

Abstract
Annexin A1 (AnxA1) is a glucocorticoid-regulated protein known for its anti-inflammatory and pro-resolving effects. We have shown previously that the cAMP-enhancing compounds rolipram (ROL; a PDE4 inhibitor) and Bt2cAMP (a cAMP mimetic) drive caspase-dependent resolution of neutrophilic inflammation. In this follow-up study, we investigated whether AnxA1 could be involved in the pro-resolving properties of these compounds using a model of LPS-induced inflammation in BALB/c mice. The treatment with ROL or Bt2cAMP at the peak of inflammation shortened resolution intervals, improved resolution indices, and increased AnxA1 expression. In vitro studies showed that ROL and Bt2cAMP induced AnxA1 expression and phosphorylation, and this effect was prevented by PKA inhibitors, suggesting the involvement of PKA in ROL-induced AnxA1 expression. Akin to these in vitro findings, H89 prevented ROL- and Bt2cAMP-induced resolution of inflammation, and it was associated with decreased levels of intact AnxA1. Moreover, two different strategies to block the AnxA1 pathway (by using N-t-Boc-Met-Leu-Phe, a nonselective AnxA1 receptor antagonist, or by using an anti-AnxA1 neutralizing antiserum) prevented ROL- and Bt2cAMP-induced resolution and neutrophil apoptosis. Likewise, the ability of ROL or Bt2cAMP to induce neutrophil apoptosis was impaired in AnxA-knock-out mice. Finally, in in vitro settings, ROL and Bt2cAMP overrode the survival-inducing effect of LPS in human neutrophils in an AnxA1-dependent manner. Our results show that AnxA1 is at least one of the endogenous determinants mediating the pro-resolving properties of cAMP-elevating agents and cAMP-mimetic drugs.
Keywords: annexin, apoptosis, cyclic AMP (cAMP), inflammation, neutrophil, protein kinase A (PKA)
Introduction
Annexin A1 (AnxA1;4 previously known as lipocortin-1) is a 37-kDa member of the annexin superfamily, which is composed of proteins that bind to cellular membranes in a calcium-dependent manner. Originally described as an endogenous mediator of the anti-inflammatory effects of glucocorticoids, over the past 20 years, AnxA1 has been shown to have a broad range of molecular and cellular actions, including modulation of leukocyte migration in acute and chronic inflammation, kinase activities in signal transduction, preservation of cytoskeleton and extracellular matrix integrity, tissue maintenance, apoptosis, cell growth, and differentiation (1–5). AnxA1 is particularly abundant in cells of the myeloid lineage, including neutrophils, eosinophils, macrophages, and mast cells (6). In resting cells, AnxA1 is by and large localized in the cytosol, and upon activation, it can be secreted and then resynthesized. Once in the extracellular medium, this protein exerts autocrine, paracrine, and juxtacrine effects that are mediated by the FPR2/ALX (formyl peptide receptor 2/lipoxin A4 receptor) receptor (7–11).
AnxA1 exerts a variety of anti-inflammatory effects, including inhibition of leukocyte migration, direct inhibition of cytosolic phospholipase A2 (cPLA2), inhibition of COX-2 and iNOS expression and stimulation of IL-10 release (12–18). AnxA1 also possesses genuine pro-resolving properties by inducing neutrophil apoptosis (10, 19, 20) and increasing the clearance of apoptotic cells by efferocytosis (10, 21–23). Both apoptosis and efferocytosis modulated by AnxA1 are crucial for resolution of inflammation (10, 20).
In the context of the discovery of new mechanisms for known drugs, production and action of AnxA1 has been shown to be involved in the pro-resolution effects of histone deacetylase (HDAC) inhibitors (24) as well as in the anti-inflammatory effects of propofol (25) and cromoglycate-like compounds (6, 26). We have shown previously that rolipram (ROL), a selective phosphodiesterase-4 (PDE4) inhibitor that increases intracellular levels of cyclic adenosine monophosphate (cAMP), induces resolution of neutrophilic inflammation, and it was associated with increased accumulation of AnxA1 in inflammatory cells (20). However, the relevance and mechanisms underlying ROL-induced AnxA1 expression remain unknown.
In addition to the known anti-inflammatory properties of cAMP-elevating agents (27–29), emerging data support a role for cAMP in some steps of the resolution process (30–37). Indeed, cAMP elevation promoted by treatment with ROL or by cAMP-mimetic drugs during established eosinophilic or neutrophilic inflammation induced resolution of inflammation via PKA, the best known cAMP effector (3, 32). Of note, modulation of cAMP may account for the pro-resolving abilities of melanocortin peptides (38) and lysophosphatidylserine (39). In this study, we investigated the ability of ROL and Bt2cAMP to modulate AnxA1 expression and wondered whether AnxA1 was involved in the pro-resolving ability of these compounds. Our results demonstrate that AnxA1 is induced by cAMP-elevating agents and is indeed involved in the pro-resolving properties of these compounds, pointing to them as potential therapeutic tools to control inflammatory diseases and induce resolution of inflammation.
Results
Rolipram and Bt2cAMP promote resolution of neutrophilic inflammation associated with increased AnxA1 expression
Initially, we evaluated whether ROL (PDE4 inhibitor) and Bt2cAMP (cAMP mimetic) would improve resolution indices during acute pleurisy. In this self-resolving model of inflammation, the intrapleural injection of LPS induces a time-dependent influx of neutrophils into the pleural cavity of mice that is detectable at 2 h and reaches a maximum at 8–24 h, decreasing thereafter with resolution occurring after 48 h, as reported previously (20, 33, 40, 41). Therefore, we quantified the resolution interval (Ri) by defining profiles of acute inflammatory parameters (42, 43). The treatment of mice with ROL or Bt2cAMP at the peak of LPS inflammation significantly reduced the number of PMNs recruited to the pleural cavity and shortened Ri in ∼12 h (Fig. 1, A and B).
Figure 1.

Effect of the treatment with rolipram and Bt2cAMP on resolution of acute inflammation. For the evaluation of the resolution indices, mice were injected with LPS (250 ng/cavity, i.pl.) or PBS and 8 h later received an injection of ROL (6 mg/kg, i.p.) or Bt2cAMP (4 mg/kg, i.pl.). Pleural washes were performed at various time points after LPS injection, and neutrophils were counted from cytospin preparations (A) to calculate resolution indices (B). In C–F, mice were injected with LPS (250 ng/cavity, i.pl.) or PBS and 4 h later received an injection of ROL or Bt2cAMP at the same dose as in A or Dexa (2 mg/kg, i.p.) as a control. The cells from the pleural cavity were harvested and processed for neutrophil count (C and D) and Western blot analysis (E and F) for detection of AnxA1 4 h after drug treatment (i.e. 8 h after LPS challenge). Two different exposure times of the cleaved band of the AnxA1 immunoblot are presented. Results are expressed as number of neutrophils/cavity and are shown as the mean ± S.E. (error bars) of at least five mice in each group. ***, p < 0.001 when compared with PBS-injected mice. ##, p < 0.01; ###, p < 0.001 when compared with LPS-challenged mice. For loading control, membranes were reprobed with anti-β-actin. Blots are representative of three independent experiments using pooled cells from at least five animals in each experiment.
To investigate the potential relationship between cAMP and AnxA1, we carried out Western blot analysis in whole-cell extracts recovered from the pleural cavity of mice treated 4 h after LPS challenge (when inflammatory cell influx was already established). Western blotting was performed to quantify the overall AnxA1 content (i.e. the sum of intracellularly localized or cell surface-bound). As seen in Fig. 1 (C and D), treatment with ROL or Bt2cAMP decreased neutrophil numbers and increased levels of intact AnxA1 (Fig. 1, E and F). AnxA1 was constitutively expressed on resident cells from the pleural cavity (PBS-injected). LPS injection induced AnxA1 cleavage (as detected by the presence of the 33-kDa breakdown product), and treatment with ROL or Bt2cAMP increased levels of intact AnxA1 (37-kDa form) and reduced AnxA1 cleavage in whole inflammatory extracts when compared with LPS.
Dexamethasone, used as an anti-inflammatory control drug, promoted resolution of neutrophilic inflammation and increased AnxA1 expression, as shown previously (20). Interestingly, ROL was able to decrease neutrophil numbers and increase intact levels of AnxA1 as early as 1 h after treatment (supplemental Fig. 1, A and B).
The N-terminal region of AnxA1 is the major effector portion responsible for the anti-inflammatory action of the protein, and its cleavage decreases the resolution-inducing effects of AnxA1 (10, 44). Here, we observed that ROL partially decreased LPS-induced AnxA1 cleavage, as evidenced by the lower levels of the 33-kDa fragment when compared with LPS alone (Fig. 1E). Because AnxA1 can be cleaved by elastase in vivo, we evaluated whether inhibition of elastase activity by ROL could account for the observed effect. We found that ROL decreased LPS-induced elastase activity and expression in whole-cell lysates recovered from pleural inflammatory cells (supplemental Fig. 1, C and D). The latter results suggest that ROL modulates AnxA1 levels at least by reducing its degradation.
Regulation of AnxA1 expression and phosphorylation by cAMP in macrophages
AnxA1 is present in many differentiated cell types (human and murine) but is particularly abundant in neutrophils, eosinophils, macrophages, and mast cells (2, 6). Protein expression can be modulated through several mechanisms, such as the mobilization of the intracellular pool of the protein to exportation and secretion, increased phosphorylation (45), or both. To explore the mechanisms underlying the effect of cAMP-elevating agents on AnxA1 expression and localization, in vitro experiments using differentiated THP-1 cells, bone marrow–derived macrophages (BMDMs), and the murine macrophage cell line RAW264.7 were carried out. THP-1 was used in this work to evaluate the expression of AnxA1, because it has been shown to be a suitable cell line to study AnxA1 modulation (45, 46). In these experimental settings, dexamethasone (Dexa) treatment induced dose-dependent induction of AnxA1 expression (data not show). As shown in Fig. 2, treatment of THP-1 cells with ROL increased AnxA1 levels in a concentration-dependent (Fig. 2A) and time-dependent (Fig. 2C) fashion. This modulatory property was also observed in BMDM and RAW264.7 cells (supplemental Fig. 2, A–C). For more quantitative data, similar experiments were performed to quantify AnxA1 message by qPCR. Significant increases in AnxA1 mRNA were observed in THP-1 cells treated with ROL, with optimal settings at 10 μm and 6-h incubation (Fig. 2, B and D, respectively). Interestingly, ROL also increased the level of phosphorylated AnxA1 in THP-1 cells (Fig. 2E) and BMDMs (supplemental Fig. 2A). In line with the ability of ROL to inhibit degradation and increase intracellular levels of cAMP, there was strong phosphorylation of the cAMP-responsive element (CRE)-binding protein (CREB), which followed the same kinetics of AnxA1 expression (Fig. 2, A and C). In accordance with the requirement of cAMP levels to induce AnxA1 expression, a cell-permeable cAMP (Bt2cAMP) induced AnxA1 accumulation and phosphorylation (Fig. 3, A and B). Densitometry data for both AnxA1 and P-AnxA1 are represented graphically (Fig. 3C). We also measured the intracellular levels of cAMP in THP-1 cells after rolipram treatment (10 μm), and we found an increase of 20% over basal levels, which return to the baseline 2 h after. As expected for a cell-permeable cAMP, Bt2cAMP greatly increased levels of intracellular cAMP 1 h after cell treatment by 150% over basal level, decreasing thereafter but still remaining high until 2 h (85% increase over basal levels).
