

Abstract
Huntington's disease (HD) is a progressive and fatal degenerative disorder that results in debilitating cognitive and motor dysfunction. Most HD studies have focused on degeneration of the CNS. We previously discovered that skeletal muscle from transgenic R6/2 HD mice is hyperexcitable due to decreased chloride and potassium conductances. The progressive and early onset of these defects suggest a primary myopathy in HD. In this study, we examined the relationship between neuromuscular transmission and skeletal muscle hyperexcitability. We used an ex vivo preparation of the levator auris longus muscle from male and female late-stage R6/2 mice and age-matched wild-type controls. Immunostaining of the synapses and molecular analyses revealed no evidence of denervation. Physiologically, we recorded spontaneous miniature endplate currents (mEPCs) and nerve-evoked EPCs (eEPCs) under voltage-clamp, which, unlike current-clamp records, were independent of the changes in muscle membrane properties. We found a reduction in the number of vesicles released per action potential (quantal content) in R6/2 muscle, which analysis of eEPC variance and morphology indicate is caused by a reduction in the number of vesicle release sites (n) rather than a change in the probability of release (prel). Furthermore, analysis of high-frequency stimulation trains suggests an impairment in vesicle mobilization. The depressed neuromuscular transmission in R6/2 muscle may help compensate for the muscle hyperexcitability and contribute to motor impersistence.
SIGNIFICANCE STATEMENT Recent evidence indicates that Huntington's disease (HD) is a multisystem disorder. Our examination of neuromuscular transmission in this study reveals defects in the motor nerve terminal that may compensate for the muscle hyperexcitability in HD. The technique we used eliminates the effects of the altered muscle membrane properties on synaptic currents and thus provides hitherto the most detailed analysis of synaptic transmission in HD. Clinically, the striking depression of neurotransmission we found may help explain the motor impersistence in HD patients. Therapies that target the highly accessible peripheral nerve and muscle system provide a promising new avenue to lessen the debilitating motor symptoms of HD.
Keywords: electrophysiology, Huntington's disease, neuromuscular transmission, synaptic transmission, trinucleotide repeat disorder
Introduction
Huntington's disease (HD) is a fatal degenerative disorder that is characterized by severe affective, cognitive, and behavioral psychiatric abnormalities, as well as numerous motor defects, including chorea, rigidity, dystonia, and bradykinesia (Wells and Ashizawa, 2006). It is caused by an expanded CAG trinucleotide repeat in exon 1 of the huntingtin gene (Huntington's Disease Collaborative Research Group, 1993) and is one of several trinucleotide repeat disorders (Wells and Ashizawa, 2006). Although the huntingtin gene is widely expressed throughout the body, including skeletal muscle, most research on the disease has focused on the CNS (Huntington's Disease Collaborative Research Group, 1993; Strand et al., 2005; Wells and Ashizawa, 2006; Lo and Hughes, 2010). Neurodegeneration in the striatum and cortex have been widely assumed to underlie the severe motor defects of HD (Strand et al., 2005; Wells and Ashizawa, 2006). However, the expression of mutated huntingtin in the skeletal muscle could cause muscle dysfunction independent of neurodegeneration.
In support of primary muscle defects, motor symptoms were shown to precede the onset of neurological symptoms in a marathon runner with HD (Kosinski et al., 2007). Moreover, a number of studies have demonstrated pathological changes in HD muscle, including metabolic and mitochondrial defects (Lodi et al., 2000; Turner et al., 2007; Mielcarek et al., 2015), atrophy (Ribchester et al., 2004; She et al., 2011; Ehrnhoefer et al., 2014), reduced muscle strength (Busse et al., 2008; Hering et al., 2016), and a reduced expression of genes necessary for normal muscle differentiation (Luthi-Carter et al., 2002; Strand et al., 2005). Additionally, we previously discovered that skeletal muscle from R6/2 transgenic HD mice is hyperexcitable due to decreased currents through chloride (ClC-1) and inwardly rectifying potassium (Kir) channels, which correlated with aberrant mRNA processing and lower levels of mature full-length mRNAs for ClC-1 and Kir channels (Waters et al., 2013). We further showed that these muscle defects begin in presymptomatic R6/2 mice, increase in parallel to disease progression, and are associated with disruptions in muscle maturation (Miranda et al., 2017).
It is also possible that defects in motor neurons could contribute to HD symptoms. It has been shown that motor neurons in the brainstem of wild-type (CD1) mice increase expression of the huntingtin gene through development into adulthood (Bhide et al., 1996). Other papers have speculated changes in HD spinal motor neurons (Ribchester et al., 2004; Mielcarek et al., 2015) and a recent study reported smaller and fewer cervical spinal motor neurons with atypical nuclei in the BACHD mice (Valadão et al., 2017).
In this report, we begin to examine the relationship between muscle and neuronal defects in HD. To determine whether denervation or motor unit loss drives the HD skeletal muscle hyperexcitability, as proposed in a study based on contraction experiments (Mielcarek et al., 2015), we examined neuromuscular morphology and the expression levels of key molecular markers for denervation in R6/2 mice. Our main focus in this report was to measure neuromuscular transmission under voltage-clamp conditions. This is important because the standard corrections used to account for nonlinear summation in current-clamp recordings of synaptic potentials (McLachlan and Martin, 1981) would not account for the changes in the R6/2 muscle membrane properties that cause hyperexcitability. Most of the previous studies of neuromuscular transmission in HD used current-clamp (Ribchester et al., 2004; Romero et al., 2008; Rozas et al., 2011; de Aragão et al., 2016) and were completed before we reported that R6/2 muscle is hyperexcitable and has a lower specific-membrane capacitance.
We found that muscle hyperexcitability in R6/2 mice was not driven by denervation. However, despite the normal appearance of the neuromuscular junctions (NMJs), we did find evidence of fewer functional active sites and impaired vesicle release in R6/2 mice, both of which may help compensate for the muscle hyperexcitability. We also show that the changes in R6/2 muscle membrane properties alter corrected synaptic potentials measured under current-clamp, which has experimental implications for many studies of synaptic transmission in disease states.
Materials and Methods
Animal care.
All animal procedures were performed in accordance with the policies of the Animal Care and Use Committee of Wright State University. We established a breeding colony in the Wright State University Laboratory Animal Resources Facility that consisted of one wild-type B6CBA female with an ovarian transplant (hemizygous for Tg(HDexon1)62Gpb) and one wild-type male (B6CBAF1/J) purchased from The Jackson Laboratory, stock #002810 (RRID: IMSR_JAX:002810). A total of 40 male and female R6/2 and 52 male and female control mice produced from 12 breeding pairs were used for this study. Tail samples cut between 7 and 14 d of age were genotyped at Laragen. Mice were housed with wild-type littermates (control) in sex-matched cages after weaning at ∼14 d of age. Beginning at 9 weeks of age, all cages housing R6/2 mice were supplied with a Petri dish containing moist chow (dry chow soaked in water) to ensure adequate nutritional intake in the symptomatic mice.
Health and phenotype severity assessment for HD mice, as described previously (Waters et al., 2013; Miranda et al., 2017), were performed weekly for mice 8–10 weeks of age, every other day for mice 10 weeks of age, and daily for mice ≥11 weeks of age. Assessment categories included physical condition, respiratory health, activity level, and weight loss. Mice were rated on a scale from 0 to 3 for each category, where 0 signified a normal condition and 3 signified a severe/poor condition. Mice were selected for experiments when the sum of all the category scores was ≥6 or when the score of any one category equaled 3. Of the behavioral measures, mouse weight has been a reliable and easy to assess measure of disease progression. We previously showed that R6/2 weight stopped increasing from 6 to 10 weeks of age and decreased after 10 weeks of age (Miranda et al., 2017). Experiment-ready mice were killed by inhalation of a saturating dose of isoflurane (∼2 g/L), followed by cervical dislocation. All R6/2 and control mice used in this study were between 11 and 14 weeks of age.
For denervation studies, SWR/J mice were anesthetized with inhaled isoflurane. The region of the left sciatic notch was shaved and sterilized, an incision was made, and a 3 mm length of the sciatic nerve was removed. The wound was closed with sutures and mice were allowed to recover. One week later mice were killed with inhaled CO2 and the right and left gastrocnemius muscles were removed and flash frozen in liquid nitrogen.
Electrophysiology.
The levator auris longus (LAL) muscle was dissected from the mice and pinned to a custom Sylgard perfusion chamber. Muscle fibers were visualized using an Olympus BX51WI microscope equipped with 10× (UMPLFLN10XW) and 40× (LUMPLFLN40XW) objectives. The extracellular buffer solution consisted of the following (in mm): 144 NaCl, 4 KCl, 1.2 CaCl2, 0.6 MgCl2, 5 glucose, 1 NaH2PO4, 10 MOPS, and pH 7.4 with NaOH. A low Ca2+ extracellular buffer was also used and consisted of the following (in mm): 140 NaCl, 4 KCl, 0.5 CaCl2, 4 MgCl2, 5 glucose, 1 NaH2PO4, 10 MOPS, and pH 7.4 with NaOH. Both the normal and low Ca2+ buffers had an osmolality of 300 ± 5 mmol/kg. To prevent muscle contraction during nerve stimulation, 60 μm N-benzyl-p-toluene sulfonamide (BTS) and 1 μm μ-conotoxin GIIIB (in normal Ca2+ only) were used. To visualize NMJs, fibers were stained with 5 μm 4-(4-diethylaminostyryl)-N-methylpyridinium iodide (4-Di-2-ASP) for 5 min. 4-Di-2-ASP is a cationic fluorescent mitochondrial dye that stains presynaptic nerve terminals with no lasting effects on their structure or function (Magrassi et al., 1987). A Thorlabs M470L3 collimated LED (470 nm) or halogen bulb and an eGFP filter cube were used to observe fluorescence. Solution changes were achieved via a custom gravity-driven perfusion system and all experiments were performed at room temperature (22°C).
Fibers within the rostral band of the LAL were impaled using two borosilicate glass microelectrodes (Sutter Instruments) positioned within 100 μm of one another and within 100 μm of the NMJ to obtain proper space clamp of the endplate region (Albuquerque and McIsaac, 1970). The distance of electrodes from one another and from the NMJ were measured with ImageJ (National Institutes of Health) from images acquired with a Santa Barbara Instruments Group (SBIG) ST-7XMEI CCD camera (SBIG). The internal solution used for the voltage-reading electrode was 3 m KCl and the internal solution of the current-passing electrode consisted of the following (in mm): 75 aspartate, 5 MgCl2, 15 Ca(OH)2, 5 ATP disodium, 5 phosphocreatine disodium, 5 glutathione, 20 MOPS, 30 EGTA, pH 7.2 with KOH, and osmolality of 300 ± 5 mmol/kg. All electrode resistances were between 10 and 15 MΩ. An Axoclamp 900A amplifier, Digidata 1550 digitizer, and the pCLAMP 10 data acquisition and analysis software (Molecular Devices) were used for synaptic current and voltage-clamp recordings. Nerve stimulation was achieved using a Master-9 Pulse Generator and an ISO-Flex stimulus isolator (AMPI) in conjunction with a CBDSH75 concentric bipolar electrode (FHC) that was positioned directly onto the nerve using a small Narishige ball-joint manipulator.
