20 randomly selected fields were counted for each mouse, and the mean values ± S.E. were plotted.
P. falciparum culturing
P. falciparum (3D7 strain), obtained from Bei Resources-ATCC (Manassas, VA), were cultured using O-positive human erythrocytes in RPMI 1640 medium containing 10–20% human O-positive plasma as described previously (64). The culture was synchronized by using 5% sorbitol and was tested for mycoplasma using a Myco Sensor Assay kit from Stratagene (La Jolla, CA). Human O-positive blood and plasma were from The Blood Bank, Hershey Medical Center, as per the protocols approved by the institutional review board of the Hershey Medical Center.
Isolation of IRBCs
P. falciparum cultures at the late trophozoite and schizont stages were harvested by centrifuging at 250 × g for 5 min (17). The cell pellets suspended in RPMI 1640 were centrifuged at 1200 × g for 15 min on 65% Percoll cushions. The IRBCs on the top of Percoll cushions were collected and washed with RPMI 1640. For isolation of PbA-IRBCs, blood from the infected mice was diluted with 2 volumes of PBS, pH 7.4, and centrifuged on Isolymph (CTL Scientific Supply, Deer Park, NY) at 1200 × g for 15 min. The cell pellets were suspended in 2 volumes of PBS and centrifuged on 70% Percoll cushions at 1200 × g for 15 min. The IRBCs on the top of Percoll cushions were collected and washed with PBS.
Lysates of the enriched PbA IRBCs were prepared by alternative freezing and thawing and then sonication in a water bath sonicator for 5 min. The lysates were centrifuged at 13,000 rpm in a microcentrifuge for 10 min. The clear supernatants were collected and protein contents were determined by using the Pierce micro-BCA protein estimation kit (Thermo Scientific, Rockford, IL). The supernatants were used for parasite-specific IgM analysis of sera by ELISA (outlined below).
Assessment of the effect of PD on P. falciparum growth
The effect of PD on P. falciparum growth was assessed by a SYBR Green assay (65). Aliquots of a 100 mm stock solution of PD were 1:2 serially diluted with DMSO, and aliquots of these solutions were further diluted with RPMI 1640 medium to obtain 62–4000 μm solutions. The ring stage parasites at 0.2% parasitemia and 2% hematocrit in 180 μl of medium containing 5% Albumax II (Life Technologies, Inc.) were dispensed into 96-well plates and treated in triplicate with 20 μl of the diluted PD solutions to obtain final concentrations ranging from 6.2 to 400 μm. The final concentration of DMSO in each well was <0.4%. Chloroquine, the drug that can completely inhibit the parasite growth, was used as a control. The plates were incubated at 37 °C, and after 72 h, to each well was added 100 μl of 20 mm Tris-HCl, pH 7.5, 5 mm EDTA, 0.008% saponin, and 0.08% Triton X-100 containing 0.2 μl/ml SYBR Green I (Life Technologies). The plates were incubated in the dark at room temperature for 1 h, and the fluorescence intensity was measured using a plate reader at excitation and emission wavelengths of 485 and 535 nm, respectively. The absorption values were expressed as relative fluorescence units.
Assessment of the effect of PD on PbA growth
Blood from PbA-infected mice was collected in a 25-gauge needle containing 50 μl of 1 mg/ml heparin and centrifuged, and erythrocyte pellets were washed with RPMI 1640. The cells (3% parasitemia) were suspended in medium containing 25% fetal calf serum incubated in 96-well plates at 2% hematocrit in the presence of 12, 24, and 48 μg/ml PD at 37 °C. Untreated cells or cells treated with 400 ng/ml chloroquine were used as a control. After 24 h, thin smears of cells on glass slides were prepared and stained with Giemsa, and parasites were assessed under light microscopy.
Analysis of spleen cells
Spleens were disrupted in ice-cold PBS or DMEM with plungers and treated with 1 mg/ml collagenase D at 37 °C for 30 min. The tissue was crushed and centrifuged at 300 × g. The pellets were suspended in RBC lysis buffer, pH 7.4, containing ammonium chloride for 3 min with occasional shaking and then diluted with incomplete DMEM. The cell suspensions were passed through 70-μm strainers and centrifuged at 300 × g. The cells were suspended in incomplete medium and used for the analysis of PMNs, Mφ, DCs, B1 B cells, NK cells, T cells, and CFSE+ phagocytosed cells by flow cytometry after staining with antibodies against appropriate cell surface markers and intracellular proteins.
