

Abstract
Bone and skeletal muscle mass are highly correlated in mammals, suggesting the existence of common anabolic signaling networks that coordinate the development of these two anatomically adjacent tissues. The activin signaling pathway is an attractive candidate to fulfill such a role. Here, we generated mice with conditional deletion of activin receptor (ACVR) type 2A, ACVR2B, or both, in osteoblasts, to determine the contribution of activin receptor signaling in regulating bone mass. Immunohistochemistry localized ACVR2A and ACVR2B to osteoblasts and osteocytes. Primary osteoblasts expressed activin signaling components, including ACVR2A, ACVR2B, and ACVR1B (ALK4) and demonstrated increased levels of phosphorylated Smad2/3 upon exposure to activin ligands. Osteoblasts lacking ACVR2B did not show significant changes in vitro. However, osteoblasts deficient in ACVR2A exhibited enhanced differentiation indicated by alkaline phosphatase activity, mineral deposition, and transcriptional expression of osterix, osteocalcin, and dentin matrix acidic phosphoprotein 1. To investigate activin signaling in osteoblasts in vivo, we analyzed the skeletal phenotypes of mice lacking these receptors in osteoblasts and osteocytes (osteocalcin-Cre). Similar to the lack of effect in vitro, ACVR2B-deficient mice demonstrated no significant change in any bone parameter. By contrast, mice lacking ACVR2A had significantly increased femoral trabecular bone volume at 6 weeks of age. Moreover, mutant mice lacking both ACVR2A and ACVR2B demonstrated sustained increases in trabecular bone volume, similar to those in ACVR2A single mutants, at 6 and 12 weeks of age. Taken together, these results indicate that activin receptor signaling, predominantly through ACVR2A, directly and negatively regulates bone mass in osteoblasts.
Keywords: activin, bone, myostatin, osteoblast, transforming growth factor beta (TGF-β)
Introduction
The musculoskeletal system evolved in mammals to perform diverse functions that include locomotion, breathing, protecting internal organs, and coordinating global energy expenditure. After the third decade of life, muscles and bones begin to lose their mass, leading to unfavorable alterations in their function. Aging is universally accompanied by the loss of bone (osteopenia) and skeletal muscle (sarcopenia), which together constitute important global medical problems. Sarcopenia results in reduced walking speed, poor balance, and instability that together predispose to falls and fractures. Thus, the coexistence of osteoporosis and sarcopenia severely compounds the problem of frailty in the elderly population (1).
During organogenesis, muscle and bone develop in close association from common mesodermal precursors and accumulate their final adult mass according to specific genetic instructions and environmental cues. Bone forms in a discrete stepwise process: mesenchymal precursors, derived from the mesoderm, first migrate to the future sites of bone where they condense. Following condensation, these precursors differentiate into chondrocytes or osteoblasts to form cartilage or bone, respectively, depending upon positional cues (2). Once formed, the skeleton is continually remodeled throughout life. These processes of modeling and remodeling are achieved by the actions of osteoblasts and osteoclasts, and are coordinated through the actions of many autocrine/paracrine factors including Wnts, Hedgehogs, Notch, bone morphogenetic protein (BMP)2 family members, transforming growth factor-β (TGF-β) family members, insulin-like growth factor-1, fibroblast growth factor-2 (FGF-2), and interleukin-6 (IL-6), among others (3). This developing skeleton becomes encased in muscle tissue, which matures along with the adjacent modeling skeleton (4–6). Similar to the regulation of bone development and maintenance of mass, skeletal muscle development and maintenance is regulated by morphogens and growth factors, many of which overlap with those involved in skeletogenesis, such as FGFs, insulin-like growth factor-1, and TGF-β (7).
Among the most important factors controlling muscle development is the activin/myostatin family of molecules. Activin, and closely related inhibin, are members of the TGF-β superfamily of molecules (8–10) first discovered over 70 years ago (8, 10–14). Like other TGF-β family members, activin and inhibin are produced as large precursors containing a signaling domain, a propeptide, and a mature C-terminal segment that possesses biological activity. Following cleavage of the propeptide, the structure of functional activin and inhibin is a disulfide linked C-terminal dimer. In the case of activin, this dimer consists of two β subunits and for inhibin, one α and one β subunit, which are encoded by distinct genes (8). Myostatin (previously GDF-8) was discovered subsequently in a screen for novel TGF-β family members (15). Although structurally similar to other TGF-β family members, its expression is restricted almost entirely to skeletal muscle, where it functions as a negative regulator of muscle growth (15). All three of these molecules function through serine/threonine kinase activin receptors that resemble other TGF-β family receptors. Type II receptors are responsible for ligand binding, which can be tempered by soluble endogenous inhibitors (e.g. follistatin), and type I receptors mediate signal transduction (16–20). Tissue specificity and activity is regulated at multiple levels in the system, including the spatiotemporal expression patterns of various components and distinct combinations of receptor/ligand binding.
Myostatin negatively regulates skeletal muscle development by activating ACVR2B and initiating Smad2/3 signaling. Smad2/3 can then activate the MAPK pathway to inhibit proliferation through the p21/Rb cascade (21, 22), or directly affect MyoD by sequestering it in the nucleus, thus halting differentiation (23). The importance of the myostatin/activin superfamily in skeletal muscle development is dramatically illustrated by the grotesque “double muscled” phenotype seen in myostatin null animals (15) or in mice carrying global mutations in Acvr2a and Acvr2b (24). These receptors appear to serve redundant functions in skeletal muscle, as heterozygous loss of Acvr2b in combination with homozygous loss of Acvr2a results in further increases in muscle mass. Additional studies using pharmacologic approaches to block access of myostatin to its receptor also support the importance of this pathway in regulating muscle development and size (24–27). In each of these genetic and pharmacologic models, the anabolic effects on muscle are achieved by inhibition of the inhibitory effect of myostatin on myoblast proliferation (21, 22) and terminal differentiation (23).
Mice and humans that develop large muscles also form large, dense bones, and the maintenance of bone and muscle mass is tightly coupled in both healthy (28–30) and disease states (31). Conversely, reduced bone mass is commonly associated with a number of myopathies including Pompe disease (32), multiple sclerosis (33), and spinal muscular atrophy (34). These observations suggest the possibility that common signaling networks control both skeletal muscle and bone development and perhaps enable these adjacent tissues to develop in synchrony. Previous studies have shown indirect evidence that the effects of activin receptor signaling are shared within the musculoskeletal system and may also affect bone mass. In support of this notion, myostatin-deficient mice not only exhibit dramatic increases in muscle mass but also significant increases in bone volume (35–38). Additionally, activin/myostatin decoy receptor administration has been shown to cause anabolic changes in both the muscle and bone compartments (39–41). Recent studies also suggest that the increases in bone volume after decoy receptor treatment are a direct effect and independent from muscle changes (42). Moreover, the anabolic effects of activin signaling blockade on the skeleton do not appear to be mediated by myostatin. Treatment with a myostatin-specific neutralizing antibody did not yield significant bone changes despite drastic increases in muscle mass (43). Additionally, decoy receptor administration in myostatin null mice demonstrated significant increases in bone volume without dramatic increases in muscle mass (43).
Taken together, studies to date unequivocally show that inhibition of activin or myostatin signaling in skeletal muscle, either by genetic or pharmacological means, increases both muscle and bone mass. However, whether the bone anabolic effects seen in these models is due to direct actions on osteoblasts or, alternatively, results indirectly through changes brought about by increased muscle mass remains unclear. It is this question that we aimed to investigate in the current study.