Figure 2.

Effect of rolipram on AnxA1 mRNA expression, protein levels, and phosphorylation in THP-1 differentiated macrophages. Cells were differentiated using PMA (20 ng/ml) and serum-deprived for 24 h. Later, the cells were untreated or treated with ROL at increasing concentrations for 6 h (A, B, and E) or for different time intervals (C–E) as indicated. Whole-cell extracts were obtained and subjected to Western blot analysis (A, C, and E) to assess for AnxA1, Ser27-phospho-AnxA1, and phospho-CREB levels (as a marker of PKA activation) or for quantitative RT-PCR (B and D). For loading control, membranes were reprobed with anti-β-actin. Blots are representative of three independent experiments. qRT-PCR data were performed in biological triplicates performed with two technical replicates. The results are presented as -fold increase of mRNA expression relative to the amount present in control samples. Data are mean ± S.E. (error bars). **, p < 0.01; ***, p < 0.001 when compared with untreated cells. ###, p < 0.001 when compared with ROL treatment at 10 μm for 6 h.
Figure 3.

Effect of Bt2cAMP and forskolin on AnxA1 expression and phosphorylation in THP-1 differentiated macrophages. Cells were differentiated using PMA (20 ng/ml) and serum-deprived for 24 h. After starvation, the cells were untreated or treated with Bt2cAMP (A and B) or forskolin (D) at different concentrations (6 h) and times as indicated in the figures. Total cell extracts were obtained and subjected to Western blot analysis to assess for AnxA1 (A, B, and D) or Ser27-phospho-AnxA1 (A). Densitometry data are presented graphically in C. For the loading control, membranes were reprobed with anti-β-actin. Blots are representative of three independent experiments. Data are mean ± S.E. (error bars). *, p < 0.05; **, p < 0.01; ***, p < 0.001 when compared with untreated cells.
Moreover, forskolin, a direct activator of adenylate cyclase, was also able to increase AnxA1 levels (Fig. 3D). It is noteworthy that physiological cAMP-elevating compounds, such as prostaglandin E2 (PGE2), norepinephrine, and adenosine (36, 47–51), were able to increase AnxA1 protein levels, as shown in supplemental Fig. 3 (A–C). Likewise, monobutyryladenosine cAMP (6MB-cAMP), a membrane-permeable analog of cAMP that activates protein kinase A and is resistant to degradation by phosphodiesterase, was able to increase AnxA1 accumulation, as did Bt2cAMP (supplemental Fig. 3D). In contrast, a naked cAMP or a cell-permeable cGMP analog (8-Br-cGMP) did not increase AnxA1 accumulation, as analyzed by Western blotting (see supplemental Fig. 3E).
Taken together, the data gathered so far suggested that ROL and other agents that elevate or mimic cAMP are acting in several ways to regulate the dynamics of AnxA1 accumulation: they increase mRNA expression, protein accumulation, and phosphorylation of AnxA1.
Rolipram and Bt2cAMP induce PKA-dependent AnxA1 expression
The promoter region of the AnxA1 gene contains one CRE, and this is functional because a CREB is required for either Dexa-induced or cAMP-induced AnxA1 synthesis (8, 52). To investigate whether the observed effects of ROL occurred via PKA, the best-known cAMP downstream effector, THP-1-differentiated macrophages were treated with two PKA inhibitors, H89 (nonselective) or cAMPS-Rp (highly selective), 30 min before ROL or Bt2cAMP treatments. As shown in supplemental Fig. 4 (A–C), the blockade of PKA with H89 or cAMPS-Rp decreased AnxA1 mRNA and protein levels induced by both ROL and Bt2cAMP. Of note, the effect of PKA inhibitors on ROL-induced AnxA1 levels was also observed in RAW264.7 murine macrophages (supplemental Fig. 2C).
Because AnxA1 expression was associated with the pro-resolving role of cAMP (Fig. 1) and the expression of AnxA1 in vitro was modulated via PKA (supplemental Figs. 2C and 4), we investigated whether such a pathway could also be engaged in vivo. In agreement with the in vitro findings, inhibition of PKA by H89 prevented ROL and Bt2cAMP-induced resolution of neutrophilic inflammation (Fig. 4, A and B), and this effect was associated with reduction of intact AnxA1 and increase of the cleaved form (Fig. 4, C and D).
Figure 4.

Rolipram and Bt2cAMP resolve neutrophilic inflammation in a PKA-dependent manner. Mice were injected with LPS (250 ng/cavity, i.pl.) or PBS and 4 h later received an injection of ROL (6 mg/kg, i.p.) or Bt2cAMP (4 mg/kg, i.pl.). Two groups of mice were pretreated for 30 min with H89 (4 mg/kg, i.pl.) before the drugs. The cells from the pleural cavity were harvested and processed to neutrophil count (A and B) and Western blot (C and D) for detection of AnxA1 4 h after drug treatment (i.e. 8 h after LPS challenge). Two different exposure times of the cleaved band of the AnxA1 immunoblot are presented. Results are expressed as the number of neutrophils/cavity and are shown as the mean ± S.E. (error bars) of at least five mice in each group. ***, p < 0.001 when compared with PBS-injected mice. #, p < 0.05; ##, p < 0.01 when compared with LPS-challenged mice. Comparison between the groups H89 and H89 + drugs are highlighted in the graphics. For loading control, membranes were reprobed with anti-β-actin. Blots are representative of three independent experiments in pools of cells from at least five animals in each experiment.
A nonselective FPR antagonist prevents rolipram and Bt2cAMP-induced resolution of neutrophilic inflammation
FPR2/ALX, a G protein–coupled member of the formyl peptide receptor (FPR) family, conveys the biological functions of a variety of ligands, including the pro-resolving mediators AnxA1 and lipoxin A4 (9). To investigate whether there was involvement of these receptors in our system, we used the nonselective antagonist N-t-Boc-Met-Leu-Phe (BOC-1), which also blocks FPR1. Administration of BOC-1, before ROL or Bt2cAMP injection, prevented resolution of inflammation induced by these cAMP-elevating agents (Fig. 5, A and B) as seen by the permanence of neutrophil and decreased apoptosis into the pleural cavity. Apoptosis was evaluated biochemically through Mcl-1, the most important Bcl-2 family protein that governs neutrophil half-life (53, 54) (Fig. 5, C and D), and annexin V staining (Fig. 5F) or by morphological criteria (Fig. 5E). Mcl-1 is a key anti-apoptotic protein of the Bcl-2 family protein known to be modulated by ROL (33). Of note, treatment of mice with BOC-1 alone had no effect on neutrophil counts (data not shown) and apoptosis (Fig. 5, E and F). Prevention of ROL-induced apoptosis by BOC-1 was associated with decreased levels of intact AnxA1 paralleled by an increase of the cleaved form in cells from pleural exudates (Fig. 5, C and D).
Figure 5.

Effect of treatment with BOC-1, a FPR/ALX antagonist, on ROL and Bt2cAMP-induced resolution of acute inflammation. Mice were injected with LPS (250 ng/cavity, i.pl.) or PBS and 4 h later received an injection of ROL (6 mg/kg, i.p.) or Bt2cAMP (4 mg/kg, i.pl.). An injection of BOC-1 (5 mg/kg, i.p.) was given 30 min before the drugs. The cells from the pleural cavity were harvested and processed to neutrophil count (A and B) and Western blot analysis (C and D) for detection of AnxA1 and Mcl-1 4 h after drug treatment (i.e. 8 h after LPS challenge). Two different exposure times of the cleaved band of AnxA1 immunoblot are presented. The number of apoptotic neutrophils was determined morphologically (E) and by flow cytometry of annexin V+ neutrophils (F) 24 h after LPS injection. Results are expressed as the number of neutrophils/cavity (A and B), percentage of neutrophils with apoptotic morphology (E), and number of apoptotic neutrophils (Ly6G+/F4/80−/AnxAV+/7AAD−) (F) and are shown as the mean ± S.E. (error bars) of at least five mice in each group. *, p < 0.05; ***, p < 0.001 when compared with PBS-injected mice. #, p < 0.05; ##, p < 0.01; ###, p < 0.001 when compared with LPS-challenged mice. Comparisons between the groups BOC-1 and BOC-1 + drugs are highlighted in the graphics. For loading control, membranes were reprobed with anti-β-actin. Blots are representative of three independent experiments using pooled cells from at least five animals in each experiment.
Neutralization of endogenous AnxA1 prevents rolipram and Bt2cAMP-induced resolution of neutrophilic inflammation
Having established the effect of AnxA1 receptor blockade on ROL- and Bt2cAMP-induced resolution (Fig. 5), we evaluated the effects of an anti-AnxA1 neutralizing strategy by using a specific antiserum. The administration of the anti-AnxA1 antiserum prevented ROL-induced resolution (Fig. 6A) and apoptosis, as assessed using either morphological criteria (Fig. 6, B and E) or biochemically by Mcl-1 (Fig. 6C) and annexin V staining (Fig. 6D). Of note, treatment of mice with a goat nonimmune serum had no effect on the resolution of LPS-induced pleurisy (data not shown), reinforcing a previous report (20). AnxA1 neutralization was also able to prevent the effect of Bt2cAMP on neutrophil numbers (Fig. 7A) and apoptosis (Fig. 7, B–E), similar to the results obtained with ROL.
Figure 6.

