

Abstract
In the adult brain, programmed death of neural stem cells is considered to be critical for tissue homeostasis and cognitive function and is dysregulated in neurodegeneration. Previously, we have reported that adult rat hippocampal neural (HCN) stem cells undergo autophagic cell death (ACD) following insulin withdrawal. Because the apoptotic capability of the HCN cells was intact, our findings suggested activation of unique molecular mechanisms linking insulin withdrawal to ACD rather than apoptosis. Here, we report that phosphorylation of autophagy-associated protein p62 by AMP-activated protein kinase (AMPK) drives ACD and mitophagy in HCN cells. Pharmacological inhibition of AMPK or genetic ablation of the AMPK α2 subunit by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 genome editing suppressed ACD, whereas AMPK activation promoted ACD in insulin-deprived HCN cells. We found that following insulin withdrawal AMPK phosphorylated p62 at a novel site, Ser-293/Ser-294 (in rat and human p62, respectively). Phosphorylated p62 translocated to mitochondria and induced mitophagy and ACD. Interestingly, p62 phosphorylation at Ser-293 was not required for staurosporine-induced apoptosis in HCN cells. To the best of our knowledge, this is the first report on the direct phosphorylation of p62 by AMPK. Our data suggest that AMPK-mediated p62 phosphorylation is an ACD-specific signaling event and provide novel mechanistic insight into the molecular mechanisms in ACD.
Keywords: AMP-activated kinase (AMPK), autophagy, mitophagy, p62 (sequestosome 1 (SQSTM1)), phosphorylation
Introduction
Autophagy is an evolutionarily conserved cellular degradation and recycling process characterized by an increased generation of autophagic vesicles (1). Autophagy plays various cytoprotective roles, including degradation of toxic proteins and damaged organelles, and provision of biochemical intermediates for the synthesis of macromolecules and metabolism (2). A growing body of evidence also suggests the involvement of autophagy in programmed cell death (PCD),2 although its role in PCD remains controversial, and the detailed mechanisms of the mediation of PCD by autophagy remain to be elucidated (3). In terms of the prodeath role of autophagy, autophagy may be a bona fide mechanism for induction and execution of cell death (autophagic cell death (ACD)) or be a prerequisite for apoptotic or necrotic cell death (autophagy-mediated cell death) (4).
Autophagosome biogenesis can be assessed by monitoring the autophagy markers microtubule-associated protein light chain 3 (LC3) and p62. LC3 is a ubiquitin-like protein; pro-LC3 is cleaved by Atg4B, resulting in the cytosolic form LC3-I (5). When autophagy is activated, LC3-I is enriched in autophagosomes and converted to LC3-II through conjugation with phosphatidylethanolamine (6). Therefore, LC3-II is a reliable biochemical marker of autophagosome formation (5, 7, 8). In a similar manner, a punctate pattern of fluorescently tagged LC3 can be indicative of autophagy induction (9). The ubiquitin-binding protein p62 interacts with LC3-II and thereby delivers cargo molecules to autophagosomes. During this process, p62 is also degraded by autophagy (10, 11). Hence, a decrease in p62 is another measure of autophagic flux (12).
However, accumulation of autophagic vesicles at one static time point can reflect either an increase (on-rate) or decrease (off-rate) of autophagic flux with diametrically opposite implication for the role of autophagy in cell death. Therefore, description of autophagy morphology in dying cells without data to clarify the functional role of autophagy in the cell death process has led to confusion about the concept of ACD. In this regard, observation of ACD in apoptosis-defective cells or cells with suppressed apoptosis, the lack of the specific pharmacological reagents for modulation of autophagy, and other difficulties and technical concerns in addressing the exact role of autophagy in relation to cell death were well discussed by Kroemer and Levine (13). Despite the controversy, an increasing number of studies support a causative role of autophagy in cell death, especially in insects and other model organisms (14). Examples of ACD in mammals are much fewer (15).
According to the criteria suggested by Shen and Codogno (16), ACD is distinguished from other modes of PCD by the lack of apoptotic features and ineffectiveness of caspase inhibition, increased autophagic flux, and dependence of cell death on autophagy-related genes (Atgs) or other key autophagy genes. We have previously demonstrated that insulin withdrawal induces ACD in adult hippocampal neural (HCN) stem cells despite their normal apoptotic capabilities. Insulin-deprived HCN cells show enhanced autophagic flux but do not display signs of apoptosis, such as caspase activation, chromosomal DNA fragmentation, and exposure of phosphatidylserine on the outer leaflet of the plasma membrane (17–20). In addition, the pan-caspase inhibitor Z-VAD failed to protect HCN cells from insulin withdrawal (17, 19, 20). In contrast, Atg7 knockdown efficiently suppresses ACD in insulin-deprived HCN cells (17, 19–21). Therefore, insulin withdrawal-induced death of HCN cells fulfills all the criteria for ACD (16, 22–26). Furthermore, we also identified several key regulators of ACD and mediators for the cross-talk between ACD and apoptosis, including glycogen synthase kinase-3β, calpain, type 3 ryanodine receptor, and p97/valosin-containing protein (19–21, 27).
AMP-activated protein kinase (AMPK) is a serine/threonine protein kinase composed of a catalytic subunit (α1 or α2) and regulatory β and γ subunits (28). AMPK regulates various cellular processes, including metabolic homeostasis, cell proliferation, and cell death (29, 30). AMPK can also induce autophagy (31–33). Given the close connection of insulin signaling with AMPK in the regulation of cell metabolism, proliferation, survival, and autophagy, we postulated that AMPK might be intimately involved in ACD of HCN cells upon insulin withdrawal. In the present study, we found that AMPK was readily activated in insulin-deprived HCN cells and served as a critical trigger of ACD. Looking further into the molecular mechanism, we identified p62 as a new molecular target of AMPK. Although many substrates connecting AMPK to metabolism regulation are known, only a few AMPK targets relevant to autophagy have been identified so far (32, 34–37). We further report that AMPK-mediated phosphorylation of p62 is required for mitophagy and ACD following insulin withdrawal but not for apoptotic death in HCN cells. Our study suggests that novel AMPK-mediated p62 phosphorylation can be an ACD-specific signaling event and that this AMPK–p62 axis drives ACD and mitophagy in HCN cells. Our findings warrant further studies on the molecular mechanisms that link autophagy to cell death; understanding these mechanisms is required to distinguish the role of autophagy in facilitation of cell death from its general protective role.
Results
AMPK activity is critical for ACD in I(−) HCN cells
To address the role of AMPK in HCN cell death, we initially assessed the activation status of AMPK in insulin-deprived HCN cells. Hereafter, we will refer to HCN cells cultured in insulin-containing and insulin-deficient media as I(+) and I(−), respectively, as in our prior reports (19, 20). We assessed Thr-172 phosphorylation of the AMPK α subunit because this post-translational modification in the kinase activation loop is associated with increased AMPK activity (38, 39). Following insulin withdrawal, AMPK was rapidly activated, and the time course of its activation was well correlated with the up-regulation of autophagic flux as revealed by an increase in LC3-II and decrease in p62 (Fig. 1A). The AMPK inhibitor compound C (CC; 0.5 μm) efficiently blocked AMPK activation and substantially reduced the autophagy level and cell death in I(−) HCN cells (Fig. 1, B–D). CC also reduced the number of GFP-LC3 puncta in these cells (Fig. 1, E and F). To corroborate these results derived from the pharmacological inhibition of AMPK, we knocked out AMPK α1 or α2 subunit using the CRISPR/Cas9 gene editing approach. The relative levels of α1 and α2 mRNAs were similar and were not changed by insulin withdrawal (supplemental Fig. S1A). Single guide RNA sequences targeting each subunit efficiently ablated the AMPK α1 or α2 gene (supplemental Fig. S1B). Of note, the deletion of the α2 but not α1 gene markedly reduced cell death (Fig. 1, G and supplemental Fig. S1C) and autophagic flux in I(−) HCN cells (Fig. 1, H and I). These data indicated that pharmacological or genetic inactivation of AMPK substantially diminished ACD in I(−) HCN cells, suggesting a critical role of AMPK in HCN cell death following insulin withdrawal.
Figure 1.

AMPK activation is required for ACD in I(−) HCN cells. A, time-course analysis of AMPK activation and autophagy induction following insulin withdrawal. Blots shown are representative of three independent experiments, which yielded similar results. B, pharmacological inhibition of AMPK with CC (0.5 μm) decreased cell death (n = 5). C, administration of CC attenuated autophagy. The levels of LC3-II and p62 were measured 5 h after insulin withdrawal. D, quantification of LC3-II and p62 after normalization to β-actin (n = 5). E, HCN cells were transfected with the GFP-LC3 plasmid, and LC3 puncta were visualized after insulin withdrawal for 5 h. F, analysis of E. Quantification of the puncta from three independent experiments (n = 40) is shown. G, AMPK α2 knock-out decreased cell death following insulin withdrawal. Cell death of AMPK α2 KO (Sg-AMPK α2) and control (Sg-con) HCN cells was measured at the indicated time points following insulin withdrawal (n = 4). H, autophagy flux following insulin withdrawal (5 h) was abrogated in AMPK α2 KO HCN cells. BafA1 (20 nm) was added 1 h before sampling. I, quantification of LC3-II and p62 after normalization to β-actin (n = 4). Error bars represent ±S.D. from independent assays. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Scale bar, 10 μm. p-AMPK, phospho-AMPK; t-AMPK, total AMPK.
