

Abstract
Background
Erdheim-Chester disease (ECD) is a rare non-Langerhans cell histiocytosis disorder that utilizes the RAS-RAF-MEK-ERK pathway. It has a highly variable clinical presentation, where virtually any organ can be involved, thus having the potential of posing a great diagnostic challenge. Over half of the reported cases have the BRAF V600E mutation and have shown a remarkable response to vemurafenib.
Case Presentation
We describe herein a patient with a history of stroke-like symptoms and retroperitoneal fibrosis that on initial pathology raised the possibility of IgG4-related disease. However, the patient was refractory to high-dose steroids and progressed further, developing an epicardial soft tissue mass and recurrent neurological symptoms. Integration of the above findings with new information at another hospital about a radiological history of symmetrical lower extremities long bone lesions raised the differential diagnosis of ECD. Molecular analysis of formalin-fixed paraffin-embedded tissue of both of the patient's retroperitoneal biopsies (the second one of which had shown a small focus of foamy histiocytes, CD68+/CD1a–) was positive for BRAF mutation, confirming the diagnosis of ECD. The patient demonstrated a dramatic and sustained metabolic response to vemurafenib on follow-up positron emission tomography scans.
Conclusion
This case highlights the need for developing a high index of suspicion for presentations of retroperitoneal fibrosis that could represent IgG4-related disease but fail to respond to steroids. When unusual multisystem involvement occurs, one should consider a diagnosis of a rare histiocytosis. Vemurafenib appears to be an effective treatment for even advanced cases of both ECD and Langerhans histiocytosis bearing the BRAF V600E mutation.
Keywords: Vemurafenib, BRAF V600E, Erdheim-Chester disease, IgG4-related disease
Introduction
Histiocytic disorders encompass a broad range of diseases with the unifying features of abnormal maturation, differentiation, and proliferation of cells derived from the monocyte-phagocyte lineage. One of them, Erdheim-Chester disease (ECD), presents with systemic dissemination, typically involving skin, bones, pituitary gland, and importantly, retroperitoneum, kidneys, and heart. Highly characteristic of ECD are the coating of the aorta and bilateral long bones sclerotic lesions [1, 2]. While previously considered to be extremely rare, incidence has dramatically risen in recent years probably due to increased recognition. Similarly, another rare pathologic entity, IgG4-related disease, is also known to form retroperitoneal masses with periaortic and pericaval involvement. Clinically, the hallmark of this disease is its excellent response to steroid therapy. Thus, the diagnosis of ECD can present a diagnostic challenge for clinicians and pathologists faced with a retroperitoneal mass encasing the great vessels [3].
We describe a case of a patient who presented with an ill-defined retroperitoneal mass and stroke-like symptoms, in which the neurological symptoms were initially believed to be unrelated to his retroperitoneal lesion, the former possibly representing a complication of his diabetes mellitus and the latter possibly representing IgG4 disease. The lack of integration of information caused a loss of precious time, with clinical deterioration. Despite a nearly 6-month delay, the patient was eventually correctly diagnosed with Erdheim-Chester disease and had a rapid response to vemurafenib.
Case Presentation
A 64-year-old man was admitted to hospital with left-sided weakness, dysarthria, and expressive aphasia after initiation of rituximab therapy for presumptive IgG4-related disease.
His past medical history included hypertension, dyslipidemia, type 2 diabetes mellitus, Crohn's disease, and dysplasia-associated lesion or mass (DALM), which had been treated with proctocolectomy and ileostomy. He had also undergone local radiation for prostate cancer 5 years prior. On admission, he appeared chronically unwell and cachectic. He had no lymphadenopathy, hepatosplenomegaly, rashes, or active joints. Neurological examination was notable for ataxia, expressive aphasia, and dysarthria. Laboratory investigations revealed marked elevation of inflammatory markers (C-reactive protein and erythrocyte sedimentation rate) and thrombocytosis.
On detailed history, the patient had had protean signs and symptoms for a number of years. Four years earlier, as part of an investigation (at another hospital) for a parastomal hernia, the patient was incidentally discovered to have bilateral sclerotic bony lesions in the femoral heads and tibial and ischial bones. Computed tomography (CT)-guided biopsy revealed nonspecific fibroinflammatory infiltrates. A year later, he was hospitalized with right-sided weakness, aphasia, and dysarthria and was diagnosed with left middle cerebral artery stroke. He had a complete spontaneous recovery only to present 2 years later with recurrence of similar albeit transient neurological signs and symptoms.