Effect of treatment with anti-AnxA1 antiserum on ROL-induced resolution of acute inflammation. Mice were injected with LPS (250 ng/cavity, i.pl.) or PBS and 4 h later received an injection of ROL (6 mg/kg, i.p.). Injections of anti-AnxA1 antiserum (αAnxA1, 200 μl, i.p.) were given 1 h before the challenge with LPS and again 1 h before ROL. Numbers of neutrophils (A), cells with distinctive apoptotic morphology (B), and Western blotting for Mcl-1 (C) were evaluated 4 h after drug treatment (i.e. 8 h after LPS challenge). The number of annexin V+ neutrophils (D) was evaluated by flow cytometry 24 h after LPS injection. Representative figures of nonapoptotic (asterisk) and apoptotic cells (arrows) and apoptotic cells inside macrophages (arrowheads) are shown in E (original magnifications, ×20). Results are expressed as the number of neutrophils/cavity (A), percentage of neutrophils with apoptotic morphology (C), and apoptotic neutrophils (Ly6G+/F4/80−/AnxAV+/7AAD− (D) and are shown as the mean ± S.E. (error bars) of at least five mice in each group. *, p < 0.05; ***, p < 0.001 when compared with PBS-injected mice. ###, p < 0.001 when compared with LPS-challenged mice. Comparisons between the groups ROL and ROL + αAnxA1 are highlighted in the graphics. For loading control, membranes were reprobed with anti-β-actin. Blots are representative of three independent experiments using pooled cells from at least five animals in each experiment.
Figure 7.

Effect of treatment with anti-AnxA1 antiserum on Bt2cAMP-induced resolution of acute inflammation. Mice were injected with LPS (250 ng/cavity, i.pl.) or PBS and 4 h later received an injection of Bt2cAMP (6 mg/kg, i.p.). Injections of anti-AnxA1 antiserum (αAnxA1, 200 μl, i.p.) were given 1 h before the challenge with LPS and again 1 h before Bt2cAMP. Numbers of neutrophils (A) and cells with distinctive apoptotic morphology (B) were evaluated 4 h after drug treatment (i.e. 8 h after LPS challenge). Representative images of nonapoptotic (asterisks) and apoptotic (arrows) and apoptotic cells inside macrophages (arrowheads) are shown in (C) (original magnifications, ×20). The number of annexin V+ neutrophils (D) with representative dot plots (E) was evaluated by flow cytometry 24 h after LPS injection. Results are expressed as the number of neutrophils/cavity (A), percentage of neutrophils with apoptotic morphology (B), and apoptotic neutrophils (Ly6G+/F4/80−/AnxAV+/7AAD−) (D) and are shown as the mean ± S.E. (error bars) of at least five mice in each group. *, p < 0.05; **, p < 0.01; ***, p < 0.001 when compared with PBS-injected mice. #, p < 0.05 when compared with LPS-challenged mice. Comparisons between the groups Bt2cAMP and Bt2cAMP + αAnxA1 are highlighted in the graphics.
Furthermore, we carried out experiments using an AnxA1-deficient mouse (24) and found results similar to those obtained by inhibition of AnxA1 actions with BOC-1 or AnxA1 neutralization. Indeed, the treatment with ROL or Bt2cAMP was able to induce neutrophil apoptosis in WT mice, and such an effect was impaired in AnxA KO mice (Fig. 8). Therefore, we have shown by pharmacological and genetic strategies the importance of AnxA1 for the pro-resolving properties of ROL and Bt2cAMP.
Figure 8.
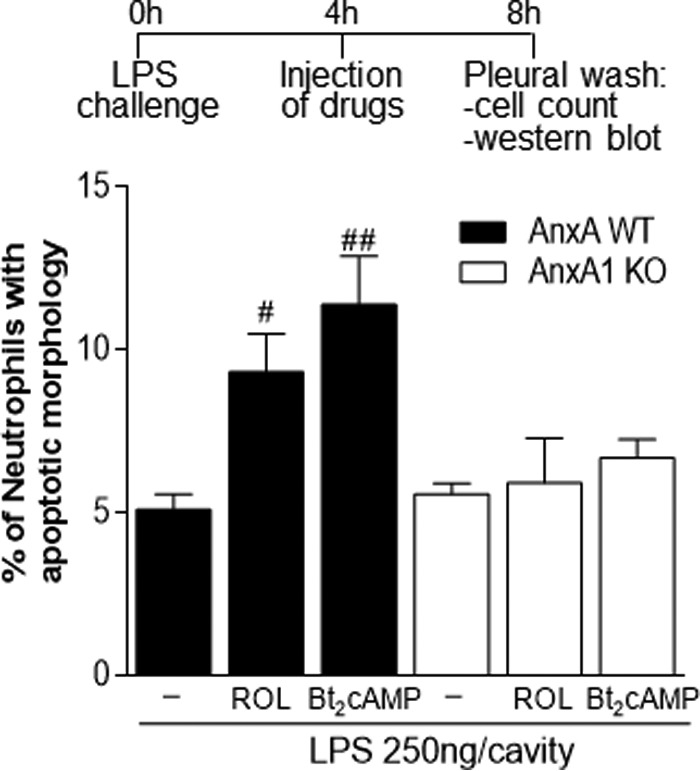
Effect of treatment with rolipram and Bt2cAMP on neutrophil apoptosis on wild-type and AnxA1-knock-out mice. WT or AnxA-KO mice were injected with LPS (250 ng/cavity, i.pl.) and 4 h later received an injection of ROL (6 mg/kg, i.p.) or Bt2cAMP (4 mg/kg, i.pl.). The cells from the pleural cavity were harvested, and numbers of cells with distinctive apoptotic morphology were evaluated 4 h after drug treatment (i.e. 8 h after LPS challenge). Results are expressed as a percentage of neutrophils with apoptotic morphology and are shown as the mean ± S.E. (error bars) of at least five mice in each group. #, p < 0.05; ##, p < 0.01 when compared with LPS-challenged mice.
Rolipram and Bt2cAMP override the survival-inducing effect of LPS in human neutrophils, and such an effect is Anxa1-dependent
Neutrophil apoptosis is an integral modulatory mechanism that constrains inflammation and contributes to its successful resolution. The fate of neutrophils inside an inflammatory milieu (i.e. whether they undergo apoptosis or remain viable) depends on the balance of pro-survival stimuli, such LPS, GM-CSF, and oxygen availability, as well as the presence of pro-apoptotic stimuli, including Fas ligand and TNF (55). Because ROL and Bt2cAMP induced neutrophil apoptosis in an inflammatory milieu in vivo, we investigated the ability of these cAMP-elevating agents to counteract the prosurvival effects of LPS in vitro. As shown previously, LPS decreased the spontaneous apoptosis of cultured human neutrophils (41), and the treatment with ROL and Bt2cAMP prevented this effect, as evaluated by the increased percentage of apoptotic neutrophils when comparing LPS-treated cells with LPS + ROL or LPS + Bt2cAMP (Fig. 9, A and G). There was no difference among the different doses used (Fig. 9, B and C). Sivelestat, a synthetic protease inhibitor, was used as a positive control for induction of neutrophil apoptosis (Fig. 9A), as reported previously (41). In accordance with our in vivo data, the ability of ROL and Bt2cAMP to decrease the prosurvival effect of LPS was abolished by pretreatment with anti-AnxA1 serum (Fig. 9, D and E) or by using WRW4, a selective FPR2 antagonist (Fig. 9F). Therefore, our data show that cAMP-elevating agents can effectively induce or accelerate a pro-apoptotic program in neutrophils, leading to resolution of inflammation.
Figure 9.

Effect of treatment with rolipram and Bt2cAMP on human neutrophil apoptosis. Neutrophils isolated from human peripheral blood (1 × 106 cells/well) were cultured with LPS (500 ng/ml) for 1 h and later with ROL (100 μm), Bt2cAMP (100 μm) (A and D–F), or different concentrations (B and C). The cells were also pretreated with anti-AnxA1 antiserum (100 μg/ml) or WRW4 (10 μm), a specific FPR2/ALXR antagonist, 1 h before LPS (D, E, and F). Sivelestat (100 μg/ml) was used as a positive control for neutrophil apoptosis (A). Neutrophils were processed for cytospin preparations for apoptosis count. Representative figures of nonapoptotic (asterisks) and apoptotic (arrows) neutrophils are shown (original magnifications, ×100). **, p < 0.01; ***, p < 0.001 when comparing the LPS-treated group with untreated (UT) neutrophils. #, p < 0.05; ##, p < 0.01; ###, p < 0.001 when comparing the LPS-treated group with drug-treated neutrophils. The experiments were performed in biological quadruplicates. Error bars, S.E.
Discussion
Cyclic AMP is a fundamental second-messenger molecule produced after adenylate cyclase activation in response to several stimuli, endowed with fundamental modulatory activities in cells involved in the inflammatory process, a property exerted primarily through PKA activity. Intracellular levels of cAMP result from a balance of modulatory pathways that involve elevation through agonist ligands (such as PGE2, adenosine, and β-adrenergic drugs) and degradation by phosphodiesterases (PDEs) (27, 28, 56). There are different families of PDEs with various roles in different cells or tissues. The PDE4 isoenzyme family plays a particularly important role in the immune system and is the predominant PDE in inflammatory cells, including mast cells, eosinophils, neutrophils, T cells, and macrophages (56). Our group has shown previously that ROL and cAMP mimetics induce resolution of an established neutrophilic or eosinophilic inflammation (32, 33) by inducing caspase-dependent apoptosis of polymorphonuclear cells.
In this follow-up study, we reveal an important role for AnxA1 in the pro-resolving properties of ROL and Bt2cAMP, the cyclic AMP mimetic of choice. This conclusion is substantiated by the following major findings. (i) ROL and Bt2cAMP promoted resolution in a model of acute inflammation in mice challenged with LPS, and this process was associated with increased levels of intact AnxA1. (ii) ROL induced AnxA1 expression and phosphorylation in macrophages, an effect associated with CREB phosphorylation. Dibutyryl-cAMP, forskolin, and physiological cAMP-elevating agents increased AnxA1 expression. (iii) The increase of AnxA1 induced by ROL was PKA-dependent in human (THP-1) and murine (RAW) macrophages. (iv) The effect of ROL and Bt2cAMP in vivo was via PKA, as shown by using PKA inhibitors. The latter drugs not only prevented cAMP-induced resolution but also prevented the increase in intact AnxA1 levels. (v) Two different pharmacological strategies were employed to inhibit the AnxA1 pathway, FPR antagonism and neutralizing AnxA1 antiserum; in both cases, there was marked reduction of the resolution properties displayed by cAMP-elevating agents. Importantly, in AnxA1-deficient mice, ROL or Bt2cAMP treatment could not induce neutrophil apoptosis. (vi) ROL and Bt2cAMP induced AnxA1-dependent apoptosis of human neutrophil in the presence of prosurvival stimulus LPS. Therefore, our results show that the effects of ROL and Bt2cAMP on resolution of inflammation are at least in part due to modulation of AnxA1 expression, stabilization, and mobilization to the cell surface. These data identify AnxA1 as a pro-resolving molecule involved in pro-resolving actions of cAMP (Fig. 10).
