

Abstract
Kluyveromyces marxianus, a non-conventional thermotolerant yeast, is potentially useful for production of ethanol and other products. This species has a strong tendency to randomly integrate transforming DNA fragments, making necessary the development of more precise methods for gene targeting. In this study, we first demonstrated that K. marxianus NBRC1777 is cold-tolerant, and then established a highly efficient and precise technique for gene editing by introducing genes encoding deaminase-mediated targeted point mutagenesis (Target-AID) and clustered regularly interspaced short palindromic repeats (CRISPR) associated proteins (CRISPR-Cas9). We used Target-AID to introduce targeted point mutations that disrupted Nej1 or Dnl4, genes that are involved in non-homologous end-joining (NHEJ). Both of the resulting mutant strains showed enhanced proportions of homology-mediated integration compared to the wild-type parent. In combination with target cleavage by CRISPR-Cas9, markerless integration was performed using short (~50 bp) flanking homologous sequences. Together, these tools render this species fully tractable for gene manipulation, permitting targeted genetic changes in the cold- and thermo-tolerant yeast K. marxianus.
Introduction
The non-conventional yeast Kluyveromyces marxianus is known to be thermotolerant and Crabtree-negative. The latter term indicates that this species produces ethanol only under anaerobic conditions1; in contrast, Crabtree-positive yeasts such as Saccharomyces cerevisiae obligately produce ethanol both aerobically and anaerobically. Thus, Crabtree-negative yeasts are more suitable for the generation of various products other than ethanol because these strains do not undergo aerobic alcoholic fermentation, which consumes carbon resources2. The thermotolerance of K. marxianus also allows faster growth and production at higher temperatures, and some volatile products can be collected during synthesis more efficiently at higher temperature. Another benefit of the faster growth of K. marxianus at 37 °C is the ability to obtain single colonies after overnight incubation, while generally used S. cerevisiae strains take 2 to 3 days at 30 °C. Recently, the complete genome sequence for K. marxianus has been reported3–5.
Unlike S. cerevisiae, which demonstrates homologous recombination (HR) of transformed DNA, K. marxianus DMKU3-1042 tends to integrate DNA fragments randomly into its genome1. Such random integration is attributed to the high activity of the non-homologous end-joining (NHEJ) pathway6. K. marxianus NBRC1777 reportedly needed relatively large homology arms (641 and 1475 bp) of flanking DNA for insertion by HR7, while S. cerevisiae requires only ~50-bp homology arms, sequence lengths that can be added at the 5′ ends of PCR primers8.
Disruption of the NHEJ pathway in yeasts has been shown to suppress random integration events and therefore increase the success rate of HR in these organisms6, 9. The core NHEJ proteins are Ku70/Ku80, Lig4/Lif1, and Nej110–13. In the first step, a stable heterodimer of Ku70/Ku80 binds to broken DNA ends and acts as a bridging complex. Lig4 and Lif1 also form a highly stable complex. Lig4 is an ATP-dependent double-strand break (DSB) repair DNA ligase and Lif1 is Lig4’s stabilizing, stimulating, and targeting co-factor10. In S. cerevisiae, deletion of NEJ1 reduces NHEJ by 100-fold9.
Clustered regularly interspaced short palindromic repeats (CRISPR) associated proteins (CRISPR-Cas9) genome-editing tools have been applied in several yeast species, facilitating NHEJ-mediated insertion/deletion formation. HR-mediated integration following CRISPR-Cas9 editing has also been demonstrated, with efficiency varying depending on the yeast species2, 14. More recently, deaminase-mediated targeted point mutagenesis (Target-AID) has been performed in S. cerevisiae; which is a hybrid system of nuclease-deficient CRISPR-Cas9 and activation-induced cytidine deaminase (AID)15. When AID ortholog PmCDA1 was fused to C-terminus of nickase Cas9(D10A), C to T/G point mutation was induced within 3–5 bases window at the 5′ end of target sequence. Target-AID also appeared to be much less toxic compared to CRISPR-Cas9.
Here, we describe the development of a comprehensive set of genome engineering tools for use with K. marxianus NBRC1777, a strain that we show to be both cold- and thermo-tolerant.