With regard to the mechanism of AMPK activation, two upstream kinases, LKB1 and Ca2+/calmodulin-dependent protein kinase kinase β (CaMKKβ), are well documented to phosphorylate Thr-172 of the AMPK α subunit (40). AMPK is activated in response to metabolic stresses that deplete the cellular ATP level, and binding of AMP to the γ subunit allosterically activates the AMPK complex by enhancing LKB1-dependent Thr-172 phosphorylation. AMPK can also be directly phosphorylated by CaMKKβ in response to increases in intracellular Ca2+ without significant change in ATP/ADP/AMP levels (40). To elucidate how insulin withdrawal activated AMPK, we first examined the cellular ATP level. Interestingly, insulin withdrawal did not induce depletion of cellular ATP compared with the substantial depletion of cellular ATP by glucose deprivation for 5 h (supplemental Fig. S2A). In contrast, a specific CaMKK inhibitor, STO-609, decreased AMPK Thr-174 phosphorylation and attenuated cell death (supplemental Fig. S2, B and C). Our previous report also demonstrated an increase in the intracellular Ca2+ level that was due to ryanodine receptor 3-mediated endoplasmic reticulum Ca2+ efflux following insulin withdrawal (27). These results suggest that insulin withdrawal activated AMPK through Ca2+–CaMKK pathway rather than ATP depletion in HCN cells.
Genetic activation of AMPK promotes ACD in I(−) HCN cells
If AMPK is critical for ACD in I(−) HCN cells, its forced activation may enhance the effects of insulin withdrawal and boost ACD. To test this assumption, we generated an AMPK construct encoding the constitutively active (CA) mutant form of the α2 subunit by introducing a point mutation of Thr-172 to Asp in combination with the deletion of C-terminal amino acid residues 313–548 containing the autoinhibitory sequence and the binding sites to the regulatory β/γ subunits (41, 42). Persistent activation of AMPK was confirmed by phosphorylation of its well known substrate, acetyl-CoA carboxylase (ACC) (supplemental Fig. S2D). As expected, constitutive activation of AMPK in I(−) HCN cells led to a more marked increase in LC3-II and decrease in p62 levels than insulin withdrawal alone but did not activate caspase-3 (Fig. 2, A and B). AMPK α2-CA also induced a higher level of cell death than insulin withdrawal (Fig. 2C). In line with the ineffectiveness of Z-VAD against insulin withdrawal-induced ACD (19, 20), Z-VAD (20 μm) failed to diminish the AMPK α2-CA-induced boost of cell death but efficiently decreased staurosporine (STS)-induced apoptosis in HCN cells (Fig. 2C).
Figure 2.

Genetic activation of AMPK enhances ACD in I(−) HCN cells. A, overexpression of AMPK-CA increased the level of LC3-II and decreased that of p62 but did not activate caspase-3 following insulin withdrawal for 5 h. STS (0.5 μm for 8 h) was used as a positive control for apoptosis. C-cas3, cleaved caspase-3. B, quantification of LC3-II and p62 after normalization to β-actin (n = 4). C, AMPK-CA increased cell death more than did I(−) alone. Z-VAD (20 μm) failed to reduce cell death induced by AMPK-CA but efficiently reduced STS-induced apoptosis (n = 5). D, AMPK-CA increased autophagic flux in I(−) HCN cells. Insulin was withdrawn for 5 h, and BafA1 (20 nm) was added 1 h before sampling. E, quantification of LC3-II and p62 after normalization to β-actin (n = 5). F, AMPK-CA (CA) increased autophagic flux. Autophagic flux was measured using tandem mRFP-GFP-LC3 after 5-h insulin withdrawal. G, analysis of F. The red and yellow puncta were counted from three independent experiments (n = 29). H, Atg7 knockdown abrogated AMPK-CA-triggered cell death. Cell death was measured 24 h after insulin withdrawal (n = 6). I, Atg7 knockdown abrogated AMPK-CA-triggered autophagic flux. AMPK-CA failed to affect LC3-II or p62 in Sh-Atg7 HCN cells analyzed 5 h after insulin withdrawal. J, quantification of LC3-II and p62 after normalization to β-actin (n = 9). Error bars represent ±S.D. from independent assays. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant. Scale bar, 10 μm. EV, empty vector; con, control.
Autophagic flux, as assessed by Western blotting of LC3-II in combination with bafilomycin A1 (BafA1), was further increased by the expression of AMPK α2-CA in I(−) HCN cells (Fig. 2, D and E). Lentiviral transduction with tandem labeled monomeric mRFP-GFP-LC3 also indicated a higher level of autophagic flux in I(−) HCN cells co-expressing AMPK α2-CA than I(−) HCN cells alone (Fig. 2, F and G). The quenching of the GFP fluorescence but not mRFP fluorescence at the acidic pH yields red puncta upon fusion of autophagosomes and lysosomes (43). Conversely, yellow puncta correspond to the immature autophagosomes before maturation. Therefore, an increase in the mRFP-only puncta of total LC3 puncta by expression of AMPK α2-CA indicates increased autophagy flux. An increase in cell death rate induced by AMPK α2-CA was effectively abrogated when Atg7 was stably knocked down using lentiviral shRNA (Fig. 2H). We have previously demonstrated that Atg7 knockdown attenuates ACD in I(−) HCN cells (17, 19). Therefore, these data indicate that the AMPK-stimulated increase in cell death is autophagy-dependent. Taken together with the lack of the effect of Z-VAD and the absence of apoptotic signs, these results indicate that ACD is the mode of cell death induced by overactivation of AMPK in I(−) HCN. Overall, these data provide the evidence supporting our hypothesis that AMPK is a critical mediator of ACD following insulin withdrawal and that forced activation of AMPK promotes ACD in HCN cells.
Novel p62 phosphorylation for ACD
To elucidate the mechanism of AMPK-induced ACD, we sought to identify the AMPK substrates. Because p62 is a signaling hub molecule that coordinates autophagy, the ubiquitin-proteasome system, and metabolism and undergoes post-translational modifications including phosphorylation (44–46), we posited p62 as a potential novel AMPK substrate. The AMPK-phosphorylated site can be predicted based on the sequence SXSXXD (35), although the optimized AMPK consensus sequence is more complex. Examination of the rat p62 amino acid sequence using the Group-based Prediction System (GPS) search engine (47) indicated several sites potentially phosphorylated by AMPK α2 (Fig. 3A). These were all novel sites, and their phosphorylation by AMPK has not yet been tested experimentally. To test whether p62 is an AMPK substrate, we first examined whether p62 knockdown would affect ACD in HCN cells. Knockdown of p62 using p62-specific shRNA reduced autophagy (Fig. 3, B and C) and attenuated cell death (Fig. 3D) in I(−) HCN cells. Furthermore, depletion of p62 abolished the AMPK α2-CA-induced increase in autophagy markers (Fig. 3, E and F), suggesting the potential role of p62 in AMPK-mediated ACD in I(−) HCN cells. To precisely determine the phosphorylation sites critical for ACD, we substituted the predicted phosphorylated Ser residues with Ala using mouse p62 cDNA and examined the effects of these phosphorylation-null p62 mutants on cell death in I(−) HCN cells. Because HCN cells were derived from an adult rat, we refer to their endogenous p62 as rp62, and to mouse and human p62 as mp62 and hp62, respectively. Among the mutants tested, we observed a significant decrease in cell death for up to 48 h only in the S296A mutant of mp62 (equivalent to Ser-293 in rp62 and Ser-294 in hp62) (supplemental Fig. S3). This site is well conserved across the mammalian species (Fig. 4A). However, a decrease in cell death rate by the mp62-S296A mutant was not robust, probably due to the presence of endogenous rp62 in HCN cells. To further validate this residue as the AMPK target site critical for ACD, we generated phosphorylation-defective or -mimicking point mutations of the hp62 gene. Before the introduction of hp62-WT or mutant forms into HCN cells, endogenous rp62 was depleted using rp62-specific shRNA to avoid its confounding effects, and HCN cells with stable knockdown of p62 (designated as Sh-rp62 HCN) and control HCN cells (transfected with control shRNA and designated as Sh-con HCN) were established after puromycin selection. The expression of hp62-WT rescued cell death in I(−) Sh-rp62 HCN cells (Fig. 4B). As expected, introduction of the phosphorylation-mimicking hp62-S294E mutant also substantially increased cell death in I(−) Sh-rp62 HCN cells; however, the expression of hp62-S294A failed to recover the cell death rate (Fig. 4B). These data indicate that this putative phosphorylation site in p62 is indispensable for ACD following insulin withdrawal in HCN cells. The Asp and Glu forms of the phosphorylation-mimicking mutation induced ACD equally (data not shown); the Glu form was used for all subsequent experiments. The effects of Ser-294 phosphodefective and phosphomimicking mutants on cell death rate were in a good agreement with those on autophagic flux because hp62-S294E restored robust LC3 turnover, but hp62-S294A did not (Fig. 4C). Interestingly, the expression level of hp62-S294E was very low despite transfection with a higher amount of DNA than hp62-WT and hp62-S294A, likely due to its active participation in autophagic degradation. Altogether, these data revealed that this novel phosphorylation of p62 is necessary and sufficient for the mediation of ACD by p62 in HCN cells following insulin withdrawal.
Figure 3.
