

Abstract
Idiopathic pulmonary fibrosis (IPF) is a rare interstitial lung disease limited to the lung with an undefined etiopathogenesis and a very short life expectancy (less than 5 years). IPF susceptibility has been associated with several genetic and environmental risk factors and the prognosis is conditioned by comorbidities such as gastro-esophageal reflux, depression, venous thromboembolism, pulmonary hypertension and lung cancer. At 5 years follow-up, 15% of IPF patients develop lung cancer, which can significantly reduce their survival. Because diagnostic or therapeutic procedures such as surgical, radiation or pharmacological treatments may induce acute exacerbations and increase mortality, the management of lung cancer in IPF patients is a very difficult task. This study discusses advantages and disadvantages of lung cancer treatments in patients with severe IPF, highlighting several controversial aspects on this topic, including potential nintedanib treatment.
Keywords: Lung cancer, idiopathic pulmonary fibrosis, nintedanib, prognosis, therapy
Idiopathic pulmonary fibrosis (IPF) is an interstitial lung disease with poor prognosis and an undefined etiopathogenesis. Several genetic and environmental risk factors for IPF have been identified including cigarette smoking, metal dust and other pollutant exposure, genetic mutations and polymorphisms (1-5). IPF prognosis is really poor (less than 5 years) and is conditioned by comorbidities negatively affecting patients’ quality of life and survival (e.g. gastro-esophageal reflux, depression, venous thromboembolism, pulmonary hypertension and other cardiovascular disorders, and lung cancer) (6-10). The incidence of pulmonary neoplasm is much higher in IPF patients than in the general population (11). Ozawa et al. (12) for instance, reported 3.3%, 15.4% and 54.7% of lung cancer incidence in IPF patients after one, five and ten years of follow-up, respectively (12). Common mechanisms shared by the two diseases remain unknown. An aberrant expression of miRNAs regulating non-small cell lung cancer (NSCLC) and IPF has been suggested together with a crucial role of tyrosine kinase inhibitor directed against growth factors (platelet-derived growth factor, vascular endothelial growth factor and fibroblast growth factor) (13,14).
Lung cancer (LC) significantly reduces survival of IPF patients, including patients with combined pulmonary emphysema and pulmonary fibrosis (15). It generally occurs in the peripheral areas of the lower lobes, where fibrotic changes are predominant. In particular, LC develops from honeycomb areas or in the border between honeycombing and non fibrotic areas and the squamous cell carcinoma is the predominant histological type (16,17). Invasive diagnostic or therapeutic procedures for neoplasms promote the development of Acute exacerbations (AE) in 80% of IPF patients and the lung surgical treatment puts about 1/5 of them at high risk of death (17,18). A perioperative Pirfenidone treatment has recently been proposed to reduce the incidence of postoperative acute events (18-20). In this context, the therapeutic management of LC is a difficult task and is still questioned, especially in IPF patients with severe fibrotic involvement, whether the tumor should be treated or not (21,22). Although Lee et al. (15) reported that radiotherapy and surgical treatments of LC result in a reduced survival of IPF patients, according to Kumar et al. (19) lung resection in selected cases can improve their survival. Furthermore, Tomassetti and colleagues (21) have suggested that in mild IPF patients the sublobar resection in stage I LC is safer than lobectomy, although this approach can increase the risk of LC recurrences. However, available knowledge indicates that the risk of AE is higher in treated than in untreated patients (21) and indications for diagnostic and therapeutic procedures for LC in IPF patients are lacking. This article aimed at contributing to the debate over pros and cons of LC treatment, reporting and discussing the clinical outcomes of patients with severe IPF and suspected lung malignant nodules.
