

Abstract
K-ras oncogene is a key factor in colorectal cancer. Based on published and our data we propose that K-ras could be the oncogene responsible for the inactivation of the tumor-suppressor gene APC, currently considered as the initial step in colorectal tumorigenesis. K-ras fulfills the criteria of the oncogene-induced DNA damage model, as it can provoke well- established causes for inactivating tumor-suppressors, i.e. DNA double-strand breaks (causing allele deletion) and ROS production (responsible for point mutation). The model we propose is a variation of the currently existing model and hypothesizes that, in a subgroup of colorectal carcinomas, K-ras mutation may precede APC inactivation, representing the earliest driving force and, probably, an early biomarker of colorectal carcinogenesis. This observation is clinically useful, since it may modify the preventive colorectal cancer strategy, restricting numerically patients undergoing colonoscopies to those bearing K-ras mutation in their colorectum, either in benign polyps or the normal accompanying mucosa.
Keywords: K-ras mutation, normal mucosa, benign polyp, biomarker, colorectal cancer, earliest mutation, review
Colorectal cancer (CRC) is the third most common cancer in men and the second most common cancer in women globally (1). Annually, the current worldwide incidence of CRC is estimated to overreach 1,200,000 patients, with a slight preponderance in males (1,2). It stands for almost 9% of the overall cancer incidence (3). Its 5-year survival rate is estimated to be 60% (1,4). Data from the USA estimated that the lifetime risk of acquiring the disease is 6% and that CRC is the third most common cause of cancer-related deaths on 2014 (4). Despite the remarkable decline of CRC incidence across the last decades, its mortality has fallen slightly, while the annual ratio of CRC-related deaths to the newly-diagnosed patients is recorded to remain unchanged during the last years, i.e. approximately 33% (3,5,6). Therefore, deep comprehension of the colorectal tumorigenesis process is indispensable both for the timely diagnosis and for the effective prevention of colorectal cancer.
Although several forms of hereditary colorectal cancer have been described, the majority of cases are sporadic (2). Sporadic colorectal cancer is an inhomogeneous disease; in fact, under the umbrella of this term several molecular genotypes are included, differing from each other according to the temporal order of events, the symptoms, the response to therapy and prognosis (2,7,8,9). Nevertheless, they share certain common genetic alterations that include, among others, mutations in the K-ras oncogene (8). As K-ras mutations are rather early events during colorectal cancer development (2), an emerging question is whether such aberrations can function as a driving force from the initial stages of development, at least in certain genotypes of this cancer. Available data will be examined and discussed to address this issue.
Current View on the Role of APC Inactivation in Colorectal Tumorigenesis
More than three decades ago, a first model attempting to describe the accumulation of genetic events that lead to colorectal carcinogenesis (the adenoma-carcinoma sequence), characterized APC aberrations as the initiating event (10). Why does the current dominant scientific thesis (11) continue to keep the same established order of these genetic events in CRC (Figure 1)?
Figure 1. The current model of colorectal cancer development: the adenoma-carcinoma sequence. The colorectal tumorigenesis process is a continuum. Colorectal cancer results from the stepwise accumulation of multiple, clonally selected, somatic mutations that force the normal crypts to transform to dysplastic epithelium and then to carcinoma. Wnt hyperactivation (APC inactivation or β-catenin mutation) usually occurs at the initial step, conferring to the initiation of the neoplastic process, whereas K-ras mutation usually occurs at an early step, contributing to the promotion of the oncogenic process. This order of events accounts for approximately 70% of sporadic colorectal cancer cases (based on references 10, 11, 13, 14, 30, 90, 93 and 99).

The Wnt/β-catenin pathway is activated in almost all human colon carcinomas (12,13). Both early and advanced stages of colorectal cancers depend on constitutive Wnt pathway activity (11). Somatic APC lesions are found even in microscopic adenomas composed of few dysplastic glands (13,14). In 70-80% of sporadic colon adenomas (11,15) and in 80-90% of sporadic colon carcinomas (8,12) biallelic APC loss is found; the remainder percentage represents functionally equivalent alterations in other components of the pathway (e.g. β-catenin) (16).
Besides this, CRC is a well-documented disease of stem cells (15,17,18); it springs from stem cells and not from their descendents (19); stemness is a characteristic of the transforming colon cells that require the Wnt activation to initiate, proliferate and settle in colon crypts (20).
In the wild-type crypt, β-catenin levels are elevated in the epithelial cells of the bottom of the crypt (due to the plethora of Wnt signaling) (20), enhancing stemness, eliminating differentiation, triggering proliferation, and facilitating the migration of colonic cells from the basis to the surface of crypts. Nevertheless, a transition from the β-catenin ON state to β-catenin OFF state occurs in the borderline of the lower and middle third of the crypt (17), which is due to the gradual decrease of Wnt signal factors and the gradual increase of APC protein from the bottom to the top of the crypt. The effects of this change to the upper part of the crypt (villous domain) are critical: cell cycle arrests, stem cells differentiate, genetic defects accumulate and cells are destined to fall in the gut lumen, via apoptosis activation (20).
In the adenomatous crypt, constant Wnt activation and reduced function of APC protein take place across the full-length of the crypt axis, resulting in the stabilization of β-catenin; β-catenin levels therefore increase; it remains in the ON state in the upper as well as in the lower parts of the crypt. The transition shifts to a higher crypt level and the mutated cells are trapped in the villous domain. Increased β-catenin levels aid these trapped, mutated cells both to retain a progenitor phenotype and to continue proliferation, since the [TCF–β-catenin] complex switches the cell programme from the differentiated intestinal cell to the proliferating progenitor cell. The mediator of these effects is c-myc, which represses the cell-cycle inhibitor p21 (21). Consequently, inside these mutated cells, differentiation process ceases, proliferation continues without cessation and apoptosis is blocked; outmigration of these cells in the gut lumen is canceled (20). In this way, clusters of aberrant cells, that carry progenitor-like phenotype, accumulate and form an aberrant crypt at the site where future polyp forms.
There exists experimental evidence of such sequence of events. Biallelic loss of APC disrupts colon crypt homeostasis; its silencing drived proliferation in the intestine, while its re-expression restored it (12). Heterozygous APC mutation at the crypt base is followed by asymmetric and symmetric division of APC wt/mt stem cells and the replacement of APC wt/wt with APC wt/mt stem cells (niche succession). APC wt/mt stem cells are at risk for acquiring a second APC mutation. In order for this 2nd hit to occur, APC mt/wt stem cell progenies move to the villous domain, undergo further division and, due to unknown genetic events, lose their heterozygosity. APC mt/mt stem cell clone eventually replaces the APC mt/wt stem cell clone and fill the crypt (further niche succession). In this way, niche conversion (monocryptal adenomas) takes place. Nonetheless, APC mutation alone is sufficient to trigger the complete niche succession (18). Monocryptal adenomas are monoclonic and are viewed as the earliest histologically-detectable precursor lesions of the colorectal tumor process. Asymmetric crypt fission follows (20), in which crypts divide laterally, providing a possible explanation to field cancerization (18).
The result is the formation of foci of aberrant crypts (aberrant crypt foci -ACF), a term equivalent to microadenoma (14). Although the oncogenic potential of ACF is uncertain (14,17,18,20,22) and despite the fact that this term is indeed endoscopic (is defined as microscopic clusters of small number of abnormal/altered crypts in unembedded colon mucosa detected in vivo with aid of dye spray using magnification endoscopy) (19), ACF are viewed as the very early lesions of the colon tumorigenesis process, preceding the formation of adenoma (13,14,20,23). In their majority they are histologically normal, but they may represent hyperplastic or dysplastic crypts (22); dysplastic crypts are those that ultimately are converted to adenomas (13,14,20).
Questions Associated with the Current Model of Colorectal Tumorigenesis
The first question to emerge regards the obscure nature of the early inactivation of APC. Tumor suppressor (ts) genes outnumber oncogenes in CRC (the ratio is 4 to 1) (24). Their inactivation is, therefore, indispensable for the initiation, the promotion and the progression of the process. Tumor suppressors’ inactivation actually is due to: i) allele deletion (loss of heterozygosity) and/or ii) point mutation. In the first case, allele deletion necessitates DNA double strand breaks (DSBs) to facilitate genomic rearrangements that eventually lead to loss of genomic segments. DSBs in the genome can occur only under oncogene-induced replication stress. In the second case, nucleotide substitutions can arise either from exogenous factors or from endogenous ones. The most frequent are endogenous causes, which arise mainly from increased ROS (reactive oxygen species) production, due to metabolic stress. Activated oncogenes are again responsible for ROS generation (25-27).
Therefore, although we are aware that the inactivation of a ts gene initiates colorectal tumorigenesis, the origin of its inactivation is unfamiliar. According to the just mentioned data, we don’t know which oncogene should be responsible for this genetic aberration; this oncogene should be activated presumably before APC aberrations, i.e. from the very early stages of the sporadic colorectal cancer. Additional questions arise: Which is the nature of the genetic event(s) that provoke the loss of heterozygosity of APC? What is the mechanistic basis of such an early inactivation?
Second, biallelic APC loss is not indispensable in all CRC patients. Genotypes 1 and 2 (CIMP-high, consisting 20% of CRC cases) may miss APC mutation; opposite to this, APC loss characterizes genotypes 3 to 5 (8). Only half of the hypermutated colon cancers hold the mutation, percentage significantly lower compared to respective in the non-hypermutated ones (80%) (28). Moreover, experimental findings from transgenic mice showed that colorectal tumorigenesis can be initiated in the absence of APC or β-catenin mutation (29).