Results
Soluble activin type II receptors increase bone mass in vivo and osteoblast differentiation in vitro
To begin to investigate the role of activin receptor signaling within the musculoskeletal system, we first determined the effects of systemic inhibition of activin receptor signaling through intraperitoneal injections of ACVR2A/Fc and ACVR2B/Fc. Consistent with previous reports, after just 4 weeks of soluble activin receptor treatment, there were significant increases in all wet muscle weights in both receptor treatment groups (Fig. 1A). ACVR2A/Fc-treated mice showed approximately double the bone volume fraction and ∼25% increase in cortical thickness (Fig. 1B). ACVR2B/Fc-treated mice nearly tripled trabecular bone volume, with no significant changes in cortical thickness (Fig. 1C). To begin to tease out whether these skeletal anabolic effects were direct or resulted through increased muscle force or due to release of excess humoral factors following the increase in muscle mass, we also examined calvarial bone volume, a skeletal site that is both non-load bearing and relatively isolated from surrounding musculature. Calvarial bone volume was significantly increased following both ACVR2A/Fc and ACVR2B/Fc treatment (Fig. 1D). To further demonstrate that soluble activin receptors affect osteoblasts directly, we then performed in vitro studies in primary osteoblasts using ACVR2B/Fc to exploit its broad binding affinity for ligands. Here, ACVR2B/Fc administration reduced basal levels of phosphorylated SMAD2 (Fig. 1E). Furthermore, ACVR2B/Fc (50 ng/ml)-treated osteoblasts demonstrated modest yet consistent increases in alkaline phosphatase and Alizarin Red staining (Fig. 1F). Taken together, these experiments strongly suggest that the anabolic effects of activin receptor signaling blockade observed in the skeleton are due to direct effects on bone cells, particularly osteoblasts.
Figure 1.

Soluble activin receptor administration increases bone volume in vivo and enhances osteoblast differentiation in vitro. ACVR2A/Fc and ACVR2B/Fc treatment demonstrates significant increases in skeletal muscle weights as compared with vehicle-treated controls (A). ACVR2A/Fc administration exhibits a near doubling in trabecular bone volume/tissue volume and a significant increase in cortical thickness (B). Similarly, ACVR2B/Fc treatment demonstrates a near tripling in trabecular bone volume/tissue volume but no significant increase in cortical thickness (C). Both ACVR2A/Fc and ACVR2B/Fc treatment demonstrate significant increases in calvarial bone volume (D). ACVR2B/Fc treatment shows a dose-dependent reduction of Smad2 phosphorylation in vitro (E). Further in vitro analysis demonstrates that ACVR2B/Fc treatment enhances alkaline phosphatase activity and mineral deposition by Alizarin Red staining in osteoblast differentiation cultures (F). *, p < 0.05.
Activin receptor signaling components are expressed and functional in osteoblasts
We next surveyed the transcriptional expression of the components of activin receptor signaling in differentiated osteoblast cultures and compared these results to skeletal muscle, a tissue in which the function of this pathway is well established. Activin type II receptors were expressed in differentiated osteoblasts, with ACVR2A expression at levels equal to skeletal muscle and ACVR2B at significantly lower, near negligible, levels in osteoblasts as compared with muscle. The cognate type I receptor to the activin type II receptors (ACVR1B/ALK4) and the monomeric components of activin ligands (InhBA and InhBB) were also highly expressed in differentiated osteoblasts (Fig. 2A). Further supporting the notion that activin signaling is important for osteoblast function, the expression of activin receptors also increased throughout the course of osteoblast differentiation, particularly for ACVR2A (Fig. 2B). It should also be noted that, despite the 2-fold increase in ACVR2B during osteoblast differentiation, its expression remained orders of magnitude lower than ACVR2A. Expression of ACVR2A and ACVR2B (Fig. 2C) was also evident in vivo and localized to osteoblasts and osteocytes within the cortical and trabecular bone by immunohistochemistry. Finally, functionality of activin receptor signaling in osteoblasts was demonstrated in vitro by phosphorylation of Smad2 following activin ligand (Activin A, AB, or B) treatment (Fig. 2D). These experiments indicate that the activin type II receptors, predominantly ACVR2A, are expressed and functional within the osteoblast in vivo and in vitro.
Figure 2.

Activin receptor signaling components are expressed and functional in osteoblasts. Transcriptional expression assays demonstrate that the activin signaling components are expressed in primary osteoblasts as compared with skeletal muscle (A). ACVR2A and ACVR2B increase transcriptional expression with osteoblast differentiation (B). Immunohistochemistry localizes ACVR2A (white arrowheads, left) and ACVR2B (white arrowheads, right) to osteoblasts and osteocytes in bone sections (C). Phosphorylation of Smad2 following activin ligand administration demonstrates activin receptor functionality in osteoblasts (D).
Disruption of ACVR2A enhances osteoblast differentiation in vitro
To next determined the role of activin receptor signaling in osteoblast function, we differentiated primary calvarial osteoblasts from either ACVR2A- or ACVR2B-floxed neonates and deleted the receptors using an adenoviral vector expressing the Cre recombinase. Proliferation, as assessed by BrdU incorporation, demonstrated a slight decrease in ΔACVR2A osteoblasts (Fig. 3A), whereas ΔACVR2B osteoblasts demonstrated no significant change (Fig. 3B). ΔACVR2B osteoblasts were unaffected in other functional assays as well, demonstrating no difference in alkaline phosphatase activity (Fig. 3E) or mineral deposition (Fig. 3F). However, differentiated osteoblasts lacking ACVR2A did show dramatic increases in alkaline phosphatase staining (Fig. 3C) as well as enhanced matrix mineralization by Alizarin Red staining (Fig. 3D). In accord with these functional assays, transcriptional expression of osteoblast markers such as Osterix, Osteocalcin, and DMP1 were significantly up-regulated at Day 7 in ΔACVR2A osteoblasts (Fig. 3G). Together, these data demonstrate that disruption of ACVR2A enhances osteoblast differentiation and maturation in vitro.
Figure 3.

Disruption of ACVR2A enhances osteoblast differentiation in vitro. ΔACVR2A osteoblasts exhibit a slight decrease in proliferation (A), whereas disruption of ACVR2B does not alter osteoblast proliferation (B). Osteoblast differentiation is significantly enhanced with ACVR2A disruption as shown by increased alkaline phosphatase activity (C) and mineral deposition (D). ACVR2B disruption does not affect alkaline phosphatase activity (E) or mineral deposition (F). Disruption of ACVR2A induces increased transcriptional expression of osteoblast differentiation markers such as Osterix, Osteocalcin, and Dmp1 at day 7 (G). *, p < 0.05.
ΔACVR2A mice demonstrate increased bone volume
To determine the consequence of osteoblast-specific disruption of ACVR2A in vivo, mice lacking ACVR2A within the osteoblast lineage were generated by crosses of Oc-Cre-Tg+; ACVR2Aflox/flox with ACVR2Aflox/flox mice. Transgenic ACVR2Aflox/flox mice carrying Oc-Cre+ (ΔACVR2A) were born within the expected Mendelian ratios and had normal lifespans. Allele-specific PCR was performed to confirm that recombination occurred only within skeletal tissues (Fig. 4A). Micro-CT analysis of the distal femur revealed that ΔACVR2A male mice at 6 weeks of age exhibited increased trabecular bone volume/tissue volume (BV/TV) (34.2 ± 4.1%) and trabecular number (24.1 ± 1.9%). Furthermore, 12-week-old male ΔACVR2A mice demonstrated sustained increases in femoral trabecular bone volume fraction (47.9 ± 5.4%), trabecular thickness (13.3 ± 0.7%), trabecular number (27.8 ± 1.7%), and decreases in trabecular spacing (−6.2 ± 0.1%) (Fig. 4, C–F). Cortical parameters within these males were not significantly changed at 6 weeks of age. However, at 12 weeks of age, the ΔACVR2A male mice demonstrated increases in tissue area (13.0 ± 0.4%), bone area (15.9 ± 0.7%), polar moment of inertia (31.6 ± 0.3%), and cortical thickness (8.6 ± 0.3%) (Fig. 4, G–J). Similar changes, albeit of lesser magnitude, were observed in female mice at 6 and 12 weeks of age (data not shown). Histomorphometric analysis of the contralateral distal femur from male mice confirmed the increase in trabecular bone volume in ΔACVR2A mice at 6 and 12 weeks of age (Table 1). Surprisingly, there were no significant changes in osteoblast or osteoclast numbers, nor dynamic parameters, suggesting the cellular changes that lead to increased bone volume in ΔACVR2A mice likely occurred much earlier in development. Overall, these findings demonstrate that ACVR2A in osteoblasts acts as an important negative regulator of skeletal mass in mice.