Case Report
A 79 year old man, ex-smoker, with bilateral glaucoma and hypovitaminosis D referred to us for severe cough and dyspnea. The patient was diagnosed with IPF, based on clinical and radiological features (definite usual interstitial pneumonia (UIP) pattern). The rheumatologic tests excluded an underlying autoimmune process and, in 2012, the patient started Pirfenidone treatment. In March 2014, High resolution computed tomography (HRCT) of the chest performed for routine follow-up revealed a pulmonary nodule in the right upper lobe, PET positive (Figure 1A). In the same period we witnessed a rapid deterioration of the interstitial lung disease with deterioration of lung function parameters, oxygen request and evident radiological progression. The multidisciplinary decision was to start Nintedanib therapy, avoiding any diagnostic or therapeutic approach to the suspected malignant nodule. Since January 2015, we shifted the patient to Nintedanib at a dosage of 150 mg twice a day. The radiological follow-up performed in the next 12 months with lung function tests, HRCT of the chest and blood gas analysis, revealed a complete stability of IPF as well as the lung nodule (lung function tests revealed forced vital capacity 50%,total lung capacity 44%, diffuse capacity for carbon monoxide 25% while, the six minute walking test and HRCT of the chest remained stable).
Figure 1. A, B: The pulmonary nodules in two patients with idiopathic pulmonary fibrosis.
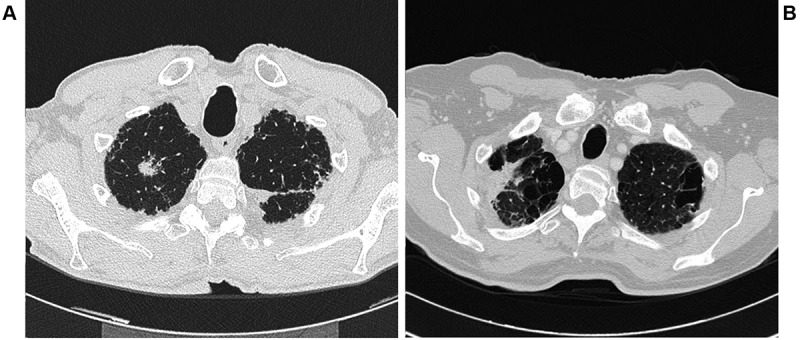
The second patient was a 74 year old male, ex-smoker (50 pack year), with no history of professional exposure. The patient had a complex clinical history that influenced our treatment choices: mellitus diabetes, chronic atrial fibrillation in treatment withan anticoagulant, arterial hypertension, abdominal aortic aneurysm, hypertrophic dilated cardiomyopathy, previous bladder cancer surgically treated. In June 2016, because of dyspnea, the patient underwent HRCT of the chest, demonstrating diffuse panlobular emphysema of the upper lobes, a bibasal definite UIP pattern with advanced peripheral honeycombing, and a nodule of 35 mm in the right upper lobe, with a fibrotic component and projected towards the hilum of the lung (Figure 1B). PET evaluation revealed high level of capitation in this area and in the mediastinal lymph nodes. Lung function tests revealed a severe reduction of DLCO (<30%), and blood gas analysis showed reduced Pa02, but in order to obtain a diagnosis a bronchoscopy with Bronchoalveolar lavage (BAL) and Transbronchial biopsy (TBNA) of lympho nodes was performed. Unfortunately no diagnosis was reached and a surgical biopsy for the suspect lung cancer was excluded because of high risk of complications related to the advanced pulmonary fibrosis. Last HRCT of the chest revealed an increased density of the mass with mediastinal lymph node involvement. Our multidisciplinary evaluation concluded in starting pharmacological treatment inthe patient with combined IPF and emphysema and suspected lung cancer. Nintedanib was started also to obtain a potential control in the evolution of the lung nodule.