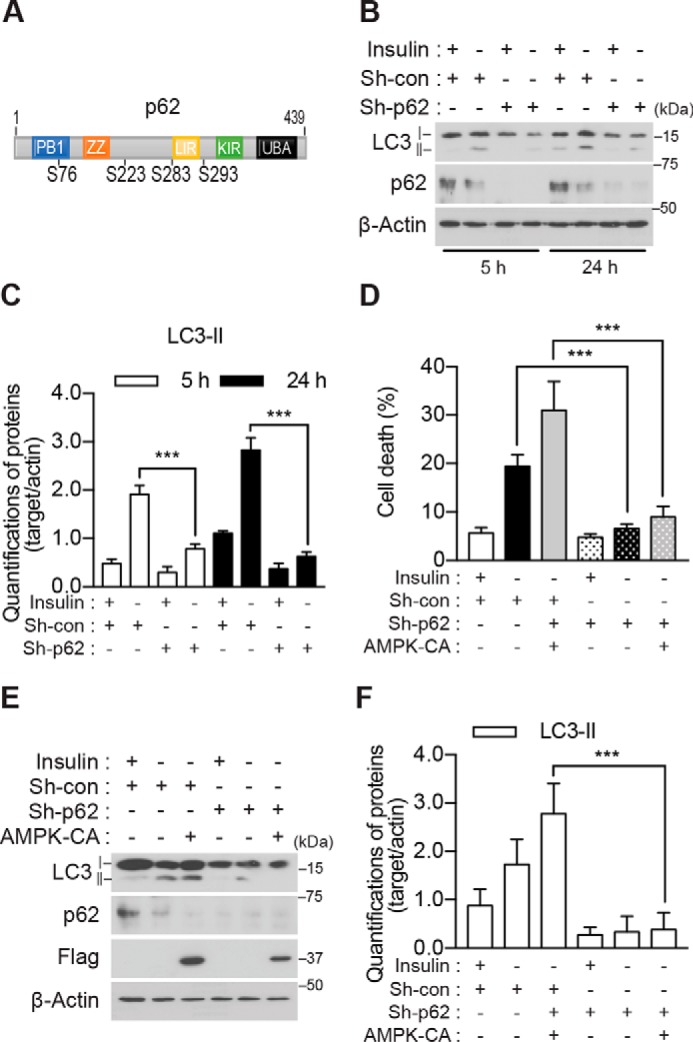
p62 is required for ACD in I(−) HCN cells. A, schematic representation of the functional domains and predicted AMPK α2 phosphorylation sites of p62. B, knockdown of p62 abrogated an increase in LC3-II following insulin withdrawal. C, quantification of LC3-II after normalization to β-actin (n = 3). D, p62 knockdown attenuated the AMPK-CA-induced increase in cell death I(−) HCN cells (n = 6). E, p62 knockdown attenuated the AMPK-CA-induced increase in LC3-II in I(−) HCN cells. F, quantification of LC3-II after normalization to β-actin (n = 6). Error bars represent ±S.D. from independent assays. ***, p < 0.001. con, control; ZZ, ZZ-type zinc finger domain; LIR, LC3-interacting region; UBA, ubiquitin-associated domain; KIR, Keap1-interacting region.
Figure 4.
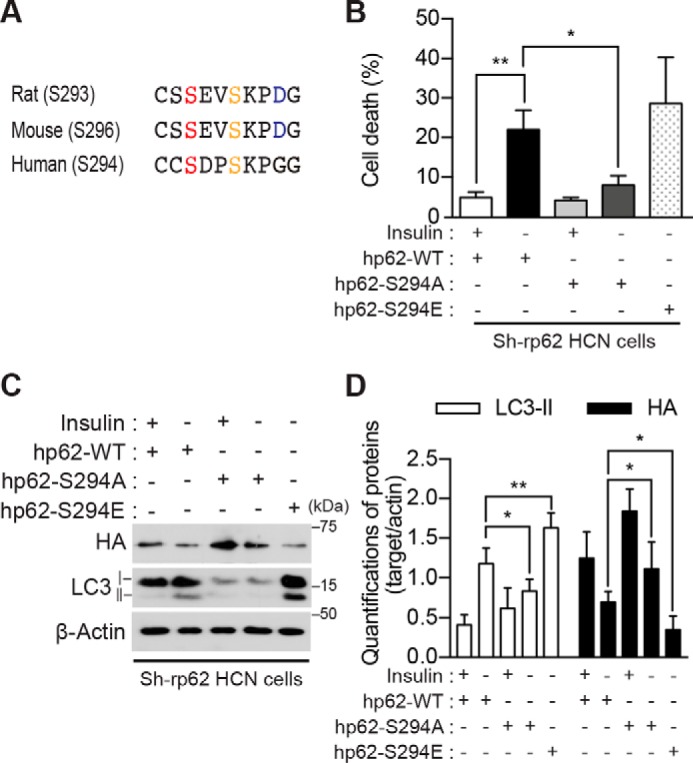
Serine phosphorylation at a novel site in p62 is required for ACD in I(−) HCN cells. A, the predicted phosphorylation motif of rat p62 Ser-293 is well conserved in mouse (Ser-296) and human (Ser-294) p62. B, expression of hp62-WT and hp62-S294E, but not hp62-S294A, restored cell death in Sh-rp62 HCN cells following insulin withdrawal. Sh-rp62 HCN cells were transfected with plasmids to express HA-tagged hp62 (WT, phosphorylation-defective mutant (S294A), or phosphomimicking mutant (S294E)). Cell death was measured 24 h after insulin withdrawal (n = 5). C, hp62-WT and hp62-S294E, but not hp62-S294A, restored autophagy flux. D, quantification of LC3-II and p62 after normalization to β-actin (n = 5). Error bars represent ±S.D. from independent assays. *, p < 0.05; **, p < 0.01.
Ser-293 phosphorylation of p62 in HCN cells is mediated by AMPK
To confirm phosphorylation of endogenous rp62 in HCN cells, we generated an antibody specific for phosphorylated Ser-293 of rp62. Western blot analysis using this antibody showed that rp62 Ser-293 phosphorylation was detectable under basal conditions and increased following insulin withdrawal (Fig. 5, A and B). Pharmacological inhibition or genetic ablation of AMPK α2 diminished rp62 Ser-293 phosphorylation in I(−) HCN cells, suggesting that AMPK is the physiological kinase for Ser-293 phosphorylation (Fig. 5, A–D). To verify that p62 is a direct substrate of AMPK and that p62 phosphorylation is not due to the action of other kinases in the wake of AMPK activation, we performed an in vitro kinase assay with GST-tagged hp62 and immunoprecipitated AMPK α2-CA. The AMPK immune complexes were enriched from HEK293 cells with a GFP antibody after overexpression of GFP-tagged AMPK α2-CA. GST-tagged hp62-WT and -S294A recombinant proteins were purified from Escherichia coli using glutathione-agarose beads and incubated with the AMPK immune complexes in the presence of ATP. Ser-294 phosphorylation of hp62 was probed with phosphospecific p62 antibody. As expected, the AMPK immune complex was able to phosphorylate hp62-WT but not the S294A mutant (Fig. 5, E and F). Recombinant AMPK protein composed of α2, β1, and γ1 also yielded the same result (supplemental Fig. S4). When the AMPK-CA immune complex was treated with the AMPK inhibitor CC, phosphorylation barely occurred, indicating that AMPK, but not another kinase, was sufficient for p62 phosphorylation (Fig. 5, G and H). These results identified Ser-293/Ser-294 (in rp62/hp62) as a novel AMPK phosphorylation site critical for ACD in HCN cells following insulin withdrawal.
Figure 5.
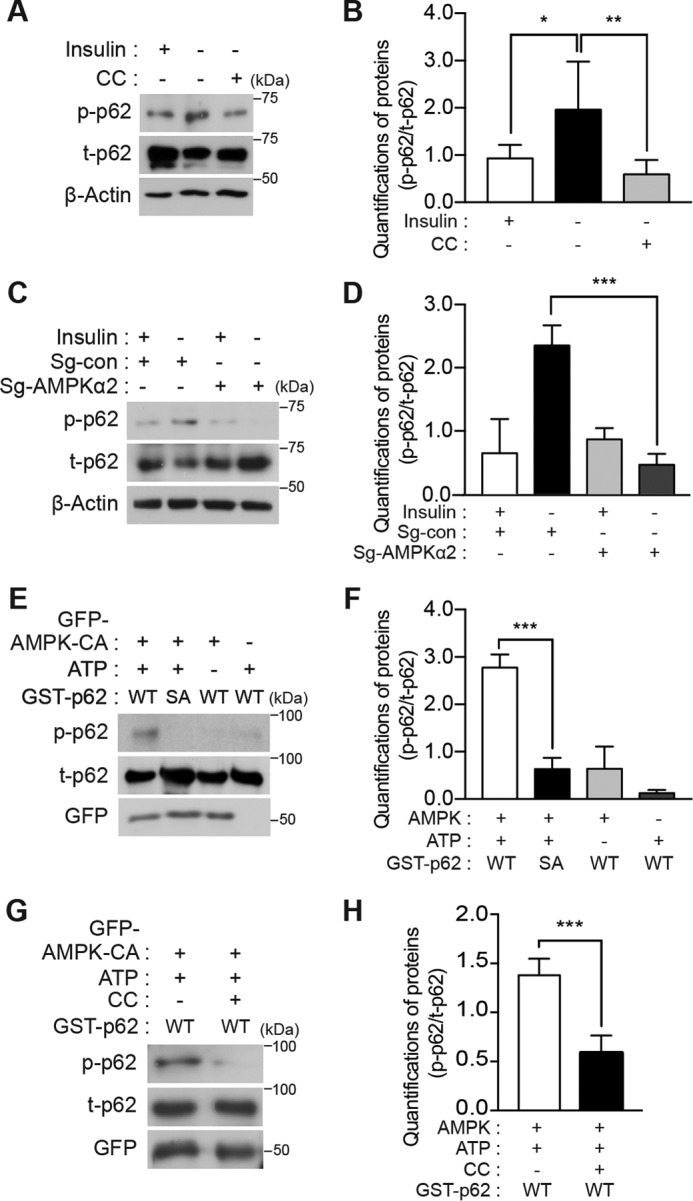
Serine phosphorylation at a novel site in p62 is mediated by AMPK. A, endogenous p62 was phosphorylated in an AMPK-dependent manner in HCN cells. Insulin withdrawal for 5 h induced phosphorylation of p62 Ser-293, which was detected by using a phosphospecific antibody. CC (0.5 μm) reduced Ser-293 phosphorylation. B, quantification of p62 Ser-293 phosphorylation (p-p62) after normalization to total p62 (t-p62) following CC treatment (n = 6). C, AMPK α2 KO reduced Ser-293 phosphorylation. D, quantification of p62 Ser-293 phosphorylation (p-p62) after normalization to total p62 (t-p62) in AMPK α2 KO HCN cells (n = 6). E, hp62-WT, but not hp62-S294A mutant (SA), was phosphorylated in vitro by the enriched AMPK immune complex. Phosphorylation was detected by Western blotting with phosphospecific antibody. F, quantification of in vitro kinase assay results (n = 4). G, treatment of the AMPK immune complex with CC (1 μm) for 1 h abolished p62 phosphorylation. H, quantification of in vitro kinase assay results with CC (n = 3). Error bars represent ±S.D. from independent assays. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
As a multidomain adaptor molecule, p62 can be modified in various ways for its signaling fidelity and has multiple phosphorylation sites. Recently, phosphorylation of hp62 Ser-403 or Ser-407 in its ubiquitin-binding domain by casein kinase 2 or Unc-51-like kinase 1 (ULK1) has been reported to increase its affinity for ubiquitin and regulate autophagic clearance of protein aggregates (46, 48). To examine whether these residues are also involved in ACD in I(−) HCN cells, we used phosphorylation-defective mutant hp62-S403A or hp62-S407A. Both mutants tended to reduce cell death in I(−) Sh-rp62 HCN cells compared with hp62-WT, although they were less effective than hp62-S294A (supplemental Fig. S5). These data suggest the potential involvement of other kinases in p62 phosphorylation during ACD of I(−) HCN cells, although they may play less important roles than AMPK. Whether other autophagy-related kinases mediate ACD in HCN cells following insulin withdrawal in an AMPK-dependent or -independent manner is currently being examined by our group. Of note, hp62-S294A had no effect on STS-triggered apoptosis in Sh-rp62 HCN cells (supplemental Fig. S6), suggesting that p62 phosphorylation by AMPK may be specific to ACD following insulin withdrawal.