Third, as Fearon and Vogelstein had made clear from the very first disclosure of their model, the progressive accumulation of genetic changes, rather than their order of occurrence is crucial for the colorectal tumor progression. They had shown that the occurrence of 5q loss was not synonymous to the initial stage of tumorigenesis. Furthermore, they had speculated that the transition from hyperplastic to neoplastic colon mucosa might not be due to a second APC allele loss, opening a window to interrogate for other events that might initiate colorectal tumorigenesis (10).
Fourth, the fundamental prototypical model for sporadic colorectal cancer development, with the order of events it premises, is valid in only 70 percent of sporadic CRC cases (30).
Fifth, the incidence of the loss of heterozygosity of APC in sporadic adenomas and carcinomas is actually lower than it is speculated, ranging from 20-63% (31-33). Despite the fact that functionally equivalent to APC loss, Wnt mutations are almost always present in colorectal tumors, initiating tumorigenesis, the possibilities of progression to cancer in case APC is normal are smallest compared to APC mutated colon adenomas (16). In such cases two issues should be enlightened: What are the alternative events that hyperactivate the Wnt pathway? Which genetic alterations play the role of the initiating events or the role of gatekeeper mutations in colorectal tumorigenesis process?
Sixth, it became experimentally evident, that, at least in some CRC cases, chromosomal instability may precede APC loss (14). Similarly, it was demonstrated that there were the changes in chromatin conformation along the villous-crypt axis that modulated APC expression and not the opposite (20). It therefore became obvious that another genetic defect, different from APC loss, was responsible for the acquisition of genomic instability.
Last, but not least, dysplastic ACF (aberrant crypt foci), which are the postulated precursor of adenomas (13,14,20,23), exhibit APC loss in a tiny fraction in non-FAP (FAP corresponds to familiar adenomatous polyposis) patients (0-6%) and in 100% of FAP patients (14). In contrast, K-ras mutations are found in 50-67% of dysplastic ACFs (14,34) and in 82% of non-dysplastic ACFs (14). It was therefore proposed that in non-FAP patients K-ras mutations might precede APC mutations and that the event that was responsible for the initiation of carcinogenesis in the development of a subset of sporadic adenomas might be an alternative genetic pathway that does not involve APC loss (14,15).
Interdependency Between Wnt/β-catenin and MAPK Pathways
Progression towards colorectal cancer needs in most cases both APC and K-ras mutations. They are not absolutely independent, though. The interplay of APC and K-ras mutations in colorectal tumorigenesis is poorly understood and extremely complicated. Early reports had shown a synergistic effect between Wnt pathway (regarding APC) and ERK pathway (regarding K-ras) (29), which are confirmed by recent ones (35).
APC monoallelic mutation confers to low cancer stem cells (CSCs) activation in the basis of colon crypts (weak β-catenin signaling). In APC mt/wt cells, the addition of K-ras mutation enhanced Wnt signaling (36), promoted β-catenin signaling (a statistical association was documented between nuclear β-catenin presence and K-ras mutation) (37), resulting in the activation of cancer stem cells, their self-renewal and the clonal expansion of their population (35). K-ras mutation by its own promotes widespread hyperplasia on the colon epithelium (38); this hyperplasia may predispose to the loss of heterozygosity (LOH) of APC. Indeed, K-ras mutation is expressed in the upper part of the crypt, where APC LOH takes also place (39). In the background of biallelic APC loss, K-ras mutation imposes a further obstacle in APC mt/mt cells to dedifferentiate, as they try to migrate towards the top of the crypt (39).
Normal APC gene can suppress tumorigenesis induced by K-ras (40), whereas homozygous APC loss, stabilizes oncogenic RAS protein (GTP-RAS), enhancing its action (41). This effect is wider, since the inhibition of degradation of the oncogenic form of Ras protein by various components of Wnt/β-catenin pathway was also proved (42). Therefore, it seems that MAPK activities (activated Ras protein included) are negatively regulated by the normal function of the APC protein, which degrades β-catenin, and is positively regulated by APC mutation or by the abundance of Wnt signaling, which increases cytoplasmic and nuclear levels of β-catenin. This regulation was indeed proved to be mediated by β-catenin (42,43). Thus, oncogenic K-ras protein augments its action in the context of Wnt/APC/β-catenin hyperactivation. To go further, it seems that high Wnt activity, though necessary, is not sufficient to initiate tumorigenesis and is regulated by MAPK signaling (41): in zebra fish, homozygous APC loss was proved to be insufficient to cause β-catenin nuclear translocation; rather, the nuclear accumulation of β-catenin required the additional activation of K-ras (43).
Moreover, K-ras mediated intestinal cell proliferation, following APC loss, required β-catenin (43,44). K-ras mutation enhances Wnt/β-catenin pathway activation, conferring to stemness and finally to cancer stem cells exaggerated activation: it was demonstrated that forced expression of mutant K-ras in colon cancer stem cells enhanced Wnt target genes and β-catenin nuclear aggregation (44), while blocking EGFR had the opposite effects (37). The enhancement of β-catenin’s action by K-ras mutation is shot up by 3 more effects: first, K-ras mutatiοn increases nuclear β-catenin translocation, via a Raf-mediated route (44), second, it increases the formation of nuclear β-catenin/TCF-4 complexes (45) and third, it augments tyrosine phosphorylation of β-catenin resulting in its dissociation from E-cadherin in cell membrane, increases its cytoplasmic pool and its translocation to cell nucleus (36).
Furthermore, few links were found between the aforementioned pathways: first, glycogen synthase kinase 3b (GSK-3β) is inhibited by mutated K-ras in a PI3K manner, i.e. through activating AKT (46); meanwhile, inhibition of GSK-3β stabilizes β-catenin. Nonetheless, this action is exerted only if Wnt signaling is active (45). Interestingly, GSK-3β is promoted via Wnt activation leading to Ras phosphorylation and Ras stabilization to RAS-GTP state, creating a vicious cycle (42). Second, LRP6 is phoshorylated by oncogenic K-ras signaling; P-LRP6 promotes canonical Wnt/β-catenin pathway and eliminates β-catenin in cell membrane, conferring to loss of cell-cell contact and epithelial-mesenchymal transition (47).
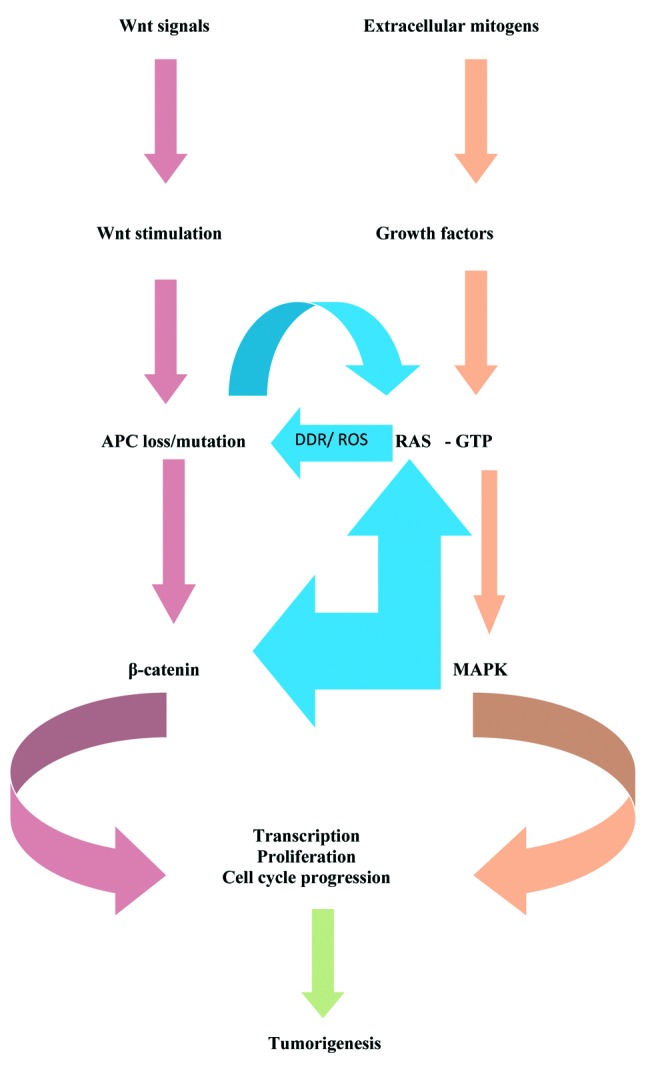
Taking into account the aforementioned data, it is attractive to speculate that oncogenic K-ras and mutagenic APC function synergistically in intestinal tumor formation and progression. They are mutually activated: K-ras mutation exerts its effects through MAPK pathway as well as the Wnt/β-catenin pathway (36), whereas APC inactivation acts via Wnt/β-catenin activation as well as MAPK pathway (41,42). To complement the synergy, some common targets, regulated by both mutations have been detected (for example c-myc is activated by mutagenic Ras, at the protein level, and by Wnt signaling, at the transcriptional level) (36) (Figure 2 and Figure 3).
Figure 2. The complex multiple mechanisms by which Wnt/β-catenin and MAPK pathways interact in the colon cell, conferring to the initiation of tumorigenesis (see text for detail). The arrows filled with red colour indicate inhibition of the target they direct; the arrows filled with green colour indicate promotion of the target they direct (based on references 25-27, 29, 35-47, 68-70, 78-80 and 95).