Figure 4.

Femurs of ΔACVR2A male mice exhibit increased bone volume. Allele-specific PCR demonstrates ACVR2A (A) and ACVR2B (B) recombination exclusively at skeletal sites. Femurs from ΔACVR2A mice exhibit increases in trabecular bone parameters (C–F), including bone volume/tissue volume (C) and trabecular number (E) at 6 weeks of age. Femurs of ΔACVR2A mice at 12 weeks of age demonstrate increases in trabecular bone volume/tissue volume (C), trabecular thickness (D), trabecular number (E), and a decrease in trabecular spacing (F). Femoral cortical parameters (G–J) were unchanged at 6 weeks of age in ΔACVR2A mice. However, cortical tissue area (G), bone area (H), and cross-sectional thickness (J) were increased, whereas cortical bone area/tissue area (I) was unchanged at 6 and 12 weeks of age. *, p < 0.05; **, p < 0.005.
Table 1.
Bone histomorphometry
| 6 weeks |
12 weeks |
|||
|---|---|---|---|---|
| Control | ΔACVR2A | Control | ΔACVR2A | |
| BV/TVa | 5.09 ± 0.28 | 6.28 ± 0.57b | 7.87 ± 0.57 | 9.99 ± 0.77b |
| Tb.Th | 18.56 ± 0.48 | 20.88 ± 1.14 | 22.83 ± 0.90 | 25.70 ± 1.14b |
| Tb.Sp | 356.18 ± 22.68 | 323.32 ± 19.59 | 275.55 ± 16.18 | 239.75 ± 14.45b |
| OV/BV | 0.99 ± 0.23 | 0.86 ± 0.07 | 0.33 ± 0.05 | 0.51 ± 0.07b |
| OS/BS | 4.17 ± 0.74 | 3.48 ± 0.27 | 2.54 ± 0.42 | 3.18 ± 0.38 |
| O.Th | 2.28 ± 0.25 | 2.53 ± 0.19 | 1.60 ± 0.15 | 2.06 ± 0.09b |
| N.Ob/BS | 254.44 ± 39.35 | 237.30 ± 79.45 | 144.80 ± 39.62 | 170.88 ± 65.61 |
| Ob.S/BS | 3.68 ± 0.51 | 2.98 ± 0.90 | 2.11 ± 0.60 | 2.44 ± 1.07 |
| ES/BS | 2.96 ± 0.61 | 3.18 ± 0.52 | 2.15 ± 0.42 | 2.83 ± 0.62 |
| E.De | 5.52 ± 0.46 | 4.23 ± 0.27b | 3.95 ± 0.64 | 4.15 ± 0.36 |
| N.Oc/BS | 95.43 ± 16.49 | 112.08 ± 19.95 | 75.73 ± 14.79 | 93.24 ± 20.41 |
| Oc.S/BS | 2.83 ± 0.64 | 3.00 ± 0.53 | 2.13 ± 0.42 | 2.74 ± 0.61 |
| MAR | 2.04 ± 0.13 | 1.71 ± 0.06b | 1.13 ± 0.07 | 1.11 ± 0.07 |
| dLS/BS | 9.77 ± 0.83 | 10.07 ± 1.17 | 10.65 ± 0.95 | 12.60 ± 1.68 |
| sLS/BS | 7.18 ± 0.61 | 8.70 ± 1.27 | 5.97 ± 0.34 | 5.55 ± 0.66 |
| MS/BS | 13.36 ± 0.96 | 14.42 ± 1.44 | 13.64 ± 1.07 | 15.37 ± 1.83 |
| BFR/BS | 3.85 ± 0.37 | 3.41 ± 0.30 | 3.21 ± 0.35 | 3.43 ± 0.46 |
| Omt | 1.17 ± 0.15 | 1.47 ± 0.08b | 1.47 ± 0.16 | 1.90 ± 0.13b |
| Mlt | 0.37 ± 0.08 | 0.37 ± 0.04 | 0.26 ± 0.04 | 0.49 ± 0.13b |
a Histomorphometry was performed in trabecular bone of the distal femur in ΔACVR2A male mice. BV/TV, bone volume/tissue Volume (%); Tb.Th, trabecular thickness (mm); Tb.Sp, trabecular spacing (mm); OV/BV, osteoid volume/bone volume (%); OS/BS, osteoid surface/bone surface (%); O.Th, osteoid thickness (mm); N.Ob/BS, osteoblast number/bone surface (no./100 mm); Ob.S/BS, osteoblast surface/bone surface (%); ES/BS, erosion surface/bone surface (%); E.De, erosion depth (mm); N.Oc/BS, osteoclast number/bone surface (no./100 mm); Oc.S/BS, osteoclast surface/bone surface (%); MAR, mineral apposition rate (day); dLS/BS, double labeled surface/bone surface (%); sLS/BS, single labeled surface/bone surface (%); MS/BS, mineralizing surface/bone surface (%); BFR/BS, bone formation rate/bone surface (μm3/μm2/day); Omt, osteoid maturation time (day); Mlt, mineralization lag time (day). Values shown are mean ± S.E.
b p < 0.05 versus age-matched control.
ΔACVR2B mice exhibit no skeletal changes
Despite exhibiting no effect on osteoblasts in vitro, we further explored whether ACVR2B may behave differently in vivo, as it was identified in osteoblasts by immunohistochemistry. We disrupted ACVR2B in the osteoblast lineage using the same osteocalcin-driven Cre by crossing Oc-Cre-Tg+; ACVR2Bflox/floxwith ACVR2Bflox/flox mice. Transgenic ACVR2Bflox/flox mice carrying Oc-Cre+ (ΔACVR2B) were born within the expected Mendelian ratios and with normal lifespans. Allele-specific PCR was performed to confirm that recombination occurred only within skeletal tissues (Fig. 4B). Micro-CT analysis of the distal femur of ΔACVR2B male mice showed virtually no changes at 6 and 12 weeks of age (Fig. 5), suggesting that ACVR2B does not function as the primary receptor for activin signaling in the osteoblast lineage.
Figure 5.

ACVR2B disruption does not affect bone volume in vivo. Femurs of ΔACVR2B male mice show no significant changes in trabecular (A–D) or cortical bone parameters (E–H) at 6 or 12 weeks of age. Trabecular bone parameters include bone volume/tissue volume (A), trabecular thickness (B), trabecular number (C), and trabecular spacing (D). Apart from a slight reduction in bone area/tissue area at 6 weeks of age (G), there are no significant changes observed in cortical parameters, including tissue area (E), bone area (F), bone area/tissue area (G), and cross-sectional thickness (H) in 6- or 12-week-old ΔACVR2B male mice. *, p < 0.05.
Compound ΔACVR2A/2B mice exhibit a skeletal phenotype similar to ΔACVR2A mice
To unequivocally determine whether ACVR2B is able to function in osteoblasts in vivo, specifically, to partially compensate for the loss of ACVR2A, we generated compound mutants of ACVR2A and ACVR2B (ΔACVR2A/2B) in the osteoblast lineage by crossing Oc-Cre-Tg+; ACVR2A/2Bflox/flox with ACVR2A/2Bflox/flox mice. ΔACVR2A/2B male mice showed significant increases in trabecular bone volume fraction (22.6 ± 1.1%), trabecular number (25.0 ± 3.9%), and decreased trabecular spacing (−15.1 ± 3.6%) at 6 weeks of age. At 12 weeks of age, ΔACVR2A/2B male mice demonstrated sustained increases in trabecular bone volume fraction (24.8 ± 2.2%) and trabecular number (18.4 ± 6.0%) (Fig. 6, A–D). However, unlike ΔACVR2A mice, the ΔACVR2A/2B mice demonstrated no significant changes in cortical parameters at 12 weeks of age (Fig. 6, E–H). As was the case for ΔACVR2A mice, female ΔACVR2A/2B mice displayed skeletal changes similar to males with slightly lesser magnitude (not shown). These data demonstrate that compound ΔACVR2A/2B mutant mice have increases in trabecular bone parameters similar to ΔACVR2A mutants, further supporting the notion that ACVR2A is the predominant activin signaling receptor in osteoblasts.