The following year, as part of surveillance CT for his metastatic prostate cancer, the patient was found to have bilateral perinephric thick rim-like soft tissue densities with significant infiltration into the parapelvic spaces and all around both kidneys. The infiltrative process encased the aorta and bilateral renal veins and caused mild compression of the inferior vena cava. Pathological analysis of a needle core re-biopsy of the retroperitoneal lesion (Fig. 1) demonstrated fibrofatty tissue with focal lymphoplasmacytic infiltrates, consisting of small lymphocytes, (polytypic) plasma cells, histiocytes, as well as number of eosinophils. CD68/PGM-1 stain highlighted a number of histiocytes, including a small focus of foamy histiocytes (which at the time was interpreted as nonspecific), negative for S-100. We counted a maximum of 27 IgG-expressing plasma cells per high-power field and a maximum of 5 IgG4+ plasma cells per high-power field (2.7 on average), with a maximum IgG4/IgG ratio of 50% (22% on average). The pathologic diagnosis of retroperitoneal fibrosis was made, raising the possibility of IgG4-related disease, and the patient was started on rituximab 375 mg/m2.
Fig. 1.
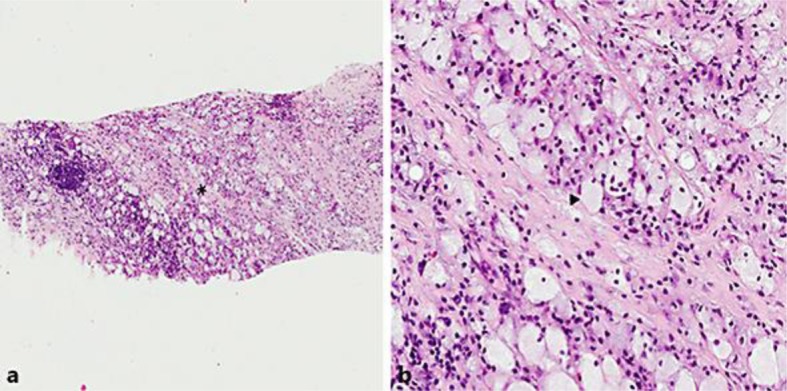
Photomicrograph of the patient's second biopsy. Hematoxylin and eosin-stained section, at 5× (a) and 20× (b) magnifications. The asterisk in a represents the magnified area seen in b; the arrowhead in b points to a representative foamy histiocyte.
During the current admission, extensive investigations were performed. Cerebral magnetic resonance imaging revealed multiple scattered punctate foci consistent with acute end-vessel infarcts in the middle cerebral artery territory. As part of the stroke workup, transthoracic echocardiography revealed intracavitary echoes, suggestive of a possible right atrial myxoma. This was further characterized by cardiac magnetic resonance imaging as a lesion in the lateroposterior wall of the right atrium, extending to the pericardium and distally surrounding the superior vena cava. Cardiac CT showed extensive epicardial soft tissue infiltration that extended around the right atrial appendage into the right atrioventricular groove, partially encasing the right coronary artery. The same tissue density was also localized adjacent to the proximal aspect of the left anterior descending and left circumflex artery. CT of the thorax revealed concentric infiltration of the aortic arch wall and origins of great vessels. The patient was discharged and continued on rituximab therapy.
He was readmitted a week later with acute coronary syndrome. During the hospitalization, his neurologic symptoms recurred. A coronary angiogram revealed triple vessel disease and the patient underwent successful angioplasty to the left anterior descending and right coronary arteries. CT angiogram of the head revealed mild atherosclerotic disease, but predominantly severe narrowing of vertebral arteries at the artery origins, bilaterally, with an appearance that was atypical for atherosclerosis. Aggressive treatment with 1 mg/kg (M1) of prednisone for presumptive IgG4-related disease was initiated. Repeat CT of the abdomen 2 weeks later revealed progression of the retroperitoneal, periaortic and perirenal infiltrative processes.
The absence of clinical and radiological response to high-dose steroids prompted us to consider diagnoses alternative to IgG4-related disease, including Erdheim-Chester disease. Suspicion for that possibility was strengthened by integration of new information about the patient's radiological history of symmetrical long bone lesions and the previously underinterpreted small focus of CD1a–/CD68+ foamy histiocytes in the second of the patient's two retroperitoneal biopsies (as aforementioned). DNA amplification (from this and the initial needle core biopsy formalin-fixed paraffin-embedded material) and reverse transcription polymerase chain reaction demonstrated the BRAF V600E mutation. Baseline fluorodeoxyglucose (FDG) positron emission tomography (PET) scan (Fig. 2a) revealed prominent FDG activity in the region of the right atrium, mild activity in the perinephric soft tissue infiltrates, multiple FDG-avid scattered sclerotic bone lesions, as well as uptake in the sella turcica.
Fig. 2.