Results and Discussions
Cold-tolerant yeast K. marxianus strain
To characterize growth rates at various temperature, K. marxianus NBRC1777 and S. cerevisiae BY4741 were cultured at 5, 10, 20, 30, 37, and 45 °C (Fig. 1). The maximum specific growth rate (µ max) was higher for K. marxianus than for S. cerevisiae at all temperatures (Fig. 1), indicating that the K. marxianus is a fast-growing strain. K. marxianus showed best growth at 37 °C and was able to grow at 45 °C. The difference in the µ max was also bigger at 10 °C (0.079/0.044) compared to the difference at 20 °C (0.23/0.20). Thus, in addition to the known thermotolerance of K. marxianus 7, strain NBRC1777 exhibited fast and robust growth at a wide range of temperatures, a feature that will be appealing for a variety of industrial applications.
Figure 1.
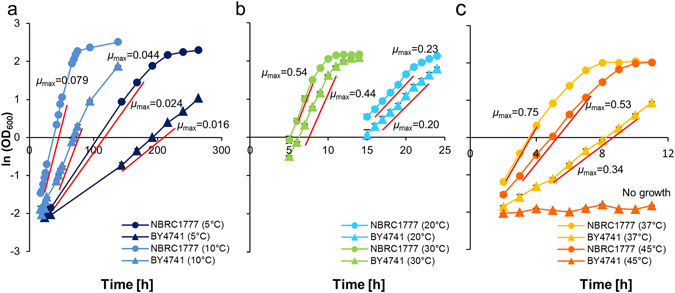
Cell growth of K. marxianus and S. cerevisiae at various temperatures. Cell growth was compared between K. marxianus NBRC1777 and S. cerevisiae BY4741 at 5, 10 °C (a), at 20, 30 °C (b), and at 37, 45 °C (c). The maximum specific growth rate (µ max) was determined as the slope of the mid-log phase of natural logarithm (ln) (OD600). Timeframes for each µ max were as follows: at 5 °C from 33 h to 168 h and 144 h to 216 h, at 10 °C from 19 h to 48 h and 42 h to 75 h, at 20 °C from 16 h to 20 h and 16 h to 22 h, at 30 °C from 6 h to 8 h and from 6 h to 10 h, at 37 °C from 2 h to 4 h and 5 h to 10 h, 45 °C from 3 h to 7 h and no growth for K. marxianus and S. cerevisiae, respectively.
Isolation of KmSNR52 promoter for expression of sgRNA for CRISPR-Cas9
To introduce the CRISPR system into K. marxianus, an efficient promoter for sgRNA expression was needed. As 5′-capped transcripts are not suitable for use as sgRNAs, PolII-driven promoters are not favoured. The SNR52 promoter has been used to express functional sgRNAs in S. cerevisiae 16, so we obtained the K. marxianus SNR52 promoter (KmPSNR52) as follows. The K. marxianus SNR52 transcript sequence was first identified using the BLASTN search program with the S. cerevisiae SNR52 transcript as a query. The region upstream of the putative KmSNR52 transcript was then aligned with the ScSNR52 promoter; a 531-bp fragment was thereby defined as the putative KmPSNR52 (Supplementary Fig. S1).
We next constructed a vector for use as a shuttle vector by combining a K. marxianus autonomously replicating sequence (KmARS7)17 with a centromere sequence (KmCEN-D)18 and demonstrated (data not shown) that this plasmid is retained as an episome in K. marxianus NBRC1777. A human-optimized Streptococcus pyogenes (Sp) cas9 16 under the control of the constitutive S. cerevisiae PDC1 promoter (ScPPDC1) was inserted into the shuttle vector, yielding a basal plasmid (Cas9_Base; Supplementary Fig. S2 and S3) capable of directing the expression of Cas9 in K. marxianus.
Inactivation of the NHEJ pathway by Target-AID point mutation of NHEJ proteins
In S. cerevisiae, the NEJ1 and DNL4 genes encode core components of the proteins mediating NHEJ. By analogy to S. cerevisiae, disruption of the K. marxianus Nej1 and Dnl4 homologs are expected to yield more genetically tractable K. marxianus hosts with higher proportions of HR. Mutation of the K. marxianus Nej1 and Dnl4 genes identified by sequence similarity to the respective budding yeast genes was performed using targeted point mutagenesis by Target-AID in K. marxianus. A basal plasmid (nCas9-CDA_Base) was generated by constructing a variant of the Cas9_Base vector that encoded a nickase mutant (D10A) SpCas9 protein fused to the Petromyzon marinus (Pm)CDA1 cytidine deaminase15 (Supplementary Fig. S2 and S4). Following a previous study in S. cerevisiae 9, target sites in the Nej1 and Dnl4 genes of K. marxianus NBRC1777 were selected so as to introduce stop codons by causing C-to-T mutagenesis at 16 to 19 bp upstream of the sequences encoding protospacer adjacent motif (PAM) sequences for the target sequences. K. marxianus NBRC1777 cells were transformed with plasmids containing each targeting sequence (nCas9-CDA_target Nej1 and nCas9-CDA_target Dnl4). G418-resistant colonies were selected; the Nej1 and Dnl4 genes of the transformants then were PCR amplified and subjected to sequence analysis to confirm the presence of the targeted lesions (Fig. 2). Both plasmids introduced stop codons as expected, providing null alleles of Nej1 (Nej1°) and Dnl4 (Dnl4°) with 1 of 8 (or 1 of 4) G418-resistant colonies contained the mutant allele, respectively. Thus, Target-AID was efficient enough to introduce targeted point mutation in K. marxianus.