Figure 6.

Femurs of ΔACVR2A/2B mice exhibit similar changes in bone volume as ΔACVR2A mice. Femurs of ΔACVR2A/2B male mice exhibit similar increases in trabecular bone parameters (A–D) as ΔACVR2A mice. ΔACVR2A/2B mice demonstrate increases in trabecular bone volume/tissue volume (A), trabecular number (C), and a decrease in trabecular spacing (D) at 6 weeks of ago. These increases were sustained in trabecular bone volume/tissue volume (A) and trabecular number (C) at 12 weeks of age. However, no significant changes were seen in trabecular thickness (B) at 6 or 12 weeks of age. Cortical bone parameters, including tissue area (E), bone area (F), bone area/tissue area (G), and cross-sectional thickness (H), were unchanged in 6- and 12-week-old ΔACVR2A/2B male mice. *, p < 0.05.
Skeletal changes in ΔACVR2A mice are accompanied by increased mechanical attributes
An important consideration for future therapeutic targeting of this pathway in bone is whether the additional bone mass observed in the mutant mice is mechanically competent. Thus, we also evaluated changes in mechanical properties of femurs from ΔACVR2A male mice by three-point bending. ΔACVR2A mutants demonstrated significant increases in the ultimate moment (24.8 ± 2.0%) and bending rigidity (30 ± 2.7%) with nonsignificant trends in ultimate stress (10.1 ± 0.5%). In addition, pre-yield parameters including strain (21.3 ± 1.6%), energy (45.8 ± 6.3%), and toughness (36.1 ± 4.0%) were also significantly increased compared with controls (Fig. 7, A–G). The ultimate bending energy and Young's modulus were, however, unchanged (Fig. 7, D and H, respectively). Overall, the mechanical testing indicates that the additional bone produced in mice with osteoblast-specific disruption of ACVR2A is of high mechanical competency and increases bone strength.
Figure 7.
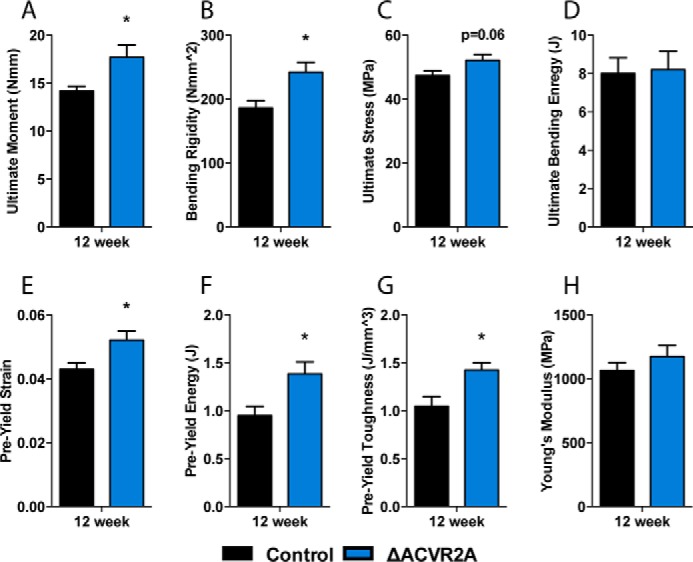
Skeletal changes in ΔACVR2A mice are accompanied by increased mechanical properties. Three-point bending analysis of femurs from ΔACVR2A mice demonstrates significant increases in mechanical properties, such as the ultimate moment (A), bending rigidity (B), pre-yield strain (E), pre-yield energy (F), and pre-yield toughness (G), and nonsignificant trends in ultimate stress (C). There were, however, no significant changes in the ultimate bending energy (D) or Young's modulus (H) with ACVR2A disruption. *, p < 0.05.
Activin receptor signaling in osteoblasts contributes significantly to the anabolic effects observed with soluble receptor administration
Finally, we returned to the question of what proportion of the anabolic effect observed from soluble activin receptor treatment results from direct effects on the osteoblast versus indirect effects from skeletal muscle, or perhaps even other secondary systems. To do so, we treated ΔACVR2A/2B mutant mice with ACVR2B/Fc for 4 weeks, as done in our initial experiments, and compared the percentage change in bone volume with that observed in wild type mice treated with the soluble receptor. In wild type animals treated with ACVR2B/Fc, the trabecular bone volume fraction increased by 114 ± 16.0%. By contrast, ΔACVR2A/2B mice treated with ACVR2B/Fc gained 62.3 ± 14.5% in trabecular BV/TV (Fig. 8A). Similarly, increases in trabecular number were differentially affected between control (82.0 ± 8.5%) and ΔACVR2A/2B (50.1 ± 11.1%) mice (Fig. 8C). In contrast to the changes observed in trabecular bone, there were no differential increases in muscle mass (gastrocnemius, tibialis anterior, and quadriceps) between control and ΔACVR2A/2B mice with ACVR2B/Fc administration (Fig. 8, E–G). Taken together, these data would suggest that a substantial portion of the effect of soluble activin receptor administration on bone volume (>50%) results from the direct actions of this pathway in osteoblasts.
Figure 8.

Activin receptor signaling is a significant contributor to anabolic changes observed with soluble receptor administration. Soluble activin receptor treatment with ACVR2B/Fc demonstrates attenuated increases in bone volume/tissue volume (A), trabecular thickness (B), and trabecular number (C) in ΔACVR2A/2B mice. There were no differential changes in trabecular bone spacing (D) or differential increases in skeletal muscle weights, including the gastrocnemius (E), tibialis anterior (F), or quadriceps (G) muscles. *, p < 0.05.
Discussion
We have described, for the first time, a role for activin receptor signaling in osteoblasts that is analogous to the known function of this pathway as a negative regulator of muscle development and mass. Curiously, early studies suggested that activin A exerted a stimulatory effect on osteoblastogenesis (44, 45), but more recent reports using human osteoblast preparations suggest that activin inhibits osteoblastogenesis and mineralization (46). Initial in vivo studies in bone were also conflicting. For example, local injection of activin A into rat fibula fractures (47) or over the calvaria of rat neonates (48) increased bone accumulation. In more recent studies, systemic infusion with a soluble ACVR2A resulted in increased bone mass in both mice (39) and monkeys (41, 49). Moreover, administration of soluble ACVR2A increased markers of bone formation in humans (50). Histomorphometric analysis of bones from mice and monkeys treated with this ACVR2A fusion peptide demonstrated that the anabolic effects were attributable to increased osteoblast activity (39, 41). Our initial studies with soluble activin receptor administration support these previous studies of pharmacologic blockade of activin signaling, even over the considerably shorter course of treatment we used. We utilized this 4-week treatment in hopes of minimizing the effect of long-term adaptation of bone mass in response to the increased mechanical forces that follow the additional muscle mass gained from systemic activin receptor blockade. Combined with the observed increases in bone mass in the calvarium, a skeletal site that is neither load bearing nor encased in skeletal muscle like the femur, we were encouraged to further investigate the direct activity of this pathway in osteoblasts.