Fibers with high miniature endplate potential (mEPP) or miniature endplate current (mEPC) frequency (>3 Hz in physiological Ca2+ and >4 Hz in low Ca2+) were discarded to avoid studying potentially damaged nerve terminals. Fibers necessitating more than −25 nA of holding current to maintain the membrane potential between −80 and −90 mV were also discarded. Before entering voltage-clamp, an equal number of small positive and negative square current pulses (between −14 and +14 nA) were injected under current-clamp to determine the membrane time constant (τm).
The motor endplates were voltage-clamped to −85 mV. Achieving adequate voltage control can be a concern. As described above, electrodes were impaled at distances from the NMJ that were within the muscle length constant. Additionally, we used the DC restore feature of the Axoclamp 900A, which sets a very high voltage-clamp gain (∼1,000,000). Under these conditions, the entire motor endplate was clamped at the holding potential (−85 mV). Even under these conditions, however, the voltage-clamp gain and lag settings influence the rate at which the amplifier will maintain the holding potential; fast currents may cause a momentary voltage deflection. Moreover, resolving mEPCs from noise required lower gain and lag settings than we previously used to voltage-clamp whole flexor digitorum brevis fibers (Waters et al., 2013; Miranda et al., 2017). To assess our voltage-clamp, we measured the voltage deflection that occurred during nerve-evoked EPCs (eEPCs) in 5 WT and R6/2 fibers (10 per fiber). These data were representative of mean eEPC values. The average voltage deflections during the peak eEPC in the WT and R6/2 fibers was only 1.4 ± 0.3 and 0.8 ± 0.2 mV, respectively. Moreover, by integrating the full mEPCs and eEPCs to obtain the total charge, we minimized the contribution of the voltage deflection. To estimate the amount of charge attributable to the voltage deflections, we converted the voltage signal to current (deflection current) using the fiber input resistance (obtained under current-clamp from the same fibers). We then integrated the deflection current over the time course of the corresponding eEPCs to obtain the deflection charge. The |deflection charge|/|eEPC charge| ratio was 3.9 × 10−4 ± 1.3 × 10−4 and 1.1 × 10−4 ± 4.0 × 10−5 for WT and R6/2 endplates, respectively. Thus, based on this ratio, the percentage of unclamped endplate in our experiments was <0.05%. Voltage-clamp settings (gain: 1500, lag: 1, and pipette capacitance neutralized) were maintained the same across all fibers. To estimate the quantal content (QC), the nerve was stimulated at 0.5 Hz with the stimulus intensity set to 1.5–2× the threshold required to elicit an eEPC and we recorded the resulting eEPCs and spontaneously occurring mEPCs for 2 min. The same protocol was used for low calcium experiments. QC was calculated using the following equation:
 |
To estimate of the probability of synaptic release (prel) and the number of available release sites (n), we used a graphical approach based on binomial vesicle release statistics (Clements and Silver, 2000; Wang et al., 2010). QC was calculated using Equation 1 and the variance of QC [Var(QC)] was calculated with the following equation:
 |
We assumed the following relationships.
To estimate n from Var(QC) versus QC plots, we substituted p = , a rearranged form of Equation 3, into Equation 4 to obtain the following parabola equation:
 |
For high-frequency stimulation, the nerve was stimulated at 50 Hz during 200 ms trains (10 eEPCs). Each fiber was stimulated with 10 such trains with a 10 s pause between each train. To assess depression in normal Ca2+, the peak amplitude of each eEPC was normalized to the peak amplitude of the largest eEPC in the train. We also used the degree of depression to compare depression severity, determined by dividing the amplitude of the last eEPC by the first eEPC in a train. In all protocols, acquisition rate was set to 20 kHz and signals were low-pass filtered at 1 kHz.
To determine the effects of the altered muscle membrane properties on corrected synaptic potentials recorded under current-clamp, we applied the 0.5 Hz stimulation protocol described above to a subset of the fibers under both voltage-clamp and current-clamp. We corrected the mEPPs and eEPPs recorded under current-clamp for differences in resting membrane potential and nonlinear summation as described previously for R6/1 HD mice (Rozas et al., 2011), which was based on work by McLachlan and Martin, 1981. Briefly, endplate potentials were normalized for changes in resting membrane potential (mEPPnorm and eEPPnorm) using mEPPnorm = mEPP(−75/Vm) and eEPPnorm = eEPP(−75/Vm), where Vm is the resting membrane potential. To correct the evoked endplate potentials for nonlinear summation, we used eEPPcorr = eEPPnorm/[1 − 0.8(eEPPnorm/E)], where E is the difference between Vm and the equilibrium potential for current through the nicotinic acetylcholine receptor (nAChR). As was done previously (Rozas et al., 2011), we assumed the equilibrium potential for nAChR currents was 0 mV. QC using current-clamp (QCIC) data was obtained from QCIC = mean eEPPcorr/mean mEPPnorm.
Resting membrane potential measurements were obtained from extensor digitorum longus fibers as previously described (Novak et al., 2015). Briefly, muscle samples were examined under an upright epifluorescence microscope (Leica DMR) and continuously perfused with a recirculating Ringer solution containing the following (in mm): 118 NaCl, 3.5 KCl, 2 CaCl2, 0.7 MgSO4, 26.2 NaHCO3, 1.7 NaH2PO4, 5.5 glucose, and equilibrated with 95% O2 and 5% CO2 to maintain the pH 7.3–7.4 at 20–22°C. Fibers were impaled with single sharp microelectrodes to minimize membrane damage. The electrodes were filled with 3 m KCl and 1 mm sulforhodamine (for visualization) and had resistances between 15 and 30 MΩ. Resting membrane potential records were acquired using a Geneclamp 500B amplifier (Molecular Devices) as well as a Power 1401 data acquisition board and Spike2 v5.13 software (Cambridge Electronics Design). The initial membrane potential upon impaling the electrode was considered the resting membrane potential.
Immunostaining.
To assess innervation, whole-mount LAL muscles were dissected and pinned out in Sylgard dishes, fixed for 15 min with 4% paraformaldehyde in PBS, and washed three times for 10 min with PBS. To identify the postsynaptic side of NMJs, we labeled nAChRs with a 30 min exposure to solution containing 20 μg/ml of rhodamine-conjugated α-bungarotoxin (Invitrogen). To label presynaptic nerve terminals, we stained the LAL for neurofilaments with monoclonal anti-mouse antibody directed against SMI 31 (Covance Research Products; 1:500 dilution) and for presynaptic vesicles with the monoclonal anti-mouse antibody against SV2 (AB_2315387, Developmental Studies Hybridoma Bank; 1 μg/ml). The LAL was exposed to both primary antibodies overnight at 4°C. A 2 h room temperature incubation with secondary fluorescein-conjugated anti-mouse antibody (Jackson ImmunoResearch Laboratories) was used to label both neurofilament and presynaptic vesicle primary antibodies. The whole-mount LAL was then washed three times in PBS and mounted using Vectashield (Vector Laboratories). We used an Olympus FV 1000 confocal microscope with a 60× oil-immersion objective (NA 1.4) to image the endplates. Stained sections were analyzed using Olympus Fluoview software.
Quantification of NMJ morphology and bassoon localization was completed in dissected, whole-mount R6/2 and control LAL muscles that were pinned out in a Sylgard dish and fixed with 4% paraformaldehyde in PBS for 1 h. The fixed LAL samples were washed in PBS, rinsed with PBS-T (0.01 m PBS containing 0.3% Triton-X, pH 7.3), and blocked with normal horse serum (10% in PBS-T) for 1 h. Samples were immunostained free-floating in 12-well plates. Primary and secondary antibodies were diluted in PBS-T with 10% normal horse serum. To detect presynaptic active zones, muscle tissue was incubated in mouse anti-bassoon antibody (Abcam AB82958 Lot GR78505; 1:1000 dilution) overnight at 4°C. Immunoreactivity (IR) was detected with AlexaFluor 488-conjugated donkey anti-mouse antibody (Jackson ImmunoResearch; 1:250 dilution) for 2 h at room temperature with gentle shaking. To detect postsynaptic membrane, muscle tissue was incubated in AlexaFluor 555-conjugated α-bungarotoxin (Invitrogen; 1:1000 dilution) for 20 min. Tissue was washed and mounted on charged microslides in Vectashield mounting medium (Vector Laboratories) and coverslipped.
Approximately 12 enface NMJs were randomly selected per mouse and imaged on a Fluoview 1000 Olympus confocal microscope with a 60× objective (NA 1.35 oil) with 2.5× zoom at 0.3 μm Z-steps through the entire NMJ. Confocal stacks were superimposed and flattened in Fluoview software (Olympus) and TIFFs analyzed in Image Pro Software (Media Cybernetics). Two complimentary features of Image Pro Software were used to quantify colocation of the bassoon-IR to α-bungarotoxin-AlexaFlour 555, in: (1) the colocalization coefficient, which estimates the contribution of one channel color (bassoon green-IR) to the overall colocalized fluorescence of the region-of-interest; and (2) the overlap coefficient of colocalization, which quantitates the overlap of the two colors. These methods are not sensitive to intensity variations in the image analysis. Quantification was always performed in original unprocessed images. Microscope images were prepared by adjusting contrast and brightness in ImagePro Plus Software (Media Cybernetics), with all information content of the images being preserved. Figures were composed using CorelDraw (v12.0). Graphs were composed in SigmaPlot 13 software (Systat Software). Some images were sharpened using a “high gauss” filter in ImagePro.
mRNA analysis.