Figure 2.
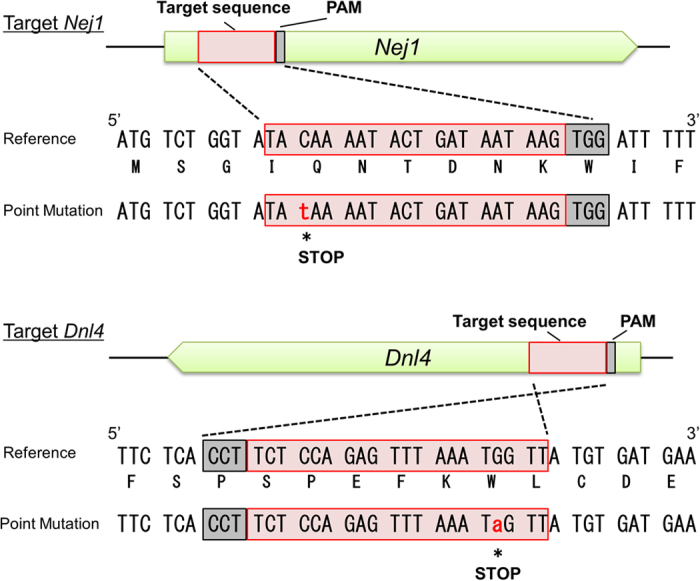
Induction of mutations by Target-AID. The Nej1 and Dnl4 target sites were selected for the introduction of stop codons by Target-AID. Red boxes indicate the target sites and grey boxes indicate the PAM sequences. Lower-case nucleotide in red font indicates the mutated base. The sequences of the induced mutations are shown in alignment with the reference sequences, with each predicted amino acid sequence below the respective nucleic acid sequence.
Conventional HR in NHEJ null mutants
To investigate if these disruptions in genes with putative NHEJ activities increased the rate of HR in K. marxianus, cells of the Nej1° or Dnl4° mutants were transformed with DNA donor fragments with homology arms. Specifically, we employed a GFP bleo cassette (i.e., encoding green fluorescent protein (GFP) fused to a protein providing a selectable marker for bleomycin/zeocin resistance) flanked by K. marxianus Ura3 sequences. Two separate constructs were used, one providing extended homology (1000-bp arms at both ends) and the second providing shorter homology (arms of 122 and 109 bp at the two ends). The fragments were expected to replace Ura3 with the GFP-bleo cassette if HR was facilitated by mutation of genes of the NHEJ pathway. The cells were transformed and selected for zeocin-resistance; the resulting transformants were then screened for 5-fluoroorotic acid (5-FOA) resistance, which would be conferred by Ura3 disruption. When using the construct providing homology arms of 1000 bp, 100% of the ZeoR transformants of the Nej1° or Dnl4° strains (17/17 and 13/13, respectively) were 5-FOAR; in comparison, only 26% (6/23) of the ZeoR transformants of the wild-type (WT) strain were 5-FOAR (Fig. 3). When using equimolar quantities of the construct providing shorter homology arms (122 bp/109 bp), 123 ZeoR transformants were obtained from the WT strain but no 5-FOAR transformants were obtained. The Nej1° or Dnl4° strains yielded one and two ZeoR transformants (respectively), of which no and one (respectively) were 5-FOAR (i.e., disrupted at the targeted Ura3 locus) (Fig. 3).
Figure 3.