Figure 3. Schematic diagram of the flow of the genetic information through the two major initiating pathways in human colorectal tumorigenesis (Wnt and MAPK), representing the assumed dipole they consist and their interdependency.
Why is K-ras Mutation a Potential Alternative Initiating Event in Colorectal Tumorigenesis?
The role of Ras proto-oncogene in colon cell physiology and homeostasis is central and important: it regulates cell motility, aids gene expression, regulates cell cycle, halts cell proliferation, enhances survival and promotes apoptosis (48).
Given the confirmed interdependency between MAPK and Wnt pathways, could K-ras mutation be the triggering event to APC inactivation? K-ras mutations are the commonest genetic alterations in CRC (39,49) and the most prevalent oncogenic driver mutation in CRC (50). It is among the earliest genetic events of the process (51) although it is well known that it can occur at any stage (52). Its mutations are present in 50-95% of dysplastic ACF (34,53), in 15-78% of colorectal adenomas (54,55) and in 36-42% of CRCs (56). K-ras mutations on codons 12/13 of exon 2 and on codon 61 of exon 3 are de novo mutations whose presence depends on the random “activation” of the protooncogene K-ras during the life-span of healthy individuals either due to carcinogen exposure or to sporadic replication errors (57). By this point of view, K-ras mutation is a kind of dosimeter of carcinogen exposure of the colorectum of the given individual in the given point of time (58).
Κ-ras mutation weaves an extended web, which confers to the polyp phenotype. It can provoke all the hallmarks and the enabling characteristics of cancer: increases local inflammation (39), promotes proliferation, suppresses apoptosis, deregulates cellular energetics, helps the cell to evade immune destruction, increases stromal remodeling (59), enhances genomic instability (60-70), induces angiogenesis and activates invasion and metastasis (59).
Ras activation appears to be vital for signaling by extracellular mitogens. K-ras mutated cells are prone to acquire additional genetic alterations, indispensable for proliferation (51). Thus, although K-ras mutation may not be sufficient to generate a neoplastic growth advantage (53,51,58,71) it may place the normal tissue at increased risk of being transformed to cancer cell (51,58,72). Indeed, K-ras mutation was shown to promote colon carcinogenesis (73). Animal studies (74) proved that mutated K-ras is a connecting link between the cause (carcinogen) and the effect (neoplasia), since it can be detected in histologically normal colorectal tissues 2-15 weeks after exposure to a carcinogen and only 2 weeks before the onset of neoplasia (57). Nevertheless, this alteration is self-limited in the absence of additional genetic events (53), probably because the wild-type K-ras allele exerts an oncosuppressive effect on its mutated counterpart (75,76) or because of the oncogene-induced senescence (48,77). Importantly, K-ras is incapable to induce senescence if APC is mutated (39).
Beyond these, and perhaps more importantly, strong indications make us claim that K-ras mutation fulfills the criteria of the oncogene-induced DNA damage model. According to this model, oncogenes cause DNA replication stress; the DNA replication forks collapse and genetic events (microdeletions, chromosomic rearrangements, DNA double-strand breaks) occur, leading to genomic instability (25). Nonetheless, cancer does not occur and the total process stops to the precancerous lesions, because apoptosis and senescence confer a barrier (25,77). This defensive mechanism has been documented in colorectal adenomas, in which the reduced proliferation activity coincides with raised incidence of apoptosis and senescence (25).
Mutated K-ras acts on S phase, where chromosomal duplication occurs. Ras increases replication stress, causing persistent mitogenic activity, which consists of increased number of active DNA replication origins and collapsed replication forks (59). Both result in DNA damage response (DDR) (59,78). DDR is tested by DNA damage sensors and may direct the transformed cell upon 4 possible directions: proliferation, senescence, apoptosis (59) or defects in the DNA replication repair mechanisms (69). The last direction is quite important, as it causes inevitably genomic instability (59) (Figure 2). K-ras mutation is a major etiological factor of genomic instability (60-70,78-80), which is absolutely necessary for the tumorigenesis process to accelerate.
Mutated K-ras generates ROS through MAPK-dependent GATA-6 phosphorylation. ROS production is furthermore augmented through a vicious cycle: ROS directly enhances oncogenic Ras activation and augments MAPK, which further generates ROS (26). Ras-induced ROS are indeed mitogenic signals and eventually cause DNA damage generation and DDR activation. They seem to be crucial for K-ras-induced cellular transformation (81) and indispensable for the tumorigenic effect of Ras mutated cells: oncogenic Ras-expressing cells depend on ROS for their proliferation. Similarly the DDR-activating factors of ROS are dependent on the ongoing replication, acting only on a proliferating cell (27) (Figure 2).
Which Established Knowledge did our Study Confirm?
We studied the status of K-ras mutation (codons 12 and 13 of the exon 2) in the entire spectrum of the intermediate stages of colorectal tumorigenesis, from the histologically normal colorectal tissue to metastatic liver colorectal tissue. 40 patients were included in the study, being grouped into cancer-bearing bowel group (24 patients with sporadic colorectal cancer, biopsied in 78 different tissue-regions, 34 normal and 44 neoplastic) and cancer-free bowel group (16 patients with sporadic colorectal benign polyps, biopsied in 50 different tissue-regions, 27 normal and 23 neoplastic, Table I). Meanwhile, we searched for K-ras mutation in a control group (13 healthy volunteers with similar age and sex characteristics with the aforementioned groups and absolutely normal total colonoscopy); in this group, we afforded three tissue-regions per bowel (i.e. from right colon, from left colon and from rectum). Globally, we tested 167 tissue-regions in all three groups (100 normal, 36 neoplastic non-cancerous, 31 neoplastic cancerous, Table I). The symbols corresponding to each distinct tissue-region, normal, benign or malignant, are listed on Table II. From each tissue-region we biopsied 8 distinct specimens, directing six of them towards histopathological diagnosis and 2 of them for molecular analysis, i.e. for detection of possible K-ras mutations. To sum up, 334 different tissue samples were tested for K-ras mutation.
Table I. Panorama of demographic features, tissue regions and tissue samples in the 3 groups of members of the study (cancer-bearing group, cancer-free group, control group).

Table II. Endoscopic features, symbols of tissue-regions and their detailed numbers in cancer-bearing group and cancer-free group.

The statistical analysis of the acquired data was performed with Stata 9.0 (StataCorp, College Station, TX, USA). Continuous variables are expressed as mean±standard deviation. Categorical variables are expressed as percentages. Chi-Square test or Fisher’s exact test were used for comparison of categorical variables. A value of p<0.05 was used as a threshold for statistical significance.
The conclusions of our study that confirmed the established knowledge from the literature are summarized as follows:
i. K-ras mutation was not associated with age, sex and the location of neoplasia (data not shown).
ii. Serrated lesions may carry K-ras mutations. Although we show that mutation rates don’t differ between serrated and non-serrated lesions (Tables III, V and VI), the small number of serrated lesions (Table II) renders the value of this observation weak.
iii. Nearly one-fifth of benign polyps carried K-ras mutations; this rate did not differ significantly comparing the two groups of patients (23.07% in the cancer-bearing group and in 17.4% in the cancer-free group, p=0.68, Tables III, V and VI).
iv. K-ras mutation was detected in more than one half of primary colorectal cancer tissues, irrespective of its staging (the rate was 54.16%, Table VI) and less than one half of metastatic liver colorectal lesions (the rate was 42.85%, Table VI). Thus, K-ras mutation does not confer to the metastatic potential of colorectal carcinomas.
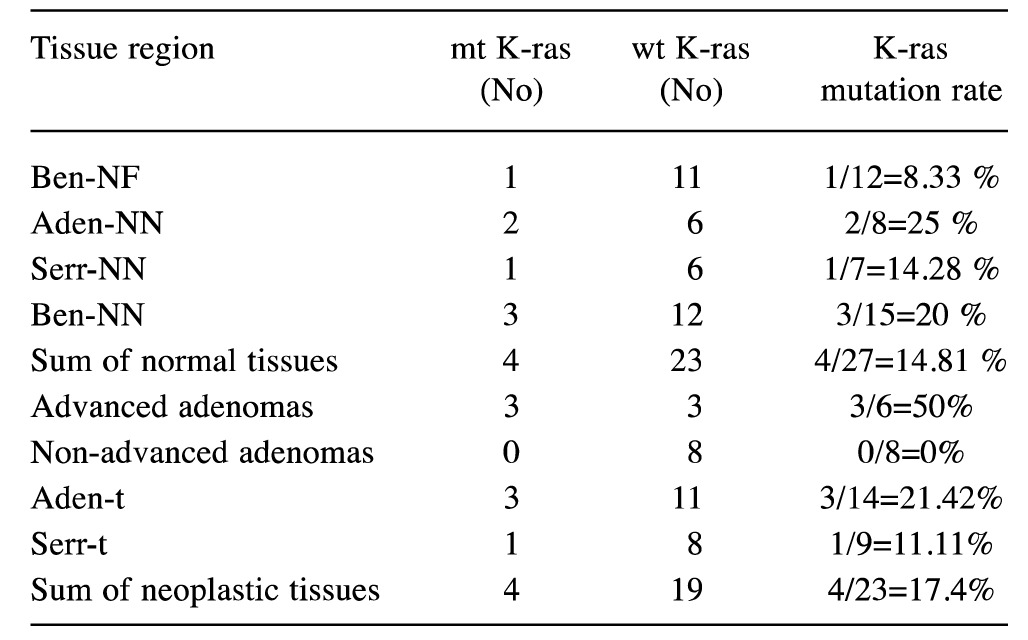
Table VI. K-ras mutation rates in different tissue-regions in the cancer-bearing group and the cancer-free group.