AMPK activation induces mitophagy
Mitophagy is selective autophagy for the elimination of dysfunctional mitochondria (49). AMPK plays a critical role in mitophagy induction upon nutrient deprivation or reduction of insulin signaling in mammals and Caenorhabditis elegans (36). To examine whether AMPK induces mitophagy in I(−) HCN cells, we first examined the overlap between mitochondria and autophagosomes after transient transfection with GFP-LC3 and DsRed2-Mito. Using super-resolution microscopy for structured illumination (SR-SIM), we observed greatly increased co-localization of LC3 puncta and mitochondria in I(−) HCN cells, comparable with their robust co-localization in HCN cells treated with carbonyl cyanide m-chlorophenylhydrazone (CCCP), a mitochondrial uncoupler and well known inducer of mitophagy (Fig. 6, A and B). CC treatment of I(−) HCN cells reduced co-localization, implying that mitophagy occurred in an AMPK-dependent manner during ACD (Fig. 6, A and B). Of note, the mitochondria of I(−)HCN cells were smaller and fragmented, similar to those of CCCP-treated cells and unlike a network of the long filamentous mitochondria in I(+) HCN cells (see insets in DsRed2-Mito images in Fig. 6A). CC treatment partially rescued the fragmented mitochondrial morphology of I(−) HCN cells. We also used a mitochondrially targeted version of monomeric Keima (mt-mKeima) to monitor mitophagy. mKeima is an acid-resistant fluorescent protein and has an emission maximum at 620 nm with a bimodal excitation spectrum peaking at 458 (neutral pH) and 561 nm (acidic pH) (50). Therefore, mt-mKeima allows the visualization of mitochondria delivered to acidic lysosomes by increasing the ratio of excitation at 561/458 nm (51). To test whether insulin withdrawal induced mitophagy, we transiently transfected HCN cells with mt-mKeima and monitored the 561/458 nm ratio using the wavelength settings of a confocal laser microscope (Carl Zeiss LSM 780). Similar to AMPK-dependent co-localization of GFP-LC3 and DsRed2-Mito, a high ratio of 561/458 nm signals was observed in I(−)HCN cells, whereas this ratio was reduced in CC-treated (Fig. 6, C and D) and I(−) AMPK α2 KO HCN cells (Fig. 6, E and F). In addition, the mitochondria were stained with MitoTracker Green, and fluorescence-activated cell sorting (FACS) analysis was performed to measure mitochondrial mass. The amount of stained mitochondria was decreased in I(−) HCN cells compared with in I(+) HCN cells as observed following CCCP treatment. Inhibition of AMPK by CC in I(−) HCN cells reversed a decrease in the labeling of mitochondria (Fig. 6, G and H), implying that mitochondrial homeostasis is modulated through AMPK-dependent mitophagy in HCN cells.
Figure 6.

AMPK induces mitophagy in I(−) HCN cells. A, transfection of HCN cells with the plasmids encoding GFP-LC3 and DsRed2-Mito. Mitophagy was assessed as the overlap of LC3 and DsRed2-Mito signals and was greatly enhanced following insulin withdrawal (5 h) and prevented by CC. Inset, a high-magnification image of the boxed area. B, analysis of A. Quantification of mitochondrial localization of GFP-LC3 (n = 8) is shown. C, fluorescence signal of mt-mKeima. Cells expressing mt-mKeima were excited (Ex.) at two wavelengths (458 and 561 nm), and the emission signal was collected at 620 nm after insulin withdrawal for 5 h. D, analysis of C. The relative intensity ratio as an index of mitophagy (n = 10) is shown. E, insulin withdrawal-induced mitophagy was abrogated in AMPK α2 KO HCN cells. F, analysis of E. The relative intensity ratio as an index of mitophagy is shown. CCCP (5 μm for 5 h) was added to I(+) HCN cells to induce mitophagy as a positive control (n = 8). ns, not significant; G, the amount of mitochondria in HCN cells following insulin withdrawal for 12 h and treatment with CC (5 μm for 12 h) was determined by FACS using MitoTracker Green (x axis). FSC, forward scatter. H, analysis of G. The relative ΔMitoTracker (%) as an index of mitophagic degradation of mitochondria is shown. CCCP (10 μm for 12 h) was used as a positive control (n = 5). Error bars represent ±S.D. from independent assays. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant. Scale bar, 10 μm.
Phosphorylation of p62 promotes its mitochondrial localization and mitophagy
To examine the localization of phosphorylated p62 during ACD, we transiently introduced hemagglutinin (HA)-tagged hp62-WT and Ser-294 mutant constructs into Sh-rp62 HCN cells and monitored the subcellular distribution of hp62 in relation to mitochondria. Immunocytochemical analysis with an antibody against the HA epitope was used to detect p62. Several hp62-WT dots were detected under basal I(+) conditions, and the number and size of the dots were markedly increased by insulin withdrawal (Fig. 7, A and B). Most p62 puncta were co-localized with mitochondria as indicated by overlap with the DsRed2-Mito signal. Mitochondrial localization of hp62-WT was inhibited by CC, indicating the requirement for AMPK activity to trigger mitochondrial translocation of p62. Phosphorylation-defective hp62-S294A lost the ability to translocate to mitochondria, whereas hp62-S294E exhibited robust co-localization with DsRed2-Mito in I(−) Sh-rp62 HCN cells, indicating that AMPK-mediated phosphorylation of p62 was critical for its localization in mitochondria (Fig. 7, A and B). Mitochondrial translocation of p62 was also monitored by live confocal imaging after transient expression of GFP-p62. Time-lapse imaging revealed that most GFP-p62 puncta were relatively small and did not move toward mitochondria in I(+) HCN cells (as indicated by arrowheads), whereas the p62 puncta were bigger and translocated to mitochondria upon insulin withdrawal (as indicated by arrows) (Fig. 7C). These large p62 puncta observed at early time points became smaller and disappeared together with the overlapping mitochondria later in I(−) HCN cells, suggesting degradation through mitophagy (Fig. 7C). Likewise, live imaging of shorter duration revealed clearly different movement of p62 between I(+) and I(−) conditions. GFP-p62 in I(+) HCN cells remained static and showed minimal interaction and overlap with mitochondria (supplemental Movies S1 and S2), which was in contrast to dynamic movement and vigorous interaction of p62 with mitochondria in the I(−) condition (supplemental Movies S3–S5). Some big p62 dots stayed with mitochondria despite continuous mitochondrial movement (supplemental Movies S4 and S5). This suggests that the p62 undergoes repeated association and dissociation with mitochondria at the early stage of mitophagy and later interacts with mitochondria more tightly during mitophagy.
Figure 7.

Phosphorylated p62 translocates to mitochondria in an AMPK-dependent manner in I(−) HCN cells. A, co-transfection of Sh-rp62 HCN cells with plasmids encoding DsRed2-Mito and HA-hp62-WT, -S294A mutant, or -S294E mutant. Translocation of p62 to mitochondria was examined by immunocytochemistry 5 h after insulin withdrawal. CC (0.5 μm) was added as indicated. B, analysis of A. Quantification of mitochondrial localization of p62 (n = 9) is shown. C, live confocal imaging for monitoring mitochondrial translocation of GFP-p62 upon insulin withdrawal. Arrowheads indicate the immobile p62 puncta in I(+) HCN cells; arrows indicate the dynamic p62 puncta that overlap with the mitochondria and change their size over time in I(−) HCN cells. D, AMPK is required for p62 translocation to mitochondria. Insulin withdrawal-induced translocation of hp62-WT, but not its phosphomimicking S294E form, was prevented in AMPK α2 KO HCN cells. E, analysis of D. Quantification of mitochondrial localization of p62 (n = 10) is shown. F, detection of phosphorylated p62 in the mitochondrial fraction following insulin withdrawal for 5 h. Subcellular fractions were analyzed by Western blotting with p62 Ser-293 phosphospecific antibody. Blots shown are representative of three independent experiments, which yielded similar results. Error bars represent ±S.D. from independent assays. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Scale bar, 10 μm. Cyto, cytosolic fraction; Mito, mitochondrial fraction; COX, cytochrome c oxidase; p-p62, phosphorylated p62; t-p62, total p62.