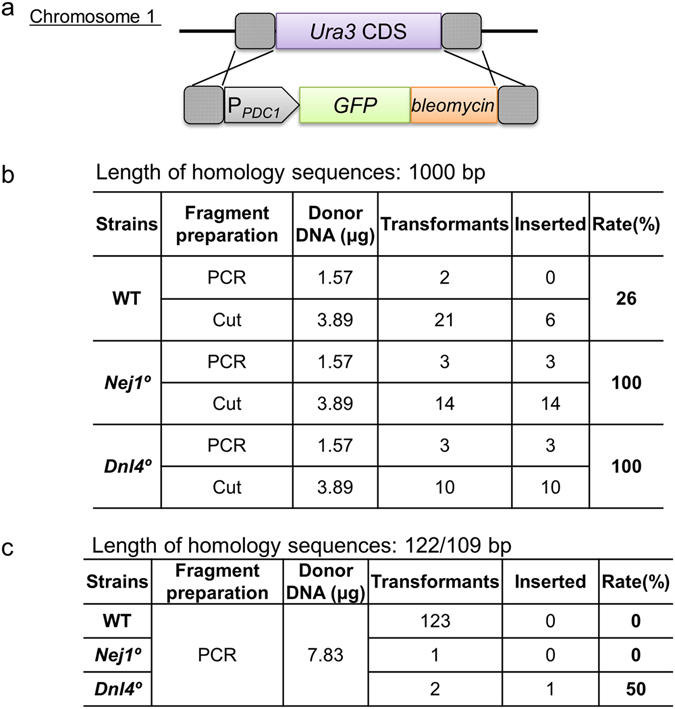
Effect of Nej1° and Dnl4° on homologous recombination. (a) Schematic of the transforming donor DNA fragment targeting the Ura3 locus. Dashed boxes indicate homologous regions. (b), (c) Homologous recombination rates (5-FOA resistant (inserted)/zeocin resistant (transformants)). Fragments were prepared either by PCR or by restriction digestion of plasmid (Cut).
Apparently, less number of transformants were obtained from the mutants of genes involved in NHEJ pathway, presumably because random integration was severely suppressed, rather than by increasing the HR frequency. This decrease in NHEJ facilitates reliable HR at the targeted (by homology) locus, although the obtained efficiency may not be sufficient to perform HR with short homology arms (i.e., of the length that can be added by PCR primers).
HR with CRISPR-Cas9
To boost the HR frequency, co-transformation with a construct encoding CRISPR-Cas9 was performed; the CRISPR system was expected to facilitate HR by providing cleavage of the targeted locus. A pair of target sites was selected at just inside of the homologous regions of Ura3. WT, Nej1°, and Dnl4° cells were transformed with the fragment with homology arms of 122 bp/109 bp (in combination with the Cas9 construct) and transformants were selected using either zeocin, (Ura3-targeting fragment), G418 (CRISPR-Cas9 vector), or both. For all three strains, a larger number of antibiotic-resistant transformants were obtained when the transformation mixes included the CRISPR-Cas9 construct. Among the antibiotic-resistant transformants, 41–79% of those derived from WT cells were 5-FOAR (i.e., disrupted at Ura3), while 95–100% of those derived from the Nej1° and Dnl4° strains were 5-FOAR (Fig. 4). These results indicated that target cleavage by Cas9 enhanced the frequency of HR sufficiently that even without selection by zeocin (i.e., without direct selection for the integration at Ura3), disruption of Ura3 was seen in virtually all transformants.
Figure 4.
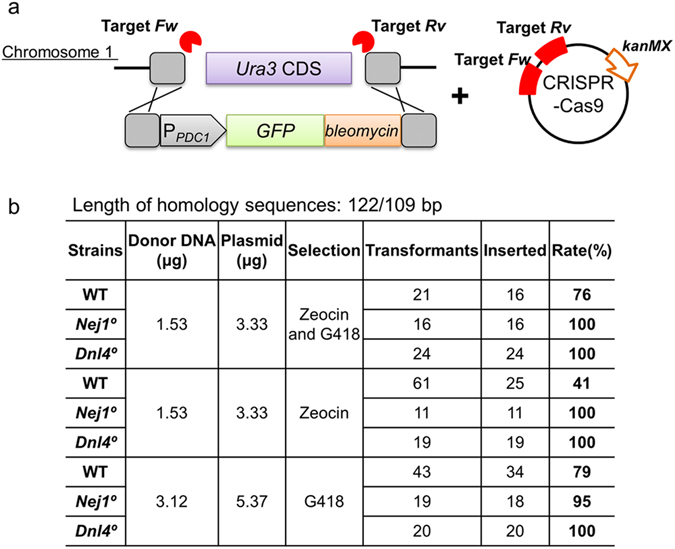
Homologous recombination facilitated by CRISPR-Cas9. (a) Schematic of the transforming donor DNA fragment targeting the Ura3 locus and CRISPR-Cas9 plasmid targeting two sites (indicated by lightning bolts) flanking the Ura3 coding sequences (CDS). Dashed boxes indicate homologous regions. Bottom panel indicates (b) Homologous recombination rate (5-FOA resistant (inserted)/zeocin and/or G418 resistant (transformants) * 100).