Tibialis anterior, soleus and LAL muscles were removed from R6/2 and control mice, snap frozen in liquid nitrogen, and stored at −80°C until analyzed. Total RNA was isolated using RNeasy kits (Qiagen) and RNA stored at −80°C. Reverse-transcriptase PCR (RT-PCR) methods were used for the quantification of the following denervation-induced mRNAs: (1) the γ-subunit of nAChR (nAChRγ; Moss et al., 1987; Witzemann et al., 1987; Gu and Hall, 1988a,b; Adams et al., 1995; Liu et al., 2016); (2) the small-conductance calcium-activated potassium channel (SK3; Pribnow et al., 1999; Neelands et al., 2001; Jacobson et al., 2002; Favero et al., 2008); and (3) the cardiac muscle isoform of the voltage-gated sodium channel, Nav1.5 (Scn5a; Rogart et al., 1989; Lupa et al., 1995; Sekiguchi et al., 2012). Reverse transcription was performed using Superscript III reverse transcriptase and random primers according to the manufacturer's instructions (Invitrogen). The levels of nAChRγ mRNA were evaluated in two ways. First, a gel-based PCR method using primers flanking the following genes: nAChRγ (upstream: 5′-GGGCCGAGTGCTGGACCG-3′, downstream: 5′-GGCGGGGGTCTCCGGGGA-3′) and SK3 (upstream: 5′-CAAGTGTCCCTGTCCATCTTC-3′, downstream: 5′-CCAAGTGGTCATTGAGATTTA-3′). The 18S ribosomal subunit was also quantified via RT-PCR for control purposes with primers from Ambion. Second, the same nAChRγ primers were used for the relative quantification of nAChRγ in a real-time SYBR green-based RT-PCR (qPCR) procedure using the Full Velocity SYBR green kit (Stratagene). The mRNA levels of SK3 were quantified in a similar manner. The mRNA levels of Scn5a were quantified using a TaqMan-based assay (Mm01342518.m1, Applied Biosystems). RNA from 14 d denervated and control mouse gastrocnemius muscles and hindlimb muscle from postnatal day 1 mice were used as positive controls for denervation-induction (and developmental expression) of the denervation-sensitive genes. For real-time RT-PCR quantification, the cycle thresholds were obtained for β-2-microglobulin as a normalizing gene using a TaqMan-based assay (Mn00437762.m1, Applied Biosystems). The ΔΔCT method was used for calculation of relative expression levels (Livak and Schmittgen, 2001).
Statistics.
The means of two normal sample populations, as per the Shapiro–Wilk test, were compared using a two-tailed Student's t test using SigmaPlot 13 software (Systat Software). Any non-normal sample population means were compared using the Mann–Whitney rank sum test. Mean values were reported as mean ± SEM. A one-way ANCOVA model generated with Stata was used to evaluate normalized eEPC charges relative to position on the 0.5 Hz train for control and R6/2 groups, as well as between-group differences between the trends. We evaluated the position along the domain of eEPCs no. 5–60 in the train, which we centered at 0 for the analysis. This model can be expressed in terms of the general linear model (GLM) equation:
 |
where b0 represents eEPC charge of the control group at position = 5, b1 represents the difference between the control and R6/2 groups at position = 5, b2 represents the average change in eEPC charge per unit of position for the control group, and b3 represents the difference in this rate of change between the R6/2 and control groups. Significance of the regression model and each parameter was evaluated at the 0.05 α level. Significance of b0 indicates that the eEPC charge for the control group at a position of 5 is significantly different from 0. A significant b1 value indicates that the eEPC charge for the R6/2 group is significantly different from the control group at this initial position. A significant b2 value indicates that there is sufficient evidence to detect a nonzero change in eEPC charge with respect to position for the control group, and a significant b3 value indicates that the rate of change for the R6/2 group is significantly different from that in the control group. All p values were calculated using the number of mice.
Results
Skeletal muscle hyperexcitability
Our previous work using two-electrode voltage-clamp demonstrated that the flexor digitorum brevis and interosseous muscles in R6/2 mice become hyperexcitable due to decreases in the resting chloride and potassium currents early in the disease progression (Waters et al., 2013; Miranda et al., 2017). Because of the reduced resting currents, the membrane time constant (τm) of the R6/2 fibers was prolonged (Waters et al., 2013). Most of the increase in τm was due to decreased currents through the muscle chloride channel, ClC-1, which constitutes ∼80% of the resting muscle conductance (Hutter and Noble, 1960; Steinmeyer et al., 1991a,b). To confirm that passive membrane properties were similarly altered in the LAL muscle, we used current-clamp to determine the τm of late-stage R6/2 (4 male and 3 female mice, 81 fibers) and age-matched control fibers from wild-type littermates (6 male and 4 female mice, 87 fibers); all mice were 11–14 weeks of age (Fig. 1). These were the same motor endplates from which we measured neuromuscular transmission in physiological extracellular Ca2+ in subsequent figures. We injected an equal number of small positive and negative square current pulses into the motor endplate and recorded the resulting changes in the membrane potential (Fig. 1A). The striking increase in R6/2 fiber input resistance, indicated by the larger voltage responses, is known to occur because of the combined effect of an increase in the membrane resistance (due to reduced chloride and potassium conductance) and muscle fiber atrophy (Ribchester et al., 2004; Waters et al., 2013). To eliminate the influence of atrophy, we measured the τm, which is independent of fiber diameter, instead of the input resistance. To help ensure passive conditions, we estimated the τm from the steady-state voltage changes (ΔV) that were <|5| mV and had linear voltage–current relationships (Voss, 2009). We excluded values of ΔV that exceeded |5| mV from our τm calculations. τm was the time it took the membrane potential to reach 1/e of ΔV (∼37% of the steady-state change in voltage) during the return to baseline after a current pulse. The τm of R6/2 fibers (1.92 ± 0.06 ms) was significantly prolonged by 42% compared with that of control (1.25 ± 0.07 ms, p = 5.94 × 10−6, t test where t = 6.805 with 15 df), indicating that, consistent with the R6/2 flexor digitorum brevis, the LAL muscle fibers we used to measure neuromuscular transmission were hyperexcitable due to a loss of ClC-1 currents (Fig. 1B). The histogram in Figure 1C shows an approximately normal distribution of R6/2 and control τm values, indicating that R6/2 muscle fibers were uniformly hyperexcitable compared with control.
Figure 1.

Increased membrane time constant (τm) indicative of hyperexcitable R6/2 skeletal muscle fibers. A, Injected current steps (top traces) and the resulting membrane potential responses in representative control and R6/2 muscle fibers (bottom traces) in normal calcium. B, Average control and R6/2 τm. C, Histogram of control (solid black bars) and R6/2 (striped gray bars) τm. *Indicates a significant difference compared with control (p = 5.94 × 10−6, t test). Regions of overlap as black striped bars. Error bars are ±SEM.
Innervation
To determine whether the skeletal muscle fibers were anatomically denervated in late-stage R6/2 mice (12–13 weeks old) compared with control (12–13 weeks old), we stained the nerve terminals and motor endplates in whole-mount LAL preparations (Fig. 2). Nerve terminals were identified by staining for the neurofilaments and synaptic vesicles (both green). We identified the motor endplates based on the binding of α-bungarotoxin-rhodamine (red) to nAChRs. In both control (276 NMJs imaged from 3 mice) and R6/2 mice (274 NMJs imaged from 3 mice), we found no denervated endplates. This strongly suggests that the R6/2 skeletal muscle hyperexcitability was not caused by denervation.
Figure 2.

Representative images of stained control and R6/2 NMJs from 12-week-old mice. Motor nerves and terminals were identified by a combination of neurofilament staining (SMI-31) of axons and SV2b staining of synaptic vesicles (both in green; nerve). Motor endplates were labeled by staining of nicotinic acetylcholine receptors with rhodamine-conjugated α-bungarotoxin (red; nAChRs). The regions of superimposed nerve and nAChRs are yellow in the merged images. In both the control and R6/2 fibers, all nAChRs were directly aligned with nerve terminals. Scale bar: bottom right, 30 μm.
To further check for denervation, we tested for increased expression of nAChRγ, which is expressed during development, repressed in adult muscle, and upregulated following denervation (Hartzell and Fambrough, 1972; Lomo and Rosenthal, 1972; Lomo and Westgaard, 1975; Moss et al., 1987; Witzemann et al., 1987; Gu and Hall, 1988a,b; Adams et al., 1995; Kues et al., 1995; Liu et al., 2016). As shown in Figure 3A, neither R6/2 nor control expressed nAChRγ mRNA at appreciable levels in the tibialis anterior (TA) or soleus muscles. In contrast, the γ-subunit was readily detected in 14 d denervated mouse gastrocnemius muscle but not in a contralateral control. We also detected nAChRγ in postnatal day 1 hindlimb muscle (Fig. 3A). Furthermore, real-time RT-PCR (qPCR) assays demonstrated that nAChRγ mRNA was not elevated in R6/2 mouse TA (Fig. 3B; control: 1.000 ± 0.146; R6/2: 0.745 ± 0.249; p = 0.398, t test where t = 0.883 with 10 df) or soleus (Fig. 3B; control: 1.000 ± 0.503; R6/2: 0.321 ± 0.180; p = 0.289, t test where t = 1.147 with 7 df) muscle relative to control. Molecular analyses of the TA were completed with two male and four female R6/2 mice as well as five male and one female controls; one TA or soleus per mouse. For that of the soleus, each RNA sample was a pool of two mouse solei of the indicated genotype without regard to sex. In contrast to the R6/2 muscle, denervation of the mouse gastrocnemius (4 mice, each with denervated sample and contralateral control) caused a significant elevation in the γ-subunit mRNA (Fig. 3B; control: 1.000 ± 0.623; denervated: 4.527 ± 0.717; p = 0.010, t test where t = −3.715 with 6 df). We also found that the levels of SK3 mRNA, which are upregulated following denervation (Pribnow et al., 1999; Neelands et al., 2001; Jacobson et al., 2002; Favero et al., 2008), were not significantly changed in the TA (control: 1.000 ± 0.136; R6/2: 1.004 ± 0.229; p = 0.991, t test where t = −0.0115 with 10 df) or soleus (control: 1.000 ± 0.406; R6/2: 0.563 ± 0.196; p = 0.403, t test where t = 0.889 with 7 df). As expected, denervation of the mouse gastrocnemius caused a significant elevation in SK3 mRNA (control: 1.000 ± 0.335; denervated: 29.092 ± 3.566; p = 2.0 × 10−4, t test where t = −7.843 with 6 df). The expression of another positive marker for denervation, Scn5a, the cardiac isoform of the voltage gated sodium channel (Rogart et al., 1989; Yang et al., 1991; Lupa et al., 1995; Rich et al., 1999; Sekiguchi et al., 2012), was significantly reduced in the TA (control 1.000 ± 0.123; R6/2 0.507 ± 0.141; p = 0.025, t test where t = 2.630 with 10 df) and unchanged in the soleus (control: 1.000 ± 0.243; R6/2: 0.422 ± 0.130; p = 0.094, t test where t = 1.934 with 7 df) of R6/2 muscle. As shown for the other denervation-sensitive genes, denervation of the mouse gastrocnemius caused a significant elevation in Scn5a mRNA (Fig. 3B; control: 1.000 ± 0.278; denervated: 44.209 ± 5.053; p = 1.0 × 10−4, t test where t = −8.538 with 6 df). There were also no molecular markers of denervation in the LAL muscle. The same qPCR examination of control (2 male and 2 female mice) and R6/2 LAL (1 male and 4 female mice) showed that nAChRγ (control: 1.000 ± 0.446; R6/2: 0.572 ± 0.228; p = 0.392, t test where t = 0.912 with 7 df), SK3 (control: 1.000 ± 0.288; R6/2: 0.554 ± 0.266; p = 0.294, t test where t = 1.133 with 7 df), and Scn5a (control: 1.000 ± 0.467; R6/2: 0.193 ± 0.085; p = 0.097, t test where t = 1.914 with 7 df) were not significantly different.