To further confirm that mitochondrial translocation of p62 depends on AMPK, we expressed HA-hp62 in AMPK α2 KO HCN cells. Similar to pharmacological inhibition of AMPK activity, ablation of the AMPK α2 subunit suppressed mitochondrial translocation of hp62-WT in the I(−) condition (Fig. 7, D and E). Similarly, hp62-S294A failed to translocate to mitochondria (Fig. 7, D and E). However, a considerable fraction of hp62-S294E was still observed in the mitochondria in I(−) AMPK α2 KO HCN cells, consistent with the ability of this phosphorylation-mimicking mutant to bypass the requirement for AMPK activation (Fig. 7, D and E). Without hp62 translocation, mitochondria retained normal filamentous morphology, but hp62-S294E-containing mitochondria were small and fragmented. Subcellular fractionation also showed a substantial increase in phosphorylated p62 in the mitochondrial fraction of I(−) HCN cells compared with the mitochondrial fraction of I(+) HCN cells (Fig. 7F). Moreover, a considerable amount of LC3, in particular LC3-II, was detected in the mitochondrial fraction of I(−) HCN cells (Fig. 7F). These results suggest the induction of mitophagy upon translocation of AMPK-phosphorylated p62. To assess whether p62 phosphorylation is required for mitophagy, we tested the ability of hp62-WT and the Ser-294 mutants to rescue mitophagy in Sh-rp62 HCN cells co-transfected with mt-mKeima. Whereas hp62-S294E and -WT rescued mitophagy and were readily detected in the mitochondria, showing a high 561/458 nm ratio in I(−) Sh-rp62 HCN cells, hp62-S294A failed to induce mitophagy (Fig. 8, A and B). These findings strongly suggest that AMPK-phosphorylated p62 mediates mitophagy in HCN cells during ACD following insulin withdrawal.
Figure 8.
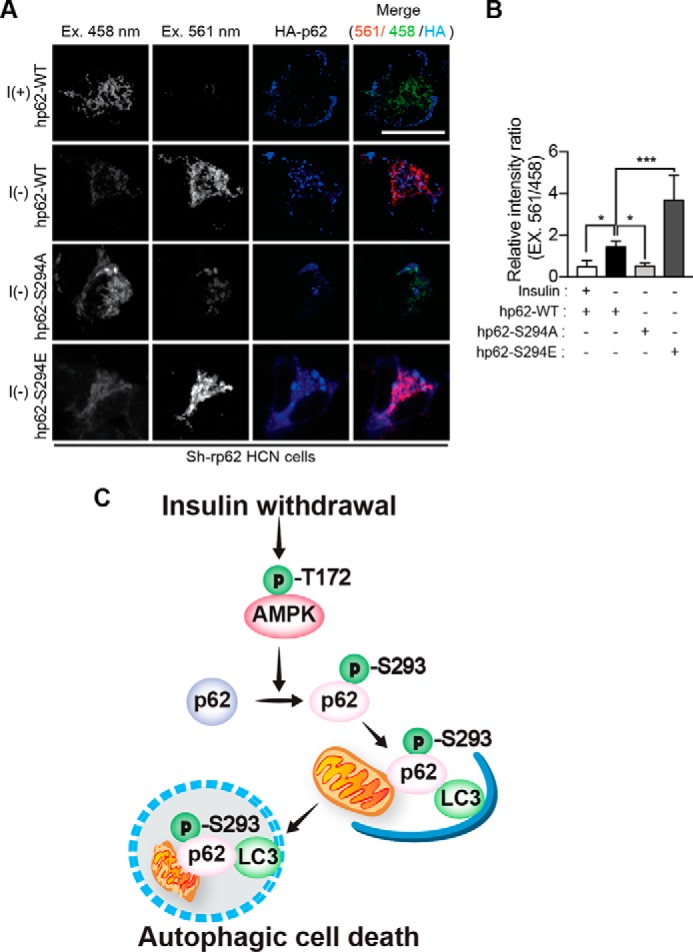
Phosphorylated p62 mediates AMPK-induced mitophagy in I(−) HCN cells. A, co-transfection of Sh-rp62 HCN cells with plasmids encoding mt-mKeima and HA-tagged hp62-WT, -S294A, or -S294E. Cells expressing hp62-WT or hp62-S294E underwent mitophagy, whereas the expression of hp62-S294A mutant prevented mitophagy. Ex, excitation. B, analysis of A. The relative intensity ratio as an index of mitophagy (n = 8) is shown. C, a schematic diagram illustrating p62 phosphorylation by AMPK and subsequent mitophagy and ACD in HCN cells following insulin withdrawal. Error bars represent ±S.D. from independent assays. *, p < 0.05; ***, p < 0.001. Scale bar, 10 μm.
Discussion
Our previous observations that insulin withdrawal drives the mode of cell death toward ACD rather than apoptosis suggest the existence of uniquely programmed cell death mechanisms in HCN cells (17–20). To elucidate the molecular mechanisms underlying this preference for ACD over apoptosis, we considered the potential role of AMPK because AMPK is regarded as more than a sensor of cellular energy status and is involved in the regulation of autophagy, proliferation, and cell death (30). Furthermore, the close relationship of AMPK activity with insulin signaling suggests that AMPK may play a role in insulin withdrawal-induced HCN cell death (52). Our findings in this study provide not only a new perspective on the role of AMPK in the induction of ACD but also a novel AMPK substrate involved for the signaling of ACD. Once we established the autophagic nature of AMPK-induced cell death based on (i) an increase of autophagy flux, (ii) the lack of apoptotic signs and ineffectiveness of Z-VAD, and (iii) abrogation of ACD in the absence of Atg7 in I(−) HCN cells, we then suspected p62 as an AMPK substrate important for the regulation of ACD given the role of p62 as a multifunctional adaptor protein in autophagy. p62 delivers the cargos to the lysosomes through its interaction with LC3-II. In addition, p62 serves as a signaling hub molecule that coordinates autophagy, the ubiquitin-proteasome system, and metabolism and undergoes post-translational modifications, including phosphorylation (44–46). Our bioinformatics analysis predicted the p62 sites potentially phosphorylated by AMPK (53). Phosphorylation of some of these sites was previously reported in large-scale phosphopeptide screening and proteomics analyses performed by others, although their functional relationship with AMPK has not yet been tested experimentally (53–55).
The phosphorylation-defective p62 mutant inhibited ACD, whereas the phosphorylation-mimicking mutant markedly increased ACD. Co-localization of p62 with mitochondria was significantly reduced when AMPK was inactivated. A non-phosphorylatable p62 mutant was defective in translocation to mitochondria and thereby failed to mediate mitophagy. Subcellular fractionation confirmed these results. In I(+) HCN cells, very little p62 was detected in the mitochondrial fraction, but after insulin withdrawal, more p62, especially its phosphorylated form, was present in the mitochondrial fraction, which also contained much more LC3-II.
AMPK can induce mitophagy by several mechanisms. One mechanism is the AMPK–ULK1 pathway. ULK1, a mammalian ortholog of the yeast kinase Atg1, is phosphorylated by AMPK during starvation-induced mitophagy in mammalian liver and C. elegans (36). Genetic deficiency in ULK1 or expression of phosphorylation-defective ULK1 with mutated AMPK phosphorylation sites leads to p62 accumulation and impairs mitophagy, but the role of p62 in this process was not further explored (36). In HepG2 cells, the activation of the AMPK–ULK1 pathway induces mitophagy to remove oxidative stress-damaged mitochondria (56). Phosphorylation of ULK1 by AMPK causes mitochondrial recruitment of ULK1 and initiates mitophagy, whereas ULK1 knockdown impairs mitophagy (56). p62 knockdown also impairs mitochondrial function and increases oxidative stress, presumably due to the failure of mitophagy (56). However, the supposed link between p62 and mitophagy was not experimentally tested. In both studies (36, 56), therefore, p62 was involved downstream of AMPK-ULK1 as a survival factor; however, the direct involvement of p62 in AMPK-induced mitophagy was not thoroughly examined. Hypoxia induces autophagic degradation of androgen receptor in prostate cancer cells via a concerted action of p62 and AMPK (57). Hypoxia promotes p62 phosphorylation on Ser-403 and thereby enhances its interaction with androgen receptor; this phosphorylation appears to require AMPK activity, although this conclusion was based only on the results of pharmacological inhibition of AMPK activity (57). Despite these reports, no biochemical evidence demonstrating direct AMPK-mediated phosphorylation of p62 in autophagy has been reported. To the best of our knowledge, our study is the first report on p62 phosphorylation directly by AMPK.
Recently, the importance of p62 in mitophagy was questioned. In Parkin-mediated mitophagy, p62 was found to be required for the aggregation but not for the degradation of dysfunctional mitochondria (58, 59). Instead, NDP52 and optineurin served as autophagy receptors in PINK1/Parkin-mediated mitophagy (60). Therefore, p62 may be involved in the subcellular redistribution of dysfunctional mitochondria to promote their clearance but may be dispensable for their autophagic degradation in some cell types. By contrast, our results demonstrate the co-localization of phosphorylated p62 with mitochondria undergoing mitophagy, whereas phosphorylation-defective p62 abolishes mitophagy even under I(−) conditions. Therefore, our data support the hypothesis that p62 phosphorylated by AMPK, at least in part, mediates mitophagy in HCN cells upon insulin withdrawal (Fig. 8C). Interestingly, p62 phosphorylation by AMPK was not required for STS-induced apoptosis, highlighting the functional significance of AMPK-mediated phosphorylation of p62 in ACD. Collectively, our findings indicate that phosphorylation of rat/human p62 by AMPK on Ser-293/Ser-294 is necessary for ACD in I(−) HCN cells and define a new pathway linking mitochondrial homeostasis and ACD with cellular stress responses coordinated by the AMPK–p62 axis in neural stem cells.