The third IPF male patient was 83 years old, no smoker, with persistent dry cough and dyspnea. The patient had undergone several investigations due to respiratory symptoms and bibasal crackles present at physical examination. Lung function tests revealed severe restrictive deficit with DLCO <35%. HRCT showed a definite UIP pattern with patchy bibasal areas of end-stage lungs and traction bronchiectasis. The concomitant presence of two nodular lesions in the right upper lobe and in the left lower lobe was observed. PET revealed-high level of capitation of lung nodules, mediastinal, pretracheal, subcarinal and hilar lymph nodes. Autoantibodies were negative and bronchoscopy with BAL excluded other interstitial lung diseases (ILDs). No cytological anomalies were documented in BAL samples, and TBNA was not diagnostic for neoplastic disease. Radiological exams were evaluated by the thoracic surgeon who confirmed the suspected malignant nature of the mass, but refused a surgical approach for diagnostic and therapeutic reasons due to the associated high risk of acute exacerbations. Nintedanib treatment was started in October 2016 at the dose of 150 mg twice a day.
The last patient was a 79-year-old man affected by advanced dysmetabolic liver cirrhosis complicated by portal hypertension. The patient referred dry recurrent cough and exertion dyspnea and he performed a HRTC of the chest showing a definite UIP pattern and the presence of a lung nodule in the right upper lobe PET positive. Lung function tests documented a restrictive deficit with severe DLCO impairment (13% of theoretical value). Management of IPF and lung cancer in such cases is even more complicated because of advanced liver disease (associated with ascites, hepatorenal syndrome and esophageal variceal hemorrhage) and doing invasive procedures or staring pharmacological therapies could have a relevant risk.
Discussion
IPF is a severe fibrotic disorder that shares common pathogenetic features with lung cancer. Several proteins up-regulated in IPF are directly involved in carcinogenesis and lung areas of active fibrosis may represent potential targets of lung neoplasms (13,23). In IPF patients, invasive diagnostic procedures, as well as surgical or pharmacological treatments may induce acute exacerbation and increased mortality (19,20,22).
In this study we described the medical histories of patients with IPF and suspected malignant lung nodules, in order to highlight the difficulties in the diagnostic and therapeutic management of these combined diseases. Lung cancer in IPF patients generally starts in peripheral areas of lung fibrosis. If fine needle aspiration biopsy (FNAB) can not be performed or is not diagnostic, the management of these patients becomes difficult (16). The international guidelines for the diagnosis and treatment of IPF do not provide a clear strategy on how to manage these patients whose survival is reduced by the presence of the highly suspected or even histologically confirmed lung cancer (1,9,10). Several studies have evaluated cancer-like outcomes in IPF (21,24,25). The most frequently used prognostic indicator in IPF (independently from lung neoplasm) is the progression-free survival (24). Unfortunately, it is unclear if it is correct to apply this indicator derived from oncologic studies to IPF. The ‘overall survival’ (OS) is the principal endpoint of oncology clinical trials, defined as “the time from randomization until death (from any cause)”. It is a universally accepted measure of direct benefit, easily and precisely detectable, but it requires large trial populations and long follow-up to identify statistical differences between different groups; it can be affected by crossover or subsequent therapies and includes deaths unrelated to cancer (26). Therefore, two other endpoints have been introduced: progression-free survival, which is the time from randomization until disease progression or death, and the time to progression, which includes the time from randomization until objective tumor progression (it does not include deaths). The latter endpoints are only applicable to limited sample sizes and shorter follow-up times than OS, they are not affected