Figure 3.

RT-PCR analyses of the expression of denervation-induced gene mRNAs in R6/2 and control mice. A, Gel-based RT-PCR for the expression of nAChRγ. Neither control nor R6/2 tibialis anterior nor soleus muscles showed expression of nAChRγ. In contrast, expression of nAChRγ was detectable in denervated (Dnv), but not control (Cont) mouse gastrocnemius (Gast) muscle. Also, expression of nAChRγ was observed in postnatal day 1 (P1) hindlimb muscles (Hind) of mice. The M designates base-pair size markers. B, Real-time RT-PCR analyses of nAChRγ, the SK3, and Scn5a mRNAs relative to β2-microglobulin in control and R6/2 mice. No significant changes in the mRNAs for nAChRγ and SK3 were observed in R6/2 mouse tibialis anterior (left; nAChRγ: p = 0.398, t test; SK3: p = 0.991, t test) and soleus (middle; nAChRγ: p = 0.289, t test; SK3: p = 0.403, t test) muscle compared with control. There was a significant decrease in the levels of Scn5a in R6/2 tibialis anterior (p = 0.025, t test) but not soleus muscle (p = 0.094, t test) relative to control. In contrast, there was a marked increase in the expression of nAChRγ (p = 0.0099, t test), SK3 (p = 0.0002, t test), and Scn5a (p = 0.0001, t test) mRNA in denervated gastrocnemius relative to the innervated contralateral muscle in control mice (n = 4 muscles/group). *Indicates a significant difference. Error bars are ±SEM.
Neuromuscular transmission in low extracellular Ca2+
Having determined that the hyperexcitability of R6/2 muscle fibers was independent of denervation, we looked for changes in late-stage R6/2 neuromuscular transmission relative to control. In ex vivo preparations of the LAL, we impaled two microelectrodes in rostral muscle fibers and voltage-clamped the motor endplate. Unlike the changes in muscle membrane potential recorded under current-clamp, voltage-clamped synaptic currents are independent of the muscle membrane properties. In a reduced extracellular Ca2+ (0.5 mm) and elevated Mg2+ (4 mm) solution, we stimulated the motor nerve at 0.5 Hz and recorded eEPCs and spontaneous mEPCs in control (3 male and 4 female mice, 46 fibers) and R6/2 (4 male and 5 female mice, fibers 80) mice (Fig. 4A). The frequency of mEPCs was significantly less in R6/2 (1.8 ± 0.3 Hz) than control (2.4 ± 0.2 Hz) NMJs (p = 0.02, t test where t = 2.703 with 14 df). Otherwise, we found no significant differences in mEPC charge (control: −2.76 ± 0.13 pC; R6/2: −2.63 ± 0.07 pC; p = 0.38, t test where t = −0.908 with 14 df) or eEPC charge (control: −3.73 ± 0.40 pC; R6/2: −4.52 ± 0.35 pC; p = 0.16, t test where t = 1.48 with 14 df) at R6/2 NMJs compared with control (Fig. 4B). We also found no significant difference in the QC, which is the number of vesicles released per action potential (eEPC charge/mEPC charge), at R6/2 NMJs (1.69 ± 0.10) compared with control (1.38 ± 0.16, p = 0.11, t test where t = 1.707 with 14 df; Fig. 4B). The findings in low Ca2+ suggest that synaptic transmission was normal in R6/2 muscle.
Figure 4.

The mEPC charge, eEPC charge, and QC in control and R6/2 NMJs in low extracellular Ca2+ (0.5 mm) and high Mg2+ (4 mm). A, Superimposed representative eEPC traces from control and R6/2 NMJs with an inset of superimposed representative mEPC traces from the same NMJs under two-electrode voltage-clamp. B, Histograms of average control (solid black bars) and R6/2 (striped gray bars) mEPC charge, eEPC charge, and QC. Regions of overlap as black striped bars.
Neuromuscular transmission in physiological extracellular Ca2+
To obtain a more complete picture of neuromuscular transmission in R6/2 skeletal muscle, we measured endplate currents in physiological extracellular Ca2+ (1.2 mm) and Mg2+ (0.6 mm) in the same fibers used to determine the membrane time constant. With muscle contractions blocked using 1 μm μ-CTX and 60 μm BTS, we stimulated the motor nerve at 0.5 Hz and recorded eEPCs and mEPCs in control and R6/2 mice for 2 min (Fig. 5A–D). The average mEPC frequency at R6/2 NMJs was significantly lower than in controls (Table 1). Additionally, there were significant reductions in the average R6/2 mEPC and eEPC charge compared with controls (Fig. 5E,F; Table 1). On average, R6/2 NMJs released 19% fewer vesicles per impulse than control terminals (QC in Fig. 5G and Table 1). That the R6/2 mEPC and eEPC charge were not significantly different from control in low Ca2+ suggests the defects in R6/2 neuromuscular transmission depend on Ca2+. Thus, it seems unlikely that impaired vesicle loading or the recently identified vesicle morphology changes (de Aragão et al., 2016) could explain the reduced R6/2 mEPC and eEPC in physiological Ca2+.
Figure 5.

The mEPC charge, eEPC charge, and QC in control and R6/2 NMJs in physiological extracellular Ca2+ (1.2 mm) and Mg2+ (0.6 mm). A, B, Representative control (A) and R6/2 (B) recordings under two-electrode voltage-clamp showing mEPCs and eEPCs (baseline adjusted to 0). C, D, Superimposed representative eEPC traces from control (C) and R6/2 (D) NMJs with an inset of superimposed mEPC traces from the same NMJs. E–G, Histograms of average control (solid black bars) and R6/2 (striped gray bars) mEPC charge (E), eEPC charge (F), and QC (G). Regions of overlap as black striped bars.
Table 1.
mEPC and eEPC properties in physiological calcium conditions
| mEPC |
eEPC |
QC | ||||||
|---|---|---|---|---|---|---|---|---|
| Charge, pC | Rise time, ms | Decay τ, ms | Frequency, Hz | Charge, pC | Rise time, ms | Decay τ, ms | ||
| Control | −4.1 ± 0.1 | 0.33 ± 0.01 | 0.94 ± 0.04 | 1.5 ± 0.1 | −261 ± 8 | 0.43 ± 0.01 | 1.24 ± 0.04 | 65 ± 6 |
| R6/2 | −3.4 ± 0.1* | 0.37 ± 0.01* | 1.16 ± 0.02* | 0.9 ± 0.1* | −179 ± 7* | 0.47 ± 0.01* | 1.50 ± 0.04* | 54 ± 5* |
| p value | 1.4 × 10−4 | 3.34 × 10−5 | 8.36 × 10−4 | 1.9 × 10−4 | 2.00 × 10−6 | 2.11 × 10−4 | 2.6 × 10−4 | 0.022 |
| t value | 5.06 | −5.83 | 4.16 | −4.92 | 7.47 | −4.85 | 4.75 | −2.61 |
| df | 15 | 15 | 15 | 15 | 15 | 15 | 15 | 15 |
Summary of NMJ data from 81 R6/2 fibers (7 mice) and 87 control fibers (10 age-matched mice). The eEPC rise time was calculated from 10% to 90% of peak.
*Indicates a significant difference compared to control. A two-tailed t test was applied in all measurements.
To determine whether the lower QC in R6/2 NMJs was related to synaptic depression, we examined the fractional amplitude of the eEPCs during the 0.5 Hz trains (Fig. 6A). The peak amplitude of each eEPC was normalized to the peak of the largest eEPC in the train. Following a transient initial depression, the fractional eEPC amplitudes in control NMJs appeared unchanging or even slightly increasing, whereas the eEPC amplitudes at R6/2 NMJs appeared to depress slightly over the span of 60 stimulations (Fig. 6A). To assess the potential synaptic modulation quantitatively, we examined eEPCs 5–60, which occurred after the initial transient depression, with a GLM (Equation 6). This model fit the data significantly better than using the mean as a predictor (F(3,108) = 125.5, p = 4.6 × 10−35, r2adj = 0.77). Figure 6B shows the linear fit of the normalized control and R6/2 eEPCs with 95% confidence intervals. The slight depression in R6/2 eEPCs (−5.2 × 10−4 ± 7.8 × 10−5 EPC amplitude/nerve stimuli) was significantly different from that of control (2.6 × 10−4 ± 5.7 × 10−5 eEPC amplitude/nerve stimuli; b3 = −7.8 × 10−4, SE = 9.7 × 10−5, p = 1.4 × 10−12, t = −8.02). To minimize the influence of this depression, we also determined the QC based on eEPCs 5–24, which occurred after the transient depression and early in the R6/2 depression. In this case, the R6/2 quantal content (53.9 ± 7.0) was still significantly lower than control (64.8 ± 3.1, p = 0.022, Mann−Whitney rank sum test where U = 11). Furthermore, the R6/2 QC based only on the first eEPC (58.5 ± 1.9) was significantly less than that of control (68.1 ± 3.3; p = 0.0097, Mann–Whitney rank sum test where U = 8).
Figure 6.

Synaptic modulation at low-frequency stimulation (0.5 Hz). A, Average control (10 mice) and R6/2 (7 mice) eEPC charges normalized to the peak eEPC charge in a train of 60 eEPCs stimulated at 0.5 Hz. B, Normalized eEPCs (postion 5–60 in train) from A showing the linear regression for control (solid black line) and R6/2 (solid gray line). Data are shown with the 95% confidence interval for each linear regression (control shaded black and R6/2 shaded gray). Error bars are ±SEM.