The functional roles of mitophagy in cell survival and death can vary. Mitophagy is thought to be associated with cell survival and maintenance of cellular homeostasis because it removes dysfunctional and damaged mitochondria (61–63), which would otherwise produce reactive oxygen species and release cytochrome c and other proapoptotic proteins leading to cell death. In contrast, mitophagy can promote cell death under some pathological conditions (64). Our data suggest that AMPK–p62-driven mitophagy is a regulatory pathway through which autophagy serves as a cell death mechanism in HCN cells.
Understanding the mechanisms of ACD is still in its infancy, and it is not clear how cell death-promoting autophagy can be distinguished from cytoprotective autophagy. There may be distinct upstream signaling pathways that determine bifurcation of autophagy between cell survival and death. Alternatively, a unique set of autophagy machinery, different from that involved in survival, may promote death. Further identification of distinct signal transduction pathways in our insulin withdrawal model of ACD in neural stem cells may help to distinguish between these intriguing possibilities.
Experimental procedures
Reagents, antibodies, and generation of phosphorylation-specific p62 antibody
Compound C (Calbiochem, 171260), BafA1 (Enzo, BML-CM110), Z-VAD (Enzo, ALX-260-020), CCCP (Sigma-Aldrich, C2759), STS (Cell Signaling Technology, 9953), puromycin (Invitrogen, NC9138068), hygromycin B (Duchefa, H0192), and STO-609 (Tocris, 1551) were purchased from the indicated companies. Antibodies against phospho-AMPK (Thr-172; 2535), total AMPK (2532), cleaved caspase-3 (9664), phospho-ACC (Ser-79; 3661), total ACC (3662), and Atg7 (8558) were purchased from Cell Signaling Technology. Antibodies for GFP (SC-9996), HA (SC-805), FLAG (SC-166355), and β-actin-horseradish peroxidase (SC-47778) were purchased from Santa Cruz Biotechnology. Antibodies for LC3 (Novus, NB100-2220), p62 (Sigma-Aldrich, P0067), AMPK α1 (Millipore, 04-323), and AMPK α2 (Abcam, ab3760) were purchased from the indicated companies. The phospho-p62 Ser-293 antibody was generated against a synthetic peptide (CSSEV(p)SKPDGAGE) corresponding to the rat p62 sequence. The purified peptide was conjugated to keyhole limpet hemocyanin and injected into rabbits over an 8-week period. The polyclonal antibody was purified by peptide-conjugated affinity chromatography.
Cell culture and cell death assay
HCN cells were obtained from a hippocampus of a 2-month-old adult rat as described previously (19, 20). Cells were seeded at 1 × 104 cells/well in a 96-well plate and stained with propidium iodide (Sigma-Aldrich, P4170) and Hoechst 33342 (Invitrogen, H3570) for 15 min at room temperature. Cell staining images were acquired with a fluorescence microscope (Carl Zeiss, Axiovert 40 CFL), and stained cells were quantified using the automated image analysis software Matlab with the CellC package (Mathworks) (65).
shRNA knockdown
The lentiviral shRNA clones targeting rat Atg7 (TRCN0000092164 and TRCN0000369085) and p62 (TRCN0000238135 and TRCN0000257058) from the MISSION library were purchased from Sigma-Aldrich, and the lentiviruses were produced following published protocols (66). For stable knockdown, HCN cells were infected with the virus for 24 h, and then the medium was replaced with fresh medium. After 72 h, HCN cells were treated with puromycin (5 μg/ml) for 6 h and then maintained in medium containing puromycin (1 μg/ml). Selected HCN cells were grown in puromycin-free medium for 24 h before conducting experiments.
CRISPR/Cas9-mediated knock-out of AMPK α subunits
HCN cells were transfected with pRGEN-cas9-Hygo-eGFP and dRGEN-AMPKα1 (CGCTGGGCGTCGGCACCTTCGGG) or -AMPKα2 (TGCCGAAGGTGCCGACGCCCAGG) single guide RNA, which were designed and purchased from ToolGen. After incubation for 48 h, cells were selected using hygromycin (100 μg/ml).
Fluorescence imaging
Cells expressing peGFP-LC3 (Addgene, 21073), pLjm1-mRFP-GFP-LC3, pMT-mKeima-red (MBL International, AM-V0251), and/or pDsRed2-Mito (a kind gift from Dr. Cheil Moon, Daegu Gyeongbuk Institute of Science and Technology) were grown on coverslips and fixed in 4% paraformaldehyde in PBS for 10 min at room temperature. After washing with PBS twice, the cells were mounted on slides with mounting solution (Dako, S3023), and images were taken with a confocal microscope (Carl Zeiss LSM 780) using a 63×/1.0 oil objective or SR-SIM (Carl Zeiss ELYRA S1) using a 63×/1.4 oil objective. LC3 puncta were measured in HCN cells transiently expressing peGFP-LC3 or stably expressing the lentiviral construct pLjm1-mRFP-GFP-LC3. Images were obtained from different fields randomly selected in each experiment. To quantify the number of puncta (diameter, 0.2–1.0 μm), the images were analyzed using the Green and Red Puncta Colocalization ImageJ plug-in (67). To monitor translocation of p62 to mitochondria, HCN cells were transiently transfected with the peGFP-p62 construct using Lipofectamine 2000 and the next day observed under live confocal microscopy (Carl Zeiss LSM 7) using a plan Apochromat 63×/1.4 oil objective upon insulin withdrawal. Mitophagy was monitored using the pMT-mKeima-red construct. To determine p62 localization, HCN cells transfected with pcDNA vector carrying HA-tagged human p62 (Addgene, 28027) were grown on coverslips in 12-well plates. The cells were fixed, permeabilized with 0.1% Triton X-100 in antibody dilution buffer (Invitrogen, 003218) for 20 min, washed twice with PBS, and then incubated overnight with anti-HA primary antibody. Cells were then washed twice with PBS and incubated for 1 h with secondary anti-rabbit DyLight 405 (Jackson ImmunoResearch, 711-475-152) or anti-rabbit Alexa Fluor 488 (Thermo Fisher Scientific, A11034) antibody followed by nuclear counterstaining with Hoechst 33342 for 10 min and imaging. Co-localization of LC3/p62 and mitochondria and the relative intensity ratio of mKeima signals were analyzed using Zen software (Carl Zeiss).
Recombinant protein purification
Human p62-WT and -S294A mutant constructs were cloned in pGEX-5X and expressed as GST-tagged forms in E. coli BL21; expression was induced with 1 mm isopropyl β-d-thiogalactopyranoside (Duchefa, I1401) overnight at 16 °C. The cells were lysed in GST binding buffer (20 mm Tris-HCl, pH 7.5, 100 mm NaCl, 0.1% Nonidet P-40, 1 mm EDTA, 1 mm phenylmethylsulfonyl fluoride) with 10 mg/ml lysozyme (Sigma-Aldrich, L6876) for 30 min at room temperature and centrifuged. To purify the recombinant proteins, supernatants were incubated with glutathione-Sepharose 4B beads (GE Healthcare, 52-2303-00AK) overnight at 4 °C, and proteins were eluted with GST elution buffer (20 mm Tris-HCl, pH 7.5, 500 mm NaCl, 0.5% Nonidet P-40, 1 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, 20 mm glutathione).
Western blot analysis
Cells were lysed in lysis buffer (50 mm Tris-HCl, pH 7.5, 250 mm sucrose, 1 mm EDTA, 1 mm EGTA, 1 mm dithiothreitol, 50 mm NaF, 1 mm phenylmethylsulfonyl fluoride, 1 mm benzamidine, 1% Triton X-100) supplemented with protease and phosphatase inhibitor mixtures (Thermo Scientific, 78429 and 1862495, respectively). Lysates were run on an SDS-polyacrylamide gel and transferred to a polyvinylidene fluoride membrane (Millipore, IPVH 00010). The membranes were blocked with 5% nonfat dry milk and 0.1% Tween 20 in Tris-buffered saline and incubated overnight with appropriate primary antibodies. The next day, the membranes were incubated with the secondary antibodies conjugated to horseradish peroxidase for 1 h and processed for detection using a chemiluminescence detection kit (Thermo Scientific, 34080).
In vitro kinase assay
HEK293FT cells transfected with peGFP-N1-AMPKα2-CA were lysed in lysis buffer for Western blotting, and the lysates were incubated with GFP antibody and protein A beads (Thermo Fisher Scientific, 20333) overnight at 4 °C. The next day, GFP-tagged AMPK was immunoprecipitated by centrifugation, washed with PBS, and suspended in lysis buffer. For the in vitro kinase reaction, immunoprecipitated AMPK complexes were incubated with recombinant human p62 substrate and 1 mm ATP for 1 h at 37 °C in kinase buffer (50 mm Tris-HCl, pH 7.5, 10 mm MgCl2, 1 mm dithiothreitol). The reaction was stopped by adding SDS sample loading buffer and boiling, and the samples were analyzed by Western blotting with phospho-p62 Ser-293 antibody. In addition, recombinant human AMPK protein (combination of α2/β1/γ1 subunits; SignalChem, P48-10H) expressed in Sf9 cells was used for the in vitro kinase assay.