Markerless integration with 50-bp homology arms and CRISPR-Cas9
Markerless integration was attempted by co-transformation using the combination of a donor DNA template containing a GFP expression cassette flanked by 50-bp homology arms along with a CRISPR-Cas9-encoding construct. Transformants were selected using G418 and then screened by both the 5-FOA test and PCR analysis. We observed successful integration (i.e., disruption of the Ura3 target locus in G418R transformants) at rates of 28%, 84%, and 92% in the WT, Nej1°, and Dnl4° backgrounds, respectively (Fig. 5). Taking advantage of the markerless integration, the GFP-encoding sequence was directly inserted into the coding sequence of the endogenous Sed1 gene so as to encode a GFP-Sed1 fusion protein preceded by the Sed1 localization peptide. A single cleavage site for CRISPR-Cas9 within this target gene was designed in such a way that the site would not be retained after successful integration. Short (50-bp) homology arms flanking the target site were added to either end of the GFP-encoding sequence by PCR. Transformants were selected by G418 and then screened by PCR, revealing gene replacement at the Sed1 target locus at 38%, 100%, and 100% success rates in the WT, Nej1°, and Dnl4° backgrounds, respectively (Fig. 6). When viewed by fluorescence microscopy, cells harboring the targeted integration exhibited peripheral localization of GFP-Sed1, consistent with the localization previously reported for the equivalent construct in S. cerevisiae 19. Thus, this method can be applied for scar-less genome editing in K. marxianus.
Figure 5.
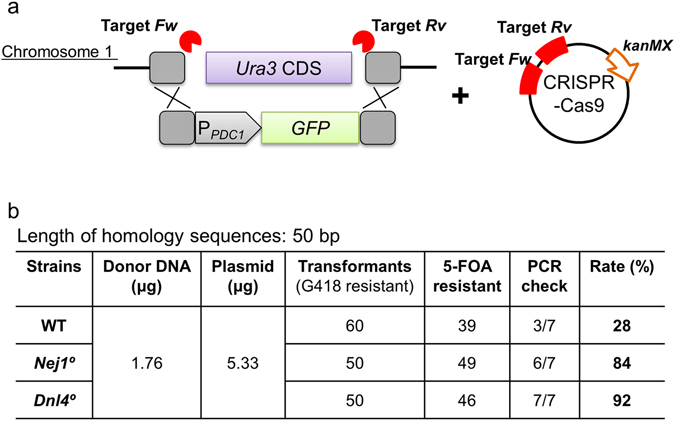
Markerless integration with short homologous arms. (a) Schematic of the transforming donor DNA fragment targeting the Ura3 locus and the CRISPR-Cas9 plasmid targeting two sites (indicated by lightning bolts) flanking the Ura3 CDS. Dashed boxes indicate homologous regions. (b) Homologous recombination rate ((5-FOA resistant * PCR check)/ transformants (G418 resistant) * 100).
Figure 6.
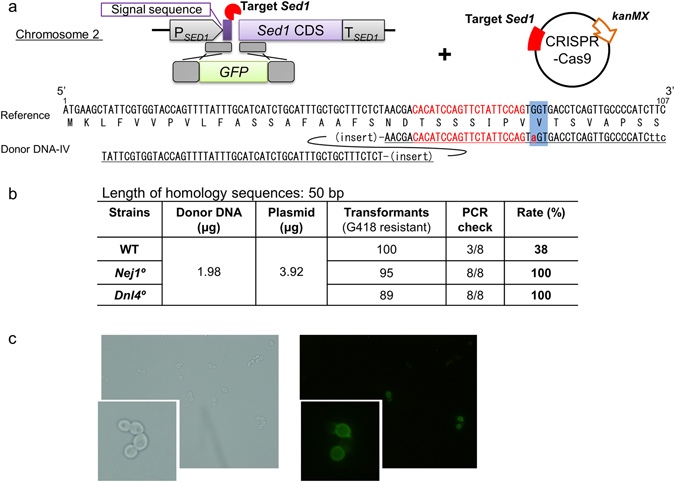
Direct fusion of GFP-coding sequence into a chromosomal locus. (a) Schematic showing integration of the GFP-coding fragment into the Sed1 CDS. The CRISPR-Cas9 plasmid targeted (indicated by lightning bolts) the end of the signal peptide-coding region of Sed1. Dashed boxes indicate homologous regions. Left arm is homologous to the end of to the signal peptide-coding sequence (bp 8 to 57 with respect to the start of the Sed1 coding sequence). Right arm is homologous to Sed1 sequence (bp 58 to 107 with respect to the start of the Sed1 coding sequence) with substitution at bp 84 (G to A; indicated by lower-case letter in red font) to eliminate a PAM. (b) The successful integration rate was calculated as PCR check * 100. (c) Representative microscopic image of positive transformants (Nej1°/GFP_Sed1) under bright-field (left) and fluorescent (right) illumination. Insets show 3-fold magnified images.