Figure 10.
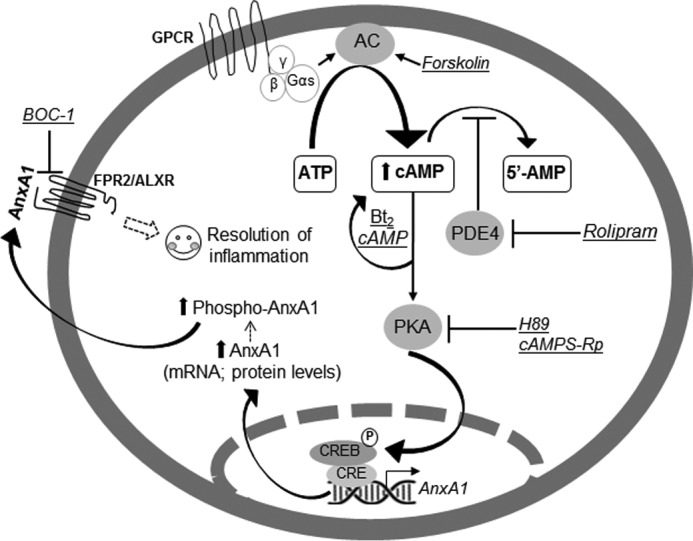
Proposed mechanism by which rolipram and Bt2cAMP modulate AnxA1 and resolution of acute inflammation. The generation of cAMP is initiated when an extracellular first messenger binds to a G protein–coupled receptor (GPCR) at the plasma membrane, which is coupled to a stimulatory G protein α subunit (Gαs). The free Gαs activates the enzyme adenylyl cyclase (AC) to convert ATP into cAMP. Forskolin directly activates adenylyl cyclase. PDEs, which degrade cAMP to 5′-AMP, are another regulator of intracellular cAMP levels. PDE inhibitors, such as rolipram, prevent cAMP degradation, resulting in accumulation of intracellular cAMP. Cyclic AMP can then bind and activate PKA, which in turn phosphorylates CREB. P-CREB binds to the CRE on the promoter region of AnxA1 gene and promotes transactivation. Bt2cAMP is a cell-permeable cAMP analog that activates PKA. H89 and cAMPS-Rp are PKA inhibitors. Both rolipram and Bt2cAMP induce AnxA1 expression and its phosphorylation. Phosphorylated AnxA accumulates on the cell membrane and is externalized. Once in the extracellular medium, this protein exerts autocrine, paracrine, and juxtacrine effects, which are mediated by the FPR2/ALXR. The peptide BOC-1 is a nonselective AnxA1 receptor antagonist. Our results shown that AnxA1 is at least one of the endogenous determinants mediating the pro-resolving properties of rolipram and Bt2cAMP.
In addition to the role of AnxA1 in mediating anti-inflammatory properties of endogenous cortisol, AnxA1 is also an important mediator of anti-inflammatory and pro-resolving properties of pharmacological doses of glucocorticoids (20, 57). During the initial steps of acute inflammation, AnxA1 limits the recruitment of leukocytes and the production of pro-inflammatory mediators (2). During the resolution phase, AnxA1 acts by promoting the apoptosis of neutrophils (10, 20) and increasing their efferocytosis by macrophages (10, 21). Recent studies indicate that modulation of AnxA1 disposition, levels, and indeed externalization in specific cell targets may represent a common mechanism evoked by anti-inflammatory agents, such as LXA4 (58) and estrogens (59). Interestingly, in this study, we have found that physiological cAMP-elevating compounds, such as adenosine, increase AnxA1 protein levels. However, it remains to be investigated whether AnxA1 accounts for the pro-resolving abilities of adenosine and which receptor is engaged to elicit this effect. Another group of drugs, HDAC inhibitors, endowed with multiple properties like the PDE4 inhibitor used here, also modulate AnxA1 expression and localization. Indeed, administration of HDAC inhibitors, such as valproic acid and sodium butyrate, at the peak of zymosan-induced peritonitis accelerated resolution in wild-type mice but much more modestly in AnxA1 null mice. These effects were a consequence of the capacity of HDAC inhibitors to elevate AnxA1 levels, which then modulated leukocyte apoptosis and efferocytosis (24). During the resolving phase of LPS inflammation, high levels of AnxA1 have been found in macrophages with resolutive phenotypes (40).
Phosphorylation and release of AnxA1 are central to the mechanism of action of the antiallergic cromoglycate-like drugs on mast cells and are essential for the inhibition of the release of histamine and PGD2. The latter effects were abolished in the presence of neutralizing anti-AnxA1 monoclonal antibody (6). In our studies, we demonstrated that ROL and Bt2cAMP promoted resolution of neutrophilic inflammation associated with high levels of intact AnxA1 and decreased levels of the cleaved form. Furthermore, ROL induced AnxA1 mRNA expression, accumulation, and phosphorylation in macrophage lineages. More importantly, the blockage of the endogenous AnxA1 prevented the pro-resolving effects of these cAMP-elevating agents, and ROL and Bt2cAMP prevented the prosurvival effect of LPS on human neutrophils.
cAMP regulates apoptosis in several cell types, inhibiting or stimulating the process, depending on the cell type and stage of differentiation (60). There are some in vitro studies using neutrophils that have shown that PDE4 inhibition or an increase of cAMP levels by other cAMP-increasing agents delays neutrophil apoptosis (61–64). In contrast with these in vitro studies, our group demonstrated that in vivo administration of ROL was clearly associated with resolution of neutrophilic inflammation by inducing caspase-3–dependent apoptosis (33). It is important to point out that our experimental settings were designed to investigate whether these drugs could interfere with neutrophil accumulation (apoptosis + efferocytosis/clearance) and not infiltration (migration), because we treated mice 4 h after LPS challenge, when inflammatory cell influx was already established. Indeed, we have shown previously that the neutrophil-active chemokines, such as CXCL1 and CXCL2, peak early in this model of inflammation (1–2 h), and their levels are similar to basal at 4 h after LPS injection (33). Also, injection of reparixin (an allosteric inhibitor of CXCR2) 4 h after LPS challenge failed to affect the accumulation of neutrophils, whereas in the same experiment, post-treatment with rolipram greatly decreased neutrophil accumulation in the cavity. Therefore, the effects of ROL and Bt2cAMP in our experimental settings are not in the migration process of the cells into the pleural cavity, because the cAMP-elevating agents were given after the stimulus and after the peak of neutrophil-active chemokine production in the cavity (33).
Here, we went further and clearly showed that cAMP-elevating agents were able to induce AnxA1-dependent resolution of inflammation and increased neutrophil apoptosis associated with loss of Mcl-1. Importantly, the blockage of the AnxA1 pathway prevented Mcl-1 loss, and it was associated with neutrophil survival. The apoptotic effect of ROL and Bt2cAMP was also observed in cultured human neutrophils exposed to the prosurvival stimuli of LPS. The apparent contradictory actions of cAMP on isolated neutrophils of the previous studies (61–64) with our study can be explained because ROL and Bt2cAMP induce apoptosis in the presence of an inflammatory milieu, when these drugs were able to counteract the prosurvival stimuli, such as LPS. The data presented here cluster with those generated with HDAC inhibitors and cromoglycate-like drugs to suggest that induction of AnxA1 may account for the anti-inflammatory and resolving mechanisms of a few known drugs. We propose that AnxA1 represents a central checkpoint mechanism regulating leukocyte survival and reactivity during on-going inflammatory reactions.
After cell activation, AnxA1 is externalized on the cell surface, and the N-terminal region is exposed and interacts with its receptor, named FPR2/ALX. Once in the extracellular medium, AnxA1 can to be cleaved at the N-terminal region by proteases, including NE and PR3, generating the 33-kDa isoform of poorly known properties. Intact AnxA1 (37 kDa) is the biologically active form of the protein (13, 44, 65). Here, we showed that compounds that we have shown previously to increase resolution of neutrophilic inflammation (33) are able to increase the levels of intact AnxA1 and partially prevent its degradation. Recent work from our group (66) showed that ROL could increase intact AnxA1 and prevent AnxA1 cleavage associated with the improvement of inflammatory parameters of pneumococcal pneumonia. Interestingly, such an effect was more efficient when ROL was combined with the antibiotic ceftriaxone. Therefore, the effect of ROL on the cleaved levels of AnxA1 may be, at least in part, due to the decreased elastase activity and expression and associated with decreased neutrophil number because elastase is an important protease present in neutrophils. In the inflammatory context, the decreased elastase levels may be important to induce resolution. During the resolving phase of LPS-induced inflammation that was associated with decreased elastase expression and activity, there was more intact AnxA1. Conversely, during the productive phase of inflammation, there were high elastase expression/activity and a higher proportion of AnxA1 cleavage. Indeed, inhibition of elastase by using synthetic (Sivelestat) or natural inhibitors (elafin or secretory leukocyte protease inhibitor) was able to promote resolution of inflammation by protection of endogenous intact levels of AnxA1 and resulting neutrophil apoptosis (13, 41, 65). Furthermore, cleavage-resistant AnxA1 exhibited a greater anti-inflammatory effect when compared with the parent protein in different animal models of inflammation (13, 41, 65).
The regulation of cAMP levels is a key feature to regulate a large number of events in the body (67). As a ubiquitous second messenger, cAMP regulates several processes in many cell types, including cells from the immune system (30–32, 34, 35). Elevated cAMP levels were reported at the resolution point in a model of resolving peritonitis, and this was important to clear PMNs and regulate monocyte-derived macrophage functions (31). Sokolowska et al. (36) demonstrated that prostaglandin E2, a potent lipid mediator involved in maintaining homeostasis, inhibits NLRP3 inflammasome activation through the EPE4 receptor and an increase in intracellular cAMP in human macrophages. In teleost fish, prostaglandin E2 promoted M2 polarization macrophages via a cAMP/CREB signaling pathway (37). Bystrom et al. (30) examined whether the macrophage phenotype was dictated by cAMP and whether this phenotype could be altered by changing intracellular levels of this potent intracellular second messenger. In M1 macrophages, TNFα production was attenuated by Bt2cAMP, whereas IL-10 production was increased, suggesting a reversion toward the anti-inflammatory or resolutive phenotype. It is noteworthy that cAMP may function as an intermediate of the effects of other pro-resolving molecules, such as melanocortin peptides (38), lysophosphatidylserine (39) and resolving D1 (68, 69). Recently, the effect of cAMP on neutrophil extracellular trap (NET) was reported. Shishikura et al. (70) have described the inhibitory action of PGE2 on PMA-induced NET formation in vitro through EP2 and EP4 Gαs-coupled receptors. Also, incubation with Bt2cAMP or inhibitors of PDE also suppressed NET formation (70). Here, we described one more immunomodulatory function for cAMP, induction of AnxA1-dependent resolution of inflammation.