Mitochondrial and cytosolic fractionation
Subcellular fractionation to separate mitochondrial and cytosolic fractions was performed according to the published protocol with a slight modification (68). HCN cells were lysed by vortexing for 15 min in lysis buffer containing 50 mm Tris-HCl, pH 7.4, 250 mm sucrose, 5 mm MgCl2 supplemented with protease and phosphatase inhibitor mixtures. The homogenate was left on ice for 30 min and then centrifuged at 800 × g for 15 min. The supernatant (S0) was separated by centrifugation at 11,000 × g for 10 min, and the supernatant (S1) and pellet (P1) were further processed for the preparation of the cytosolic and mitochondrial fractions, respectively. S1 was precipitated by adding an equal volume of cold 100% acetone overnight at −20 °C. After centrifugation at 12,000 × g for 5 min, the precipitate was resuspended in 100 μl of lysis buffer and used as the cytosolic fraction. To increase the purity of mitochondria, P1 was resuspended in lysis buffer and centrifuged again at 11,000 × g for 10 min at 4 °C. The final pellet (P2) was resuspended in 40 μl of extraction buffer (50 mm Tris-HCl, pH 6.8, 1 mm EDTA, 0.5% Triton X-100, protease and phosphatase inhibitor mixtures) and sonicated by a Bioruptor KRB-01(CosmoBio) on ice five times for 5–10 s at a high setting with 30-s pauses and used as the mitochondrial fraction.
FACS analysis
Cells were harvested in the medium and centrifuged at 500 × g for 3 min. Cell pellets were resuspended in PBS containing MitoTracker Green (20 nm; Thermo Fisher Scientific, M7514) and stained for 8 min. The stained cells were subjected to analysis using a BD Accuri flow cytometer (BD Biosciences). The data were further analyzed using Accuri C6 software (BD Biosciences).
Statistical analysis
All values are presented as mean ± standard deviation (S.D.) and were obtained by averaging the data from at least three independent experiments. Statistical significance was determined by one-way analysis of variance followed by Holm-Sidak multiple comparison tests using GraphPad Prism (GraphPad Software).
Author contributions
S.-W. Y. contributed to conception and design, data analysis and interpretation, manuscript writing, and final approval of manuscript. S. H. contributed to conception and design, collection and assembly of data, data analysis and interpretation, and manuscript writing. S.-H. J., K. Y., K. M. C., and C. J. H. collected and assembled data. S. W. K. and E.-K. K. contributed to conception, design, and data analysis and interpretation. All authors commented on the manuscript and approved the final version.
Supplementary Material
Acknowledgment
We thank the Center for Core Research Facilities at Daegu Gyeongbuk Institute of Science and Technology for support for the use of SR-SIM (Carl Zeiss ELYRA S1).
This work was supported by Bio and Medical Technology Development Program of the National Research Foundation of Korea Grants (2012M3A9C6049935 and 2013M3C7A1056099), Korea Brain Research Institute Basic Research Program (17-BR-04), and Daegu Gyeongbuk Institute of Science and Technology Convergence Science Center Program (17-BD-04) of the Ministry of Science, ICT and Future Planning of Korea. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S6 and Movies S1–S5.
- PCD
- programmed cell death
- ACC
- acetyl-CoA carboxylase
- ACD
- autophagic cell death
- AMPK
- AMP-activated protein kinase
- Atg
- autophagy-related gene
- BafA1
- bafilomycin A1
- CA
- constitutively active
- CC
- compound C
- CCCP
- carbonyl cyanide m-chlorophenylhydrazone
- CRISPR
- clustered regularly interspaced short palindromic repeats
- HCN
- hippocampal neural stem cells
- LC3
- microtubule-associated protein light chain 3
- STS
- staurosporine
- ULK1
- Unc-51-like kinase 1
- Z-VAD
- benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone
- I
- insulin
- CaMKK
- Ca2+/calmodulin-dependent protein kinase kinase
- mRFP
- monomeric red fluorescent protein
- rp62
- rat p62
- mp62
- mouse p62
- hp62
- human p62
- SR-SIM
- super-resolution microscopy for structured illumination
- mt-mKeima
- mitochondrially targeted version of monomeric Keima.
References
- 1. Klionsky D. J., and Emr S. D. (2000) Cell biology—autophagy as a regulated pathway of cellular degradation. Science 290, 1717–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lum J. J., DeBerardinis R. J., and Thompson C. B. (2005) Autophagy in metazoans: cell survival in the land of plenty. Nat. Rev. Mol. Cell Biol. 6, 439–448 [DOI] [PubMed] [Google Scholar]
- 3. Codogno P., and Meijer A. (2005) Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 12, 1509–1518 [DOI] [PubMed] [Google Scholar]
- 4. Puyal J., Ginet V., and Clarke P. G. (2013) Multiple interacting cell death mechanisms in the mediation of excitotoxicity and ischemic brain damage: a challenge for neuroprotection. Prog. Neurobiol. 105, 24–48 [DOI] [PubMed] [Google Scholar]
- 5. Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., and Yoshimori T. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ichimura Y., Kirisako T., Takao T., Satomi Y., Shimonishi Y., Ishihara N., Mizushima N., Tanida I., Kominami E., Ohsumi M., Noda T., and Ohsumi Y. (2000) A ubiquitin-like system mediates protein lipidation. Nature 408, 488–492 [DOI] [PubMed] [Google Scholar]
- 7. Suzuki K., Kirisako T., Kamada Y., Mizushima N., Noda T., and Ohsumi Y. (2001) The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 20, 5971–5981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mizushima N., Yamamoto A., Hatano M., Kobayashi Y., Kabeya Y., Suzuki K., Tokuhisa T., Ohsumi Y., and Yoshimori T. (2001) Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 152, 657–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mizushima N., Yamamoto A., Matsui M., Yoshimori T., and Ohsumi Y. (2004) In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 15, 1101–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bjørkøy G., Lamark T., Brech A., Outzen H., Perander M., Overvatn A., Stenmark H., and Johansen T. (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171, 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pankiv S., Clausen T. H., Lamark T., Brech A., Bruun J.-A., Outzen H., Øvervatn A., Bjørkøy G., and Johansen T. (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145 [DOI] [PubMed] [Google Scholar]
- 12. Ichimura Y., Kominami E., Tanaka K., and Komatsu M. (2008) Selective turnover of p62/A170/SQSTM1 by autophagy. Autophagy 4, 1063–1066 [DOI] [PubMed] [Google Scholar]
- 13. Kroemer G., and Levine B. (2008) Autophagic cell death: the story of a misnomer. Nat. Rev. Mol. Cell Biol. 9, 1004–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kourtis N., and Tavernarakis N. (2009) Autophagy and cell death in model organisms. Cell Death Differ. 16, 21–30 [DOI] [PubMed] [Google Scholar]
- 15. Kim N.-Y., and Lee M. (2014) The pro-death role of autophagy and apoptosis in cell death induced by the BH3 mimetic gossypol. Animal Cells Syst. 18, 183–189 [Google Scholar]
- 16. Shen H. M., and Codogno P. (2011) Autophagic cell death: Loch Ness monster or endangered species? Autophagy 7, 457–465 [DOI] [PubMed] [Google Scholar]
- 17. Yu S. W., Baek S. H., Brennan R. T., Bradley C. J., Park S. K., Lee Y. S., Jun E. J., Lookingland K. J., Kim E. K., Lee H., Goudreau J. L., and Kim S. W. (2008) Autophagic death of adult hippocampal neural stem cells following insulin withdrawal. Stem Cells 26, 2602–2610 [DOI] [PubMed] [Google Scholar]
- 18. Baek S.-H., Kim E.-K., Goudreau J. L., Lookingland K. J., Kim S. W., and Yu S.-W. (2009) Insulin withdrawal-induced cell death in adult hippocampal neural stem cells as a model of autophagic cell death. Autophagy 5, 277–279 [DOI] [PubMed] [Google Scholar]
- 19. Ha S., Ryu H. Y., Chung K. M., Baek S.-H., Kim E.-K., and Yu S.-W. (2015) Regulation of autophagic cell death by glycogen synthase kinase-3β in adult hippocampal neural stem cells following insulin withdrawal. Mol. Brain 8, 30–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chung K. M., Park H., Jung S., Ha S., Yoo S.-J., Woo H., Lee H. J., Kim S. W., Kim E.-K., Moon C., and Yu S.-W. (2015) Calpain determines the propensity of adult hippocampal neural stem cells to autophagic cell death following insulin withdrawal. Stem Cells 33, 3052–3064 [DOI] [PubMed] [Google Scholar]
- 21. Yeo B. K., Hong C. J., Chung K. M., Woo H., Kim K., Jung S., Kim E.-K., and Yu S.-W. (2016) Valosin-containing protein is a key mediator between autophagic cell death and apoptosis in adult hippocampal neural stem cells following insulin withdrawal. Mol. Brain 9, 31–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hong C. J., Park H., and Yu S.-W. (2016) Autophagy for the quality control of adult hippocampal neural stem cells. Brain Res. 1649, 166–172 [DOI] [PubMed] [Google Scholar]
- 23. Denton D., Nicolson S., and Kumar S. (2012) Cell death by autophagy: facts and apparent artefacts. Cell Death Differ. 19, 87–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chung K. M., and Yu S.-W. (2013) Interplay between autophagy and programmed cell death in mammalian neural stem cells. BMB Rep. 46, 383–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ryu J. R., Hong C. J., Kim J. Y., Kim E.-K., Sun W., and Yu S.-W. (2016) Control of adult neurogenesis by programmed cell death in the mammalian brain. Mol. Brain 9, 43–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Clarke P. G., and Puyal J. (2012) Autophagic cell death exists. Autophagy 8, 867–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chung K. M., Jeong E. J., Park H., An H. K., and Yu S. W. (2016) Mediation of autophagic cell death by type 3 ryanodine receptor (RyR3) in adult hippocampal neural stem cells. Front. Cell. Neurosci. 10, 116–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stapleton D., Mitchelhill K. I., Gao G., Widmer J., Michell B. J., Teh T., House C. M., Fernandez C. S., Cox T., Witters L. A., and Kemp B. E. (1996) Mammalian AMP-activated protein kinase subfamily. J. Biol. Chem. 271, 611–614 [DOI] [PubMed] [Google Scholar]