Further interrogation of this pathway in osteoblasts, both through pharmacologic and genetic blockade of activin receptor activity, demonstrated that activin receptor signaling is, indeed, a negative regulator of osteoblast function. Interestingly, both in vitro and in vivo, this effect appears to be nearly entirely dependent on ACVR2A. Previous studies have suggested that myostatin and/or ACRV2B may also play a role in regulating bone mass. For example, myostatin null mice have increased bone mass (35), and infusion of a soluble ACVR2B fusion molecule increased bone mass in a mouse model of androgen deprivation (51). In our hands, as well, administration of soluble ACVR2B resulted in profound increases in trabecular bone volume. However, we consider it unlikely that ACVR2B and myostatin are physiologically relevant in osteoblasts to regulate bone mass for the following reasons. In the case of the myostatin null mice, increased bone mass was only observed in the regions of long bones immediately adjacent to entheses (35), strongly suggesting that the effect is secondary to increased mechanical force generated as a result of the double-muscled phenotype in myostatin null mice. Additionally, we observed no detectable expression of myostatin in primary mouse osteoblasts, and ACVR2B expression was orders of magnitude lower than ACVR2A expression (Fig. 2A). Finally, whereas ACVR2B and various soluble forms of this receptor do predominantly bind and sequester myostatin, ACVR2B is the more promiscuous of the two activin receptors and also binds many ligands including activins with nearly as high affinity as it binds myostatin (52).
Our studies with genetic disruption of ACVR2A and ACVR2B in osteoblasts bear out the notion that ACVR2A functions as a negative regulator of osteoblast function. Both in vitro and in vivo, disruption of ACVR2B had no discernable effect on osteoblast function. Moreover, compound mutants that lacked both activin receptors in osteoblasts displayed changes in trabecular bone that were strikingly similar to ΔACVR2A mice. Of interest, however, was the fact that the increased cortical bone phenotype observed in ΔACVR2A mice at 12 weeks of age (Fig. 4, G–J) was not observed in the double ΔACVR2A/2B mice (Fig. 6, E–H). One possible explanation for this change, which presumably, is exactly the opposite of what one would expect if ACVR2B were able to partially compensate for ACVR2A loss, is that complete loss of type II activin receptors from osteoblasts alters other TGF-β family and/or BMP signaling in osteoblasts, as these receptor families are well known to be promiscuous in ligand-binding and signaling activity. In support of this notion, Lowery and colleagues (53) have recently demonstrated that selective disruption of BMPR2 in osteoprogenitor cells actually impaired activin signaling, with no effect on BMP signaling, resulting in a high bone mass phenotype. Their result also indirectly supports our overall finding that activin receptor signaling in osteoblasts acts as a negative regulator of their function.
Despite the clear skeletal phenotype observed in ΔACVR2A mice, we were unable to detect any significant changes in bone cell numbers at either time point observed, in either sex. We believe that these changes must have occurred much earlier in development for a number of reasons. Both genetic and pharmacologic blockade of activin signaling in vitro resulted in rapid and robust differentiation of osteoblasts. We presume it is likely that the same effect would have taken place in vivo with an early burst of osteoblast differentiation and bone formation that was then damped by yet unknown compensatory mechanisms. In support of this idea, the only statistically significant histomorphometric parameters observed were a slightly repressed mineral apposition rate at 6 weeks of age, increased osteoid maturation time at both 6 and 12 weeks of age, and increased mineralization lag time at 12 weeks of age (Table 1); all opposite of what one would typically expect in mice with higher bone mass, but consistent with a repression of osteoblast activity. Future studies are already underway to examine the role of activin receptor signaling at varying points in the osteoblast lineage, the current study having focused on the mature osteoblast/osteocyte using a human osteocalcin Cre driver, and will explore these early developmental changes in greater detail. Regardless, our current study provides clear evidence of the importance of ACVR2A in osteoblasts as a negative regulator of bone development and mass.
In an attempt to begin to understand the precise contribution of activin receptor signaling in osteoblasts to the observed anabolic effects of soluble activin receptor administration in the skeleton, we also treated the double ΔACVR2A/2B mice with soluble ACVR2B. The results of this experiment suggest that over half of the observed increase in bone mass following administration of soluble ACVR2B/Fc is due to the direct activity of this pathway in osteoblasts (Fig. 8A). Interestingly, the absolute final trabecular bone volume fraction following soluble receptor treatment was nearly the same between treated wild type mice and double mutants, suggesting a sort of ceiling by which modulating this pathway can influence bone volume in mice (not shown). Interestingly, we observed no difference in skeletal muscle wet weights between ACVR2B/Fc-treated wild type and ΔACVR2A/2B mice, hinting that changes in activin receptor signaling in osteoblasts do not seem to reciprocally affect skeletal muscle. The additional increase in bone volume in ΔACVR2A/2B mice following ACVR2B/Fc treatment is likely to occur from multiple secondary mechanisms, given the established functions of activin signaling in other systems known to affect bone, e.g. the pituitary and immune system (54, 55). Other possible mechanisms also include soluble activin receptors binding to ligands apart from the activins, GDF molecules, or myostatin, such as the BMP ligands (56, 57). It is possible that soluble activin receptor administration may also be affecting BMP signaling, either directly or indirectly through the removal of ligand competition. Future studies are already underway to determine the precise contribution of skeletal muscle to this effect by observing changes in the skeleton following muscle-specific disruption of activin receptor signaling.
Our study is the first to conclusively demonstrate the activity of activin receptor signaling in osteoblasts as a negative regulator of bone development, analogous to the well established function of this pathway in skeletal muscle. This effect appears to be mediated predominantly through ACVR2A and its associated ligands, suggesting an evolutionary split in the control of musculoskeletal development; with ACVR2A and its ligands controlling the skeleton, whereas ACVR2B and myostatin exert control over skeletal muscle. Such an arrangement would allow for precise, concerted control of the development and maintenance of the musculoskeletal system, and also provides a particularly attractive therapeutic area to modulate cross-talk between both tissues.
Experimental procedures
Soluble activin type II receptor animal studies
All animal protocols were approved by the Johns Hopkins University Institutional Animal Care and Use Committee. Systemic suppression of activin ligands was accomplished by intraperitoneal injections (10 mg/kg body weight) of a soluble chimeric fusion protein consisting of either the extracellular ACVR2A or ACVR2B domain conjugated to the Fc domain of a murine IgG antibody (ACVR2A/Fc or ACVR2B/Fc, respectively). Injections were administered weekly for 4 weeks in 12-week-old female mice (n = 5). After soluble activin receptor treatment, skeletal tissues were harvested and wet muscle weights were measured.
Transgenic animal studies
Osteoblast-specific disruption of ACVR2A, ACVR2B, or both was accomplished by crossing ACVR2Aflox/flox or ACVR2Bflox/flox mice (58), with mice expressing the Cre recombinase under the direction of the human osteocalcin promoter (Oc-Cre), in which Cre recombinase expression is restricted to mature osteoblasts (59). Recombination within the skeletal tissues was confirmed using allele-specific PCR and details are available upon request. Control (ACVR2Aflox/flox or ACVR2Bflox/flox) and mutant (ACVR2Aflox/flox or ACVR2Bflox/flox; Oc-Cre+) male and female mice were sacrificed at 6 and 12 weeks of age (n = 10). Skeletal tissues then were harvested and fixed in formalin overnight and stored until analysis in 70% ethanol. Prior to harvest, 6-week-old mice were double labeled by two sequential intraperitoneal calcein injections (10 mg/kg body weight) at 7 and 2 days prior to sacrifice for dynamic histomorphometric analysis. Twelve-week-old mice were double labeled with calcein injections at 10 and 3 days before sacrifice. Mice were maintained on a pure C57Bl/6 background.
Microcomputed tomography (μCT)
Images of skeletal tissues were obtained using a Skyscan 1172 desktop imaging system (Skyscan-Bruker). Scans were performed with an isotropic voxel size of 10 μm at 65 kV and 200 μA through a 0.5-mm aluminum filter. Femoral trabecular parameters were measured in a 200-μm region of interest located ∼50 μm proximal to the distal growth plate. Femoral cortical parameters were measured within a 500-μm region of interest centered at the midshaft. All bone analysis was performed in accordance with the American Society of Bone and Mineral Research (ASBMR) recommended parameters (60).
Bone histomorphometry
For histomorphometric analysis, femurs were fixed in 10% neutral buffered formalin, dehydrated, and embedded in polymethylmethacrylate. Three-μm sections were cut through the primary spongiosium and stained using the Mason-Goldner Trichrome stain. Additional serial sections were cut for fluorescent microscopy. Osteoblast and osteoclast measures were assessed at standardized sites beneath the growth plate using Osteoplan II (Kontron). Static and dynamic parameters were calculated in accordance with the ASBMR guidelines (61).