We also analyzed the kinetics of mEPC and eEPC recorded in physiological Ca2+ and Mg2+ (Fig. 7; Table 1). Figure 7 shows the average mEPC (Fig. 7A) and eEPC (Fig. 7B) from representative control (solid lines) and R6/2 (dashed lines) fibers, each normalized to the peak. The average rise and decay times of the R6/2 mEPCs and eEPCs were prolonged compared with control (Fig. 7C,D; Table 1).
Figure 7.

Kinetics of control and R6/2 mEPCs and eEPCs. A, B, Average mEPC (A) and eEPC (B) from representative control (solid lines) and R6/2 (dashed lines) NMJs, each normalized to peak (peak value set to 1 for each endplate current). C, D, Histograms of the control (solid black bars) and R6/2 (striped gray bars) mEPC decay constants (C) and eEPC decay constants (D). Regions of overlap as black striped bars.
Understanding the contribution of changes in neuromuscular transmission to motor function will be aided by knowing the resting membrane potential. Previous studies have shown that the resting membrane potential of R6/2 muscle fibers was depolarized relative to control (Ribchester et al., 2004), but that of R6/1 fibers, a different HD mouse model, was not significantly different from control (Rozas et al., 2011). Our use of relatively blunt electrodes was required for voltage-clamp experiments but prevented reliable estimates of the resting membrane potential. Thus, as we have done previously (Novak et al., 2015), we used single sharp electrodes that minimize membrane damage to record the resting membrane potential of extensor digitorum longus fibers from control (1 male and 2 female mice, 31 fibers) and R6/2 (1 male and 2 female mice, 30 fibers) mice. The resting membrane potential of R6/2 (−76.6 ± 0.7 mV) was significantly depolarized compared with control (−82.4 ± 0.5 mV) fibers (p = 0.007, t test where t = −5.078 with 4 df).
Neuromuscular transmission under voltage- and current-clamp
Our data showing depressed neuromuscular transmission in HD differs from other studies that used current-clamp recordings (Romero et al., 2008; Rozas et al., 2011). The previous studies were completed before our reports describing the reduced membrane conductances and capacitance in HD skeletal muscle (Waters et al., 2013; Miranda et al., 2017). Given our work on the hyperexcitability of R6/2 fibers, we hypothesized that current-clamping the endplate in HD would yield different and potentially misleading results compared with voltage-clamp. To test this, we measured mEPPs and eEPPs from a subset of R6/2 (3 male and 3 female mice, 64 fibers) and control NMJs (2 male and 2 female mice, 36 fibers) that we also used to obtain voltage-clamp measurements. The mEPPs and eEPPs were normalized (mEPPnorm and eEPPnorm) for changes in the resting membrane potential and the evoked potentials were further corrected for nonlinear summation (eEPPcorr) by replicating the method used for studying the NMJ in R6/1 mice (Rozas et al., 2011). We found that the mEPPnorm from R6/2 fibers (1.29 ± 0.07 mV) tended to be larger than in control fibers (1.08 ± 0.04 mV), but did not quite reach our cutoff for statistical significance (Fig. 8A; p = 0.071, t test where t = −2.08 with 8 df). The R6/2 eEPPcorr (58.1 ± 3.2 mV) were not significantly different from control (54.7 ± 1.4 mV; Fig. 8B; p = 0.45, t test where t = −0.08 with 8 df). These results were in stark contrast to the voltage-clamp measurements obtained from the same fibers, in which the average R6/2 mEPC charge and eEPC charge were significantly smaller compared with control (Fig. 8A,B). Estimates of quantal content tended to be lower from current-clamp recordings (QCIC) than voltage-clamp (QC), which is consistent with previous work that examined neuromuscular transmission under both conditions (Wood and Slater, 1997). It happened that the estimates of QC were lower in R6/2 fibers compared with control in both the current and voltage-clamp recordings. These results indicate that the nonlinearity corrections (McLachlan and Martin, 1981) commonly used in current-clamp studies of synaptic transmission are not easily applicable to disease models with disrupted postsynaptic membrane properties.
Figure 8.

Comparison of NMJ transmission data collected under voltage-clamp (mEPC and eEPC) and current-clamp (mEPPnorm and eEPPcorr) from the same control (cntrl) and R6/2 fibers. A, mEPCs (p = 0.003, t test), but not mEPPnorm (p = 0.071, t test), were significantly lower in R6/2 compared with control fibers. B, Similarly, eEPCs (p = 6.4 × 10−5, t test), but not eEPPcorr (p = 0.45, t test), were significantly lower in R6/2 compared with control fibers. C, Estimates of quantal content from voltage-clamp (QC) and current-clamp (QCIC) were lower for R6/2 compared with control (p = 0.008 and p = 0.02, respectively, t test). *Indicates a significant difference compared with control NMJs. Error bars are ±SEM.
Estimates of prel and n
To determine whether the decrease in QC was due to a reduction in the probability of vesicle release (prel) or the number of functional vesicle release sites (n), we used a previously described graphical method based on binomial statistics (Clements and Silver, 2000; Wang et al., 2010) to estimate prel and n with data collected in physiological Ca2+ and Mg2+(Fig. 9). To minimize the contribution of depression in the R6/2 NMJs, we determined prel and n from evoked potentials 5–24 elicited at 0.5 Hz (Fig. 6). Briefly, we plotted experimental values of QC and Var(QC) on two superimposed models of QC and Var(QC). One model shows QC and Var(QC) if prel was varied and n was held constant (parabolas). In this model, Var(QC) = 0 if no vesicles are released (prel = 0) or if vesicles are released from all available sites (prel = 1); the maximum Var(QC) occurs when prel = 0.5. For reference, we show parabolas for n = 5, 10, 20, 40, 60, and 80. We estimated n using experimental QC and Var(QC) values and Equation 5 in Materials and Methods. The intersecting straight lines of the other model show QC and Var(QC) if n was varied and prel was constant. We determined prel using experimental QC values and estimates of n using the relationship of QC = np. As shown in Figure 9, the estimated n for R6/2 NMJs (52.3 ± 3.0 release sites) was trending lower than control (58.9 ± 2.6 release sites, p = 0.12, t test where t = 1.662 with 15 df). Estimates of prel were similar for R6/2 (0.81 ± 0.03) and control (0.84 ± 0.01) NMJs (p = 0.31, Mann–Whitney rank sum test, where U = 24). These results suggest that reductions in n contribute more to the lower R6/2 QC than reductions in p.
Figure 9.

Var(QC) versus QC plot for estimating the probability of release (prel) and number of vesicle release sites (n). Experimental Var(QC) and QC data superimposed on two theoretical models (parabolas and straight lines). The parabolas model Var(QC) and QC while varying prel and holding n constant. Straight lines model Var(QC) and QC while varying n and holding prel constant. The lower QC (vesicle release per impulse) in R6/2 compared with control fibers was primarily due to a lower n. Error bars are ±SEM.
Neuromuscular morphology
A reduction in n could arise from alterations in NMJ morphology. For example, a loss of active sites could be caused by atrophy of R6/2 NMJs, which has been shown to occur in proportion to the muscle atrophy (Ribchester et al., 2004). We examined NMJ morphology in whole-mount LAL preparations from one male and four female late-stage R6/2 mice (63 fibers) and three male and three female controls (70 fibers). Motor endplates were visualized based on the binding of AlexaFluor 555-conjugated α-bungarotoxin to nAChRs. Vesicle release sites were identified by immunostaining of the protein bassoon, a widely used marker for active zones in the synapses of central and peripheral tissues (Dieck et al., 1998; Nishimune et al., 2004; Spaulding et al., 2016). Nearly every R6/2 NMJ and all control NMJs examined had a normal “pretzel-like” morphology with bassoon punctae in apposition to nAChRs and no obvious fragmentation (Fig. 10A,B). One exception was an R6/2 NMJ with clear bassoon immunoreactivity that juxtaposed an endplate with only partial coverage of nAChRs. Other studies have shown abnormalities in a small fraction of HD NMJs (Ribchester et al., 2004; de Aragão et al., 2016). However, the decrease in average R6/2 n would need to be explained by a consistent or uniform NMJ change.
Figure 10.

Comparison of R6/2 and control NMJs stained for postsynaptic nAChRs and presynaptic bassoon proteins. A, B, Representative control (A) and R6/2 (B) NMJs stained for nAChRs on the motor endplate (red, left), bassoon in the motor nerve terminals (green, middle), and the merge image of the nAChR and bassoon staining (right). nAChRs were stained with α-bungarotoxin-AlexaFluor 555 and bassoon was detected by immunofluorescence. C, Total endplate area for control (black bar) and R6/2 (gray bar) NMJs determined by circumscribing the nAChR staining region. D, The percentage of the control (black bar) and R6/2 (gray bar) endplate occupied by nAChR. This estimate of synaptic area was significantly less in R6/2 compared with control NMJs (p = 0.01, t test). E, The overlap coefficient of bassoon to nAChR in control (black bar) and R6/2 (gray bar) NMJs. A value of 1 indicates that each pixel of bassoon staining (green) colocalized with red pixels (nAChR). *Indicates a significant difference compared with control NMJs. Error bars are ±SEM.
To quantitate NMJ morphology, the endplate area was determined by circumscribing the semicircular/elliptical nAChR staining region (Fig. 10A,B, left). The average R6/2 endplate area (357 ± 45 μm2) trended 23% smaller than that of control (463 ± 37 μm2) (Fig. 10C; p = 0.10, t test where t = 1.82 with 9 df). Additionally, the fraction of the endplate occupied by nAChR (% synaptic area) was significantly less in R6/2 (50.9 ± 3.9%) than control (63.2 ± 1.4%) fibers (Fig. 10D; p = 0.01, t test where t = 3.20 with 9 df). These percentages correspond to a 38% reduction in actual synaptic area of R6/2 endplates (182 μm2) compared with control (293 μm2). To assess the relationship between bassoon and nAChRs, we estimated the overlap coefficient, an intensity independent parameter that was 1.0 if every green pixel (bassoon) colocalized with a red pixel (nAChR). The overlap coefficients of R6/2 (0.98 ± 0.01) and control (0.99 ± 0.01) NMJs were not significantly different (Fig. 10E; p = 0.66, Mann–Whitney rank sum test with U = 12). Also, the colocalization coefficient, which indicates the contribution of bassoon to the colocalized regions, was not significantly different in R6/2 (0.39 ± 0.13) compared with control (0.42 ± 0.12) NMJs (p = 0.86, Mann–Whitney rank sum test with U = 12). In summary, bassoon tightly colocalized with nAChR in control and R6/2 NMJs and the decrease in vesicle release sites suggested by the binomial statistics appears to be explained by a reduction in the synaptic area of R6/2 NMJs.