by crossover or subsequent therapies, and are generally based on objective and quantitative assessment (26). However, these endpoints are useful only if based on widely accepted and validated criteria including anatomical tumor burden. Such criteria, known as RECIST (Response Evaluation Criteria in Solid Tumors), were published 17 years ago and established the minimal size of measurable lesions, how to manage the follow-up of malignant lesions up to 10 (a maximum five/organ site), and the use of unidimensional, rather than bidimensional, measures for overall evaluation of tumor burden (26). The RECIST criteria were revised in 2009 and are still adopted by academic institutions, cooperative groups, and industry for trials where the primary endpoints are objective response or progression (26). These parameters are helpful to evaluate the extension of lung solid cancers (with some limits when evaluating semi-solid, multifocal lesions or ground glass opacities). This experience with prognostic bioindicators in the oncological field has been applied to IPF straightaway, in a mechanistic way, considering the progression-free survival rather than the overall survival to evaluate clinical response and IPF progression after the beginning of a treatment. King et al. (27) reported many individual clinical variables predictive of IPF survival which can be divided into clinical predictors (age, sex, ethnicity, smoking status, dyspnea, physical findings, comorbidities), radiographical predictors (number of parenchymal abnormalities and extension of fibrosis), physiologic predictors (changes in FVC and DLCO), biomarkers from BAL and pathologic predictors (25). These clinical variables may be useful to derive clinical prediction models, but larger population sizes and longer follow-ups are required for their development (27). Therefore, it is complicated to evaluate the prognosis of a patient with advanced IPF and lung cancer and to establish a correct diagnostic and prognostic approach, also considering the potential negative effects of radio- and chemotherapies in these patients (9,10,28,29). As reported in our case series in the subpopulation of IPF patients with suspected lung cancer, the decision to start nintedanib therapy is commonly taken in order to treat the advanced fibrotic lung disease and the lung cancer (29). Nintedanib is a potent triple angiokinase inhibitor that suppresses the proangiogenic pathways mediated by vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF) and platelet derived growth factor (PDGF) with a high degree of specificity (30-32). It competitively binds to the ATP binding-pocket of these receptors and also inhibits some non-receptor kinases such as the proto-oncogene RET (31). This approved therapy for IPF is capable to reduce FVC decline reducing disease progression. It can be orally administered at the dosages of 100 or 150 mg twice a day according to drug tolerability in IPF (30). Nintedanib is approved for the second line treatment of patients with NSCLC adenocarcinoma subtype, at the dosage of 200 mg twice a day. In the phase III trials LUME-lung 1, Nintedanib plus Docetaxel showed a significantly longer progression-free survival with more benefits in patients with adenocarcinoma subtype (33,34). Unfortunately, no data are available on the potential effects of nintedanib in monotherapy (without docetaxel) at the dosage of 150 mg twice a day for the treatment of lung cancer associated with severe IPF, and it would be a really relevant goal to understand if these patients have any benefits from this treatment (35,37). The open questions on this topic include: may nintedanib stabilize both pathologies increasing patient’s survival? May nintedanib be used in squamous lung tumors without previous chemotherapy regimen as first line? Should we use nintedanib alone, without docetaxel, as chemotherapy treatment in IPF and lung cancer without a histological diagnosis?
These open questions require further evaluations and need properly designed clinical trials to help patients with severe IPF and associated lung neoplasms and to establish if it is correct to treat them with nintedanib following an ‘a priori’ approach.