Isolation of spleen myeloid and T cells
To isolate CD11c+ and CD11b+ cells, total spleen cells were stained with anti-mouse CD11c and anti-human/mouse CD11b microbeads, respectively, and then passed through magnetic columns. The bound cells were recovered. T cells were similarly isolated using anti-CD90.2 antibody-conjugated microbeads. The CD11chiMHCIIhiNK1.1−CD3ϵ−CD19− DCs were purified from CD11c+ spleen cells by FACS sorting.
Isolation of liver cells
Mouse livers were flushed with 10 ml of ice-cold PBS, crushed, and treated with 1 mg/ml of collagenase D at 37 °C. After 30 min, the cells were suspended in PBS, filtered through a 70-μm strainer, and centrifuged at 300 × g for 15 min. The cell pellets suspended in 40% Percoll in PBS were layered on 70% Percoll cushions, and centrifuged at 1200 × g for 15 min. The cell layers on the top of 70% Percoll were collected and washed with PBS, pH 7.2, containing 1% BSA. PMNs, Mφs, Kupffer cells, and CFSE+ phagocytosed cells were analyzed by flow cytometry after staining with antibodies against cell surface marker proteins. The total liver cells isolated as above were also used for the isolation of NK and phagocytic cells by using the mouse NK cell isolation kit and anti-human/mouse CD11b microbeads from Miltenyi Biotec, respectively.
Analysis of brain-infiltrated immune cells
Brains were flushed with ice-cold PBS, crushed, suspended in PBS, and treated with 1 mg/ml collagenase D at 37 °C for 30 min. The cell suspensions were centrifuged at 300 × g, and the cell pellets were suspended in 40% Percoll in PBS and layered on 70% Percoll cushions. After centrifugation at 1200 × g, the cell layers on the top 70% Percoll were collected, washed with PBS, and used for the analysis of PMNs, inflammatory Mφs, and T cells by flow cytometry after staining with antibodies against cell surface marker and intracellular proteins.
Analysis of IRBC phagocytosis
The purified PbA IRBCs, suspended in 1 ml of PBS, were stained with either 2 μm CFSE (in vivo phagocytosis analysis) or 0.5 μm CFSE (in vitro phagocytosis assay) at room temperature. After 10 min, 200 μl of FBS was added, and cells were washed 3 times with PBS. The CFSE-labeled IRBCs (∼2.5 × 107) were injected i.v. into mice at 4 days pi. After overnight, spleen and liver cells, prepared as above, were stained with antibodies against surface markers of PMNs and Mφs, and CFSE+ cells were analyzed by flow cytometry for IRBC uptake.
For the analysis of IRBC phagocytosis in vitro, the CFSE-labeled PbA IRBCs were treated with 1:10 diluted serum from vehicle-treated (control) and PD-treated mice at 5 days pi. The IRBCs were incubated with CD11b+ cells (isolated by using CD11b-conjugated magnetic beads) from spleen and liver of mice at 4 days pi at 1:10 ratios. To access the receptor-mediated phagocytosis, liver CD11b+ cells were treated with 5 μg/ml of either anti-Fcα/μR or anti-CR1/CR2 antibody for 15 min before they were cocultured with CFSE-labeled IRBCs. After 2 h, cells were stained with antibodies against marker proteins of PMN and Mφs, and CFSE+ cells were analyzed by flow cytometry.
Flow cytometry
Total spleen, liver, and brain cells from infected or uninfected mice were treated with Fc block (anti-mouse CD16/32 antibody) for 10 min. The cells were then stained at 4 °C with antibodies against surface marker proteins for 20 min and analyzed by flow cytometry. For analyzing FcγII/IIIR expression, cells were first stained with FITC-labeled anti-mouse CD16/32 antibody and then with antibodies against marker proteins and analyzed by flow cytometry.
For analysis of intracellular proteins, cells were first surface-stained for marker proteins, fixed with 2% paraformaldehyde for 20 min at room temperature, and stained with antibodies against the proteins in PBS containing 1% BSA and 0.5% saponin at room temperature for 35 min. For cytokine analysis, the total spleen cells (1 × 107/well) were cultured in 24-well plates for 6 h in the presence of GolgiPlug and then treated with Fc Block. The cells were then surface-stained with marker proteins, fixed with 2% paraformaldehyde, stained with antibodies against indicated cytokines, and analyzed by flow cytometry. In all cases, flow cytometry analyses were performed with BD LSRII, and the data were analyzed by using FlowJo software.