The biology of cAMP is mediated by downstream effector molecules, and the most important one is PKA; cAMP binds directly to PKA, provoking a functional rearrangement with enzymatic activity. PKA was shown to mediate, for example, the inhibition on macrophage inflammatory mediator generation induced by cAMP (71), and according to (32), the inhibition of PKA by H89 was able to limit the cAMP-mediated neutrophilic resolution. EPAC is another protein that, together with PKA, is the major binding partner of cAMP (72). In our model, we investigated whether inhibition of PKA with H89 could inhibit the resolution induced by ROL and Bt2cAMP. In agreement with these previous studies, H89 reverted resolution induced by both ROL and Bt2cAMP in vivo, and these events were followed by a decrease in the levels of intact AnxA1. Moreover, our in vitro experiments confirmed the non-redundant function of PKA because pretreatment with H89 or a more selective cAMP-Rp thethylammonium reduced cellular levels of AnxA1 below those measured in RAW264.7 or THP-1 cells treated with ROL alone. When this antagonist is used, the levels of AnxA1 are lower, which proves once again the importance of cAMP to the action of ROL in this system. Altogether, these experiments led us to conclude that PKA is the major effector for cAMP in the processes evoked by ROL.
Obviously, by interfering with cAMP levels, we may alter cGMP levels (because some PDEs hydrolyze both cAMP and cGMP, so PDEs that metabolize cGMP may be altered by intracellular cAMP levels) (73, 74). For this reason, it will be important in the future to study the cross-talk between cAMP and cGMP.
In conclusion, our study showed that cAMP-elevating agents increase levels of AnxA1, and this is functionally involved in the pro-resolving abilities of cAMP. These results reinforce the hypothesis that AnxA1 acts at multiple regulatory levels to promote resolution of inflammation and may be a common mechanism that accounts for the pro-resolving actions of pro-resolving molecules. cAMP-elevating drugs may represent a useful therapeutic strategy not only to block inflammatory processes (during onset of inflammation), but also, equally importantly, to actively induce the mechanisms underlying the resolution of inflammation.
Experimental procedures
Animals
Male BALB/c mice (8–10 weeks) obtained from the Bioscience Unit of Instituto de Ciências Biológicas (Belo Horizonte, Brazil) were housed under standard conditions of optimum light, temperature, and humidity (12-h/12-h light/dark cycle, 22 ± 1 °C, 50–60%) with food and water provided ad libitum. Annexin A1-knock-out (BALB/c background) mice were generated as described previously (57) and bred at the Universidade Federal de Minas Gerais. All described procedures had prior approval from the Animal Ethics Committee of Universidade Federal de Minas Gerais (CEUA/UFMG, protocol number 15/2011).
Drugs, reagents, and antibodies
Bt2cAMP, 6MB-cAMP, cAMP, forskolin, dexamethasone, adenosine, (−)-norepinephrine, sodium butyrate, cGMP, anti-β-actin, and LPS (from Escherichia coli serotype O:111:B4) were from Sigma-Aldrich. 8-Br-cGMP was from Calbiochem. Rolipram was purchased from Enzo Life Sciences. H89 dihydrochloride and cAMPS-Rp triethylammonium salt were from Tocris. PGE2 was from Cayman Chemical Co. Anti-AnxA1 antiserum was a kind gift from Dr. Steve Poole (Biotherapeutics Group, National Institute for Biological Standards and Control, Potters Bar, UK). Anti-AnxA1 (Sc-11387), anti-elastase (Sc-9521), and secondary anti-mouse (Sc-2005) peroxidase conjugate antibody were from Santa Cruz Biotechnology, Inc. (Dallas, TX). Anti-P-CREB (catalog no. 9191) and secondary anti-rabbit peroxidase conjugate antibody (catalog no. 7074) were from Cell Signaling Technology (Danvers, MA). We also used anti-AnxA1 (catalog no. 713400) from Invitrogen. BOC-1 was from MP Biomedicals. Polyclonal anti-Ser27-AnxA1 antibody was generated as described previously (75).
LPS-induced pleurisy model and treatment with drugs
Mice received an intrapleural (i.pl.) injection of LPS (250 ng/cavity) or PBS as described previously (20, 33). Four hours later, mice were treated with rolipram (6 mg/kg, i.p.), Dexa (2 mg/kg, i.p.), or Bt2cAMP (4 mg/kg, i.pl.). These doses and the route of administration were validated in our previous studies (20, 32). AnxA1-knock-out mice were also treated with these drug doses. To prevent the action of AnxA1, mice were treated with BOC-1, a nonselective AnxA1 receptor antagonist (5 mg/kg, i.p.) 30 min before the drugs or with anti-AnxA1 antiserum (0.1 ml of hyperimmune serum diluted in 100 μl of PBS/mouse, i.p.) given 1 h before the challenge with LPS and again 1 h before ROL. In other cases, the PKA inhibitor H89 (4 mg/kg, i.pl.) was used. Compounds were diluted in DMSO or ethanol and further in PBS. Bt2cAMP was only diluted in PBS. Control mice received the respective vehicle only. Mice were euthanized by inhalation of CO2. Cells in the pleural cavity were harvested by washing the cavity with 2 ml of PBS, and total cell counts were performed in a modified Neubauer chamber using Turk's stain. Differential cell counts were performed on cytocentrifuge preparations (Shandon Cytospin III), and the slides were stained with May-Grünwald-Giemsa using standard morphological criteria to identify cell types (20, 33, 57). Results are presented as the number of cells per cavity.
Calculation of resolution indices
We quantified the resolution indices as described previously (42, 43). Murine pleural exudates were collected at 8-, 24-, 36-, and 48-h time points after LPS challenge. The number of PMNs and mononuclear cells was determined by total and differential leukocyte counting. The resolution of acute inflammation was defined in quantitative terms by the following resolution indices: (i) magnitude (ψmax (the maximum PMN numbers in the exudates) and Tmax (time point when PMN numbers reach maximum)); (ii) duration (T50; time point when PMN numbers reduce to 50% of maximum); and (iii) resolution interval Ri (the time period when 50% PMNs are lost from the pleural cavity; i.e. T50 − Tmax).
Cell culture and in vitro assays
The human promonocytoid cell line THP-1 and murine macrophages RAW264.7 were obtained from the American Type Culture Collection (ATCC, Manassas, VA). THP-1 cells were cultured in RPMI 1640 medium (Cultilab, São Paulo, Brazil) supplemented with 8% heat-inactivated FBS and antibiotics (Cultilab), and RAW264.7 cells were cultured in DMEM (Cultilab) in the same conditions. Cell cultures were maintained at 37 °C and 5% CO2, and cell viability was determined using a trypan blue dye exclusion assay. THP-1 cells were differentiated using 4α-phorbol 12-myristate 13-acetate (PMA; 20 ng/ml, Sigma-Aldrich) and serum-deprived with 1% FBS for 24 h; subsequently, cells were treated with drugs at different time intervals and concentrations, as indicated in the specific figures. Dexa was used as a positive control for AnxA1 induction.
cAMP measurements
cAMP levels in cellular extracts were measure using the cAMP direct immunoassay kit, as described by the manufacturer (catalog no. ab65355, Abcam, Cambridge, UK). Briefly, THP-1 cells were lysed completely with 0.1 m HCl, followed by centrifugation at 10,000 rpm for 10 min. The supernatant was collected as the testing sample. To be ready for quantification, cAMP standards and samples were neutralized and acetylated using the neutralizing buffer and acetylating reagent supplied in the kit, respectively. During the quantification, standard cAMP and testing samples were added to the protein G-coated 96-well plate. After blending with anti-cAMP antibody, the suspension was incubated for 1 h at room temperature with gentle agitation and for another 1 h with the addition of cAMP-HRP. Then the plate was washed five times, followed by incubation with HRP developer for 1 h. The reaction was stopped by 1 m HCl, and the absorbance was detected by a microtiter plate reader (Spectra Max 190, Molecular Devices) at 450 nm. The molar concentration of cAMP in cells was determined from standard curves generated using standard preparation. The cAMP levels are expressed as percentage above the control untreated cells.
BMDMs
Bone marrow cell suspensions were isolated by flushing femurs and tibias of 8–10-week BALB/c mice with complete DMEM (+10% FCS, + 1% penicillin/streptomycin) and 20% L929 cell-conditioned medium as a source of M-CSF (76). Aggregates were dislodged by gentle pipetting, and debris was removed by passaging the suspension through a cell strainer (BD Biosciences). Cells were seeded on 6-well plates and incubated at 37 °C in a 5% CO2 atmosphere. Five days after seeding, another 2 ml of DMEM containing 10% FBS and 20% L929 cell-conditioned medium was added. On the seventh day, cells were completely differentiated into macrophages. Cells were seeded on 24-well plates (5 × 105 cells/well) and later were preincubated with rolipram (10 μm) for 1 h and further stimulated with LPS (100 ng/ml) for 24 h.
In vitro assay to evaluate neutrophil apoptosis
Neutrophils were isolated from human peripheral blood from healthy donors (Ethics Committee of the Universidade Federal de Minas Gerais, Brazil; institutional review board project number 0319.0.203.000-11) by using histopaque gradient (Histopaque 1119 and 1077; Sigma) as described previously (41, 77). Neutrophils (1 × 106 cells/well) were resuspended in RPMI 1640 medium, seeded in 96-well culture plates (BD Biosciences), and incubated at 37 °C in a 5% CO2 atmosphere. Cell viability was determined using a trypan blue dye exclusion assay, and the purity of preparations was 95%. To evaluate the effect of ROL or Bt2cAMP on LPS-induced prosurvival/delayed apoptosis of neutrophils, isolated neutrophils were cultured in the presence of LPS (500 ng/ml) and 1 h later were treated with the drugs for a further 5 h as indicated in the figure legends. In some experiments, neutrophils were pretreated with an anti-AnxA1 antiserum (αAnxA1, 100 μg/ml) or a selective antagonist of FPR2, WRW4 (10 μm) (catalog no. 344220, Calbiochem) before the addition of LPS. Sivelestat (100 μg/ml) (catalog no. S7198, Sigma-Aldrich) was used as a positive control for neutrophil apoptosis (41). Apoptosis was evaluated morphologically (as described above), and the experiments were performed in biological quadruplicates.