Osteoblast isolation and culture
Osteoblasts were isolated from neonatal calvaria of ACVR2Aflox/flox and ACVR2Bflox/flox mice by serial digestion in a 1.8 mg/ml of collagenase type I (Worthington Biochemical) solution. Calvaria were incubated in 10 ml of collagenase solution at 37 °C for 15 min with constant agitation per digestion cycle. The digestion solutions were collected and fresh collagenase was added to the remaining digestion cycles an additional four times. Digestions 3–5, containing osteoblasts, were pooled and cultured in α-minimum essential medium (α-MEM) containing 10% FBS and 1% penicillin/streptomycin. Osteoblasts were cultured at 37 °C at 5% carbon dioxide in a humidified incubator. Osteoblasts were grown to 70% confluence and then transduced in PBS with either a control adenovirus encoding green fluorescent protein (GFP) or an adenovirus encoding Cre recombinase (Vector Biolabs) at a titer of 100 multiplicity of infection. After 1 h of incubation, osteoblasts were supplemented with α-MEM containing 10% fetal bovine serum and 1% penicillin/streptomysin and then allowed to recover for 48 h. Osteoblasts were then harvested to assess ACVR2A or ACVR2B deletion efficiency by quantitative PCR and re-plated for proliferation studies and differentiation studies.
Osteoblast proliferation and differentiation
Osteoblast proliferation was assessed using BrdU incorporation and flow cytometry. Osteoblasts were seeded at a low density (5 × 104 cells per well) and cultured in α-MEM containing 0.5% FBS for 24 h to arrest cell cycle. BrdU (10 μm) (BD Biosciences) was then added to the medium for an additional 24 h prior to harvest to allow incorporation. Osteoblasts were then fixed, stained with anti-BrdU antibody and 7-aminoactinomycin D, and analyzed by FACS Calibur flow cytometry (BD Biosciences). 10,000 events were collected for each sample and the results were processed using Flowing Software version 2.5. For differentiation experiments, control and ΔACVR2A or ΔACVR2B osteoblasts were cultured to confluence and then cultured in medium supplemented with 10 mm β-glycerol phosphate and 50 μg/ml of ascorbic acid. For alkaline phosphatase and mineral deposition assays, osteoblasts deficient in activin receptor were seeded at confluence and subsequently differentiated in complete medium supplemented with 10 mm β-glycerol phosphate and 50 μg/ml of ascorbic acid. Cultures were then fixed at days 7 and 14 using 100% ethanol and assessed for alkaline phosphatase activity and mineral deposition with Fast Red TR/Naphthol AS-MX phosphate (Sigma) or 40 mm Alizarin Red (Sigma) staining, respectively. Image analysis was performed using FIJI (62).
Quantitative real-time PCR
Total RNA was collected from cells and tissue samples using TRIzol (Invitrogen) following the manufacturer's protocol. One μg of RNA was then reversely transcribed using an iScript cDNA synthesis kit (Bio-Rad). Two microliters of cDNA were amplified under standard PCR conditions using iQ SYBR Green Supermix (Bio-Rad). All cDNA samples were run in triplicate, averaged, and normalized to endogenous β-actin expression levels. Primer sequences were designed using Primer-BLAST (NCBI) and are available upon request.
Cell lysis and immunoblotting
For signaling experiments, osteoblasts were cultured to near confluence in α-MEM containing 10% FBS. Cells were then starved in α-MEM containing 0.5% FBS overnight to reduce endogenous cellular activity prior to stimulation. Activin A, AB, and B (R&D Systems) were added to the medium for a final concentration of 20 ng/ml. ACVR2B/Fc was supplemented to a final concentration of 50 ng/ml. Whole cell lysates were then collected at appropriate time points in lysis buffer (50 mm Tris (pH 7.4), 150 mm NaCl, 1 mm MgCl2, 1 mm EDTA, 1% Triton X-100, and 10% glycerol). Protein concentrations were measured by BCA Protein Assay (Pierce) and 15 μg of total protein were loaded into each lane using a mini-SDS-PAGE system (Bio-Rad). Following electrophoresis, proteins were transferred to a PDVF membrane using a Mini Trans-blot Cell System (Bio-Rad). Membranes were then blocked using 5% BSA at room temperature for 1 h and then incubated with the antibody of interest (Cell Signaling) at 4 °C overnight. Protein signal was detected using secondary antibodies conjugated with horseradish peroxidase. Chemiluminescence of bound antibody was produced by Supersignal West Femto Substrate (Pierce) and captured by Gel Doc XR+ System (Bio-Rad). Blot analysis was performed using Image Lab (Bio-Rad).
Mechanical testing
Femurs were harvested from 12-week-old control and ΔACVR2A male mice. Soft tissue was then cleared from the femur and placed on a custom three-point bending apparatus with a span of 5.3 mm. After a preload of 0.3 newton, the femur was subjected to force in displacement control at 0.1 mm/s until fracture was achieved (Bose Electroforce 3100). Bone hydration was maintained throughout the testing period using PBS. Force-displacement data and micro-CT imaging was analyzed using a custom MATLAB (MathWorks) program similar to others (63).
Statistical analysis
All values are shown as mean ± S.E. Statistical significance between comparable groups was assessed using Student's t test with an assigned significance level (α) of 0.05.
Author contributions
D. J. D. conceived of the hypothesis, designed experiments, and supervised studies; B. C. G., V. S., A. J. H., R. E. T., S. K., and M. C. F. conducted experiments; E. L. L. and T. L. C. provided intellectual input; S. J. L. provided intellectual input and reagents; D. J. D. and B. C. G. wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by the NIAMS National Institutes of Health Grants R01AR062074 (to D. J. D.) and R01AR060636 (to S.-J. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- BMP
- bone morphogenetic protein
- ACVR
- activin receptor
- BV
- bone volume
- TV
- tissue volume
- CT
- computed tomography
- MEM
- minimal essential medium.