Analysis of brain chemokines
The brains were suspended and disrupted in ice-cold PBS containing 10 μl/ml of HaltTMProtease and Phosphatase mixture (Pierce/Thermo Scientific) and homogenized by alternatively freezing and thawing three times. The homogenates were centrifuged at 20,800 × g, and supernatants were analyzed for chemokines by ELISA.
Preparation of FL-DCs
The DCs were prepared by culturing mouse bone marrow cells in 6-well plates with DMEM complete medium containing 15% of FLT3 ligand-containing DMEM for 7 or 8 days (17). The FLT3 ligand-containing conditioned medium was prepared by culturing B16 cells expressing retrovirus-encoded FLT3 ligand (66) in DMEM supplemented with 10% FBS, 1% penicillin-streptomycin, non-essential amino acids, 1 mm sodium pyruvate, and 50 μm 2-mercaptoethanol in roller flasks at 37 °C for 4–5 days.
Cell stimulation
To determine malaria parasite-induced DC activation and cytokine production, FL-DCs in 24-well plates (5 × 105 cells/well), either untreated or pretreated with 20 μm PD for 1 h, were stimulated with P. falciparum IRBCs (2 × 106/ml). After 24 h, DCs were stained with antibodies against cell surface markers and analyzed for co-stimulatory molecules by flow cytometry. To assess cytokine responses, FL-DCs in 96-well plates (1 × 105 cells/well) were similarly treated with PD and IRBCs. Cytokines secreted into the culture supernatants were analyzed by ELISA.
To analyze the effect of PD on ex vivo DC-induced production of cytokines by NK cells, spleen DCs (1 × 105 cells/well) from PD-treated and vehicle-treated infected mice at 4 days pi were cocultured with NK cells (5 × 104 cells/well) from uninfected mice in 96-well U-bottomed plates. The cells were stimulated with 2 μg/ml CpG ODN. Wells containing either DCs or NK cells alone were used as controls. After 36 h of culturing, cytokines in the culture supernatants were measured by ELISA.
To determine the effect of PD on the ability of IRBC-stimulated DCs to induce the production of cytokines by NK cells, FL-DCs (1 × 105 cells/well) and NK cells (5 × 104/well) from uninfected mice, either untreated or pretreated with 20 μm PD for 1 h, were cocultured in 96-well U-bottomed plates, stimulated with P. falciparum IRBCs (2 × 106/ml). Wells containing either DCs or NK cells alone were used as controls. After 36 h of culturing, cytokines in the culture supernatants were measured by ELISA.
To determine the effect of PD on stimulus-induced responses, spleen T cells from uninfected mice were either untreated or treated with PD and then stimulated with anti-CD3ϵ antibody coated on plates at 5 μg/ml and with soluble anti-CD28 antibody at 2 μg/ml for 72 h. The cells were surface-stained with anti-CD69 antibody and analyzed by flow cytometry. For cytokine analysis, the antibody-stimulated cells were treated with 20 ng/ml PMA, 500 ng/ml ionomycin in the presence of Golgi block for 6 h, fixed, stained with anti-cytokine antibodies, and analyzed by flow cytometry. To determine T cell proliferation by the dye dilution method, CFSE-labeled T cells were similarly activated and analyzed by flow cytometry.
Serum IgM analysis
The sera from PD- and vehicle-treated mice were analyzed for total IgM and parasite-specific IgM by using a mouse Ig-isotyping ELISA kit from BD Biosciences. To measure total IgM levels, 96-well plates were coated with 1:5-diluted rat anti-mouse IgM monoclonal antibody, blocked with 1% BSA in PBS, pH 7.2, and incubated with 1:10,000-diluted sera from control and PD-treated mice. The bound IgM was measured by using 1:100-diluted HRP-conjugated rat anti-mouse Ig and SureBlueTM TMB peroxidase substrate solution (KPL, Gaithersburg, MD). The reaction was stopped by adding 2 n H2SO4, and absorbance at 450 nm was measured. To measure parasite-specific IgM, 96-well plates were coated with a lysate (20 μg/ml protein) of PbA IRBCs, blocked with 1% BSA, and incubated with 1:100-diluted sera from control and PD-treated mice. The bound IgM was measured by using rat anti-mouse IgM monoclonal antibody followed by HRP-conjugated goat anti-rat IgG (KPL, Inc., Gaithersburg, MD). The color development was as outlined above. Analysis was performed in duplicate.