Assessment of leukocyte apoptosis
Apoptosis was assessed as reported previously (20, 33). Briefly, cells (5 × 104) collected after LPS challenge or from in vitro experiments were cyto-centrifuged, fixed, stained with May-Grünwald-Giemsa, and counted using oil immersion microscopy (×100 objective) to determine the proportion of cells with distinctive apoptotic morphology (cells with chromatin condensation, nuclear fragmentation, and formation of apoptotic bodies inside of macrophages). At least 500 cells were counted per slide, and results are expressed as the mean ± S.E. of the percentage of cells with apoptotic morphology. Assessment of neutrophil apoptosis (Ly6G+/F4/80−/AnxV+/7AAD−) was also performed by flow cytometry using FITC-labeled annexin V and 7-aminoactinomycin D (BD Biosciences) as reported previously (40, 41). Antibodies used were F4/80 (PEcy7, eBioscience, San Diego, CA) and Ly6G (V450, BD Biosciences). Stained cells were acquired in a BD FACSCanto II cell analyzer (BD Biosciences) and analyzed using FlowJo software (Tree Star, Ashland, OR).
Western blot analysis
Inflammatory cells harvested from the pleural cavity, THP-1, RAW264.7, or BMDMs were washed with PBS, and whole-cell extracts were prepared as described (32, 78, 79). The protein content of the lysate was determined by Bradford assay reagent (Bio-Rad). Extracts (20 μg) were separated by electrophoresis on 10% SDS-PAGE and electrotransferred to nitrocellulose membranes, as described (78). Membranes were blocked overnight at 4 °C with PBS containing 5% (w/v) nonfat dry milk and 0.1% Tween 20, washed three times with PBS containing 0.1% Tween 20, and then incubated with anti-AnxA1 (Santa Cruz Biotechnology (1:1000) or Invitrogen (1:3000)), polyclonal anti-Ser27-AnxA1 (1:1000), anti-P-CREB (1:1000), anti-Mcl-1 (1:1000), anti-elastase (1:1000), and anti-β-actin (1:5000) antibodies in PBS containing 5% (w/v) BSA and 0.1% Tween 20. After washing, membranes were incubated with appropriate horseradish peroxidase-conjugated secondary antibody (1:3000). Immunoreactive bands were visualized by using an ECL detection system, as described by the manufacturer (GE Healthcare). The values of AnxA1 or P-AnxA1 were quantified by using densitometric analysis software (ImageJ, National Institutes of Health, Bethesda, MD). Changes in protein levels were estimated by the control (untreated cells), and the results were expressed as -fold increase of the arbitrary units of AnxA1 or P-AnxA1 normalized to the values of β-actin in the same sample.
RNA extraction and quantitative RT-PCR
Total RNA was extracted using the RNeasy minikit (Qiagen, Crawley, UK) according to the manufacturer's instructions. cDNA was synthesized from 1 μg of RNA with SuperScript III reverse transcriptase (Invitrogen), following the manufacturer's recommended protocol. Synthesized cDNA was added to the relevant forward and reverse primer together with Power SYBR Green PCR Master Mix (Applied Biosystems, Warrington, UK). Real-time PCR was performed in duplicate, with 1 μl of cDNA at a concentration of 100 ng, 0.5 mm primers, and Power SYBR Green PCR Master Mix (Applied Biosystems, Warrington, UK) using StepOne (Applied Biosystems, Foster City, CA). The data were analyzed using StepOne Detection System software with a cycle threshold (Ct) in the linear range of amplification and then processed by the 2−ΔΔCt method. Reactions were run in duplicates. Primers (IDT) used were as follows: human ANXA1 (5′-ATCAGCGGTGAGCCCCTATC-3′/5′-TTCATCCAGGGGGCTTTCCTG-3′) and human GAPDH (5′-AGAAGACTGTGGATGGCCCC-3′/5′-TGACCTTGCCCACAGCCTT-3′). A dissociation step was always included to confirm the absence of unspecific products. Samples of all groups were run on one plate with two technical replicates. Gapdh was used as an endogenous control to normalize the variability in expression levels, and results were expressed as -fold increase.
Elastase activity assay
The elastase activity was measured in cell extracts prepared in the absence of protease inhibitors by using an in-house procedure that relies on the use of MeO-succinyl-AA-Pro-Val-p-nitroanilide (M4765, Sigma-Aldrich) as substrate. Cells obtained from the pleural cavity of mice were lysed on appropriate buffer (200 mm NaCl, 20 mm Tris-HCl, 1% Triton X-100, pH 8.0). The lysate was centrifuged at 12,000 rpm in a microcentrifuge for 15 min at 4 °C, and supernatant (30 μl) was added to 20 μl of TBS (Tris-HCl, pH 8.0) and 50 μl of the substrate (1 mm) in a 96-well microplate. Following incubation for 2 h at 37 °C, the absorbance of samples was analyzed in a spectrophotometer (Spectra Max 190, Molecular Devices) at 405 nm. A standard curve was performed with p-nitroanilide in accordance with the procedures supplied by the manufacturer (BioVision Inc.). The results are presented as elastase activity absorbance.
Statistical analysis
All results are presented as the mean ± S.E. Data were analyzed by one-way analysis of variance, and differences between groups were assessed using the Student-Newman-Keuls post-test. A p value < 0.05 was considered significant. Calculations were performed using Prism version 5.0 software for Windows (GraphPad Software, La Jolla, CA).
Author contributions
L. P. S. and M. M. T. designed the research, analyzed data, and wrote the paper. K. M. L. and J. P. V. performed the main experiments, analyzed data, and helped to write the paper. R. G. A., B. R. C. C., A. A. F. C., K. M. L., M. A. S., and I. G. performed in vitro experiments. K. M. L., T. R. C., and F. M. S. carried out PCR analyses. G. L. N.-L. and L. P. T. performed some in vivo experiments. V. P. provided expertise. E. S. provided the P-AnxA1 antibody and contributed to manuscript revision. M. P. provided guidance on experimental design and contributed to manuscript writing. All authors approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Frankcinéia Assis and Ilma Marçal for technical assistance.
This work was supported by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil), Fundação de Amparo a Pesquisa do Estado de Minas Gerais (FAPEMIG, Brazil), Pró-Reitoria de Pesquisa da Universidade Federal de Minas Gerais-PRPq, Brazil, and the European Community's Seventh Framework Programme (FP7-2007-2013) under Grant Agreement HEALTH-F4–2011-281608. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. 1–4.
- AnxA1
- annexin A1
- P-AnxA1
- phospho-AnxA1
- αAnxA1
- anti-AnxA1 antiserum
- BOC-1
- N-t-Boc-Met-Leu-Phe
- CRE
- cAMP-responsive element
- CREB
- cAMP-responsive element-binding protein
- P-CREB
- phospho-CREB
- Bt2cAMP
- dibutyryl cyclic AMP
- Dexa
- dexamethasone
- i.pl.
- intrapleural
- PMA
- 4α-phorbol 12-myristate 13-acetate
- ROL
- rolipram
- WRW4
- Trp-Arg-Trp-Trp-Trp-NH2
- HDAC
- histone deacetylase
- PDE
- phosphodiesterase
- 6MB-cAMP
- monobutyryladenosine cAMP
- 8-Br-cGMP
- 8-bromo-cyclic GMP
- FPR
- formyl peptide receptor
- NET
- neutrophil extracellular trap
- BMDM
- bone marrow–derived macrophage.
References
- 1. Alessandri A. L., Sousa L. P., Lucas C. D., Rossi A. G., Pinho V., and Teixeira M. M. (2013) Resolution of inflammation: mechanisms and opportunity for drug development. Pharmacol. Ther. 139, 189–212 [DOI] [PubMed] [Google Scholar]
- 2. Perretti M., and D'Acquisto F. (2009) Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat. Rev. Immunol. 9, 62–70 [DOI] [PubMed] [Google Scholar]
- 3. Sousa L. P., Alessandri A. L., Pinho V., and Teixeira M. M. (2013) Pharmacological strategies to resolve acute inflammation. Curr. Opin. Pharmacol. 13, 625–631 [DOI] [PubMed] [Google Scholar]
- 4. Gavins F. N., and Hickey M. J. (2012) Annexin A1 and the regulation of innate and adaptive immunity. Front. Immunol. 3, 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bizzarro V., Belvedere R., Dal Piaz F., Parente L., and Petrella A. (2012) Annexin A1 induces skeletal muscle cell migration acting through formyl peptide receptors. PLoS One 7, e48246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yazid S., Sinniah A., Solito E., Calder V., and Flower R. J. (2013) Anti-allergic cromones inhibit histamine and eicosanoid release from activated human and murine mast cells by releasing Annexin A1. PLoS One 8, e58963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Taylor A. D., Philip J. G., John C. D., Cover P. O., Morris J. F., Flower R. J., and Buckingham J. C. (2000) Annexin 1 (lipocortin 1) mediates the glucocorticoid inhibition of cyclic adenosine 3′,5′-monophosphate-stimulated prolactin secretion. Endocrinology 141, 2209–2219 [DOI] [PubMed] [Google Scholar]
- 8. Castro-Caldas M., Mendes A. F., Duarte C. B., and Lopes M. C. (2003) Dexamethasone-induced and estradiol-induced CREB activation and annexin 1 expression in CCRF-CEM lymphoblastic cells: evidence for the involvement of cAMP and p38 MAPK. Mediators Inflamm. 12, 329–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bena S., Brancaleone V., Wang J. M., Perretti M., and Flower R. J. (2012) Annexin A1 interaction with the FPR2/ALX receptor: identification of distinct domains and downstream associated signaling. J. Biol. Chem. 287, 24690–24697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dalli J., Consalvo A. P., Ray V., Di Filippo C., D'Amico M., Mehta N., and Perretti M. (2013) Proresolving and tissue-protective actions of annexin A1-based cleavage-resistant peptides are mediated by formyl peptide receptor 2/lipoxin A4 receptor. J. Immunol. 190, 6478–6487 [DOI] [PubMed] [Google Scholar]