v. K-ras mutation does not confer to the formation of early adenomas (confirming other researchers) (32,55,82), but it rather aids the evolution of the non-advanced adenoma to the advanced one (data not shown).
vi. The molecular behavior of advanced adenomas resembles that of colorectal cancer; the rate of K-ras mutation is approximately the same between them (50% and 54.16%, p=0.82), they both carry the mutation more often than non-advanced adenomas (p=0.009 vs. 0.02 respectively) and serrated lesions (p=0.08 and 0.019) and they both have more mutations compared to total normal tissues of the study (p=0.02 vs. p<0.001 respectively). Furthermore, both neoplasms have insignificant differences with the corresponding metastases (p=0.65 vs. p=0.65). Thus, K-ras mutation does not promote the transformation of advanced adenomas to carcinomas.
vii. Considering separately the criteria of whether an adenoma is categorized as advanced or not (55,83,84) (i.e. size 1 cm, villous component over 80%, low or high grade dysplasia), we confirmed that neither of them had an etiological relationship to K-ras mutation incidence (p-value was 0.11, 0.36 and 0.54, respectively – data not shown).
viii. Normal tissues adjacent to colorectal carcinomas may carry K-ras mutations (Table IV and Table VI); the status of K-ras of this normal mucosa does not necessarily agree with K-ras status of cancer (data not shown).
Table IV. K-ras mutation rates are identical between the various categories, subcategories and supercategories of normal tissue-regions inside the cancer-bearing group and the cancer-free group.
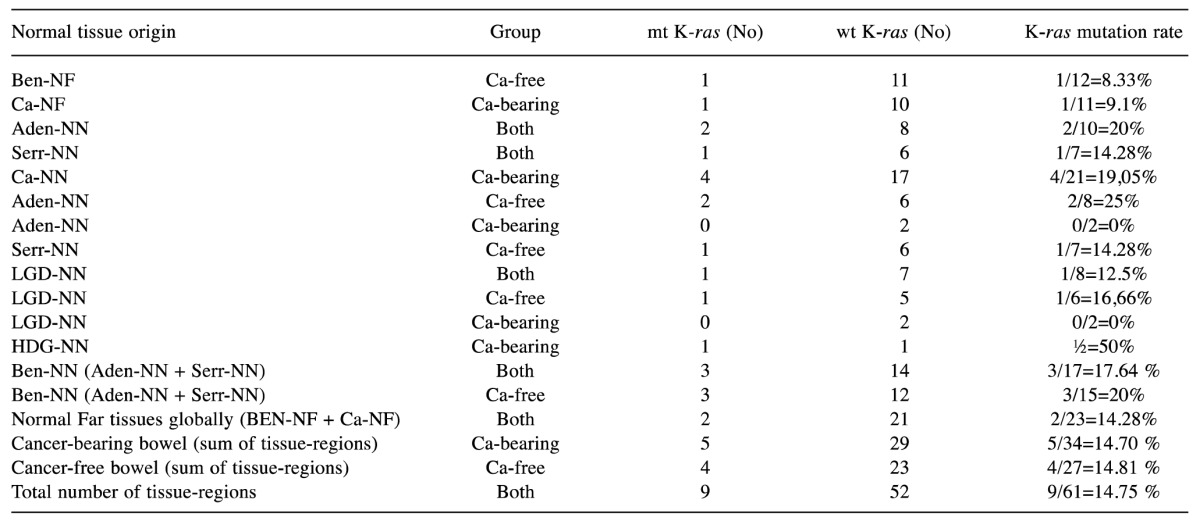
HGD-NN: normal tissue adjacent to adenoma with high-grade dysplasia; LGD-NN: normal tissue adjacent to adenoma with low-grade dysplasia.
Novel Aspects from our Study
The present study revealed several data concerning colorectal carcinogenesis, that are reported for the first time. In brief, the novelties of our study are the following:
i. It is the first study, to the best of our knowledge, to analyze for possible K-ras mutations the endoscopicaly and histologically normal colorectal mucosa belonging to a totally normal bowel; it was proved that the mutation was totally absent inside this bowel group (K-ras mutation rate was 0% inside the 78 normal specimens from the control group).
ii. It was the first time to demonstrate that normal (endoscopicaly and histologically) colorectal tissue from a non-cancerous bowel with benign polyps may carry K-ras mutations.
iii. All categories, subcategories and supercategories of normal tissues belonging in ca-free group, in ca-bearing group, or in combined groups, displayed the same possibility of carrying a K-ras mutation (p>0.05 in all possible comparisons); this possibility (approximately 15%) was significantly higher, comparing the normal mucosa of Ca-bearing group to the control group (p=0.018) and the normal mucosa of ca-free group to the control group (p=0.024) (Table IV). This observation indicated that K-ras mutated normal colon mucosa carries equal risk for developing cancer, regardless of the presence of cancer.
iv. Benign colorectal polyps had the same possibility to carry K-ras mutations with their co-existing normal colorectal mucosa; this fact was not dependent on the presence of cancer (p=0.66 for the ca-bearing group, p=1 for the Ca-free group).
v. Ca-free group displayed homogeneity, concerning K-ras status (Table V); the possibility for a tissue-region from this bowel group to bear the mutation was not dependent on its nature (i.e. neoplastic or normal) and was greater than the control group (p=0.008)
Table V. Regarding K-ras mutation, cancer-free group constitutes a homogenous entity: the proportions K-ras mutations inside normal tissues of the group are almost the same compared to the proportions of K-ras mutations inside neoplastic tissues of the group.
vi. K-ras mutation status remarkably agreed between the neoplastic and the non-neoplastic tissues, inside the same group of patients (detailed data not shown). Despite agreement (agreement rate=68.80%), no correspondence was evident in the cancer-bearing group between cancerous and normal tissue (p=0.039, i.e. the percentages differ significantly), meaning that K-ras status of the neoplastic cancerous tissue cannot be predicted by K-ras status in the co-existing normal colorectal tissue. On the contrary, in the absence of cancer, the agreement rate was even greater (82.60%) and correspondence was evident between polypoid and normal tissue (p=0.12, i.e. the percentages don’t differ significantly), meaning that K-ras status of the benign neoplastic tissue can be reliably predicted by K-ras status in the co-existing normal colorectal tissue.
vii. Cancer-bearing group had greater possibility to carry K-ras mutations compared to control group (p<0.001), but only modestly higher possibility, compared to cancer-free group (p=0.059, denoting a strong trend).
viii. Normal colorectal tissues far from benign or malignant neoplasm have the same possibility to carry the mutation compared to the control group (p=0.23 for the ca-free group and 0.22 for the ca-bearing group); instead, normal colorectal tissues adjacent to benign or malignant neoplasms had more K-ras mutations related to the control group (p<0.05 in all possible comparisons, data not shown).
ix. Collectively, taking into consideration the last 2 observations (viii and ix) and the well-documented knowledge that K-ras mutation does not confer to the formation of early adenoma, we can conclude that K-ras mutation is a very early event in the colorectal carcinogenesis process.
Future Implications of our Findings
To the best of our knowledge, this is the first study that explores simultaneously all the successive intermediate stages implicated in human colon tumorigenesis. Also, to the best of our knowledge, this study demonstrates that the endoscopicaly and simultaneously histologically normal mucosa from a cancer-free bowel with sporadic benign polyps may carry K-ras mutations. Two previous works (81,85) showed conflicting evidence concerning K-ras mutations rates in non-neoplastic bowel, whereas others detected K-ras mutation in colonic effluent samples (86) and in stool (87) from persons with colorectal adenomas. Besides these data, K-ras mutations’ presence in histologically normal mucosa from cancer-bearing bowel has gained plethora of proofs (58,71,81), ranging the mutation percentage from 5 to 25% in this category (88).
Many researchers have demonstrated that (32,72,74,81,82,85,89): 1st, in cancer-carrying bowel, K-ras mutated normal mucosa represents an early event of the successive steps that potentially end up to cancer, and, 2nd, this mutation predicts future colorectal cancer formation in the same bowel (81). We now support the expansion of both conclusions to the cancer-free bowel for the following reasons: First, we demonstrated that normal colorectal tissues adjacent to neoplasms carry the mutation more often than the control bowel, whereas normal colorectal mucosa far from neoplasms does not. Second, we detected the presence of K-ras mutation in the normal mucosa of polyp-bearing bowel, but not in the normal mucosa of the absolutely normal bowel. Third, we confirmed that K-ras mutated normal colon mucosa carries an increased risk to transform to cancerous tissue; this risk does not depend on the presence of concomitant cancer. Fourth, we demonstrated that K-ras de novo somatic mutation is not age-dependent; its acquisition is rather random, takes place at any age and is due to exposure of colon cells to environmental mitogens. Fifth, we proved that cancer-bearing bowel carried a moderate only (statistically insignificant) higher K-ras mutation rate.