References
- 1. DiGirolamo D. J., Kiel D. P., and Esser K. A. (2013) Bone and skeletal muscle: neighbors with close ties. J. Bone Miner. Res. 28, 1509–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Olsen B. R., Reginato A. M., and Wang W. (2000) Bone development. Annu. Rev. Cell Dev. Biol. 16, 191–220 [DOI] [PubMed] [Google Scholar]
- 3. Jilka R. L. (2007) Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone 40, 1434–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kablar B., Asakura A., Krastel K., Ying C., May L. L., Goldhamer D. J., and Rudnicki M. A. (1998) MyoD and Myf-5 define the specification of musculature of distinct embryonic origin. Biochem. Cell. Biol. 76, 1079–1091 [PubMed] [Google Scholar]
- 5. Hasty P., Bradley A., Morris J. H., Edmondson D. G., Venuti J. M., Olson E. N., and Klein W. H. (1993) Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 364, 501–506 [DOI] [PubMed] [Google Scholar]
- 6. Nabeshima Y., Hanaoka K., Hayasaka M., Esumi E., Li S., Nonaka I., and Nabeshima Y. (1993) Myogenin gene disruption results in perinatal lethality because of severe muscle defect. Nature 364, 532–535 [DOI] [PubMed] [Google Scholar]
- 7. Allen R. E., and Rankin L. L. (1990) Regulation of satellite cells during skeletal muscle growth and development. Proc. Soc. Exp. Biol. Med. 194, 81–86 [DOI] [PubMed] [Google Scholar]
- 8. Mayo K. E. (1994) Inhibin and activin: molecular aspects of regulation and function. Trends Endocrinol. Metab. 5, 407–415 [DOI] [PubMed] [Google Scholar]
- 9. Kingsley D. M. (1994) The TGF-β superfamily: new members, new receptors, and new genetic tests of function in different organisms. Genes Dev. 8, 133–146 [DOI] [PubMed] [Google Scholar]
- 10. Vale W. (1990) The Inhibin/Activin Family of Hormones and Growth Factors, Springer-Verlag, Berlin [Google Scholar]
- 11. McCullagh D. R. (1932) Dual endocrine activity of the testes. Science 76, 19–20 [DOI] [PubMed] [Google Scholar]
- 12. Vale W., Rivier C., Hsueh A., Campen C., Meunier H., Bicsak T., Vaughan J., Corrigan A., Bardin W., and Sawchenko P. (1988) Chemical and biological characterization of the inhibin family of protein hormones. Recent Prog. Horm. Res. 44, 1–34 [DOI] [PubMed] [Google Scholar]
- 13. Ling N., Ying S. Y., Ueno N., Shimasaki S., Esch F., Hotta M., and Guillemin R. (1986) Pituitary FSH is released by a heterodimer of the β-subunits from the two forms of inhibin. Nature 321, 779–782 [DOI] [PubMed] [Google Scholar]
- 14. Vale W., Rivier J., Vaughan J., McClintock R., Corrigan A., Woo W., Karr D., and Spiess J. (1986) Purification and characterization of an FSH releasing protein from porcine ovarian follicular fluid. Nature 321, 776–779 [DOI] [PubMed] [Google Scholar]
- 15. McPherron A. C., Lawler A. M., and Lee S. J. (1997) Regulation of skeletal muscle mass in mice by a new TGF-β superfamily member. Nature 387, 83–90 [DOI] [PubMed] [Google Scholar]
- 16. Krummen L. A., Woodruff T. K., DeGuzman G., Cox E. T., Baly D. L., Mann E., Garg S., Wong W. L., Cossum P., and Mather J. P. (1993) Identification and characterization of binding proteins for inhibin and activin in human serum and follicular fluids. Endocrinology 132, 431–443 [DOI] [PubMed] [Google Scholar]
- 17. Hill C. S. (1996) Signalling to the nucleus by members of the transforming growth factor-β (TGF-β) superfamily. Cell Signal. 8, 533–544 [DOI] [PubMed] [Google Scholar]
- 18. Attisano L., Wrana J. L., López-Casillas F., and Massagué J. (1994) TGF-beta receptors and actions. Biochim. Biophys. Acta 1222, 71–80 [DOI] [PubMed] [Google Scholar]
- 19. ten Dijke P., Miyazono K., and Heldin C. H. (1996) Signaling via hetero-oligomeric complexes of type I and type II serine/threonine kinase receptors. Curr. Opin. Cell Biol. 8, 139–145 [DOI] [PubMed] [Google Scholar]
- 20. McCarthy S. A., and Bicknell R. (1993) Inhibition of vascular endothelial cell growth by activin-A. J. Biol. Chem. 268, 23066–23071 [PubMed] [Google Scholar]
- 21. Thomas M., Langley B., Berry C., Sharma M., Kirk S., Bass J., and Kambadur R. (2000) Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J. Biol. Chem. 275, 40235–40243 [DOI] [PubMed] [Google Scholar]
- 22. Joulia D., Bernardi H., Garandel V., Rabenoelina F., Vernus B., and Cabello G. (2003) Mechanisms involved in the inhibition of myoblast proliferation and differentiation by myostatin. Exp. Cell Res. 286, 263–275 [DOI] [PubMed] [Google Scholar]
- 23. Langley B., Thomas M., Bishop A., Sharma M., Gilmour S., and Kambadur R. (2002) Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J. Biol. Chem. 277, 49831–49840 [DOI] [PubMed] [Google Scholar]
- 24. Lee S. J., Reed L. A., Davies M. V., Girgenrath S., Goad M. E., Tomkinson K. N., Wright J. F., Barker C., Ehrmantraut G., Holmstrom J., Trowell B., Gertz B., Jiang M. S., Sebald S. M., Matzuk M., et al. (2005) Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc. Natl. Acad. Sci. U.S.A. 102, 18117–18122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bogdanovich S., Krag T. O., Barton E. R., Morris L. D., Whittemore L. A., Ahima R. S., and Khurana T. S. (2002) Functional improvement of dystrophic muscle by myostatin blockade. Nature 420, 418–421 [DOI] [PubMed] [Google Scholar]
- 26. Whittemore L. A., Song K., Li X., Aghajanian J., Davies M., Girgenrath S., Hill J. J., Jalenak M., Kelley P., Knight A., Maylor R., O'Hara D., Pearson A., Quazi A., Ryerson S., et al. (2003) Inhibition of myostatin in adult mice increases skeletal muscle mass and strength. Biochem. Biophys. Res. Commun. 300, 965–971 [DOI] [PubMed] [Google Scholar]
- 27. Wolfman N. M., McPherron A. C., Pappano W. N., Davies M. V., Song K., Tomkinson K. N., Wright J. F., Zhao L., Sebald S. M., Greenspan D. S., and Lee S. J. (2003) Activation of latent myostatin by the BMP-1/tolloid family of metalloproteinases. Proc. Natl. Acad. Sci. U.S.A. 100, 15842–15846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blain H., Jaussent A., Thomas E., Micallef J. P., Dupuy A. M., Bernard P. L., Mariano-Goulart D., Cristol J. P., Sultan C., Rossi M., and Picot M. C. (2010) Appendicular skeletal muscle mass is the strongest independent factor associated with femoral neck bone mineral density in adult and older men. Exp. Gerontol. 45, 679–684 [DOI] [PubMed] [Google Scholar]
- 29. Rantalainen T., Heinonen A., Komi P. V., and Linnamo V. (2008) Neuromuscular performance and bone structural characteristics in young healthy men and women. Eur. J. Appl. Physiol. 102, 215–222 [DOI] [PubMed] [Google Scholar]
- 30. Marin R. V., Pedrosa M. A., Moreira-Pfrimer L. D., Matsudo S. M., and Lazaretti-Castro M. (2010) Association between lean mass and handgrip strength with bone mineral density in physically active postmenopausal women. J. Clin. Densitom. 13, 96–101 [DOI] [PubMed] [Google Scholar]
- 31. Montgomery E., Pennington C., Isales C. M., and Hamrick M. W. (2005) Muscle-bone interactions in dystrophin-deficient and myostatin-deficient mice. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 286, 814–822 [DOI] [PubMed] [Google Scholar]
- 32. van den Berg L. E., Zandbergen A. A., van Capelle C. I., de Vries J. M., Hop W. C., van den Hout J. M., Reuser A. J., Zillikens M. C., and van der Ploeg A. T. (2010) Low bone mass in Pompe disease: muscular strength as a predictor of bone mineral density. Bone 47, 643–649 [DOI] [PubMed] [Google Scholar]
- 33. Formica C. A., Cosman F., Nieves J., Herbert J., and Lindsay R. (1997) Reduced bone mass and fat-free mass in women with multiple sclerosis: effects of ambulatory status and glucocorticoid use. Calcif. Tissue Int. 61, 129–133 [DOI] [PubMed] [Google Scholar]
- 34. Khatri I. A., Chaudhry U. S., Seikaly M. G., Browne R. H., and Iannaccone S. T. (2008) Low bone mineral density in spinal muscular atrophy. J. Clin. Neuromuscul. Dis. 10, 11–17 [DOI] [PubMed] [Google Scholar]
- 35. Hamrick M. W. (2003) Increased bone mineral density in the femora of GDF8 knockout mice. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 272, 388–391 [DOI] [PubMed] [Google Scholar]