Western blotting
Spleens from uninfected and vehicle-, PD-, and trametinib-treated mice at 1, 3, and 5 days pi were lysed with radioimmune precipitation buffer and centrifuged, and the protein content in the supernatants was estimated by the BCA method. Aliquots of lysates containing 30 μg of protein/gel lane were electrophoresed on 1.5-mm-thick 10% SDS-polyacrylamide gels under reducing conditions. The protein bands on gels were electroblotted onto nitrocellulose membranes, and the membranes were blocked with 5% nonfat dry milk and probed with antibodies against ERK, p38 JNK, c-Jun, and c-Fos and phospho-specific ERK, p38, and JNK.
Statistical analysis
Statistical analyses of data were performed using GraphPad Prism version 6.01. Statistical significance for the differences in the data from the control and experimental groups was determined by one-way ANOVA followed by the Newman–Keuls test (for results shown in Figs. 1D, 3, 5 (A, C, and D), 6, 8 (B, D, and E), 9, and 11D), by two-way ANOVA followed by Newman-Keuls test (Fig. 5E), or unpaired two-tailed t test (Figs. 2 (C and F), 4, 7, 8C, 10, and 11 (A, B, E, and F)). Mouse survival data were analyzed by log–rank (Mantel–Cox) test (Fig. 2, B and E). The results are presented as the mean ± S.D. or S.E. p values < 0.05 were considered statistically significant. The number of animals assigned to each group and the number of independent repetitions of experiments are given in the figure legends.
Author contributions
D. C. G. and X. W. conceptualized the project. D. C. G., X. W., and C. C. N. designed the experiments and interpreted the results. X. W., K. K. D., and R. P. T. performed the experiments. D. C. G., X. W., and C. C. N. wrote the manuscript.
Supplementary Material
Acknowledgments
The malaria parasites used in this study were obtained through BEI Resources, NIAID, National Institutes of Health. We thank Dr. Glenn Dranoff (Dana-Farber Cancer Institute, Harvard University Medical School) for providing FLT3 ligand-expressing B16 cells.
This work was supported in part by NIAID, National Institutes of Health, Grant R01 AI41139. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S5.
- IRBC
- infected red blood cell
- ICAM-1
- intercellular cell adhesion molecule 1
- CM
- cerebral malaria
- ECM
- experimental CM
- TLR
- Toll-like receptor
- BBB
- blood–brain barrier
- DC
- dendritic cell
- Th
- helper T cell
- ODN
- oligodeoxynucleotide(s)
- PbA
- P. berghei ANKA strain
- FL-DCs
- DCs obtained by the differentiation of mouse bone marrow cells with FLT3 ligand
- FLT3
- Fms-like tyrosine kinase 3
- PMN
- neutrophil
- Mφ
- macrophage
- MM
- marginal zone Mφs
- MMM
- marginal zone metallophilic Mφs
- RPM
- red pulp Mφs
- NK
- natural killer
- pi
- postinfection
- PD
- PD98059
- CFSE
- carboxy fluorescein succinimidyl ester
- CR
- complement receptor
- PE
- phycoerythrin
- APC
- allophycocyanin.