Thus, in a subset of sporadic colorectal adenomas, K-ras mutation could fulfill the role of the earliest activated oncogene that drives colorectal tumorigenesis and predicts colon cancer formation: its presence in a benign polyp or in its adjacent or far normal colorectal mucosa implicates that colorectal tumorigenesis process has already been initiated by a driver mutation, that an irreversible stage of the entire procedure has been established at the host mucosa, and, consequently, that the patient is at risk of developing cancer (51,90). The quite possible mechanistic basis for this phenomenon is the inactivation of the ts gene APC.
In general, we identify a probable window in the current model, which may challenge the absolute domination of APC inactivation as the initiation event of sporadic colorectal tumorigenesis, but we don’t dispute the predominant role of APC in the tumorigenesis process. We propose an alternative model, a possible variation of the current one, which may be valid in a subset of sporadic colorectal tumors. In this model, K-ras mutation may represent the driving force to cause APC loss or inactivation to occur and may be the prerequisite by which APC mutation exerts its action (Figure 3 and Figure 4). Since K-ras mutation is very early and implies an irreversible step of the process, it might represent the springboard to accelerate it. Alternatively, since K-ras mutation and APC loss share the activation of common routes (i.e. Wnt/β-catenin pathway is activated by K-ras (36) and the MAPK pathway is enhanced by APC) (43), K-ras and APC mutations may need each other to fully unfold their action. In other words, they exhibit interdependency (Figure 3). They may constitute a dipole; whichever member of the pair is destined to occur first, it activates inevitably the second pole, which acts complementary to the initial event and is a prerequisite for the complete action of the dipole to fulfill (Figures 3, 4). The fact that K-ras mutation may precede APC inactivation agrees with recent reviews that have placed K-ras as the initiating event of the process (14,91).
Figure 4. An alternative model proposed to occur in a fraction of sporadic colorectal cancers, a variation of the currently valid adenoma-carcinoma sequence: K-ras mutation, the first mutation in the Ras/Raf/MAPK cascade, takes place very early in the colorectal tumorigenesis process, aiding in the abnormal niche succession, the crypt fission, the formation of microadenoma or dysplastic aberrant crypt foci (ACF) and, therefore, contributes in the initiation of colorectal tumorigenesis. In parallel, by causing DNA damage and DNA damage response (DDR), by generating ROS and by unfolding the full arsenal of its tumorigenic capabilities, it hyperactivates the Wnt pathway, mainly by inactivating the tumor suppressor gene APC. In this way K-ras mutation is the triggering factor, the driving force that enhances sharply the proliferative effects of the Wnt pathway, being indispensable to Wnt pathway to fully exert its own actions. The hyperactivation of Wnt/β-catenin has two major results: first, it forces the conversion of ACFs (always monoclonal) to early adenomas (polyclonal in some 70-80%), promoting the benign, precancerous, stages of colon carcinogenesis process (low-grade dysplasia) and, second, it reinforces the MAPK pathway (being already locked on the RAS-GTP state) to rocket up its effects. The MAPK pathway needs actually this boom to accomplish its effects. In fact, the monoclonal adenoma would never follow its fate to become a real adenoma, in case APC inactivation (or equivalent mutations) should never occur; instead, it should abolish itself, representing an extra possible defensive mechanism by which human bowel is discharged by such a common, potentially dangerous, somatic mutation. By achieving their mutual activation, each of the 2 pathways needs one another to succeed; they use one another to act oncogenetically. Thus, they may constitute a theoretical dipole. The somatic mutation of the protooncogene K-ras is the initiating event, a prerequisite for APC loss/inactivation (which is the predominant genetic alteration is the whole process) and absolutely necessary for the building of this dipole. Even though monoallelic APC loss is the starting point, this event is insufficient to be the initiating point; it rather helps the migration of stem cells to the field where mutated K-ras acts, i.e. from the bottom to the villous domain of colon crypts, giving K-ras the chance to drive APC LOH, absolutely necessary for the initiation of the tumorigenesis. This model explains the higher proportion of K-ras mutation in the dysplastic ACF compared to colon carcinomas and the extreme variability of K-ras mutation rates observed in colon adenomas. In the subset of colorectal adenomas where this model might prove true, it would not be an exaggeration to claim that K-ras mutation is indeed necessary, and, probably, sufficient for the tumorigenesis to accelerate, opposite to the common belief, that it is neither sufficient nor necessary for the colorectal tumorigenesis process (based on references 12-15, 17-18, 20, 22, 36-40, 53-56, 91 and 98).

In the multiple pathways with different starting points or initiators leading to the common end called sporadic colorectal cancer (14), we assume that the dipole [MAPK–Wnt/β-catenin] is virtually always the entrance gate by which the normal colorectum epithelium is transformed to adenoma. Since APC mutations are spread over a very large protein and, hence, quite difficult to identify (14), the easy-to-detect K-ras mutations are the obvious target to aim in our effort to decide whether a given bowel has initiated its course to cancer.
Why Detecting K-ras Mutation in the Non-cancerous Colorectal Tissue is Clinically Significant?
People with benign colorectal polyps are “obliged” to undergo frequent follow-up colonoscopies, in order to have detected and eradicated possible index benign polyps, since every adenoma, serrated or not, has the capacity for malignant transformation (54,92). This strategy results in unnecessary colonoscopies; most adenomas don’t progress to cancer (54), (the conversion rate is below 5%) (50), whilst a great gap is evident between the incidence of polyps (approximately 40%) (93) and that of CRC (nearly 6%) (1,4). Trying to fill the gap, we propose the detection of K-ras mutation in benign polyps: its presence implies either the carcinogenesis process has been initiated, or its progress to an advanced state has started; whatever eventuality is true, K-ras mutation reveals that a non-reversible process is in evolution and this polyp carries increased risk for being transformed to cancer. Thus, K-ras mutation’s detection inside benign polyps might prove a valuable biomarker for clinicians to select the population to be referred for frequent surveillance.
Unfortunately, it is sometimes difficult or even impossible to afford polyp tissue for molecular study, because of the inherent shortcomings of the process of colonoscopy (94). For instance, it may be hard to obtain tissue for molecular diagnosis from tiny polyps, because its restricted material is destined exclusively for histological diagnosis or because small polyps may not be visible in an inadequately prepared bowel. Moreover, polyps, especially small, flat and depressed, may be located in bowel curves, where it is hard to detect or approach. A subset of flat or laterally-spreading adenomas are endoscopicaly visible only in case advanced endoscopic techniques (e.g. narrow-band imaging) are used; advanced endoscopic technology is not available, however, in every Endoscopy Unit. The adenoma detection rate ranges from 7 to 44%, depending on the endoscopist’s experience (94) and a recent study showed that 5%-32% of polyps are missed during colonoscopy, depending on their morphology (pedunculated or sessile), the preparation status of the bowel, the picture analysis of the colonoscope and the operator’s characteristics (95). In these conditions, a precancerous lesion, possibly K-ras mutated, remain intact after a colonoscopy, where “all polyps were removed”. Last, but not least, enormous polyps that are placed in a strangled bowel may be located beyond our reach. Whatever the exact cause of “failure” of detection, polyps may be overlooked during colonoscopy, making their total removal an elusive target.
In other cases, a polyp cut by polypectomy snare may become invisible, covered by the enteric content, and remain inside the bowel. Rarely, polyps are located in close proximity or in direct contact with diverticular openings; this condition deters endoscopists from removal or even biopsying them, because the risk of danger is high. Endoscopists are also obliged not to biopsy polyps that show tendency to bleed or polyps found in a bowel with active bleeding. In all these cases, molecular analysis of the normal colorectal mucosa from the same bowel which carries polyp/polyps that appear non-cancerous by endoscopic criteria is of the same value, since we demonstrated that K-ras status of the normal colon tissue could credibly predetermine the K-ras status of the coexisting benign polyp in case no cancer is present.
All K-ras mutated individuals, carrying the mutation inside their non-cancerous colorectal tissue, should be offered conventional endoscopic surveillance with increased frequency. They should be first referred for high-definition white-light colonoscopy. In case endoscopic suspicions for a malignant or premalignant lesion are raised, advanced endoscopy techniques, as chromoendoscopy, narrow-band-imaging (NBI) - magnification endoscopy, cap-assisted colonoscopy or colonoscopy with the “third-eye retroscope” may be performed (94). They should de subjected to additional biopsies, targeted or not, to enable detection of other crucial mutations, e.g. on the ts genes APC or TP53. In case local experience is available, these patients may be offered probe-based confocal laser endomicroscopy (p-CLE) a method of in vivo histology mapping of the colorectal mucosa, as distinct morphological patterns which distinguish normal from malignant networks in colorectal mucosa are described (96). Equally important, life-style of these subjects should be modified in a very strict way (smoking cessation, avoidance of red meat-especially smoked, restriction of sugar and fat, increasing physical activity, losing weight, increasing the intake of dietary fibers, fruits, vegetables, fish and milk and complying with Mediterranean diet) (53).
Conversely, K-ras wild-type patients should follow an alternative strategy, because, as K-ras is not a germ-line mutation, its absence in the normal mucosa demonstrates that the process of carcinogenesis has not probably started yet (71). Nevertheless, follow-up of such persons is still necessary, depending on patient’s clinical profile and risk factors, since K-ras mutation, although very important, is not necessary for colorectal tumorigenesis initiation (53,74).