Conflicts of Interest
Elena Bargagli has received speaker honorarium from Roche, Boheringer and Novartis, Viola Bont has no conflict of interest to declare, Katia Ferrari has received speaker honorarium from Roche and Boheringer, Elisabetta Rosi from Roche and Boheringer, Bindi Alessandra from Roche and Boheringer, Bartolucci Maurizio has not received grants, Moroni Chiara speaker honorarium from Roche and Boheringer, Massimo Pistolesi has received research grants from Roche, Boheringer, Novartis, Luca Voltolini from Roche and Boheringer.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
References
- 1.King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949–1961. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 2.Landi C, Bargagli E, Bianchi L, Gagliardi A, Carleo A, Bennett D, Perari MG, Armini A, Prasse A, Rottoli P, Bini L. Towards a functional proteomics approach to the comprehension of idiopathic pulmonary fibrosis, sarcoidosis, systemic sclerosis and pulmonary Langerhans cell histiocytosis. J Proteomics. 2013;83:60–75. doi: 10.1016/j.jprot.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 3.Cameli P, Bargagli E, Refini RM, Pieroni MG, Bennett D, Rottoli P. Exhaled nitric oxide in interstitial lung diseases. Respir Physiol Neurobiol. 2014;197:46–52. doi: 10.1016/j.resp.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 4.Bargagli E, Olivieri C, Bennett D, Prasse A, Muller-Quernheim J, Rottoli P. Oxidative stress in the pathogenesis of diffuse lung diseases: a review. Respir Med. 2009;103(9):1245–1256. doi: 10.1016/j.rmed.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 5.Mushiroda T, Wattanapokayakit S, Takahashi A, Nukiwa T, Kudoh S, Ogura T, Taniguchi H, Kubo M, Kamatani N, Nakamura Y, Pirfenidone Clinical Study Group A genome-wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J Med Genet. 2008;45:654–656. doi: 10.1136/jmg.2008.057356. [DOI] [PubMed] [Google Scholar]
- 6.Ryu JH, Moua T, Daniels CE, Hartman TE, Yi ES, Utz JP, Limper AH. Idiopathic pulmonary fibrosis: evolving concepts. Mayo Clin Proc. 2014;89:1130–1142. doi: 10.1016/j.mayocp.2014.03.016. [DOI] [PubMed] [Google Scholar]
- 7.Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–151. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- 8.Raghu G, Amatto V, Behr J, Stowasser S. Comorbidities in idiopathic pulmonary fibrosis patients: a systematic literature review. Eur Respir J. 2015;46(4):1113–1130. doi: 10.1183/13993003.02316-2014. [DOI] [PubMed] [Google Scholar]
- 9.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE Jr, Kondoh Y, Myers J, Müller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schünemann HJ, ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Travis WD, Costabel U, Hansell DM, King TE Jr., Lynch DA, Nicholson AG, Ryerson CJ, Ryu JH, Selman M, Wells AU, Behr J, Bouros D, Brown KK, Colby TV, Collard HR, Cordeiro CR, Cottin V, Crestani B, Drent M, Dudden RF, Egan J, Flaherty K, Hogaboam C, Inoue Y, Johkoh T, Kim DS, Kitaichi M, Loyd J, Martinez FJ, Myers J, Protzko S, Raghu G, Richeldi L, Sverzellati N, Swigris J, Valeyre D, ATS/ERS Committee on Idiopathic Interstitial Pneumonias An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–748. doi: 10.1164/rccm.201308-1483ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le Jeune I, Gribbin J, West J, Smith C, Cullinan P, Hubbard R. The incidence of cancer in patients with Idiopathic Pulmonary Fibrosis and Sarcoidosis in the UK. Respir Med. 2007;101(12):2534–2540. doi: 10.1016/j.rmed.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 12.Ozawa Y, Suda T, Naito T, Enomoto N, Hashimoto D, Fujisawa T, Nakamura Y, Inui N, Nakamura H, Chida K. Cumulative incidence of and predictive factors for lung cancer in IPF. Respirology. 2009;14(5):723–728. doi: 10.1111/j.1440-1843.2009.01547.x. [DOI] [PubMed] [Google Scholar]