References
- 1. World Health Organization (2015) World Malaria Report, World Health Organization, Geneva [Google Scholar]
- 2. Murray C. J., Rosenfeld L. C., Lim S. S., Andrews K. G., Foreman K. J., Haring D., Fullman N., Naghavi M., Lozano R., and Lopez A. D. (2012) Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet 379, 413–431 [DOI] [PubMed] [Google Scholar]
- 3. Neafsey D. E., Juraska M., Bedford T., Benkeser D., Valim C., Griggs A., Lievens M., Abdulla S., Adjei S., Agbenyega T., Agnandji S. T., Aide P., Anderson S., Ansong D., Aponte J. J., et al. (2015) Genetic diversity and protective efficacy of the RTS,S/AS01 malaria vaccine. N. Engl. J. Med. 373, 2025–2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stanisic D. I., and Good M. F. (2015) Whole organism blood stage vaccines against malaria. Vaccine 33, 7469–7475 [DOI] [PubMed] [Google Scholar]
- 5. Farooq U., and Mahajan R. C. (2004) Drug resistance in malaria. J. Vector Borne Dis. 41, 45–53 [PubMed] [Google Scholar]
- 6. Ashley E. A., Dhorda M., Fairhurst R. M., Amaratunga C., Lim P., Suon S., Sreng S., Anderson J. M., Mao S., Sam B., Sopha C., Chuor C. M., Nguon C., Sovannaroth S., Pukrittayakamee S., et al. (2014) Spread of artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 371, 411–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kwiatkowski D., and Nowak M. (1991) Periodic and chaotic host-parasite interactions in human malaria. Proc. Natl. Acad. Sci. U.S.A. 88, 5111–5113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stevenson M. M., and Riley E. M. (2004) Innate immunity to malaria. Nat. Rev. Immunol. 4, 169–180 [DOI] [PubMed] [Google Scholar]
- 9. van der Heyde H. C., Nolan J., Combes V., Gramaglia I., and Grau G. E. (2006) A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends Parasitol. 22, 503–508 [DOI] [PubMed] [Google Scholar]
- 10. Storm J., and Craig A. G. (2014) Pathogenesis of cerebral malaria: inflammation and cytoadherence. Front. Cell Infect. Microbiol. 4, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miller L. H., Baruch D. I., Marsh K., and Doumbo O. K. (2002) The pathogenic basis of malaria. Nature 415, 673–679 [DOI] [PubMed] [Google Scholar]
- 12. Rowe J. A., Claessens A., Corrigan R. A., and Arman M. (2009) Adhesion of Plasmodium falciparum-infected erythrocytes to human cells: molecular mechanisms and therapeutic implications. Expert Rev. Mol. Med. 11, e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schofield L., and Grau G. E. (2005) Immunological processes in malaria pathogenesis. Nat. Rev. Immunol. 5, 722–735 [DOI] [PubMed] [Google Scholar]
- 14. Campanella G. S., Tager A. M., El Khoury J. K., Thomas S. Y., Abrazinski T. A., Manice L. A., Colvin R. A., and Luster A. D. (2008) Chemokine receptor CXCR3 and its ligands CXCL9 and CXCL10 are required for the development of murine cerebral malaria. Proc. Natl. Acad. Sci. U.S.A. 105, 4814–4819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Howland S. W., Claser C., Poh C. M., Gun S. Y., and Rénia L. (2015) Pathogenic CD8+ T cells in experimental cerebral malaria. Semin. Immunopathol. 37, 221–231 [DOI] [PubMed] [Google Scholar]
- 16. Gazzinelli R. T., Kalantari P., Fitzgerald K. A., and Golenbock D. T. (2014) Innate sensing of malaria parasites. Nat. Rev. Immunol. 14, 744–757 [DOI] [PubMed] [Google Scholar]
- 17. Wu X., Gowda N. M., Kumar S., and Gowda D. C. (2010) Protein-DNA complex is the exclusive malaria parasite component that activates dendritic cells and triggers innate immune responses. J. Immunol. 