High-frequency stimulation
Normal muscle function requires that motor neurons release vesicles during trains of stimulation. To assess the ability of R6/2 NMJs to maintain higher frequency trains of activity, we stimulated the motor nerve with 10 trains at 10 s intervals. Each train consisted of 10 stimulations in 200 ms (50 Hz). We measured the trains in physiological Ca2+ and Mg2+ in the same fibers we used to assess muscle hyperexcitability and QC. Figure 11 shows eEPCs recorded during single trains in control (Fig. 11A) and R6/2 (Fig. 11B) fibers. To assess the modulation of neuromuscular transmission during activity, we divided each eEPC amplitude by the maximum eEPC in a train to obtain a normalized eEPC amplitude (Fig. 11C). During high-frequency trains, the overall degree of depression (last eEPC/first eEPC in a train) was significantly greater in R6/2 NMJs (0.71 ± 0.02) compared with control (0.83 ± 0.01, p = 2.6 × 10−4, t test where t = 4.74 with 14 df); note, a lower value is indicative of greater depression.
Figure 11.
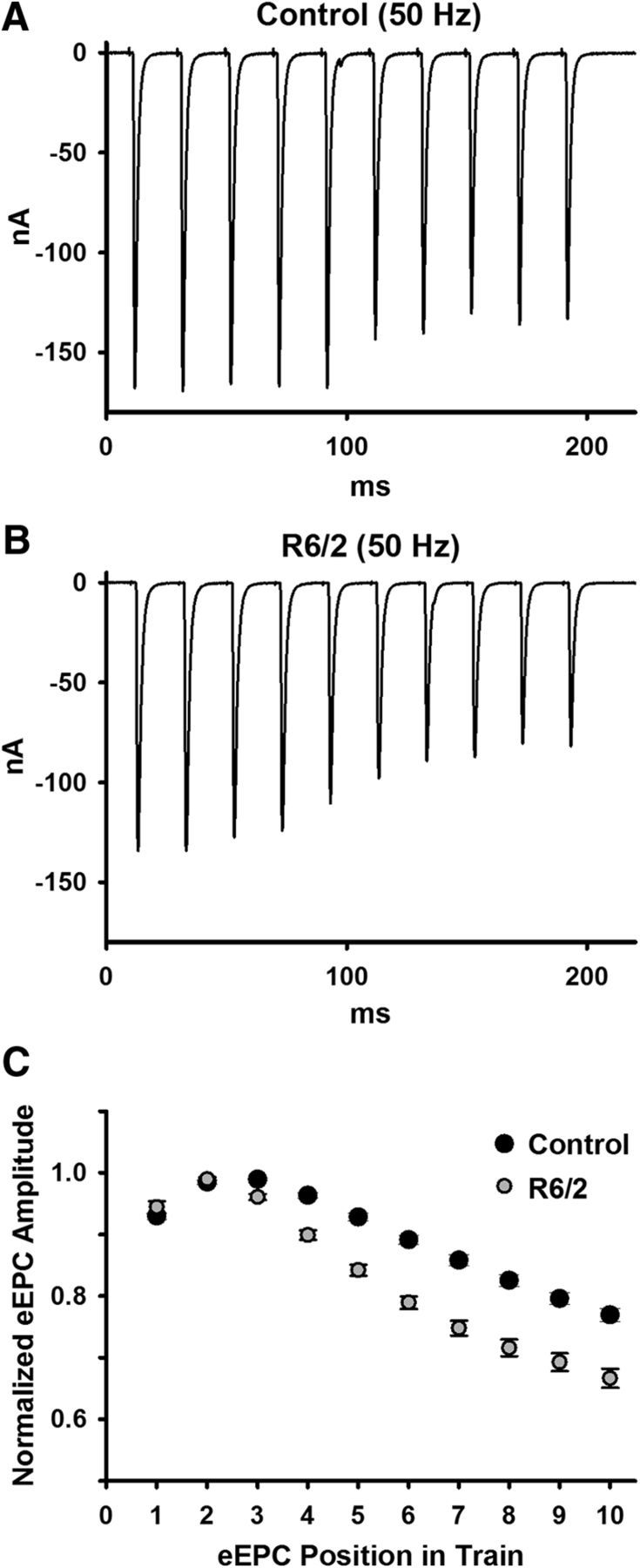
eEPC depression during high-frequency (50 Hz) stimulation trains. A, B, Representative raw traces for control (A) and R6/2 (B) NMJs. C, Average control (10 mice) and R6/2 (7 mice) eEPC amplitudes normalized to the peak eEPC amplitude in a train of 10 eEPCs at 50 Hz stimulation. Error bars are ±SEM.
Discussion
R6/2 muscle innervation
Previous work reported that atrophied R6/2 skeletal muscle had an increased sensitivity to acetylcholine and anode-break action potentials, which are characteristics of denervation (Ribchester et al., 2004). They described the phenotype as “denervation-like” because they found no evidence of physical denervation and observed minor morphological changes in only a small fraction of R6/2 NMJs. Nonetheless, a more recent study reported motor unit loss and denervation in R6/2 muscle based on changes in twitch tension in response to stimuli of increasing strength (Mielcarek et al., 2015). It was further proposed in the same study that the hyperexcitability in R6/2 skeletal muscle was the result of denervation. Considering that denervation is known to cause skeletal muscle atrophy and hyperexcitability (Albuquerque and McIsaac, 1970; Thesleff and Ward, 1975), it was important to further examine whether denervation occurs in HD.
We found that all of the postsynaptic sites were adjoined to presynaptic terminals in R6/2 muscle, tight bassoon and nAChR colocalization, and that the expression level of nAChRγ, SK3, and Scn5a, all of which upregulate upon denervation, were normal or downregulated in R6/2 muscle. Also, if R6/2 muscle were denervated, we would expect to find a mixed population of fibers with distinct excitability and NMJ characteristics, as is seen in other motor neuron diseases that display denervation/reinnervation patterns, such as ALS (Rocha et al., 2013). However, we found that the hyperexcitability of the fibers and the characteristics of neuromuscular transmission were normally distributed.
Perhaps the examination of R6/2 motor unit number (Mielcarek et al., 2015) was influenced by motor neuron hyperexcitability; their method of quantifying motor units assumes that motor nerve excitability is the same in disease and control muscle. Supporting this possibility, hyperexcitability or decreased potassium currents have been reported in medium spiny neurons and astrocytes in mouse models of HD (Ariano et al., 2005; Rocha et al., 2013; Tong et al., 2014).
This study and the results from Ribchester et al. (2004) show that denervation is not occurring or only develops in an extremely small number of R6/2 muscle fibers. The “denervation-like” effects found in R6/2 muscle likely arise from decreases in the resting chloride and potassium currents that coincide with aberrant mRNA processing (Waters et al., 2013; Miranda et al., 2017).
Voltage-clamp analysis of neuromuscular transmission
Because the muscle membrane potential does not change during our voltage-clamp recordings, mEPCs and eEPCs are independent of the muscle membrane properties. Previous studies of HD neuromuscular transmission, mostly completed before we showed the disrupted R6/2 muscle membrane properties, used current-clamp recordings (Ribchester et al., 2004; Romero et al., 2008; Rozas et al., 2011; de Aragão et al., 2016). One study corrected for nonlinearities using methods that assume the postsynaptic membrane properties are unaltered, including a correction factor for membrane capacitance (Rozas et al., 2011). To assess the effect of the altered R6/2 muscle membrane conductances and capacitance (Waters et al., 2013; Miranda et al., 2017) on the corrected data, we measured neuromuscular transmission in the same fibers under current and voltage-clamp. In voltage-clamp, the spontaneous and evoked responses in R6/2 fibers were decreased relative to control, whereas under current-clamp they were larger. Thus, because of the altered muscle membrane properties, reduced synaptic transmission may actually appear enhanced in current-clamp records. These results highlight the need to account for the postsynaptic membrane properties in synaptic studies and, if possible, record the endplate currents in voltage-clamp. Such voltage-clamp studies may be exceedingly difficult or impossible with central synapses, but can be achieved in large monosynaptic muscle fibers.
Neuromuscular transmission in HD
Under conditions of low Ca2+ and high Mg2+, we found no clear impairments in R6/2 neuromuscular transmission. If anything, the trend was for larger R6/2 eEPCs and QC compared with control in the low Ca2+ conditions. Thus, under conditions in which vesicle release and the need for vesicle mobilization are minimized, synaptic transmission in R6/2 muscle is normal.
However, in normal extracellular Ca2+ and Mg2+, the mEPC and eEPC charge are reduced in R6/2 compared with controls NMJs. Additionally, the mEPCs and eEPCs are prolonged in R6/2 fibers. A decrease in acetylcholinesterase function would explain the prolonged mEPC and eEPC kinetics but is contradicted by the decrease in mEPC and eEPC charge. A change in the nAChR isoform seems unlikely because we found no change in the expression level of nAChRγ. Moreover, a previous study found no change in the density of nAChRs in R6/2 NMJs (Ribchester et al., 2004). Possible explanations for the mEPC and eEPC properties are impaired vesicle fusion and posttranslational modification of nAChRs.
Our analysis of eEPC variance suggests a reduction in the number of R6/2 vesicle release sites (n), which we found coincides with a decrease in the synaptic area of R6/2 NMJ. At first pass, a decrease in the physical number of active sites may not seem compatible with our observed dependence of QC on Ca2+ (normal QC in low but reduced in normal Ca2+). However, recent reports show that n is dependent on Ca2+ (Wang et al., 2010), indicating that n also depends on physiological factors such as vesicle mobilization.
Our data showing depression in neuromuscular transmission under trains of stimulation, even at 0.5 Hz stimulation, indicate impaired vesicle mobilization in HD. Other neuromuscular studies using FM styryl dyes have produced conflicting results, with no changes in vesicle recycling reported in R6/1 mice (Rozas et al., 2011) and depressed vesicle exocytosis reported for BACHD mice (de Aragão et al., 2016). Supporting defects in vesicle mobilization, the huntingtin protein is thought to interact with clathrin-coated vesicles and proteins involved in vesicle trafficking (Velier et al., 1998). Furthermore, a study on CNS synapses in HD found that R6/2 mice demonstrated a progressive loss of complexin II, a known SNARE complex-binding protein believed to be involved in exocytosis modulation and vesicle release synchronization (Morton and Edwardson, 2001; Morton et al., 2001; Freeman and Morton, 2004; Lin et al., 2013). Thus, a decrease in complexin activity could result in an apparent decrease in functional active sites as vesicle fusion is stimulated.