- 36. Hamrick M. W., McPherron A. C., and Lovejoy C. O. (2002) Bone mineral content and density in the humerus of adult myostatin-deficient mice. Calcif. Tissue Int. 71, 63–68 [DOI] [PubMed] [Google Scholar]
- 37. Hamrick M. W., McPherron A. C., Lovejoy C. O., and Hudson J. (2000) Femoral morphology and cross-sectional geometry of adult myostatin-deficient mice. Bone 27, 343–349 [DOI] [PubMed] [Google Scholar]
- 38. Hamrick M. W., Pennington C., and Byron C. D. (2003) Bone architecture and disc degeneration in the lumbar spine of mice lacking GDF-8 (myostatin). J. Orthop. Res. 21, 1025–1032 [DOI] [PubMed] [Google Scholar]
- 39. Pearsall R. S., Canalis E., Cornwall-Brady M., Underwood K. W., Haigis B., Ucran J., Kumar R., Pobre E., Grinberg A., Werner E. D., Glatt V., Stadmeyer L., Smith D., Seehra J., and Bouxsein M. L. (2008) A soluble activin type IIA receptor induces bone formation and improves skeletal integrity. Proc. Natl. Acad. Sci. U.S.A. 105, 7082–7087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cadena S. M., Tomkinson K. N., Monnell T. E., Spaits M. S., Kumar R., Underwood K. W., Pearsall R. S., and Lachey J. L. (2010) Administration of a soluble activin type IIB receptor promotes skeletal muscle growth independent of fiber type. J. Appl. Physiol. 109, 635–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lotinun S., Pearsall R. S., Davies M. V., Marvell T. H., Monnell T. E., Ucran J., Fajardo R. J., Kumar R., Underwood K. W., Seehra J., Bouxsein M. L., and Baron R. (2010) A soluble activin receptor Type IIA fusion protein (ACE-011) increases bone mass via a dual anabolic-antiresorptive effect in Cynomolgus monkeys. Bone 46, 1082–1088 [DOI] [PubMed] [Google Scholar]
- 42. Di Girolamo D. J., Singhal V., Clemens T. L., and Lee S. J. (2011) Systemic administration of soluble activin receptors produces differential anabolic effects in muscle and bone in mice. Abstracts of the 2011 Annual Meeting of the American Society for Bone and Mineral Research San Diego, CA, September 16–20, 2011 J. Bone Miner. Res. 26, Abstr. 1167 [Google Scholar]
- 43. Bialek P., Parkington J., Li X., Gavin D., Wallace C., Zhang J., Root A., Yan G., Warner L., Seeherman H. J., and Yaworsky P. J. (2014) A myostatin and activin decoy receptor enhances bone formation in mice. Bone 60, 162–171 [DOI] [PubMed] [Google Scholar]
- 44. Centrella M., McCarthy T. L., and Canalis E. (1991) Activin-A binding and biochemical effects in osteoblast-enriched cultures from fetal-rat parietal bone. Mol. Cell. Biol. 11, 250–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Centrella M., McCarthy T. L., and Canalis E. (1991) Glucocorticoid regulation of transforming growth factor β1 activity and binding in osteoblast-enriched cultures from fetal rat bone. Mol. Cell. Biol. 11, 4490–4496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Eijken M., Swagemakers S., Koedam M., Steenbergen C., Derkx P., Uitterlinden A. G., van der Spek P. J., Visser J. A., de Jong F. H., Pols H. A., and van Leeuwen J. P. (2007) The activin A-follistatin system: potent regulator of human extracellular matrix mineralization. FASEB J. 21, 2949–2960 [DOI] [PubMed] [Google Scholar]
- 47. Sakai R., Miwa K., and Eto Y. (1999) Local administration of activin promotes fracture healing in the rat fibula fracture model. Bone 25, 191–196 [DOI] [PubMed] [Google Scholar]
- 48. Oue Y., Kanatani H., Kiyoki M., Eto Y., Ogata E., and Matsumoto T. (1994) Effect of local injection of activin A on bone formation in newborn rats. Bone 15, 361–366 [DOI] [PubMed] [Google Scholar]
- 49. Fajardo R. J., Manoharan R. K., Pearsall R. S., Davies M. V., Marvell T., Monnell T. E., Ucran J. A., Pearsall A. E., Khanzode D., Kumar R., Underwood K. W., Roberts B., Seehra J., and Bouxsein M. L. (2010) Treatment with a soluble receptor for activin improves bone mass and structure in the axial and appendicular skeleton of female cynomolgus macaques (Macaca fascicularis). Bone 46, 64–71 [DOI] [PubMed] [Google Scholar]
- 50. Ruckle J., Jacobs M., Kramer W., Pearsall A. E., Kumar R., Underwood K. W., Seehra J., Yang Y., Condon C. H., and Sherman M. L. (2009) Single-dose, randomized, double-blind, placebo-controlled study of ACE-011 (ActRIIA-IgG1) in postmenopausal women. J. Bone Miner. Res. 24, 744–752 [DOI] [PubMed] [Google Scholar]
- 51. Koncarevic A., Cornwall-Brady M., Pullen A., Davies M., Sako D., Liu J., Kumar R., Tomkinson K., Baker T., Umiker B., Monnell T., Grinberg A. V., Liharska K., Underwood K. W., Ucran J. A., et al. (2010) A soluble activin receptor type IIb prevents the effects of androgen deprivation on body composition and bone health. Endocrinology 151, 4289–4300 [DOI] [PubMed] [Google Scholar]
- 52. Sako D., Grinberg A. V., Liu J., Davies M. V., Castonguay R., Maniatis S., Andreucci A. J., Pobre E. G., Tomkinson K. N., Monnell T. E., Ucran J. A., Martinez-Hackert E., Pearsall R. S., Underwood K. W., Seehra J., and Kumar R. (2010) Characterization of the ligand binding functionality of the extracellular domain of activin receptor type IIb. J. Biol. Chem. 285, 21037–21048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lowery J. W., Intini G., Gamer L., Lotinun S., Salazar V. S., Ote S., Cox K., Baron R., and Rosen V. (2015) Loss of BMPR2 leads to high bone mass due to increased osteoblast activity. J. Cell Sci. 128, 1308–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bilezikjian L. M., Justice N. J., Blackler A. N., Wiater E., and Vale W. W. (2012) Cell-type specific modulation of pituitary cells by activin, inhibin and follistatin. Mol. Cell. Endocrinol. 359, 43–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Licona P., Chimal-Monroy J., and Soldevila G. (2006) Inhibins are the major activin ligands expressed during early thymocyte development. Dev. Dyn. 235, 1124–1132 [DOI] [PubMed] [Google Scholar]
- 56. Aykul S., and Martinez-Hackert E. (2016) Transforming growth factor-β family ligands can function as antagonists by competing for type II receptor binding. J. Biol. Chem. 291, 10792–10804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Aykul S., Ni W., Mutatu W., and Martinez-Hackert E. (2015) Human Cerberus prevents nodal-receptor binding, inhibits nodal signaling, and suppresses nodal-mediated phenotypes. PloS One 10, e0114954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lee S. J., Huynh T. V., Lee Y. S., Sebald S. M., Wilcox-Adelman S. A., Iwamori N., Lepper C., Matzuk M. M., and Fan C. M. (2012) Role of satellite cells versus myofibers in muscle hypertrophy induced by inhibition of the myostatin/activin signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 109, E2353–E2360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang M., Xuan S., Bouxsein M. L., von Stechow D., Akeno N., Faugere M. C., Malluche H., Zhao G., Rosen C. J., Efstratiadis A., and Clemens T. L. (2002) Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J. Biol. Chem. 277, 44005–44012 [DOI] [PubMed] [Google Scholar]
- 60. Bouxsein M. L., Boyd S. K., Christiansen B. A., Guldberg R. E., Jepsen K. J., and Müller R. (2010) Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J. Bone Miner. Res. 25, 1468–1486 [DOI] [PubMed] [Google Scholar]
- 61. Parfitt A. M., Drezner M. K., Glorieux F. H., Kanis J. A., Malluche H., Meunier P. J., Ott S. M., and Recker R. R. (1987) Bone histomorphometry: standardization of nomenclature, symbols, and units: report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 2, 595–610 [DOI] [PubMed] [Google Scholar]
- 62. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J. Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., and Cardona A. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schriefer J. L., Robling A. G., Warden S. J., Fournier A. J., Mason J. J., and Turner C. H. (2005) A comparison of mechanical properties derived from multiple skeletal sites in mice. J. Biomech. 38, 467–475 [DOI] [PubMed] [Google Scholar]