184, 4338–4348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhu J., Krishnegowda G., and Gowda D. C. (2005) Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of Plasmodium falciparum: the requirement of extracellular signal-regulated kinase, p38, c-Jun N-terminal kinase and NF-κB pathways for the expression of proinflammatory cytokines and nitric oxide. J. Biol. Chem. 280, 8617–8627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhu J., Wu X., Goel S., Gowda N. M., Kumar S., Krishnegowda G., Mishra G., Weinberg R., Li G., Gaestel M., Muta T., and Gowda D. C. (2009) MAPK-activated protein kinase 2 differentially regulates Plasmodium falciparum glycosylphosphatidylinositol-induced production of tumor necrosis factor-α and interleukin-12 in macrophages. J. Biol. Chem. 284, 15750–15761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Coban C., Ishii K. J., Uematsu S., Arisue N., Sato S., Yamamoto M., Kawai T., Takeuchi O., Hisaeda H., Horii T., and Akira S. (2007) Pathological role of Toll-like receptor signaling in cerebral malaria. Int. Immunol. 19, 67–79 [DOI] [PubMed] [Google Scholar]
- 21. Akira S. (2009) Innate immunity to pathogens: diversity in receptors for microbial recognition. Immunol. Rev. 227, 5–8 [DOI] [PubMed] [Google Scholar]
- 22. Kim E. K., and Choi E. J. (2010) Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 1802, 396–405 [DOI] [PubMed] [Google Scholar]
- 23. de Souza J. B., Hafalla J. C., Riley E. M., and Couper K. N. (2010) Cerebral malaria: why experimental murine models are required to understand the pathogenesis of disease. Parasitology 137, 755–772 [DOI] [PubMed] [Google Scholar]
- 24. de Oca M. M., Engwerda C., and Haque A. (2013) Plasmodium berghei ANKA (PbA) infection of C57BL/6J mice: a model of severe malaria. Methods Mol. Biol. 1031, 203–213 [DOI] [PubMed] [Google Scholar]
- 25. Hora R., Kapoor P., Thind K. K., and Mishra P. C. (2016) Cerebral malaria: clinical manifestations and pathogenesis. Metab. Brain. Dis. 31, 225–237 [DOI] [PubMed] [Google Scholar]
- 26. Seydel K. B., Kampondeni S. D., Valim C., Potchen M. J., Milner D. A., Muwalo F. W., Birbeck G. L., Bradley W. G., Fox L. L., Glover S. J., Hammond C. A., Heyderman R. S., Chilingulo C. A., Molyneux M. E., and Taylor T. E. (2015) Brain swelling and death in children with cerebral malaria. N. Engl. J. Med. 372, 1126–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Taylor T. E., Fu W. J., Carr R. A., Whitten R. O., Mueller J. S., Fosiko N. G., Lewallen S., Liomba N. G., and Molyneux M. E., and Mueller J. G. (2004) Differentiating the pathologies of cerebral malaria by postmortem parasite counts. Nat. Med. 10, 143–145 [DOI] [PubMed] [Google Scholar]
- 28. Yamaguchi T., Kakefuda R., Tajima N., Sowa Y., and Sakai T. (2011) Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int. J. Oncol. 39, 23–31 [DOI] [PubMed] [Google Scholar]
- 29. Cabrera A., Neculai D., and Kain K. C. (2014) CD36 and malaria: friends or foes? A decade of data provides some answers. Trends Parasitol. 30, 436–444 [DOI] [PubMed] [Google Scholar]
- 30. Anstey N. M., Russell B., Yeo T. W., and Price R. N. (2009) The pathophysiology of vivax malaria. Trends Parasitol. 25, 220–227 [DOI] [PubMed] [Google Scholar]
- 31. Amante F. H., Haque A., Stanley A. C., Rivera Fde L., Randall L. M., Wilson Y. A., Yeo G., Pieper C., Crabb B. S., de Koning-Ward T. F., Lundie R. J., Good M. F., Pinzon-Charry A., Pearson M. S., Duke M. G., et al. (2010) Immune-mediated mechanisms of parasite tissue sequestration during experimental cerebral malaria. J. Immunol. 185, 3632–3642 [DOI] [PubMed] [Google Scholar]
- 32. Hashimoto D., Miller J., and Merad M. (2011) Dendritic cell and macrophage heterogeneity in vivo. Immunity 35, 323–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Areschoug T., and Gordon S. (2009) Scavenger receptors: role in innate immunity and microbial pathogenesis. Cell Microbiol. 11, 1160–1169 [DOI] [PubMed] [Google Scholar]
- 34. Turrini F., Ginsburg H., Bussolino F., Pescarmona G. P., Serra M. V., and Arese P. (1992) Phagocytosis of Plasmodium falciparum-infected human red blood cells by human monocytes: involvement of immune and nonimmune determinants and dependence on parasite developmental stage. Blood 80, 801–808 [PubMed] [Google Scholar]