Assembly and integration of multiple fragments without markers
In vivo assembly and integration of multiple fragments was performed using three DNA fragments, each containing 50 bp overlap with the adjacent fragments or target sites, such that the entire construct was designed to replace Ura3 (Fig. 7). Compared to single PCR product donor constructs, a smaller number of transformants was obtained by G418 selection when using the multiple-fragment DNA as the donor. Nonetheless, the WT, Nej1°, and Dnl4° strains showed integration efficiencies of 18% (2/11), 100% (3/3), and 100% (4/4), respectively. This assembly and integration method is expected to allow combinatorial integration of gene cassettes using (for example) combinations of various promoters and coding regions.
Figure 7.
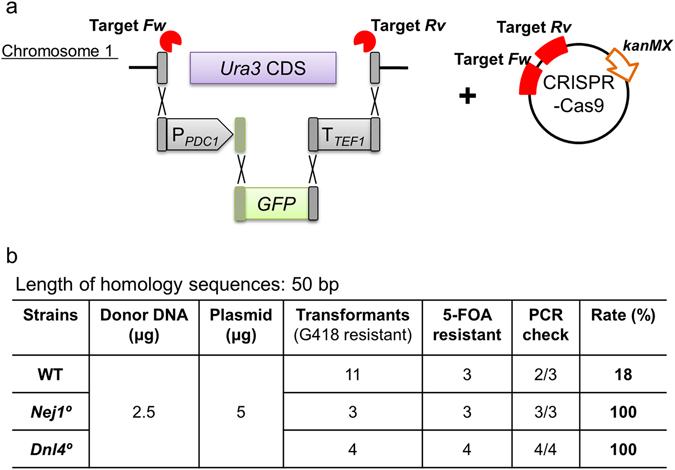
In vivo assembly and integration of three DNA fragments with 50 bp overlaps. (a) Schematic of the three transforming donor DNA fragments replacing the Ura3 locus along with the CRISPR-Cas9 plasmid targeting two sites (indicated by lightning bolts) flanking the Ura3 CDS. Dashed boxes indicate homologous regions. Bottom panel shows (b) Homologous recombination rate ((5-FOA resistant * PCR check)/ transformants (G418 resistant) * 100).
In summary, targeted point mutagenesis (by Target-AID) and markerless CRISPR-mediated integration were demonstrated in K. marxianus using a fast-growing strain that was shown to exhibit both thermo- and cold-tolerance. Mutation of Nej1 or Dnl4 loci encoding homologs of NHEJ proteins, enhanced the proportion of HR events. These genome-engineering tools are expected to greatly facilitate industrial use of K. marxianus, a prototype for the class of underexploited non-conventional yeasts.
Methods
Strains and culture conditions
S. cerevisiae and K. marxianus strains used in this study are listed in Table 1. Yeast cells were grown in YPD medium (10 g/L yeast extract, 20 g/L peptone, and 20 g/L glucose) at 30 °C unless otherwise noted. Escherichia coli strain DH5α (Toyobo, Osaka, Japan) which was used for cloning, was grown in Luria-Bertani (LB) medium (10 g/L peptone, 5 g/L yeast extract, and 5 g/L sodium chloride) supplemented with 100 mg/L ampicillin at 37 °C.
Table 1.
Plasmids and strains used in this study.