- 13.Vancheri C. Common pathways in idiopathic pulmonary fibrosis and cancer. Eur Resp Rev. 2013;22:265–272. doi: 10.1183/09059180.00003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Antoniou K, Tomassetti S, Tsitoura E, Vancheri C. Idiopathic pulmonary fibrosis and lung cancer: a clinical and pathogenesis update. Curr Opin Pulm Med. 2015;21:626–633. doi: 10.1097/MCP.0000000000000217. [DOI] [PubMed] [Google Scholar]
- 15.Lee T, Park JY, Lee HY, Cho YJ, Yoon HI, Lee JH, Jheon S, Lee CT, Park JS. Lung cancer in patients with idiopathic pulmonary fibrosis: clinical characteristics and impact on survival. Respir Med. 2014;108:1549–1555. doi: 10.1016/j.rmed.2014.07.020. [DOI] [PubMed] [Google Scholar]
- 16.Khan KA, Kennedy MP, Moore E, Crush L, Prendeville S, Maher MM, Burke L, Henry MT. Radiological characteristics, histological features and clinical outcomes of lung cancer patients with coexistent idiopathic pulmonary fibrosis. Lung. 2015;193:71–77. doi: 10.1007/s00408-014-9664-8. [DOI] [PubMed] [Google Scholar]
- 17.Vancheri C. Idiopathic pulmonary fibrosis and cancer: do they really look similar. BMC Med. 2015;13:220–228. doi: 10.1186/s12916-015-0478-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Junyao, Yang Ming, Li Ping, Su Z, Gao P, Zhang J. Idiopathic pulmonary fibrosis will increase the risk of lung cancer. Clin Med J. 2014;127(17):3142–3149. [PubMed] [Google Scholar]
- 19.Kumar P, Goldstraw P, Yamada K, Nicholson A andWells A. Pulmonary fibrosis and lung cancer: risk and benefit analysis of pulmonary resection. J Thorac Cardiovasc Surg. 2013;125:1321–1327. doi: 10.1016/s0022-5223(03)00028-x. [DOI] [PubMed] [Google Scholar]
- 20.Iwata T, Yoshino I, Yoshida S, Ikeda N, Tsuboi M, Asato Y, Katakami N, Sakamoto K, Yamashita Y, Okami J, Mitsudomi T, Yamashita M, Yokouchi H, Okubo K, Okada M, Takenoyama M, Chida M, Tomii K, Matsuura M, Azuma A, Iwasawa T, Kuwano K, Sakai S, Hiroshima K, Fukuoka J, Yoshimura K, Tada H, Nakagawa K, Nakanishi Y, West Japan Oncology Group A phase III trial evaluating the efficacy and safety of perioperative pirfenidone for prevention of acute exacerbation of idiopathic pulmonary fibrosis in lung cancer patients undergoing pulmonary resection: West Japan Oncology Group 6711 L (PEOPLE study) Resp Research. 2016;17:90–99. doi: 10.1186/s12931-016-0398-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tomassetti S, Gurioli C, Ryu JH, Decker PA, Ravaglia C, Tantalocco P, Buccioli M, Piciucchi S, Sverzellati N, Dubini A, Gavelli G, Chilosi M, Poletti V. The impact of lung cancer on survival of idiopathic pulmonary fibrosis. Chest. 2015;147(1):157–164. doi: 10.1378/chest.14-0359. [DOI] [PubMed] [Google Scholar]
- 22.Voltolini L, Bongiolatti S, Luzzi L, Bargagli E, Fossi A, Ghiribelli C, Rottoli P, Gotti G. Impact of interstitial lung disease on short-term and long-term survival of patients undergoing surgery for non-small-cell lung cancer: analysis of risk factors. Eur J Cardiothorac Surg. 2013;43(1):17–23. doi: 10.1093/ejcts/ezs560. [DOI] [PubMed] [Google Scholar]
- 23.Lehtonen ST, Veijola A, Karvonen H, Lappi-Blanco E, Sormunen R, Korpela S, Zagai U, Sköld MC, Kaarteenaho R. Pirfenidone and nintedanib modulate properties of fibroblasts and myofibroblasts in idiopathic pulmonary fibrosis. Resp Res. 2016;17:14–18. doi: 10.1186/s12931-016-0328-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vancheri C, Du Bois MR. A progression-free end-point for idiopathic pulmonary fibrosis trials: lessons from cancer. Eur Resp J. 2013;41:262–269. doi: 10.1183/09031936.00115112. [DOI] [PubMed] [Google Scholar]