- 35. Osier F. H., Feng G., Boyle M. J., Langer C., Zhou J., Richards J. S., McCallum F. J., Reiling L., Jaworowski A., Anders R. F., Marsh K., and Beeson J. G. (2014) Opsonic phagocytosis of Plasmodium falciparum merozoites: mechanism in human immunity and a correlate of protection against malaria. BMC Med. 12, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Couper K. N., Phillips R. S., Brombacher F., and Alexander J. (2005) Parasite-specific IgM plays a significant role in the protective immune response to asexual erythrocytic stage Plasmodium chabaudi AS infection. Parasite Immunol. 27, 171–180 [DOI] [PubMed] [Google Scholar]
- 37. Pleass R. J., Moore S. C., Stevenson L., and Hviid L. (2016) Immunoglobulin M: restrainer of inflammation and mediator of immune evasion by Plasmodium falciparum malaria. Trends Parasitol. 32, 108–119 [DOI] [PubMed] [Google Scholar]
- 38. Notley C. A., Brown M. A., Wright G. P., and Ehrenstein M. R. (2011) Natural IgM is required for suppression of inflammatory arthritis by apoptotic cells. J. Immunol. 186, 4967–4972 [DOI] [PubMed] [Google Scholar]
- 39. Grönwall C., Vas J., and Silverman G. J. (2012) Protective roles of natural IgM antibodies. Front. Immunol. 3, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kyaw T., Tipping P., Bobik A., and Toh B. H. (2012) Protective role of natural IgM-producing B1a cells in atherosclerosis. Trends Cardiovasc. Med. 22, 48–53 [DOI] [PubMed] [Google Scholar]
- 41. Racine R., and Winslow G. M. (2009) IgM in microbial infections: taken for granted? Immunol. Lett. 125, 79–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Baumgarth N. (2011) The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat. Rev. Immunol. 11, 34–46 [DOI] [PubMed] [Google Scholar]
- 43. Contreras C. E., June C. H., Perrin L. H., and Lambert P. H. (1980) Immunopathological aspects of Plasmodium berghei infection in five strains of mice. I. Immune complexes and other serological features during the infection. Clin. Exp. Immunol. 42, 403–411 [PMC free article] [PubMed] [Google Scholar]
- 44. Shibuya A., Sakamoto N., Shimizu Y., Shibuya K., Osawa M., Hiroyama T., Eyre H. J., Sutherland G. R., Endo Y., Fujita T., Miyabayashi T., Sakano S., Tsuji T., Nakayama E., Phillips J. H., Lanier L. L., and Nakauchi H. (2000) Fc α/μ receptor mediates endocytosis of IgM-coated microbes. Nat. Immunol. 1, 441–446 [DOI] [PubMed] [Google Scholar]
- 45. Teo A., Feng G., Brown G. V., Beeson J. G., and Rogerson S. J. (2016) Functional antibodies and protection against blood-stage malaria. Trends Parasitol. 32, 887–898 [DOI] [PubMed] [Google Scholar]
- 46. Artavanis-Tsakonas K., Tongren J. E., and Riley E. M. (2003) The war between the malaria parasite and the immune system: immunity, immunoregulation and immunopathology. Clin. Exp. Immunol. 133, 145–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Perry J. A., Olver C. S., Burnett R. C., and Avery A. C. (2005) Cutting edge: the acquisition of TLR tolerance during malaria infection impacts T cell activation. J. Immunol. 174, 5921–5925 [DOI] [PubMed] [Google Scholar]
- 48. Deroost K., Pham T.-T., Opdenakker G., and Van den Steen P. E. (2016) The immunological balance between host and parasite in malaria. FEMS Microbiol. Rev. 40, 208–257 [DOI] [PubMed] [Google Scholar]
- 49. Gowda N. M., Wu X., and Gowda D. C. (2012) TLR9 and MyD88 are crucial for the development of protective immunity to malaria. J. Immunol. 188, 5073–5085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wu X., Gowda N. M., and Gowda D. C. (2015) Phagosomal acidification prevents macrophage inflammatory cytokine production to malaria, and dendritic cells are the major source at the early stages of infection: implication for malaria protective immunity development. J. Biol. Chem. 290, 23135–23147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Villegas-Mendez A., Greig R., Shaw T. N., de Souza J. B., Gwyer Findlay E., Stumhofer J. S., Hafalla J. C., Blount D. G., Hunter C. A., Riley E. M., and Couper K. N. (2012) IFN-γ-producing CD4+ T cells promote experimental cerebral malaria by modulating CD8+ T cell accumulation within the brain. J. Immunol. 189, 968–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Haque A., Best S. E., Unosson K., Amante F. H., de Labastida F., Anstey N. M., Karupiah G., Smyth M. J., Heath W. R., and Engwerda C. R. (2011) Granzyme B expression by CD8+ T cells is required for the development of experimental cerebral malaria. J. Immunol. 186, 6148–6156 [DOI] [PubMed] [Google Scholar]