- 11. Cooray S. N., Gobbetti T., Montero-Melendez T., McArthur S., Thompson D., Clark A. J., Flower R. J., and Perretti M. (2013) Ligand-specific conformational change of the G-protein-coupled receptor ALX/FPR2 determines proresolving functional responses. Proc. Natl. Acad. Sci. U.S.A. 110, 18232–18237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Getting S. J., Flower R. J., and Perretti M. (1997) Inhibition of neutrophil and monocyte recruitment by endogenous and exogenous lipocortin 1. Br. J. Pharmacol. 120, 1075–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Oliani S. M., Paul-Clark M. J., Christian H. C., Flower R. J., and Perretti M. (2001) Neutrophil interaction with inflamed postcapillary venule endothelium alters annexin 1 expression. Am. J. Pathol. 158, 603–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oh J., Rhee H. J., Kim S., Kim S. B., You H., Kim J. H., and Na D. S. (2000) Annexin-I inhibits PMA-induced c-fos SRE activation by suppressing cytosolic phospholipase A2 signal. FEBS Lett. 477, 244–248 [DOI] [PubMed] [Google Scholar]
- 15. Wu C. C., Croxtall J. D., Perretti M., Bryant C. E., Thiemermann C., Flower R. J., and Vane J. R. (1995) Lipocortin 1 mediates the inhibition by dexamethasone of the induction by endotoxin of nitric oxide synthase in the rat. Proc. Natl. Acad. Sci. U.S.A. 92, 3473–3477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Minghetti L., Nicolini A., Polazzi E., Greco A., Perretti M., Parente L., and Levi G. (1999) Down-regulation of microglial cyclo-oxygenase-2 and inducible nitric oxide synthase expression by lipocortin 1. Br. J. Pharmacol. 126, 1307–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ferlazzo V., D'Agostino P., Milano S., Caruso R., Feo S., Cillari E., and Parente L. (2003) Anti-inflammatory effects of annexin-1: stimulation of IL-10 release and inhibition of nitric oxide synthesis. Int. Immunopharmacol. 3, 1363–1369 [DOI] [PubMed] [Google Scholar]
- 18. Parente L., and Solito E. (2004) Annexin 1: more than an anti-phospholipase protein. Inflamm. Res. 53, 125–132 [DOI] [PubMed] [Google Scholar]
- 19. Solito E., Kamal A., Russo-Marie F., Buckingham J. C., Marullo S., and Perretti M. (2003) A novel calcium-dependent proapoptotic effect of annexin 1 on human neutrophils. FASEB J. 17, 1544–1546 [DOI] [PubMed] [Google Scholar]
- 20. Vago J. P., Nogueira C. R., Tavares L. P., Soriani F. M., Lopes F., Russo R. C., Pinho V., Teixeira M. M., and Sousa L. P. (2012) Annexin A1 modulates natural and glucocorticoid-induced resolution of inflammation by enhancing neutrophil apoptosis. J. Leukocyte Biol. 92, 249–258 [DOI] [PubMed] [Google Scholar]
- 21. Maderna P., Yona S., Perretti M., and Godson C. (2005) Modulation of phagocytosis of apoptotic neutrophils by supernatant from dexamethasone-treated macrophages and annexin-derived peptide Ac(2–26). J. Immunol. 174, 3727–3733 [DOI] [PubMed] [Google Scholar]
- 22. Scannell M., Flanagan M. B., deStefani A., Wynne K. J., Cagney G., Godson C., and Maderna P. (2007) Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J. Immunol. 178, 4595–4605 [DOI] [PubMed] [Google Scholar]
- 23. Dalli J., Jones C. P., Cavalcanti D. M., Farsky S. H., Perretti M., and Rankin S. M. (2012) Annexin A1 regulates neutrophil clearance by macrophages in the mouse bone marrow. FASEB J. 26, 387–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Montero-Melendez T., Dalli J., and Perretti M. (2013) Gene expression signature-based approach identifies a pro-resolving mechanism of action for histone deacetylase inhibitors. Cell Death Differ. 20, 567–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tang J., Chen X., Tu W., Guo Y., Zhao Z., Xue Q., Lin C., Xiao J., Sun X., Tao T., Gu M., and Liu Y. (2011) Propofol inhibits the activation of p38 through up-regulating the expression of annexin A1 to exert its anti-inflammation effect. PLoS One 6, e27890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yazid S., Solito E., Christian H., McArthur S., Goulding N., and Flower R. (2009) Cromoglycate drugs suppress eicosanoid generation in U937 cells by promoting the release of Anx-A1. Biochem. Pharmacol. 77, 1814–1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Teixeira M. M., al-Rashed S., Rossi A. G., and Hellewell P. G. (1997) Characterization of the prostanoid receptors mediating inhibition of PAF-induced aggregation of guinea-pig eosinophils. Br. J. Pharmacol. 121, 77–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Soderling S. H., and Beavo J. A. (2000) Regulation of cAMP and cGMP signaling: new phosphodiesterases and new functions. Curr. Opin. Cell Biol. 12, 174–179 [DOI] [PubMed] [Google Scholar]
- 29. Schudt C., Hatzelmann A., Beume R., and Tenor H. (2011) Phosphodiesterase inhibitors: history of pharmacology. Handb. Exp. Pharmacol. 10.1007/978-3-642-17969-3_1 [DOI] [PubMed] [Google Scholar]
- 30. Bystrom J., Evans I., Newson J., Stables M., Toor I., van Rooijen N., Crawford M., Colville-Nash P., Farrow S., and Gilroy D. W. (2008) Resolution-phase macrophages possess a unique inflammatory phenotype that is controlled by cAMP. Blood 112, 4117–4127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rajakariar R., Newson J., Jackson E. K., Sawmynaden P., Smith A., Rahman F., Yaqoob M. M., and Gilroy D. W. (2009) Nonresolving inflammation in gp91phox−/− mice, a model of human chronic granulomatous disease, has lower adenosine and cyclic adenosine 5′-monophosphate. J. Immunol. 182, 3262–3269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sousa L. P., Carmo A. F., Rezende B. M., Lopes F., Silva D. M., Alessandri A. L., Bonjardim C. A., Rossi A. G., Teixeira M. M., and Pinho V. (2009) Cyclic AMP enhances resolution of allergic pleurisy by promoting inflammatory cell apoptosis via inhibition of PI3K/Akt and NF-κB. Biochem. Pharmacol. 78, 396–405 [DOI] [PubMed] [Google Scholar]
- 33. Sousa L. P., Lopes F., Silva D. M., Tavares L. P., Vieira A. T., Rezende B. M., Carmo A. F., Russo R. C., Garcia C. C., Bonjardim C. A., Alessandri A. L., Rossi A. G., Pinho V., and Teixeira M. M. (2010) PDE4 inhibition drives resolution of neutrophilic inflammation by inducing apoptosis in a PKA-PI3K/Akt-dependent and NF-κB-independent manner. J. Leukoc. Biol. 87, 895–904 [DOI] [PubMed] [Google Scholar]
- 34. Lee H. N., and Surh Y. J. (2013) Resolvin D1-mediated NOX2 inactivation rescues macrophages undertaking efferocytosis from oxidative stress-induced apoptosis. Biochem. Pharmacol. 86, 759–769 [DOI] [PubMed] [Google Scholar]
- 35. Sheldon K. E., Shandilya H., Kepka-Lenhart D., Poljakovic M., Ghosh A., and Morris S. M. Jr. (2013) Shaping the murine macrophage phenotype: IL-4 and cyclic AMP synergistically activate the arginase I promoter. J. Immunol. 191, 2290–2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sokolowska M., Chen L. Y., Liu Y., Martinez-Anton A., Qi H. Y., Logun C., Alsaaty S., Park Y. H., Kastner D. L., Chae J. J., and Shelhamer J. H. (2015) Prostaglandin E2 inhibits NLRP3 inflammasome activation through EP4 receptor and intracellular cyclic AMP in human macrophages. J. Immunol. 194, 5472–5487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Montero J., Gómez-Abellán V., Arizcun M., Mulero V., and Sepulcre M. P. (2016) Prostaglandin E2 promotes M2 polarization of macrophages via a cAMP/CREB signaling pathway and deactivates granulocytes in teleost fish. Fish Shellfish Immunol. 55, 632–641 [DOI] [PubMed] [Google Scholar]
- 38. Montero-Melendez T., Patel H. B., Seed M., Nielsen S., Jonassen T. E., and Perretti M. (2011) The melanocortin agonist AP214 exerts anti-inflammatory and proresolving properties. Am. J. Pathol. 179, 259–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Frasch S. C., and Bratton D. L. (2012) Emerging roles for lysophosphatidylserine in resolution of inflammation. Prog. Lipid Res. 51, 199–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vago J. P., Tavares L. P., Garcia C. C., Lima K. M., Perucci L. O., Vieira É. L., Nogueira C. R., Soriani F. M., Martins J. O., Silva P. M., Gomes K. B., Pinho V., Bruscoli S., Riccardi C., Beaulieu E., et al. (2015) The role and effects of glucocorticoid-induced leucine zipper in the context of inflammation resolution. J. Immunol. 194, 4940–4950 [DOI] [PubMed] [Google Scholar]
- 41. Vago J. P., Tavares L. P., Sugimoto M. A., Lima G. L., Galvão I., de Caux T. R., Lima K. M., Ribeiro A. L., Carneiro F. S., Nunes F. F., Pinho V., Perretti M., Teixeira M. M., and Sousa L. P. (2016) Proresolving actions of synthetic and natural protease inhibitors are mediated by annexin A1. J. Immunol. 196, 1922–1932 [DOI] [PubMed] [Google Scholar]
- 42. Bannenberg G. L., Chiang N., Ariel A., Arita M., Tjonahen E., Gotlinger K. H., Hong S., and Serhan C. N. (2005) Molecular circuits of resolution: formation and actions of resolvins and protectins. J. Immunol. 174, 4345–4355 [DOI] [PubMed] [Google Scholar]
- 43. Chiang N., Shinohara M., Dalli J., Mirakaj V., Kibi M., Choi A. M., and Serhan C. N. (2013) Inhaled carbon monoxide accelerates resolution of inflammation via unique proresolving mediator-heme oxygenase-1 circuits. J. Immunol. 190, 6378–6388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pederzoli-Ribeil M., Maione F., Cooper D., Al-Kashi A., Dalli J., Perretti M., and D'Acquisto F. (2010) Design and characterization of a cleavage-resistant Annexin A1 mutant to control inflammation in the microvasculature. Blood 116, 4288–4296 [DOI] [PubMed] [Google Scholar]