Our study has several limitations. Although lots of samples were examined, the number of patients is not large. The size of the control group is small, too. The absolute number of K-ras mutations in all normal tissue categories and the mean size of benign lesions are restricted. There was a small number of cases where simultaneous samples to cover the majority of the sequential stages of colorectal carcinogenesis process in the same patient were available. Last, our results are limited by the fact that, since many bowels are dirty during colonoscopy, there is always a considerable risk to “lose” a small or flat cancer. We should therefore not only look into the increased number of the future bowels that need to be examined, but also to the quality of the bowel preparation as well.
Conclusions and Future Perspectives
In a nutshell, K-ras mutation’s value could stand beyond its established role to guide oncologists for advanced therapies or not (97). In a subset of sporadic colorectal carcinomas this mutation represents a very early critical genetic event, responsible for the initiation of tumorigenesis in normal colon epithelium. It might prove the “unknown factor” by which the tumor suppressor gene APC is inactivated; K-ras mutation probably achieves this goal, either by generation of ROS or by causing DNA double strand breaks. Nevertheless, whatever mutation (APC or K-ras) comes first, both oncogenic pathways (Wnt and MAPK) are hyperactivated ultimately, behaving genetically as a dipole : Wnt/β-catenin pathway needs the MAPK pathway components to fully unfold its effects and, therefore takes advantage of them and MAPK pathway exploits analogously β-catenin and other components of the Wnt pathway. Since APC mutation or loss is not so feasible to detect due to technical limitations, it sounds sensible to search for the other pole of the dipole, i.e. mutation in K-ras, since the access to the normal or/and to neoplastic colorectal tissue is easy via colonoscopy. Its presence in benign polyps and/or in normal tissues reveals that an irreversible process is in progress. Therefore, K-ras mutation can be seen as a prelude to sporadic colorectal cancer, although neither obligatory nor sufficient for colon carcinogenesis.
Searching for K-ras mutation in the benign colorectal polyps and/or their concomitant normal colorectal mucosa during colonoscopy may prove a useful strategy to aid clinicians, endoscopists and health systems to restrict the surveillance group in the population who really needs it. Benign polyps and their concomitant normal tissues, which carry K-ras mutation and received from a bowel without cancer, hold a high risk to transform to colorectal cancer equal as if the bowel carried cancer. Clinicians might advise for conventional or advanced endoscopic surveillance those whose benign polyps are K-ras mutated. If benign polyp tissue sampling is ineffective, endoscopists should advise those whose normal colorectal mucosa is K-ras mutated to adopt a strict follow-up programme, since K-ras status in the normal colorectal tissue reliably predicts K-ras status in the coexisting polyp or polyps. K-ras mutation, albeit some decades in the forefront of colorectal tumorigenesis, may become nowadays an important biomarker, clinically useful, capable to motivate doctors and patients to the necessary follow-up and, therefore, endoscopists should be encouraged to be alert for searching this mutation. Future studies with greater numbers of patients may confirm our data and should investigate the specificity, the sensitivity and the diagnostic accuracy of such a strategy. Last, they should study whether this strategy reduces the rate of interval cancers and whether it prevents colorectal cancer, at least in a proportion of patients.
Conflicts of Interest
The Authors declare no conflicts of interest.
Acknowledgements
This work was supported by materials and reagents of the Histology-Embryology Department of the Medical School of the National and Kapodistrian University of Athens, Athens, Greece.
Table III. K-ras mutation rates are identical between the various categories and subcategories of benign polyps inside the cancer-bearing group and the cancer-free group.

References
- 1.Arvelo F, Sojo F, Cotte C. Biology of colorectal cancer. Ecancermedicalscience. 2015;9:520. doi: 10.3332/ecancer.2015.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bogaert J, Prenen H. Molecular genetics of colorectal cancer. Ann Gastroenterol. 2014;27:9–14. [PMC free article] [PubMed] [Google Scholar]
- 3.Haggar FA, Boushey RP. Colorectal cancer epidemiology: Incidence, mortality, survival, and risk factors. Clin. Colon Rectal Surg. 2009;22:191–197. doi: 10.1055/s-0029-1242458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin. 2014;64:104–117. doi: 10.3322/caac.21220. [DOI] [PubMed] [Google Scholar]
- 5.Edwards BK, Ward E, Kohler BA, Eheman C, Zauber AG, Anderson RN, Jemal A, Schymura MJ, Lansdorp-Vogelaar I, Seeff LC, van Ballegooijen M, Goede SL, Ries LA. Annual report to the nation on the status of cancer, 1975-2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010;116:544–573. doi: 10.1002/cncr.24760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas J, Ohtsuka M, Pichler M, Ling H. MicroRNAs: Clinical relevance in colorectal cancer. Int J Mol Sci. 2015;16:28063–28076. doi: 10.3390/ijms161226080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phipps AI, Limburg PJ, Baron JA, Burnett-Hartman AN, Weisenberger DJ, Laird PW, Sinicrope FA, Rosty C, Buchanan DD, Potter JD, Newcomb PA. Association between molecular subtypes of colorectal cancer and patient survival. Gastroenterology. 2015;148:77–87.e2. doi: 10.1053/j.gastro.2014.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology. 2007;50(1):113–130. doi: 10.1111/j.1365-2559.2006.02549.x. [DOI] [PubMed] [Google Scholar]
- 9.Bettington M, Walker N, Clouston A, Brown I, Leggett B, Whitehall V. The serrated pathway to colorectal carcinoma: current concepts and challenges. Histopathology. 2013;62:367–386. doi: 10.1111/his.12055. [DOI] [PubMed] [Google Scholar]
- 10.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 11.Fearon ER. Molecular genetics of colorectal cancer. Annual Rev Pathol. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 12.Dow LE, O’Rourke KP, Simon J, Tschaharganeh DF, van Es JH, Clevers H, Lowe SW. Apc restoration promotes cellular differentiation and reestablishes crypt homeostasis in colorectal cancer. Cell. 2015;161:1539–1552. doi: 10.1016/j.cell.2015.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology. 2010;138:2059–2072. doi: 10.1053/j.gastro.2009.12.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pretlow TP, Pretlow TG. Mutant KRAS in aberrant crypt foci (ACF): initiation of colorectal cancer. Biochim Biophys Acta. 2005;1756:83–96. doi: 10.1016/j.bbcan.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 15.Humphries A, Wright NA. Colonic crypt organization and tumorigenesis. Nat Rev Cancer. 2008;8:415–424. doi: 10.1038/nrc2392. [DOI] [PubMed] [Google Scholar]
- 16.Samowitz WS, Powers MD, Spirio LN, Nollet F, van Roy F, Slattery ML. Beta-catenin mutations are more frequent in small colorectal adenomas than in larger adenomas and invasive carcinomas. Cancer Res. 1999;59:1442–1444. [PubMed] [Google Scholar]
- 17.Cui S, Chang PY. Current understanding concerning intestinal stem cells. World J Gastroenterol. 2016;22:7099–7110. doi: 10.3748/wjg.v22.i31.7099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeki SS, Graham TA, Wright NA. Stem cells and their implications for colorectal cancer. Nat Rev Gastroenterol Hepatol. 2011;8:90–100. doi: 10.1038/nrgastro.2010.211. [DOI] [PubMed] [Google Scholar]
- 19.Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- 20.Boman BM, Fields JZ. An APC: WNT counter-current-like mechanism regulates cell division along the human colonic crypt axis: a mechanism that explains how APC mutations induce proliferative abnormalities that drive colon cancer development. Front Oncol. 2013;3:244. doi: 10.3389/fonc.2013.00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D, Haramis AP, Tjon-Pon-Fong M, Moerer P, van den Born M, Soete G, Pals S, Eilers M, Medema R, Clevers H. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241–250. doi: 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 22.Perse M, Cerar A. The role, significance and applicability of aberrant crypt foci in clinical practice, chapter 18 (pp. 467-478), In: Colorectal Cancer – Surgery, Diagnostics and Treatment, edited by Jim Khan. InTech. 2014:467–478. [Google Scholar]
- 23.Smith AJ, Stern HS, Penner M, Hay K, Mitri A, Bapat BV, Gallinger S. Somatic APC and K-ras codon 12 mutations in aberrant crypt foci from human colons. Cancer Res. 1994;54:5527–5530. [PubMed] [Google Scholar]