- 29. Mihaylova M. M., and Shaw R. J. (2011) The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 13, 1016–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Motoshima H., Goldstein B. J., Igata M., and Araki E. (2006) AMPK and cell proliferation—AMPK as a therapeutic target for atherosclerosis and cancer. J. Physiol. 574, 63–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Han D., Yang B., Olson L. K., Greenstein A., Baek S. H., Claycombe K. J., Goudreau J. L., Yu S. W., and Kim E. K. (2010) Activation of autophagy through modulation of 5′-AMP-activated protein kinase protects pancreatic β-cells from high glucose. Biochem. J. 425, 541–551 [DOI] [PubMed] [Google Scholar]
- 32. Liang J., Shao S. H., Xu Z.-X., Hennessy B., Ding Z., Larrea M., Kondo S., Dumont D. J., Gutterman J. U., Walker C. L., Slingerland J. M., and Mills G. B. (2007) The energy sensing LKB1–AMPK pathway regulates p27kip1 phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 9, 218–224 [DOI] [PubMed] [Google Scholar]
- 33. Matsui Y., Takagi H., Qu X., Abdellatif M., Sakoda H., Asano T., Levine B., and Sadoshima J. (2007) Distinct roles of autophagy in the heart during ischemia and reperfusion roles of AMP-activated protein kinase and beclin 1 in mediating autophagy. Circ. Res. 100, 914–922 [DOI] [PubMed] [Google Scholar]
- 34. Kim J., Kundu M., Viollet B., and Guan K.-L. (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gwinn D. M., Shackelford D. B., Egan D. F., Mihaylova M. M., Mery A., Vasquez D. S., Turk B. E., and Shaw R. J. (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Egan D. F., Shackelford D. B., Mihaylova M. M., Gelino S., Kohnz R. A., Mair W., Vasquez D. S., Joshi A., Gwinn D. M., Taylor R., Asara J. M., Fitzpatrick J., Dillin A., Viollet B., Kundu M., et al. (2011) Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim J., Kim Y. C., Fang C., Russell R. C., Kim J. H., Fan W., Liu R., Zhong Q., and Guan K.-L. (2013) Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152, 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hawley S. A., Davison M., Woods A., Davies S. P., Beri R. K., Carling D., and Hardie D. G. (1996) Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem. 271, 27879–27887 [DOI] [PubMed] [Google Scholar]
- 39. Stein S. C., Woods A., Jones N. A., Davison M. D., and Carling D. (2000) The regulation of AMP-activated protein kinase by phosphorylation. Biochem. J. 345, 437–443 [PMC free article] [PubMed] [Google Scholar]
- 40. Kim J., Yang G., Kim Y., Kim J., and Ha J. (2016) AMPK activators: mechanisms of action and physiological activities. Exp. Mol. Med. 48, e224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Crute B. E., Seefeld K., Gamble J., Kemp B. E., and Witters L. A. (1998) Functional domains of the α1 catalytic subunit of the AMP-activated protein kinase. J. Biol. Chem. 273, 35347–35354 [DOI] [PubMed] [Google Scholar]
- 42. Woods A., Azzout-Marniche D., Foretz M., Stein S. C., Lemarchand P., Ferré P., Foufelle F., and Carling D. (2000) Characterization of the role of AMP-activated protein kinase in the regulation of glucose-activated gene expression using constitutively active and dominant negative forms of the kinase. Mol. Cell. Biol. 20, 6704–6711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kimura S., Noda T., and Yoshimori T. (2007) Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3, 452–460 [DOI] [PubMed] [Google Scholar]
- 44. Ichimura Y., Waguri S., Sou Y.-S., Kageyama S., Hasegawa J., Ishimura R., Saito T., Yang Y., Kouno T., Fukutomi T., Hoshii T., Hirao A., Takagi K., Mizushima T., Motohashi H., et al. (2013) Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 51, 618–631 [DOI] [PubMed] [Google Scholar]
- 45. Linares J. F., Amanchy R., Greis K., Diaz-Meco M. T., and Moscat J. (2011) Phosphorylation of p62 by cdk1 controls the timely transit of cells through mitosis and tumor cell proliferation. Mol. Cell. Biol. 31, 105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lim J., Lachenmayer M. L., Wu S., Liu W., Kundu M., Wang R., Komatsu M., Oh Y. J., Zhao Y., and Yue Z. (2015) Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet. 11, e1004987–e1005014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xue Y., Liu Z., Cao J., Ma Q., Gao X., Wang Q., Jin C., Zhou Y., Wen L., and Ren J. (2011) GPS 2.1: enhanced prediction of kinase-specific phosphorylation sites with an algorithm of motif length selection. Protein Eng. Des. Sel. 24, 255–260 [DOI] [PubMed] [Google Scholar]
- 48. Matsumoto G., Wada K., Okuno M., Kurosawa M., and Nukina N. (2011) Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol. Cell 44, 279–289 [DOI] [PubMed] [Google Scholar]
- 49. Kim I., Rodriguez-Enriquez S., and Lemasters J. J. (2007) Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 462, 245–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kogure T., Karasawa S., Araki T., Saito K., Kinjo M., and Miyawaki A. (2006) A fluorescent variant of a protein from the stony coral Montipora facilitates dual-color single-laser fluorescence cross-correlation spectroscopy. Nat. Biotechnol. 24, 577–581 [DOI] [PubMed] [Google Scholar]
- 51. Katayama H., Kogure T., Mizushima N., Yoshimori T., and Miyawaki A. (2011) A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem. Biol. 18, 1042–1052 [DOI] [PubMed] [Google Scholar]
- 52. Towler M. C., and Hardie D. G. (2007) AMP-activated protein kinase in metabolic control and insulin signaling. Circ. Res. 100, 328–341 [DOI] [PubMed] [Google Scholar]
- 53. Hoffman N. J., Parker B. L., Chaudhuri R., Fisher-Wellman K. H., Kleinert M., Humphrey S. J., Yang P., Holliday M., Trefely S., Fazakerley D. J., Stöckli J., Burchfield J. G., Jensen T. E., Jothi R., Kiens B., et al. (2015) Global phosphoproteomic analysis of human skeletal muscle reveals a network of exercise-regulated kinases and AMPK substrates. Cell Metab. 22, 922–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wu X., Tian L., Li J., Zhang Y., Han V., Li Y., Xu X., Li H., Chen X., Chen J., Jin W., Xie Y., Han J., and Zhong C. Q. (2012) Investigation of receptor interacting protein (RIP3)-dependent protein phosphorylation by quantitative phosphoproteomics. Mol. Cell. Proteomics 11, 1640–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kettenbach A. N., Schweppe D. K., Faherty B. K., Pechenick D., Pletnev A. A., and Gerber S. A. (2011) Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci. Signal. 4, rs5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sinha R. A., Singh B. K., Zhou J., Wu Y., Farah B. L., Ohba K., Lesmana R., Gooding J., Bay B.-H., and Yen P. M. (2015) Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ROS-AMPK-ULK1 signaling. Autophagy 11, 1341–1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mitani T., Minami M., Harada N., Ashida H., and Yamaji R. (2015) Autophagic degradation of the androgen receptor mediated by increased phosphorylation of p62 suppresses apoptosis in hypoxia. Cell. Signal. 27, 1994–2001 [DOI] [PubMed] [Google Scholar]
- 58. Narendra D., Kane L. A., Hauser D. N., Fearnley I. M., and Youle R. J. (2010) p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6, 1090–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Okatsu K., Saisho K., Shimanuki M., Nakada K., Shitara H., Sou Y. S., Kimura M., Sato S., Hattori N., Komatsu M., Tanaka K., and Matsuda N. (2010) p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 15, 887–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lazarou M., Sliter D. A., Kane L. A., Sarraf S. A., Wang C., Burman J. L., Sideris D. P., Fogel A. I., and Youle R. J. (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Elmore S. P., Qian T., Grissom S. F., and Lemasters J. J. (2001) The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 15, 2286–2287 [DOI] [PubMed] [Google Scholar]
- 62. Tal R., Winter G., Ecker N., Klionsky D. J., and Abeliovich H. (2007) Aup1p, a yeast mitochondrial protein phosphatase homolog, is required for efficient stationary phase mitophagy and cell survival. J. Biol. Chem. 282, 5617–5624 [DOI] [PubMed] [Google Scholar]
- 63. Youle R. J., and Narendra D. P. (2011) Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dagda R. K., Zhu J., Kulich S. M., and Chu C. T. (2008) Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress. Autophagy 4, 770–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Selinummi J., Seppälä J., Yli-Harja O., and Puhakka J. A. (2005) Software for quantification of labeled bacteria from digital microscope images by automated image analysis. BioTechniques 39, 859–863 [DOI] [PubMed] [Google Scholar]
- 66. Tiscornia G., Singer O., and Verma I. M. (2006) Production and purification of lentiviral vectors. Nat. Protoc. 1, 241–245 [DOI] [PubMed] [Google Scholar]
- 67. Pampliega O., Orhon I., Patel B., Sridhar S., Díaz-Carretero A., Beau I., Codogno P., Satir B. H., Satir P., and Cuervo A. M. (2013) Functional interaction between autophagy and ciliogenesis. Nature 502, 194–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dimauro I., Pearson T., Caporossi D., and Jackson M. J. (2012) A simple protocol for the subcellular fractionation of skeletal muscle cells and tissue. BMC Res. Notes 5, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.