- 45. Yazid S., Leoni G., Getting S. J., Cooper D., Solito E., Perretti M., and Flower R. J. (2010) Antiallergic cromones inhibit neutrophil recruitment onto vascular endothelium via annexin-A1 mobilization. Arterioscler. Thromb. Vasc. Biol. 30, 1718–1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tabe Y., Jin L., Contractor R., Gold D., Ruvolo P., Radke S., Xu Y., Tsutusmi-Ishii Y., Miyake K., Miyake N., Kondo S., Ohsaka A., Nagaoka I., Andreeff M., and Konopleva M. (2007) Novel role of HDAC inhibitors in AML1/ETO AML cells: activation of apoptosis and phagocytosis through induction of annexin A1. Cell Death Differ. 14, 1443–1456 [DOI] [PubMed] [Google Scholar]
- 47. Rich T. C., Xin W., Mehats C., Hassell K. A., Piggott L. A., Le X., Karpen J. W., and Conti M. (2007) Cellular mechanisms underlying prostaglandin-induced transient cAMP signals near the plasma membrane of HEK-293 cells. Am. J. Physiol. Cell Physiol. 292, C319–C331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ding L., Zhang F., Zhao M. X., Ren X. S., Chen Q., Li Y. H., Kang Y. M., and Zhu G. Q. (2016) Reduced lipolysis response to adipose afferent reflex involved in impaired activation of adrenoceptor-cAMP-PKA-hormone sensitive lipase pathway in obesity. Sci. Rep. 6, 34374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yang J. H., Lee E. O., Kim S. E., Suh Y. H., and Chong Y. H. (2012) Norepinephrine differentially modulates the innate inflammatory response provoked by amyloid-β peptide via action at β-adrenoceptors and activation of cAMP/PKA pathway in human THP-1 macrophages. Exp. Neurol. 236, 199–206 [DOI] [PubMed] [Google Scholar]
- 50. Takahashi H. K., Liu K., Wake H., Mori S., Zhang J., Liu R., Yoshino T., and Nishibori M. (2009) Prostaglandin E2 inhibits advanced glycation end product-induced adhesion molecule expression, cytokine production, and lymphocyte proliferation in human peripheral blood mononuclear cells. J. Pharmacol. Exp. Ther. 331, 656–670 [DOI] [PubMed] [Google Scholar]
- 51. Minguet S., Huber M., Rosenkranz L., Schamel W. W., Reth M., and Brummer T. (2005) Adenosine and cAMP are potent inhibitors of the NF-κB pathway downstream of immunoreceptors. Eur. J. Immunol. 35, 31–41 [DOI] [PubMed] [Google Scholar]
- 52. Antonicelli F., De Coupade C., Russo-Marie F., and Le Garrec Y. (2001) CREB is involved in mouse annexin A1 regulation by cAMP and glucocorticoids. Eur. J. Biochem. 268, 62–69 [DOI] [PubMed] [Google Scholar]
- 53. Murphy M. P., and Caraher E. (2015) Mcl-1 is vital for neutrophil survival. Immunol. Res. 62, 225–233 [DOI] [PubMed] [Google Scholar]
- 54. Milot E., and Filep J. G. (2011) Regulation of neutrophil survival/apoptosis by Mcl-1. ScientificWorldJournal 11, 1948–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jones H. R., Robb C. T., Perretti M., and Rossi A. G. (2016) The role of neutrophils in inflammation resolution. Semin. Immunol. 28, 137–145 [DOI] [PubMed] [Google Scholar]
- 56. Page C. P., and Spina D. (2011) Phosphodiesterase inhibitors in the treatment of inflammatory diseases. Handb. Exp. Pharmacol. 10.1007/978-3-642-17969-3_17 [DOI] [PubMed] [Google Scholar]
- 57. Hannon R., Croxtall J. D., Getting S. J., Roviezzo F., Yona S., Paul-Clark M. J., Gavins F. N., Perretti M., Morris J. F., Buckingham J. C., and Flower R. J. (2003) Aberrant inflammation and resistance to glucocorticoids in annexin 1−/− mouse. FASEB J. 17, 253–255 [DOI] [PubMed] [Google Scholar]
- 58. Brancaleone V., Dalli J., Bena S., Flower R. J., Cirino G., and Perretti M. (2011) Evidence for an anti-inflammatory loop centered on polymorphonuclear leukocyte formyl peptide receptor 2/lipoxin A4 receptor and operative in the inflamed microvasculature. J. Immunol. 186, 4905–4914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nadkarni S., Cooper D., Brancaleone V., Bena S., and Perretti M. (2011) Activation of the annexin A1 pathway underlies the protective effects exerted by estrogen in polymorphonuclear leukocytes. Arterioscler. Thromb. Vasc. Biol. 31, 2749–2759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Parvathenani L. K., Buescher E. S., Chacon-Cruz E., and Beebe S. J. (1998) Type I cAMP-dependent protein kinase delays apoptosis in human neutrophils at a site upstream of caspase-3. J. Biol. Chem. 273, 6736–6743 [DOI] [PubMed] [Google Scholar]
- 61. Rossi A. G., Cousin J. M., Dransfield I., Lawson M. F., Chilvers E. R., and Haslett C. (1995) Agents that elevate cAMP inhibit human neutrophil apoptosis. Biochem. Biophys. Res. Commun. 217, 892–899 [DOI] [PubMed] [Google Scholar]
- 62. Martin M. C., Dransfield I., Haslett C., and Rossi A. G. (2001) Cyclic AMP regulation of neutrophil apoptosis occurs via a novel protein kinase A-independent signaling pathway. J. Biol. Chem. 276, 45041–45050 [DOI] [PubMed] [Google Scholar]
- 63. Krakstad C., Christensen A. E., and Døskeland S. O. (2004) cAMP protects neutrophils against TNF-α-induced apoptosis by activation of cAMP-dependent protein kinase, independently of exchange protein directly activated by cAMP (Epac). J. Leukoc. Biol. 76, 641–647 [DOI] [PubMed] [Google Scholar]
- 64. Parkkonen J., Hasala H., Moilanen E., Giembycz M. A., and Kankaanranta H. (2008) Phosphodiesterase 4 inhibitors delay human eosinophil and neutrophil apoptosis in the absence and presence of salbutamol. Pulm. Pharmacol. Ther. 21, 499–506 [DOI] [PubMed] [Google Scholar]
- 65. Vong L., D'Acquisto F., Pederzoli-Ribeil M., Lavagno L., Flower R. J., Witko-Sarsat V., and Perretti M. (2007) Annexin 1 cleavage in activated neutrophils: a pivotal role for proteinase 3. J. Biol. Chem. 282, 29998–30004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tavares L. P., Garcia C. C., Vago J. P., Queiroz-Junior C. M., Galvão I., David B. A., Rachid M. A., Silva P. M., Russo R. C., Teixeira M. M., and Sousa L. P. (2016) Inhibition of phosphodiesterase-4 during pneumococcal pneumonia reduces inflammation and lung injury in mice. Am. J. Respir. Cell Mol. Biol. 55, 24–34 [DOI] [PubMed] [Google Scholar]
- 67. Lefkimmiatis K., and Zaccolo M. (2014) cAMP signaling in subcellular compartments. Pharmacol. Ther. 143, 295–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hsiao H. M., Sapinoro R. E., Thatcher T. H., Croasdell A., Levy E. P., Fulton R. A., Olsen K. C., Pollock S. J., Serhan C. N., Phipps R. P., and Sime P. J. (2013) A novel anti-inflammatory and pro-resolving role for resolvin D1 in acute cigarette smoke-induced lung inflammation. PLoS One 8, e58258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Titos E., Rius B., González-Périz A., López-Vicario C., Morán-Salvador E., Martínez-Clemente M., Arroyo V., and Clària J. (2011) Resolvin D1 and its precursor docosahexaenoic acid promote resolution of adipose tissue inflammation by eliciting macrophage polarization toward an M2-like phenotype. J. Immunol. 187, 5408–5418 [DOI] [PubMed] [Google Scholar]
- 70. Shishikura K., Horiuchi T., Sakata N., Trinh D. A., Shirakawa R., Kimura T., Asada Y., and Horiuchi H. (2016) Prostaglandin E2 inhibits neutrophil extracellular trap formation through production of cyclic AMP. Br. J. Pharmacol. 173, 319–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Aronoff D. M., Carstens J. K., Chen G. H., Toews G. B., and Peters-Golden M. (2006) Short communication: differences between macrophages and dendritic cells in the cyclic AMP-dependent regulation of lipopolysaccharide-induced cytokine and chemokine synthesis. J. Interferon Cytokine Res. 26, 827–833 [DOI] [PubMed] [Google Scholar]
- 72. Tiwari S., Felekkis K., Moon E. Y., Flies A., Sherr D. H., and Lerner A. (2004) Among circulating hematopoietic cells, B-CLL uniquely expresses functional EPAC1, but EPAC1-mediated Rap1 activation does not account for PDE4 inhibitor-induced apoptosis. Blood 103, 2661–2667 [DOI] [PubMed] [Google Scholar]
- 73. Maurice D. H., Ke H., Ahmad F., Wang Y., Chung J., and Manganiello V. C. (2014) Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 13, 290–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ahmad F., Murata T., Shimizu K., Degerman E., Maurice D., and Manganiello V. (2015) Cyclic nucleotide phosphodiesterases: important signaling modulators and therapeutic targets. Oral Dis. 21, e25–e50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Solito E., Mulla A., Morris J. F., Christian H. C., Flower R. J., and Buckingham J. C. (2003) Dexamethasone induces rapid serine-phosphorylation and membrane translocation of annexin 1 in a human folliculostellate cell line via a novel nongenomic mechanism involving the glucocorticoid receptor, protein kinase C, phosphatidylinositol 3-kinase, and mitogen-activated protein kinase. Endocrinology 144, 1164–1174 [DOI] [PubMed] [Google Scholar]
- 76. Englen M. D., Valdez Y. E., Lehnert N. M., and Lehnert B. E. (1995) Granulocyte/macrophage colony-stimulating factor is expressed and secreted in cultures of murine L929 cells. J. Immunol. Methods 184, 281–283 [DOI] [PubMed] [Google Scholar]
- 77. Lucas C. D., Allen K. C., Dorward D. A., Hoodless L. J., Melrose L. A., Marwick J. A., Tucker C. S., Haslett C., Duffin R., and Rossi A. G. (2013) Flavones induce neutrophil apoptosis by down-regulation of Mcl-1 via a proteasomal-dependent pathway. FASEB J. 27, 1084–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sousa L. P., Silva B. M., Brasil B. S., Nogueira S. V., Ferreira P. C., Kroon E. G., Kato K., and Bonjardim C. A. (2005) Plasminogen/plasmin regulates α-enolase expression through the MEK/ERK pathway. Biochem. Biophys. Res. Commun. 337, 1065–1071 [DOI] [PubMed] [Google Scholar]
- 79. Souza D. G., Vieira A. T., Pinho V., Sousa L. P., Andrade A. A., Bonjardim C. A., McMillan M., Kahn M., and Teixeira M. M. (2005) NF-κB plays a major role during the systemic and local acute inflammatory response following intestinal reperfusion injury. Br. J. Pharmacol. 145, 246–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.