The neuromuscular depression in late-stage HD muscle likely depends on disease progression and may compensate for the muscle hyperexcitability. Similarly, the depolarized resting membrane potential may further counter the muscle hyperexcitability. Previous studies using the R6/1 mouse and Drosophila models of HD at an earlier stage of the disease show increased neuromuscular transmission (Romero et al., 2008; Rozas et al., 2011). These results cannot be fully explained by their use of current-clamp instead of voltage-clamp. For example, we found a significantly lower mEPC frequency in late-stage R6/2 muscle, which is consistent with a previous current-clamp study (Ribchester et al., 2004) and is independent of using voltage- or current-clamp. In comparison, the frequency of mEPPs was not significantly different in early-stage HD muscle from R6/1, R6/2, and BACHD mice as well as Drosophila (Ribchester et al., 2004; Romero et al., 2008; Rozas et al., 2011; de Aragão et al., 2016). A time-course study of the NMJ, with voltage-clamp recordings, will be necessary to determine whether HD neuromuscular transmission transitions from elevated to depressed during disease progression. Such studies can help us determine whether the depressed neuromuscular transmission we observed in late-stage R6/2 compensates for the muscle hyperexcitability. This would be supported by NMJ depression beginning after the onset of the muscle membrane defects, which we have shown begins at or before 5 weeks of age in R6/2 mice (Miranda et al., 2017). Similarly, enhanced neuromuscular transmission in young R6/2 muscle, as observed in Drosophila larva (Romero et al., 2008), would support nerve-to-muscle effects.
It has become increasingly accepted that HD is a multisystem disorder. Defects in both the brain and skeletal muscle likely contribute to the motor symptoms, perhaps in a vicious cycle. Adding to this, the defects we found in neuromuscular transmission could arise from mutant huntingtin expression in motor neurons. The NMJ is an excellent model to examine the interplay between neuronal and muscle defects in HD. Furthermore, our use of voltage-clamp to measure neuromuscular transmission provides the most detailed analysis of synaptic transmission in HD to date and can serve as a model for the less accessible multisynaptic networks of the CNS. Clinically, the striking depression we observed in R6/2 neurotransmission, even at 0.5 Hz stimulation, could help explain the motor impersistence experienced by many HD patients (Gordon et al., 2000; Reilmann et al., 2001). Time-course studies, although resource-intensive, could help reveal an underlying primary defect in the muscle, motor neuron, or CNS that drives secondary defects in the other systems. Perhaps more likely, the widespread expression of mutant huntingtin causes independent and subsequently interacting defects in multiple tissues. Regardless, therapies that enhance neuromuscular transmission or reduce muscle hyperexcitability represent a largely unconsidered class of agents that may lessen the motor symptoms in late-stage HD patients.
Footnotes
This work was supported by NIH/NIGMS Grant 1SC3GM096945 and Wright State University startup funds (A.A.V.) as well as NIH/NINDS Grant R01082354 (M.M.R.). We thank Lori Goss for surgical assistance with the denervation studies, Dr. Kevin Novak for work with the extensor digitorum longus muscle, Steven Burke for assistance with the levator auris longus dissections, and Dr. Xueyong Wang for technical advice. Postnatal day 1 muscle samples were kindly provided by Dr. Theodore Garland Jr of the University of California, Riverside.
The authors declare no competing financial interests.
References
- Adams L, Carlson BM, Henderson L, Goldman D (1995) Adaptation of nicotinic acetylcholine receptor, myogenin, and MRF4 gene expression to long-term muscle denervation. J Cell Biol 131:1341–1349. 10.1083/jcb.131.5.1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albuquerque EX, McIsaac RJ (1970) Fast and slow mammalian muscles after denervation. Exp Neurol 26:183–202. 10.1016/0014-4886(70)90099-3 [DOI] [PubMed] [Google Scholar]
- Ariano MA, Cepeda C, Calvert CR, Flores-Hernández J, Hernández-Echeagaray E, Klapstein GJ, Chandler SH, Aronin N, DiFiglia M, Levine MS (2005) Striatal potassium channel dysfunction in Huntington's disease transgenic mice. J Neurophysiol 93:2565–2574. 10.1152/jn.00791.2004 [DOI] [PubMed] [Google Scholar]
- Bhide PG, Day M, Sapp E, Schwarz C, Sheth A, Kim J, Young AB, Penney J, Golden J, Aronin N, DiFiglia M (1996) Expression of normal and mutant huntingtin in the developing brain. J Neurosci 16:5523–5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse ME, Hughes G, Wiles CM, Rosser AE (2008) Use of hand-held dynamometry in the evaluation of lower limb muscle strength in people with Huntington's disease. J Neurol 255:1534–1540. 10.1007/s00415-008-0964-x [DOI] [PubMed] [Google Scholar]
- Clements JD, Silver RA (2000) Unveiling synaptic plasticity: a new graphical and analytical approach. Trend Neurosci 23:105–113. 10.1016/S0166-2236(99)01520-9 [DOI] [PubMed] [Google Scholar]
- de Aragão BC, Rodrigues HA, Valadão PA, Camargo W, Naves LA, Ribeiro FM, Guatimosim C (2016) Changes in structure and function of diaphragm neuromuscular junctions from BACHD mouse model for Huntington's disease. Neurochem Int 93:64–72. 10.1016/j.neuint.2015.12.013 [DOI] [PubMed] [Google Scholar]
- Ehrnhoefer DE, Skotte NH, Ladha S, Nguyen YT, Qiu X, Deng Y, Huynh KT, Engemann S, Nielsen SM, Becanovic K, Leavitt BR, Hasholt L, Hayden MR (2014) p53 increases caspase-6 expression and activation in muscle tissue expressing mutant huntingtin. Hum Mol Genet 23:717–729. 10.1093/hmg/ddt458 [DOI] [PubMed] [Google Scholar]
- Favero M, Jiang DJ, Chiamulera C, Cangiano A, Fumagalli GF (2008) Expression of small-conductance calcium-activated potassium channels (SK3) in skeletal muscle: regulation by muscle activity. J Physiol 586:4763–4774. 10.1113/jphysiol.2008.156588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman W, Morton AJ (2004) Regional and progressive changes in brain expression of complexin II in a mouse transgenic for the Huntington's disease mutation. Brain Res Bull 63:45–55. 10.1016/j.brainresbull.2003.12.004 [DOI] [PubMed] [Google Scholar]
- Gordon AM, Quinn L, Reilmann R, Marder K (2000) Coordination of prehensile forces during precision grip in Huntington's disease. Exp Neurol 163:136–148. 10.1006/exnr.2000.7348 [DOI] [PubMed] [Google Scholar]
- Huntington's Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72:971–983. 10.1016/0092-8674(93)90585-E [DOI] [PubMed] [Google Scholar]
- Gu Y, Hall ZW (1988a) Characterization of acetylcholine receptor subunits in developing and in denervated mammalian muscle. J Biol Chem 263:12878–12885. [PubMed] [Google Scholar]
- Gu Y, Hall ZW (1988b) Immunological evidence for a change in subunits of the acetylcholine receptor in developing and denervated rat muscle. Neuron 1:117–125. 10.1016/0896-6273(88)90195-X [DOI] [PubMed] [Google Scholar]
- Hartzell HC, Fambrough DM (1972) Acetylcholine receptors. Distribution and extrajunctional density in rat diaphragm after denervation correlated with acetylcholine sensitivity. J Gen Physiol 60:248–262. 10.1085/jgp.60.3.248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering T, Braubach P, Landwehrmeyer GB, Lindenberg KS, Melzer W (2016) Fast-to-slow transition of skeletal muscle contractile function and corresponding changes in myosin heavy and light chain formation in the R6/2 mouse model of Huntington's disease. PLoS One 11:e0166106. 10.1371/journal.pone.0166106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter OF, Noble D (1960) The chloride conductance of frog skeletal muscle. J Physiol 151:89–102. [PMC free article] [PubMed] [Google Scholar]
- Jacobson D, Herson PS, Neelands TR, Maylie J, Adelman JP (2002) SK channels are necessary but not sufficient for denervation-induced hyperexcitability. Muscle Nerve 26:817–822. 10.1002/mus.10280 [DOI] [PubMed] [Google Scholar]
- Kosinski CM, Schlangen C, Gellerich FN, Gizatullina Z, Deschauer M, Schiefer J, Young AB, Landwehrmeyer GB, Toyka KV, Sellhaus B, Lindenberg KS (2007) Myopathy as a first symptom of Huntington's disease in a marathon runner. Mov Disord 22:1637–1640. 10.1002/mds.21550 [DOI] [PubMed] [Google Scholar]
- Kues WA, Brenner HR, Sakmann B, Witzemann V (1995) Local neurotrophic repression of gene transcripts encoding fetal AChRs at rat neuromuscular synapses. J Cell Biol 130:949–957. 10.1083/jcb.130.4.949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MY, Rohan JG, Cai H, Reim K, Ko CP, Chow RH (2013) Complexin facilitates exocytosis and synchronizes vesicle release in two secretory model systems. J Physiol 591:2463–2473. 10.1113/jphysiol.2012.244517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Xie F, Wei K, Hao XC, Li P, Cao J, Min S (2016) Sepsis induced denervation-like changes at the neuromuscular junction. J Surg Res 200:523–532. 10.1016/j.jss.2015.09.012 [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Lo DC, Hughes RE (2010) Neurobiology of Huntington's disease: applications to drug discovery. Boca Raton, FL: CRC/Taylor and Francis. [PubMed] [Google Scholar]
- Lodi R, Schapira AH, Manners D, Styles P, Wood NW, Taylor DJ, Warner TT (2000) Abnormal in vivo skeletal muscle energy metabolism in Huntington's disease and dentatorubropallidoluysian atrophy. Ann Neurol 48:72–76. 10.1002/1531-8249(200007)48:1%3C72::AID-ANA11%3E3.0.CO;2-I [DOI] [PubMed] [Google Scholar]