- 53. Stevenson M. M., and Urban B. C. (2006) Antigen presentation and dendritic cell biology in malaria. Parasite Immunol. 28, 5–14 [DOI] [PubMed] [Google Scholar]
- 54. Ing R., and Stevenson M. M. (2009) Dendritic cell and NK cell reciprocal cross talk promotes γ interferon-dependent immunity to blood-stage Plasmodium chabaudi AS infection in mice. Infect. Immun. 77, 770–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schumak B., Klocke K., Kuepper J. M., Biswas A., Djie-Maletz A., Limmer A., van Rooijen N., Mack M., Hoerauf A., and Dunay I. R. (2015) Specific depletion of Ly6Chi inflammatory monocytes prevents immunopathology in experimental cerebral malaria. PLoS One 10, e0124080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ryg-Cornejo V., Nie C. Q., Bernard N. J., Lundie R. J., Evans K. J., Crabb B. S., Schofield L., and Hansen D. S. (2013) NK cells and conventional dendritic cells engage in reciprocal activation for the induction of inflammatory responses during Plasmodium berghei ANKA infection. Immunobiology 218, 263–271 [DOI] [PubMed] [Google Scholar]
- 57. Denucci C. C., Mitchell J. S., and Shimizu Y. (2009) Integrin function in T cell homing to lyphpoid and non-lymphoid sites: Getting there and staying there. Crit. Rev. Immunol. 29, 87–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Parroche P., Lauw F. N., Goutagny N., Latz E., Monks B. G., Visintin A., Halmen K. A., Lamphier M., Olivier M., Bartholomeu D. C., Gazzinelli R. T., and Golenbock D. T. (2007) Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc. Natl. Acad. Sci. U.S.A. 104, 1919–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Weiss G. E., Crabb B. S., and Gilson P. R. (2016) Overlaying molecular and temporal aspects of malaria parasite invasion. Trends Parasitol. 32, 284–295 [DOI] [PubMed] [Google Scholar]
- 60. Hentati B., Sato M. N., Payelle-Brogard B., Avrameas S., and Ternynck T. (1994) Beneficial effect of polyclonal immunoglobulins from malaria-infected BALB/c mice on the lupus-like syndrome of (NZB × NZW)F1 mice. Eur. J. Immunol. 24, 8–15 [DOI] [PubMed] [Google Scholar]
- 61. Lobo P. I., Schlegel K. H., Bajwa A., Huang L., Kurmaeva E., Wang B., Ye H., Tedder T. F., Kinsey G. R., and Okusa M. D. (2015) Natural IgM switches the function of lipopolysaccharide-activated murine bone marrow-derived dendritic cells to a regulatory dendritic cell that suppresses innate inflammation. J. Immunol. 195, 5215–5226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Clark C. J., and Phillips R. S. (2011) Cerebral malaria protection in mice by species-specific Plasmodium coinfection is associated with reduced CC chemokine levels in the brain. Parasite Immunol. 33, 637–641 [DOI] [PubMed] [Google Scholar]
- 63. Nacer A., Movila A., Baer K., Mikolajczak S. A., Kappe S. H., and Frevert U. (2012) Neuroimmunological blood brain barrier opening in experimental cerebral malaria. PLoS Pathog. 8, e1002982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Alkhalil A., Achur R. N., Valiyaveettil M., Ockenhouse C. F., and Gowda D. C. (2000) Structural requirements for the adherence of Plasmodium falciparum-infected erythrocytes to chondroitin sulfate proteoglycans of human placenta. J. Biol. Chem. 275, 40357–40364 [DOI] [PubMed] [Google Scholar]
- 65. Vinayaka A. C., Sadashiva M. P., Wu X., Biryukov S. S., Stoute J. A., Rangappa K. S., and Gowda D. C. (2014) Facile synthesis of antimalarial 1,2-disubstituted 4-quinolones from 1,3-bisaryl-monothio-1,3-diketones. Org. Biomol. Chem. 12, 8555–8561 [DOI] [PubMed] [Google Scholar]
- 66. Brasel K., De Smedt T., Smith J. L., and Maliszewski C. R. (2000) Generation of murine dendritic cells from flt3-ligand-supplemented bone marrow cultures. Blood 96, 3029–3039 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.