- 25.Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in Idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183:431–440. doi: 10.1164/rccm.201006-0894CI. [DOI] [PubMed] [Google Scholar]
- 26.Eisenhauera EA, Therasseb P, Bogaertsc J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J. New response evaluation criteria in solid tumours: revised RECIST guideline. Eur J of Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 27.King TE Jr., Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM, Kardatzke D, Lancaster L, Lederer DJ, Nathan SD, Pereira CA, Sahn SA, Sussman R, Swigris JJ, Noble PW, ASCEND Study Group A Phase 3 Trial of Pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–2092. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 28.Puglisi S, Torrisi SE, Vindigni V, Giuliano R, Palmucci S, Mulè M, Vancheri C. New perspectives on management of idiopathic pulmonary fibrosis. Ther Adv Chronic Dis. 2016;7(2):108–120. doi: 10.1177/2040622315624276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richeldi L. Clinical trials of investigational agents for IPF: a review of a Cochrane report. Respir Res. 2013;14(Suppl 1):S4. doi: 10.1186/1465-9921-14-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mazzei ME, Richeldi HR, Collard HR. Nintedanib in the treatment of idiopathic pulmonary fibrosis. Ther Adv Resp Dis. 2015;9(3):121–129. doi: 10.1177/1753465815579365. [DOI] [PubMed] [Google Scholar]
- 31.Chaudhary N, Roth G, Hilberg F, Müller-Quernheim J, Prasse A, Zissel G, Schnapp A, Park JE. Inhibition of PDGF, VEGF and FGF signalling attenuates fibrosis. Eur Respir J. 2007;29:976–985. doi: 10.1183/09031936.00152106. [DOI] [PubMed] [Google Scholar]
- 32.Richeldi L, Cottin V, Flaherty KR, Kolb M, Inoue Y, Raghu G, Taniguchi H, Hansell DM, Nicholson AG, Le Maulf F, Stowasser S, Collard HR. Design of the INPULSIS trials: two phase 3 trials of nintedanib in patients with idiopathic pulmonary fibrosis. Respir Med. 2014;108:1023–1030. doi: 10.1016/j.rmed.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 33.Bronte G, Passiglia F, Galvano A, Barraco N, Listì A, Castiglia M, Rizzo S, Fiorentino E, Bazan V, Russo A. Nintedanib in NSCLC: evidence to date and place in therapy. Ther Adv Med Oncol. 2016;8(3):188–197. doi: 10.1177/1758834016630976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cortinovis D, Gregorc V, Migliorino MR, Abate MI, Manzo A, Malapelle U, Morabito A, Italian Lung Cancer Working Group New perspectives in the second line treatment of non squamous NSCLC patients: results from a large Italian Lung Cancer Working Group. Crit Rev Oncol Ematol. 2017;109:35–41. doi: 10.1016/j.critrevonc.2016.11.015. [DOI] [PubMed] [Google Scholar]
- 35.Reck M, Kaiser R, Mellengaard A, Douillard JY, Orlov S, Krzakowski M, von Pawel J, Gottfried M, Bondarenko I, Liao M, Gann CN, Barrueco J, Gaschler-Markefski B, Novello S. LUME-Lung 1 Study Group: Docetaxel plus Nintedanib versus docetaxel plus placebo in patients with previously treated non small cell lung cancer (LUME- Lung 1): a phase 3, double bind, randomised controlled trial. Lancet Oncol. 2014;1: – . doi: 10.1016/S1470-2045(13)70586-2. [DOI] [PubMed] [Google Scholar]
- 36.Wollin L, Wex E, Pautsch A, Schnapp G, Hostettler KE, Stowasser S, Kolb M . Mode of action of Nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Resp J. 2015;45:1434–1445. doi: 10.1183/09031936.00174914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Awasthi N, Schwarz RE. Profile of Nintedanib in the treatment of solid tumors: the evidence to date. Onco Targets Ther. 2015;8:3691–3701. doi: 10.2147/OTT.S78805. [DOI] [PMC free article] [PubMed] [Google Scholar]