- Lomo T, Rosenthal J (1972) Control of ACh sensitivity by muscle activity in the rat. J Physiol 221:493–513. 10.1113/jphysiol.1972.sp009764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomo T, Westgaard RH (1975) Further studies on the control of ACh sensitivity by muscle activity in the rat. J Physiol 252:603–626. 10.1113/jphysiol.1975.sp011161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupa MT, Krzemien DM, Schaller KL, Caldwell JH (1995) Expression and distribution of sodium channels in short- and long-term denervated rodent skeletal muscles. J Physiol 483:109–118. 10.1113/jphysiol.1995.sp020571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthi-Carter R, Hanson SA, Strand AD, Bergstrom DA, Chun W, Peters NL, Woods AM, Chan EY, Kooperberg C, Krainc D, Young AB, Tapscott SJ, Olson JM (2002) Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum Mol Genet 11:1911–1926. 10.1093/hmg/11.17.1911 [DOI] [PubMed] [Google Scholar]
- Magrassi L, Purves D, Lichtman JW (1987) Fluorescent probes that stain living nerve terminals. J Neurosci 7:1207–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan EM, Martin AR (1981) Non linear summation of end plate potentials in the frog and mouse. J Physiol 311:307–324. 10.1113/jphysiol.1981.sp013586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielcarek M, Toczek M, Smeets CJ, Franklin SA, Bondulich MK, Jolinon N, Muller T, Ahmed M, Dick JR, Piotrowska I, Greensmith L, Smolenski RT, Bates GP (2015) HDAC4-myogenin axis as an important marker of HD-related skeletal muscle atrophy. PLoS Genet 11:e1005021. 10.1371/journal.pgen.1005021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda DR, Wong M, Romer SH, McKee C, Garza-Vasquez G, Medina AC, Bahn V, Steele AD, Talmadge RJ, Voss AA (2017) Progressive Cl− channel defects reveal disrupted skeletal muscle maturation in R6/2 Huntington's mice. J Gen Physiol 149:55–74. 10.1085/jgp.201611603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton AJ, Edwardson JM (2001) Progressive depletion of complexin II in a transgenic mouse model of Huntington's disease. J Neurochem 76:166–172. 10.1046/j.1471-4159.2001.00059.x [DOI] [PubMed] [Google Scholar]
- Morton AJ, Faull RL, Edwardson JM (2001) Abnormalities in the synaptic vesicle fusion machinery in Huntington's disease. Brain Res Bull 56:111–117. 10.1016/S0361-9230(01)00611-6 [DOI] [PubMed] [Google Scholar]
- Moss SJ, Beeson DM, Jackson JF, Darlison MG, Barnard EA (1987) Differential expression of nicotinic acetylcholine receptor genes in innervated and denervated chicken muscle. EMBO J 6:3917–3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelands TR, Herson PS, Jacobson D, Adelman JP, Maylie J (2001) Small-conductance calcium-activated potassium currents in mouse hyperexcitable denervated skeletal muscle. J Physiol 536:397–407. 10.1111/j.1469-7793.2001.0397c.xd [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimune H, Sanes JR, Carlson SS (2004) A synaptic laminin-calcium channel interaction organizes active zones in motor nerve terminals. Nature 432:580–587. 10.1038/nature03112 [DOI] [PubMed] [Google Scholar]
- Novak KR, Norman J, Mitchell JR, Pinter MJ, Rich MM (2015) Sodium channel slow inactivation as a therapeutic target for myotonia congenita. Ann Neurol 77:320–332. 10.1002/ana.24331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribnow D, Johnson-Pais T, Bond CT, Keen J, Johnson RA, Janowsky A, Silvia C, Thayer M, Maylie J, Adelman JP (1999) Skeletal muscle and small-conductance calcium-activated potassium channels. Muscle Nerve 22:742–750. 10.1002/(SICI)1097-4598(199906)22:6%3C742::AID-MUS11%3E3.0.CO;2-1 [DOI] [PubMed] [Google Scholar]
- Reilmann R, Kirsten F, Quinn L, Henningsen H, Marder K, Gordon AM (2001) Objective assessment of progression in Huntington's disease: a 3-year follow-up study. Neurology 57:920–924. 10.1212/WNL.57.5.920 [DOI] [PubMed] [Google Scholar]
- Ribchester RR, Thomson D, Wood NI, Hinks T, Gillingwater TH, Wishart TM, Court FA, Morton AJ (2004) Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the Huntington's disease mutation. Eur J Neurosci 20:3092–3114. 10.1111/j.1460-9568.2004.03783.x [DOI] [PubMed] [Google Scholar]
- Rich MM, Kraner SD, Barchi RL (1999) Altered gene expression in steroid-treated denervated muscle. Neurobiol Dis 6:515–522. 10.1006/nbdi.1999.0257 [DOI] [PubMed] [Google Scholar]
- Rocha MC, Pousinha PA, Correia AM, Sebastião AM, Ribeiro JA (2013) Early changes of neuromuscular transmission in the SOD1(G93A) mice model of ALS start long before motor symptoms onset. PLoS One 8:e73846. 10.1371/journal.pone.0073846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogart RB, Cribbs LL, Muglia LK, Kephart DD, Kaiser MW (1989) Molecular cloning of a putative tetrodotoxin-resistant rat heart Na+ channel isoform. Proc Natl Acad Sci U S A 86:8170–8174. 10.1073/pnas.86.20.8170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero E, Cha GH, Verstreken P, Ly CV, Hughes RE, Bellen HJ, Botas J (2008) Suppression of neurodegeneration and increased neurotransmission caused by expanded full-length huntingtin accumulating in the cytoplasm. Neuron 57:27–40. 10.1016/j.neuron.2007.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas JL, Gómez-Sánchez L, Tomás-Zapico C, Lucas JJ, Fernández-Chacón R (2011) Increased neurotransmitter release at the neuromuscular junction in a mouse model of polyglutamine disease. J Neurosci 31:1106–1113. 10.1523/JNEUROSCI.2011-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi K, Kanda F, Mitsui S, Kohara N, Chihara K (2012) Fibrillation potentials of denervated rat skeletal muscle are associated with expression of cardiac-type voltage-gated sodium channel isoform Nav1.5. Clin Neurophysiol 123:1650–1655. 10.1016/j.clinph.2012.01.002 [DOI] [PubMed] [Google Scholar]
- She P, Zhang Z, Marchionini D, Diaz WC, Jetton TJ, Kimball SR, Vary TC, Lang CH, Lynch CJ (2011) Molecular characterization of skeletal muscle atrophy in the R6/2 mouse model of Huntington's disease. Am J Physiol Endocrinol Metab 301:E49–E61. 10.1152/ajpendo.00630.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaulding EL, Sleigh JN, Morelli KH, Pinter MJ, Burgess RW, Seburn KL (2016) Synaptic deficits at neuromuscular junctions in two mouse models of Charcot–Marie–Tooth type 2d. J Neurosci 36:3254–3267. 10.1523/JNEUROSCI.1762-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmeyer K, Ortland C, Jentsch TJ (1991a) Primary structure and functional expression of a developmentally regulated skeletal muscle chloride channel. Nature 354:301–304. 10.1038/354301a0 [DOI] [PubMed] [Google Scholar]
- Steinmeyer K, Klocke R, Ortland C, Gronemeier M, Jockusch H, Gründer S, Jentsch TJ (1991b) Inactivation of muscle chloride channel by transposon insertion in myotonic mice. Nature 354:304–308. 10.1038/354304a0 [DOI] [PubMed] [Google Scholar]
- Strand AD, Aragaki AK, Shaw D, Bird T, Holton J, Turner C, Tapscott SJ, Tabrizi SJ, Schapira AH, Kooperberg C, Olson JM (2005) Gene expression in Huntington's disease skeletal muscle: a potential biomarker. Hum Mol Genet 14:1863–1876. 10.1093/hmg/ddi192 [DOI] [PubMed] [Google Scholar]
- Thesleff S, Ward MR (1975) Studies on the mechanism of fibrillation potentials in denervated muscle. J Physiol 244:313–323. 10.1113/jphysiol.1975.sp010800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- tom Dieck S, Sanmartí-Vila L, Langnaese K, Richter K, Kindler S, Soyke A, Wex H, Smalla KH, Kämpf U, Fränzer JT, Stumm M, Garner CC, Gundelfinger ED (1998) Bassoon, a novel zinc-finger CAG/glutamine-repeat protein selectively localized at the active zone of presynaptic nerve terminals. J Cell Biol 142:499–509. 10.1083/jcb.142.2.499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV, Khakh BS (2014) Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington's disease model mice. Nat Neurosci 17:694–703. 10.1038/nn.3691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner C, Cooper JM, Schapira AH (2007) Clinical correlates of mitochondrial function in Huntington's disease muscle. Mov Disord 22:1715–1721. 10.1002/mds.21540 [DOI] [PubMed] [Google Scholar]
- Valadão PA, de Aragão BC, Andrade JN, Magalhães-Gomes MP, Foureaux G, Joviano-Santos JV, Nogueira JC, Ribeiro FM, Tapia JC, Guatimosim C (2017) Muscle atrophy is associated with cervical spinal motoneuron loss in BACHD mouse model for Huntington's disease. Eur J Neurosci 45:785–796. 10.1111/ejn.13510 [DOI] [PubMed] [Google Scholar]
- Velier J, Kim M, Schwarz C, Kim TW, Sapp E, Chase K, Aronin N, DiFiglia M (1998) Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways. Exp Neurol 152:34–40. 10.1006/exnr.1998.6832 [DOI] [PubMed] [Google Scholar]
- Voss AA. (2009) Extracellular ATP inhibits chloride channels in mature mammalian skeletal muscle by activating P2Y(1) receptors. J Physiol 587:5739–5752. 10.1113/jphysiol.2009.179275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Pinter MJ, Rich MM (2010) Ca2+ dependence of the binomial parameters p and n at the mouse neuromuscular junction. J Neurophysiol 103:659–666. 10.1152/jn.00708.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters CW, Varuzhanyan G, Talmadge RJ, Voss AA (2013) Huntington disease skeletal muscle is hyperexcitable owing to chloride and potassium channel dysfunction. Proc Natl Acad Sci U S A 110:9160–9165. 10.1073/pnas.1220068110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells RD, Ashizawa T (2006) Genetic instabilities and neurological diseases, Ed 2 Amsterdam; Boston: Elsevier. [Google Scholar]
- Witzemann V, Barg B, Nishikawa Y, Sakmann B, Numa S (1987) Differential regulation of muscle acetylcholine receptor gamma- and epsilon-subunit mRNAs. FEBS Lett 223:104–112. 10.1016/0014-5793(87)80518-5 [DOI] [PubMed] [Google Scholar]
- Wood SJ, Slater CR (1997) The contribution of postsynaptic folds to the safety factor for neuromuscular transmission in rat fast- and slow-twitch muscles. J Physiol 500:165–176. 10.1113/jphysiol.1997.sp022007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, Sladky JT, Kallen RG, Barchi RL (1991) TTX-sensitive and TTX-insensitive sodium channel mRNA transcripts are independently regulated in adult skeletal muscle after denervation. Neuron 7:421–427. 10.1016/0896-6273(91)90294-A [DOI] [PubMed] [Google Scholar]