- 24.The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 26.Pan JS, Hong MZ, Ren JL. Reactive oxygen species: a double-edged sword in oncogenesis. World J Gastroenterol. 2009;15:1702–1707. doi: 10.3748/wjg.15.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogrunc M, Di Micco R, Liontos M, Bombardelli L, Mione M, Fumagalli M, Gorgoulis VG, d’Adda di Fagagna F. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ. 2014;21:998–1012. doi: 10.1038/cdd.2014.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carethers JM, Jung BH. Genetics and Genetic Biomarkers in Sporadic Colorectal Cancer. Gastroenterology. 2015;149:1177–1190.e3. doi: 10.1053/j.gastro.2015.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.D’ Abaco GM, Whitehead RH, Burgess AW. Synergy between Apc min and an activated ras mutation is sufficient to induce colon carcinomas. Mol Cell Biol. 1996;16:884–891. doi: 10.1128/mcb.16.3.884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Obuch JC, Ahnen DJ. Colorectal cancer: genetics is changing everything. Gastroenterol Clin North Am. 2016;45:459–476. doi: 10.1016/j.gtc.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 31.Zauber NP, Sabbath-Solitare M, Marotta SP, Bishop DT. K-ras mutation and loss of heterozygosity of the adenomatous polyposis coli gene in patients with colorectal adenomas with in situ carcinoma. Cancer. 1999;86:31–36. doi: 10.1002/(sici)1097-0142(19990701)86:1<31::aid-cncr6>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 32.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 33.Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–237. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- 34.Takayama T, Ohi M, Hayashi T, Miyanishi K, Nobuoka A, Nakajima T, Satoh T, Takimoto R, Kato J, Sakamaki S, Niitsu Y. Analysis of K-ras, APC, and beta-catenin in aberrant crypt foci in sporadic adenoma, cancer, and familial adenomatous polyposis. Gastroenterology. 2001;121:599–611. doi: 10.1053/gast.2001.27203. [DOI] [PubMed] [Google Scholar]
- 35.Moon BS, Jeong WJ, Park J, Kim TI, Min do S, Choi KY. Role of oncogenic K-Ras in cancer stem cell activation by aberrant Wnt/β-catenin signaling. J Natl Cancer Inst. 2014;106:djt373. doi: 10.1093/jnci/djt373. [DOI] [PubMed] [Google Scholar]
- 36.Janssen KP, Alberici P, Fsihi H, Gaspar C, Breukel C, Franken P, Rosty C, Abal M, El Marjou F, Smits R, Louvard D, Fodde R, Robine S. APC and oncogenic KRAS are synergistic in enhancing Wnt signaling in intestinal tumor formation and progression. Gastroenterology. 2006;131:1096–1109. doi: 10.1053/j.gastro.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 37.Horst D, Chen J, Morikawa T, Ogino S, Kirchner T, Shivdasani RA. Differential WNT activity in colorectal cancer confers limited tumorigenic potential and is regulated by MAPK signaling. Cancer Res. 2012;72:1547–1556. doi: 10.1158/0008-5472.CAN-11-3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feng Y, Bommer GT, Zhao J, Green M, Sands E, Zhai Y, Brown K, Burberry A, Cho KR, Fearon ER. Mutant KRAS promotes hyperplasia and alters differentiation in the colon epithelium but does not expand the presumptive stem cell pool. Gastroenterology. 2011;141:1003–1013.e1-10. doi: 10.1053/j.gastro.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Velho S, Moutinho C, Cirnes L, Albuquerque C, Hamelin R, Schmitt F, Carneiro F, Oliveira C, Seruca R. BRAF, KRAS and PIK3CA mutations in colorectal serrated polyps and cancer: primary or secondary genetic events in colorectal carcinogenesis. BMC Cancer. 2008;8:255. doi: 10.1186/1471-2407-8-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 41.Fearon ER, Wicha MS. KRAS and cancer stem cells in APC-mutant colorectal cancer. J Natl Cancer Inst. 2014;106(2):djt444. doi: 10.1093/jnci/djt444. [DOI] [PubMed] [Google Scholar]
- 42.Jeong WJ, Yoon J, Park JC, Lee SH, Lee SH, Kaduwal S, Kim H, Yoon JB, Choi KY. Ras stabilization through aberrant activation of Wnt/β-catenin signaling promotes intestinal tumorigenesis. Sci Signal. 2012;5(219):ra30. doi: 10.1126/scisignal.2002242. [DOI] [PubMed] [Google Scholar]
- 43.Park KS, Jeon SH, Kim SE, Bahk YY, Holmen SL, Williams BO, Chung KC, Surh YJ, Choi KY. APC inhibits ERK pathway activation and cellular proliferation induced by RAS. J Cell Sci. 2006;119:819–827. doi: 10.1242/jcs.02779. [DOI] [PubMed] [Google Scholar]
- 44.Phelps RA, Chidester S, Dehghanizadeh S, Phelps J, Sandoval IT, Rai K, Broadbent T, Sarkar S, Burt RW, Jones DA. A two-step model for colon adenoma initiation and progression caused by APC loss. Cell. 2009;137:623–634. doi: 10.1016/j.cell.2009.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li J, Mizukami Y, Zhang X, Jo WS, Chung DC. Oncogenic K-ras stimulates Wnt signaling in colon cancer through inhibition of GSK-3beta. Gastroenterology. 2005;128:1907–1918. doi: 10.1053/j.gastro.2005.02.067. [DOI] [PubMed] [Google Scholar]
- 46.Castellano E, Downward J. RAS Interaction with PI3K: more than just another effector pathway. Genes Cancer. 2011;2:261–274. doi: 10.1177/1947601911408079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lemieux E, Cagnol S, Beaudry K, Carrier J, Rivard N. Oncogenic KRAS signalling promotes the Wnt/β-catenin pathway through LRP6 in colorectal cancer. Oncogene. 2015;34:4914–4927. doi: 10.1038/onc.2014.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ahn DH, Ciombor KK, Mikhail S, Bekaii-Saab T. Genomic diversity of colorectal cancer: Changing landscape and emerging targets. World J Gastroenterol. 2016;22:5668–5677. doi: 10.3748/wjg.v22.i25.5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Le Rolle AF, Chiu TK, Zeng Z, Shia J, Weiser MR, Paty PB, Chiu VK. Oncogenic KRAS activates an embryonic stem cell-like program in human colon cancer initiation. Oncotarget. 2016;7:2159–2174. doi: 10.18632/oncotarget.6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ronai Z. Ras oncogene detection in pre-neoplastic lesions: possible applications for diagnosis and prevention. Oncol Res. 1992;4:45–48. [PubMed] [Google Scholar]
- 52.Ishii M, Sugai T, Habano W, Nakamura S. Analysis of Ki-ras gene mutations within the same tumor using a single tumor crypt in colorectal carcinomas. J Gastroenterol. 2004;39:544–549. doi: 10.1007/s00535-003-1340-3. [DOI] [PubMed] [Google Scholar]
- 53.Raskov H, Pommergaard HC, Burcharth J, Rosenberg J. Colorectal carcinogenesis-update and perspectives. World J Gastroenterol. 2014;20:18151–18164. doi: 10.3748/wjg.v20.i48.18151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Einspahr JG, Martinez ME, Jiang R, Hsu CH, Rashid A, Bhattacharrya AK, Ahnen DJ, Jacobs ET, Houlihan PS, Webb CR, Alberts DS, Hamilton SR. Associations of Ki-ras proto-oncogene mutation and p53 gene overexpression in sporadic colorectal adenomas with demographic and clinicopathologic characteristics. Cancer Epidemiol Biomarkers Prev. 2006;15:1443–1450. doi: 10.1158/1055-9965.EPI-06-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yadamsuren EA, Nagy S, Pajor L, Lacza A, Bogner B. Characteristics of advanced- and non advanced sporadic polypoid colorectal adenomas: correlation to KRAS mutations. Pathol Oncol Res. 2012;18:1077–1084. doi: 10.1007/s12253-012-9547-3. [DOI] [PubMed] [Google Scholar]
- 56.Morkel M, Riemer P, Bläker H, Sers C. Similar but different: distinct roles for KRAS and BRAF oncogenes in colorectal cancer development and therapy resistance. Oncotarget. 2015;6:20785–20800. doi: 10.18632/oncotarget.4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kumar R, Sukumar S, Barbacid M. Activation of ras oncogenes preceding the onset of neoplasia. Science. 1990;248:1101–1104. doi: 10.1126/science.2188364. [DOI] [PubMed] [Google Scholar]
- 58.Zhu D, Keohavong P, Finkelstein SD, Swalsky P, Bakker A, Weissfeld J, Srivastava S, Whiteside TL. K-ras gene mutations in normal colorectal tissues from K-ras mutation-positive colorectal cancer patients. Cancer Res. 1997;57:2485–2492. [PubMed] [Google Scholar]
- 59.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hagag N, Diamond L, Palermo R, Lyubsky S. High expression of ras p21 correlates with increased rate of abnormal mitosis in NIH3T3 cells. Oncogene. 1990;5:1481–1489. [PubMed] [Google Scholar]
- 61.Denko N, Stringer J, Wani M, Stambrook P. Mitotic and post mitotic consequences of genomic instability induced by oncogenic Ha-ras. Somat Cell Mol Genet. 1995;21:241–253. doi: 10.1007/BF02255779. [DOI] [PubMed] [Google Scholar]
- 62.Denko NC, Giaccia AJ, Stringer JR and, Stambrook PJ. The human Ha-ras oncogene induces genomic instability in murine fibroblasts within one cell cycle. Proc Natl Acad Sci USA 1994; 1994;91:5124–5128. doi: 10.1073/pnas.91.11.5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wani MA, Denko NC, Stambrook PJ. Expression of Rap 1 suppresses genomic instability of H-ras transformed mouse fibroblasts. Somat Cell Mol Genet. 1997;23:123–133. doi: 10.1007/BF02679971. [DOI] [PubMed] [Google Scholar]