| Plasmids and strains | Genotype | Reference |
|---|---|---|
| Plasmids | ||
| nCas9-CDA_Base | ScPPDC1_nCas9_TTDH3, KmARS7, Km CEN D, kanMX, ori, and AmpR | This study |
| nCas9-CDA_target Nej1 | KmPSNR52_target_Nej1_sgRNA_Tsup4 cassette in nCas9-CDA_Base | This study |
| nCas9-CDA_target Dnl4 | KmPSNR52_target_Dnl4_sgRNA_Tsup4 cassette in nCas9-CDA_Base | This study |
| Cas9_Base | ScPPDC1_Cas9_TTDH3, KmARS7, Km CEN D, kanMX, ori, and AmpR | This study |
| Cas9_Base_target_Ura3_Fw | KmPSNR52_target_Ura3_Fw_sgRNA_Tsup4 cassette in Cas9_Base | This study |
| Cas9_Base_target _Ura3_Fw + Rv | KmPSNR52_target_Ura3_Fw_sgRNA_Tsup4 cassette and KmPSNR52_target_Ura3_Rv_sgRNA_Tsup4 cassette in Cas9_Base | This study |
| Cas9_Base_target Sed1 | KmPSNR52_target_Sed1_sgRNA_Tsup4 cassette in Cas9_Base | This study |
| TOPO/F7 | ScPPDC1_EGFP_bleomycin on both sides Ura3 homologous sequence cassette in pCRTM4Blunt-TOPO® | This study |
| Strains | ||
| S. cerevisiae | ||
| BY4741 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | ATCC |
| K. marxianus | ||
| NBRC1777 | Wild-type | NITE Biological Resource Center, Japan |
| Nej1° | Nej1disrupted by C to T point mutation at position 13 | This study |
| Dnl4° | Dnl4 disrupted by G to A point mutation at position 44 | This study |
Growth measurements
S. cerevisiae BY4741 and K. marxianus NBRC1777 strains were grown in YPD medium overnight at 30 °C by shaking in test tubes. The cells were inoculated into 5 mL of YPD medium at a starting optical density at 600 nm (OD600) of 0.05, and cultures were cultivated at 5, 10, 20, 30, 37, or 45 °C. Cell growth was tracked by measuring OD600 using a UV mini spectrophotometer (Shimadzu, Kyoto, Japan). The µ max was determined as the slope of the log phase of natural logarithm (ln) transformation of the linear region.
Construction of basal genome-editing plasmids
The vector sequences are provided in Supplementary Fig. S3 and S4. Vector assembly was performed by PCR using the In-fusion cloning method (Takara Bio, Shiga, Japan). The vector backbone was designed to contain KmARS7, KmCEN-D and selectable marker kanMX, which encodes G418 resistance. For CRISPR-Cas9 (Cas9_Base) and Target-AID (nCas9-CDA_Base) vectors, a gene encoding human-optimized nCas9-PmCDA1 or Cas915, 16 (respectively) was placed between ScPPDC1 and the TDH3 terminator (ScTTDH3) on the vector backbone.
Construction of target sequence-containing plasmids
To generate the sgRNA expression casettes, the K. marxianus SNR52 promoter (KmPSNR52) (531 bp; identified by alignment with the S. cerevisiae gene) was amplified by PCR using the combination of the forward primer (P_Km01-013) and each reverse primer (5′-CTAGCTCTAAAAC- reverse complement of target sequence-GATTCGAACTGCGGACGTTG-3′). The sgRNA scaffold and the SUP4 terminator were amplified by PCR from plasmid pRS426-SNR52p-gRNA.CAN1.Y-SUP4t16 using the primers P_Km01-014 and P_Km01-015 (Supplementary Table S1). The two fragments were fused by overlap extension PCR using the primers P_Km01-013 and P_Km01-015 to yield each target-containing sgRNA cassette (Supplementary Fig. S1). Each cassette was digested with restriction enzymes NotI and NheI and ligated into NotI-, SpeI-digested nCas9-CDA_Base or Cas9_Base plasmid.
Transformation
K. marxianus cells were transformed by the lithium acetate method20. After transformation, cells were plated on YPD containing appropriate selection reagents (100 μg/mL G418 and/or 50 μg/mL zeocin) and grown for one day. For the 5-FOA test, cells were streaked onto YPD containing 3 mg/mL 5-FOA, incubated overnight, and assessed for growth. Colony PCR was performed to check fragment size and to obtain DNA for Sanger sequencing using a 3130xL Genetic Analyzer (Applied Biosystems, CA, USA).
Donor DNA templates
Each donor DNA template was constructed by PCR and cloned into the plasmid TOPO/F7 using the Zero Blunt® TOPO® PCR Cloning Kit (Invitrogen, CA, USA) (Fig. 5). Each cloned donor DNA was either excised by restriction enzymes or amplified by PCR using the respective primer pair (Supplementary Table S1), and the resulting fragment was purified by agarose gel electrophoresis before use in transformation.
Electronic supplementary material
Acknowledgements
This research was supported by the Project Focused on Developing Key Technology for Discovering and Manufacturing Drugs for Next-Generation Treatment and Diagnosis from the Japan Agency for Medical Research and Development (AMED), Japan.