- 64.Guerra C, Mijimolle N, Dhawahir A, Dubious, Barradas M, Serrano M, Campuzano V, Barbacid M. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell. 2003;4:111–120. doi: 10.1016/s1535-6108(03)00191-0. [DOI] [PubMed] [Google Scholar]
- 65.Tsunoda T, Takashima Y, Fujimoto T, Koyanagi M, Yoshida Y, Doi K, Tanaka Y, Kuroki M, Sasazuki T, Shirasawa S. Three-dimensionally specific inhibition of DNA repair-related genes by activated KRAS in colon crypt model. Neoplasia. 2010;12:397–404. doi: 10.1593/neo.10170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zauber P, Marotta S, Sabbath-Solitare M. Colorectal cancers with the uncommon findings of KRAS mutation and microsatellite instability. Cytogenet Genome Res. 2015;146:261–267. doi: 10.1159/000441086. [DOI] [PubMed] [Google Scholar]
- 67.Yang G, Mercado-Uribe I, Multani AS, Sen S, Shih IeM, Wong KK, Gershenson DM, Liu J. RAS promotes tumorigenesis through genomic instability induced by imbalanced expression of Aurora-A and BRCA2 in midbody during cytokinesis. Int J Cancer. 2013;133:275–285. doi: 10.1002/ijc.28032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Abulaiti A, Fikaris AJ, Tsygankova OM, Meinkoth JL. Ras induces chromosome instability and abrogation of the DNA damage response. Cancer Res. 2006;66:10505–10512. doi: 10.1158/0008-5472.CAN-06-2351. [DOI] [PubMed] [Google Scholar]
- 69.Rao CV, Yamada HY. Genomic instability and colon carcinogenesis: from the perspective of genes. Front Oncol. 2013;3:130. doi: 10.3389/fonc.2013.00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kamata T, Pritchard C. Mechanisms of aneuploidy induction by RAS and RAF oncogenes. Am J Cancer Res. 2011;1:955–971. [PMC free article] [PubMed] [Google Scholar]
- 71.Minamoto T, Yamashita N, Ochiai A, Mai M, Sugimura T, Ronai Z, Esumi H. Mutant K-ras in apparently normal mucosa of colorectal cancer patients. Its potential as a biomarker of colorectal tumorigenesis. Cancer. 1995;75:1520–1526. doi: 10.1002/1097-0142(19950315)75:6+<1520::aid-cncr2820751523>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 72.Minamoto T, Ronai Z, Yamashita N, Ochiai A, Sugimura T, Mai M, Esumi H. Detection of Ki-ras mutation in non-neoplastic mucosa of Japanese patients with colorectal cancers. Int J Oncol. 1994;4:397–401. doi: 10.3892/ijo.4.2.397. [DOI] [PubMed] [Google Scholar]
- 73.Luo F, Poulogiannis G, Ye H, Hamoudi R, Zhang W, Dong G, Arends MJ. Mutant K-ras promotes carcinogen-induced murine colorectal tumourigenesis, but does not alter tumour chromosome stability. J Pathol. 2011;223:390–399. doi: 10.1002/path.2790. [DOI] [PubMed] [Google Scholar]
- 74.Jacoby RF, Llor X, Teng BB, Davidson NO, Brasitus TA. Mutations in the K-ras oncogene induced by 1, 2-dimethylhydrazine in preneoplastic and neoplastic rat colonic mucosa. J Clin Invest. 1991;87:624–630. doi: 10.1172/JCI115039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Luo F, Poulogiannis G, Ye H, Hamoudi R, Dong G, Zhang W, Ibrahim AE, Arends MJ. Wild-type K-ras has a tumour suppressor effect on carcinogen-induced murine colorectal adenoma formation. Int J Exp Pathol. 2014;95:8–15. doi: 10.1111/iep.12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Diaz R, Lue J, Mathews J, Yoon A, Ahn D, Garcia-España A, Leonardi P, Vargas MP, Pellicer A. Inhibition of Ras oncogenic activity by Ras protooncogenes. Int J Cancer. 2005;113:241–248. doi: 10.1002/ijc.20563. [DOI] [PubMed] [Google Scholar]
- 77.Gorgoulis VG, Halazonetis TD. Oncogene-induced senescence: the bright and dark side of the response. Curr Opin Cell Biol. 2010;22:816–827. doi: 10.1016/j.ceb.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 78.Grabocka E, Commisso C, Bar-Sagi D. Molecular pathways: targeting the dependence of mutant RAS cancers on the DNA damage response. Clin Cancer Res. 2015;21:1243–1247. doi: 10.1158/1078-0432.CCR-14-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Park MT, Kim MJ, Suh Y, Kim RK, Kim H, Lim EJ, Yoo KC, Lee GH, Kim YH, Hwang SG, Yi JM, Lee SJ. Novel signaling axis for ROS generation during K-Ras-induced cellular transformation. Cell Death Differ. 2014;21:1185–1197. doi: 10.1038/cdd.2014.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grabocka E, Pylayeva-Gupta Y, Jones MJ, Lubkov V, Yemanaberhan E, Taylor L, Jeng HH, Bar-Sagi D. Wild-type H- and N-Ras promote mutant K-Ras-driven tumorigenesis by modulating the DNA damage response. Cancer Cell. 2014;25:243–256. doi: 10.1016/j.ccr.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yamada S, Yashiro M, Maeda K, Nishiguchi Y, Hirakawa K. A novel high-specificity approach for colorectal neoplasia: Detection of K ras2 oncogene mutation in normal mucosa. Int J Cancer. 2005;113:1015–1021. doi: 10.1002/ijc.20666. [DOI] [PubMed] [Google Scholar]
- 82.Poulogiannis G, Luo F, Arends MJ. RAS signalling in the colorectum in health and disease. Cell Commun Adhes. 2012;19:1–9. doi: 10.3109/15419061.2011.649380. [DOI] [PubMed] [Google Scholar]
- 83.Vieth M, Quirke P, Lambert R, von Karsa L, Risio M. Quality assurance in pathology in colorectal cancer screening and diagnosis: annotations of colorectal lesions. VirchowsArch. 2011;458:21–30. doi: 10.1007/s00428-010-0997-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nagorni A, Katic V, Zivkovic V, Stanojevic G. Advanced colorectal adenoma. Arch Oncol. 2004;12:59–60. [Google Scholar]
- 85.Ronai Z, Luo F, Gradia S, Hart W, Butler R. Detection of K-ras mutation in normal and malignant colonic tissues by an enriched PCR method. Int J Oncol. 1994;4:391–396. doi: 10.3892/ijo.4.2.391. [DOI] [PubMed] [Google Scholar]
- 86.Tobi M, Luo FC, Ronai Z. Detection of K-ras mutation in colonic effluent samples from patients without evidence of colorectal carcinoma. J Natl Cancer Inst. 1994;86:1007–1010. doi: 10.1093/jnci/86.13.1007. [DOI] [PubMed] [Google Scholar]
- 87.Sidransky D, Tokino T, Hamilton SR, Kinzler KW, Levin B, Frost P, Vogelstein B. Identification of ras oncogene mutations in the stool of patients with curable colorectal tumors. Science. 1992;256:102–105. doi: 10.1126/science.1566048. [DOI] [PubMed] [Google Scholar]
- 88.Ronai Z, Minamoto T, Butler R, Tobi M, Luo FQ, Zang E, Esumi H, Sugimura T. Sampling method as a key factor in identifying K-ras oncogene mutations in preneoplastic colorectal lesions. Cancer Detect Prev. 1995;19:512–517. [PubMed] [Google Scholar]
- 89.Hamilton SR. Molecular genetics of colorectal carcinoma. Cancer. 1992;70:1216–1221. doi: 10.1002/1097-0142(19920901)70:3+<1216::aid-cncr2820701505>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 90.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Worthley DL, Whitehall VL, Spring KJ, Leggett BA. Colorectal carcinogenesis: road maps to cancer. World J Gastroenterol. 2007;13:3784–3791. doi: 10.3748/wjg.v13.i28.3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Langner C. Serrated and non-serrated precursor lesions of colorectal cancer. Dig Dis. 2015;33:28–37. doi: 10.1159/000366032. [DOI] [PubMed] [Google Scholar]
- 93.Leslie A, Carey FA, Pratt NR, Steele RJ. The colorectal adenoma - carcinoma sequence. Br J Surg. 2002;89:845–860. doi: 10.1046/j.1365-2168.2002.02120.x. [DOI] [PubMed] [Google Scholar]
- 94.Sovich JL, Sartor Z, Misra S. Developments in Screening Tests and Strategies for Colorectal Cancer. Biomed Res Int. 2015;2015:326728. doi: 10.1155/2015/326728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Heresbach D, Barrioz T, Lapalus MG, Coumaros D, Bauret P, Potier P, Sautereau D, Boustière C, Grimaud JC, Barthélémy C, Sée J, Serraj I, D’Halluin PN, Branger B, Ponchon T. Miss rate for colorectal neoplastic polyps: a prospective multicenter study of back-to-back video colonoscopies. Endoscopy. 2008;40:284–290. doi: 10.1055/s-2007-995618. [DOI] [PubMed] [Google Scholar]
- 96.De Palma GD, Maione F, Esposito D, Luglio G, Giglio MC, Siciliano S, Gennarelli N, Cassese G, Campione S, D’Armiento FP, Bucci L. In vivo assessment of tumour angiogenesis in colorectal cancer: the role of confocal laser endomicroscopy. Colorectal Dis. 2016;18(2):O66–73. doi: 10.1111/codi.13222. [DOI] [PubMed] [Google Scholar]
- 97.Al-Shamsi HO, Alhazzani W, Wolff RA. Extended RAS testing in metastatic colorectal cancer-Refining the predictive molecular biomarkers. J Gastrointest Oncol. 2015;6:314–321. doi: 10.3978/j.issn.2078-6891.2015.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bosman F, Yan P. Molecular pathology of colorectal cancer. Pol J Pathol. 2014;65:257–266. doi: 10.5114/pjp.2014.48094. [DOI] [PubMed] [Google Scholar]
- 99.Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008;135:1079–1099. doi: 10.1053/j.gastro.2008.07.076. [DOI] [PMC free article] [PubMed] [Google Scholar]