Author Contributions
Y.N. and K.N. designed the experiment and contributed to interpretation of data. Y.N. wrote the initial draft of the manuscript. K.N. and T.H. assisted in the preparation of the manuscript. A.K. supervised the manuscript. All authors reviewed the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-08356-5
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Nonklang S, et al. High-temperature ethanol fermentation and transformation with linear DNA in the thermotolerant yeast Kluyveromyces marxianus DMKU3-1042. Appl. Environ. Microbiol. 2008;74:7514–7521. doi: 10.1128/AEM.01854-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wagner JM, Alper HS. Synthetic biology and molecular genetics in non-conventional yeasts: Current tools and future advances. Fungal Genet. Biol. 2016;89:126–36. doi: 10.1016/j.fgb.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 3.Jeong H, et al. Genome sequence of the thermotolerant yeast Kluyveromyces marxianus var. marxianus KCTC 17555. Eukaryot. Cell. 2012;11:1584–1585. doi: 10.1128/EC.00260-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suzuki T, Hoshino T, Matsushika A. Draft Genome Sequence of Kluyveromyces marxianus Strain DMB1, Isolated from Sugarcane Bagasse Hydrolysate. Genome Announc. 2014;2:e00733–14. doi: 10.1128/genomeA.00733-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Inokuma K, Ishii J, Hara KY, Mochizuki M, Hasunuma T. Complete Genome Sequence of Kluyveromyces marxianus NBRC1777, a Nonconventional Thermotolerant yeast. Yeast. 2015;3:e00389–15. doi: 10.1128/genomeA.00389-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abdel-Banat BMA, Nonklang S, Hoshida H, Akada R. Random and targeted gene integrations through the control of non-homologous end joining in the yeast Kluyveromyces marxianus. Yeast. 2009;27:29–39. doi: 10.1002/yea.1729. [DOI] [PubMed] [Google Scholar]
- 7.Hong J, Wang Y, Kumagai H, Tamaki H. Construction of thermotolerant yeast expressing thermostable cellulase genes. J. Biotechnol. 2007;130:114–123. doi: 10.1016/j.jbiotec.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 8.Da Silva N. a & Srikrishnan, S. Introduction and expression of genes for metabolic engineering applications in Saccharomyces cerevisiae. FEMS Yeast Res. 2012;12:197–214. doi: 10.1111/j.1567-1364.2011.00769.x. [DOI] [PubMed] [Google Scholar]
- 9.Valencia M, et al. NEJ1 controls non-homologous end joining in Saccharomyces cerevisiae. Nature. 2001;414:666–669. doi: 10.1038/414666a. [DOI] [PubMed] [Google Scholar]
- 10.Dudášová Z, Dudáš A, Chovanec M. Non-homologous end-joining factors of Saccharomyces cerevisiae. FEMS Microbiol. Rev. 2004;28:581–601. doi: 10.1016/j.femsre.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 11.Feldmann H, Winnacker EL. A putative homologue of the human autoantigen Ku from Saccharomyces cerevisiae. J. Biol. Chem. 1993;268:12895–12900. [PubMed] [Google Scholar]
- 12.Feldmann H, et al. HDF2, the second subunit of the Ku homologue from Saccharomyces cerevisiae. J. Biol. Chem. 1996;271:27765–27769. doi: 10.1074/jbc.271.44.27765. [DOI] [PubMed] [Google Scholar]
- 13.Moore JK, Haber JE. Cell cycle and genetic requirements of two pathways of nonhomologous endjoining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:2164–2173. doi: 10.1128/MCB.16.5.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jakočiūnas T, Jensen MK, Keasling JD. CRISPR/Cas9 advances engineering of microbial cell factories. Metab. Eng. 2016;34:44–59. doi: 10.1016/j.ymben.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 15.Nishida, K. et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science353, aaf8729-1-aaf8729-8 (2016). [DOI] [PubMed]
- 16.Dicarlo JE, et al. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 2013;41:4336–4343. doi: 10.1093/nar/gkt135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoshida, H., Murakami, N., Suzuki, A., Tamura, R. & Asakawa, J. Non-homologous end joining-mediated functional marker selection for DNA cloning in the yeast Kluyveromyces marxianus. Yeast31, 29–46 (2014). [DOI] [PubMed]
- 18.Yarimizu T, Nakamura M, Hoshida H, Akada R. Screening of accurate clones for gene synthesis in yeast. J. Biosci. Bioeng. 2015;119:251–259. doi: 10.1016/j.jbiosc.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 19.Huh W-K, et al. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–91. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- 20.Gietz D, St Jean A, Woods RA, Schiestl RH. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992;20:1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.