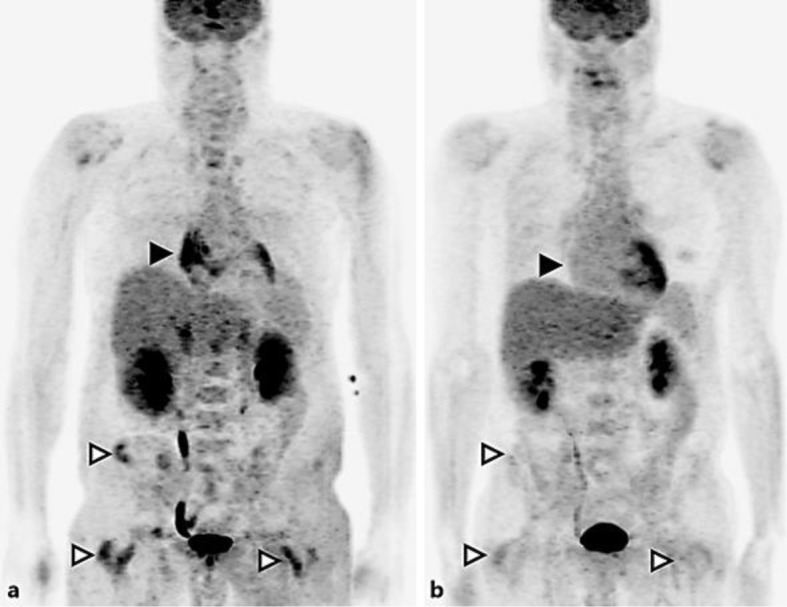
FDG PET maximum intensity projection images at baseline (a) and 3 months after treatment (b). Black arrowheads show the right atrium, white arrowheads show the pelvic and femoral bone lesions.
Given the identification of BRAF mutation, the patient was started on vemurafenib, 960 mg twice daily. Steroids were slowly tapered off. After 1 month of therapy, the patient showed dramatic clinical recovery in functional status, with normalization of gait and speech. Follow-up FDG PET/CT scan demonstrated excellent metabolic response at 1 month. Considering the efficacy of this treatment, vemurafenib was reduced to 480 mg twice daily.
The patient tolerated therapy extremely well, with only minor grade 1 cutaneous toxicity, affecting his feet. Another FDG PET/CT, performed at 3 months into targeted therapy, confirmed ongoing remission (Fig. 2b). For comparison, note the intense uptake near the right atrium, which resolved with treatment (Fig. 2a, 2b lack arrowheads), and the osseous uptake in the bones of the pelvis and femurs (white arrowheads), which also resolved. Follow-up cardiac CT showed a substantial decrease in the size of the cardiac pseudotumor, while CT of the abdomen revealed improvement of the perinephric fat stranding. A third, fourth, and fifth PET/CT after 9, 15, and 21 months on vemurafenib (reduced to 720 mg daily after 6 months and to 420 mg daily after 15 months) confirmed sustained complete metabolic response. Publication of these results occurs with written consent of the patient.
Discussion
Histopathological analysis of biopsy tissue specimens remains the cornerstone of diagnosis of IgG4-related disease. However, it should be borne in mind that although increased IgG4+ plasma cells (with or without elevated serum IgG4) is characteristic of IgG4-related disease, it is not diagnostic thereof. Thus, it is now known that many medical conditions such as repeated infections, autoimmune disease, lymphomas, as well as ECD have been reported to be associated with elevated serum IgG4 levels [3]. Semiquantitative analysis of IgG4 immunostaining helps distinguish IgG4-related from other diseases: a cell count of over 10–50 IgG4+ plasma cells as well as a ratio of IgG4-bearing plasma cells to IgG-bearing plasma cells of at least 50% are highly predictive of IgG4-related disease [3]. Further, particularly in the case of needle core biopsies (in which the complete histological picture may not be represented), the presence of (i) a dense lymphoplasmacytic infiltrate and (ii) fibrosis (although insufficient for definitive diagnosis) might place our case somewhere between the “histologically highly suggestive of IgG4-related disease” and “probable histologic features of IgG4-related disease” categories [4]. Nonetheless, the unsatisfactory clinical response to classic therapies for IgG4-related disease (particularly steroids) should raise a high index of suspicion for an alternative diagnosis. Although flare-ups of IgG4-related disease are common, glucocorticoids are effective as initial therapy in the majority of cases, unless the disease is at a late fibrotic stage [5]. For patients with refractory disease, a rapid response to B-cell depletion has also been observed [6].
Like IgG4-related disease, histiocytoses can have a myriad of clinical presentations, linked by the prominent histopathological finding of marked fibrosis. Within this group of diseases, ECD can present variably from asymptomatic bony infiltration to life-threatening multiorgan involvement. The clinical course of the disease is highly dependent on the extent of systemic involvement. Recently, the detection of the BRAF V600E gain-of-function mutation in ECD biopsies, as well as in the early myelomonocytic precursors of patients with Langerhans cell histiocytosis has revolutionized our thinking about these entities and generated the hypothesis that histiocytoses could, in fact, be neoplasms of myeloid lineage [3]. While traditional treatment for ECD has been interferon alpha, therapy is limited by its poor tolerance and potential resistance in cases of central nervous system (CNS) and/or cardiovascular involvement [7, 8]. Results of a case series of eight ECD patients with CNS and/or cardiac involvement treated with first-line vemurafenib were published [9]. All patients carried the BRAF V600E mutation. At least partial or better responses were attained in 7 patients and responses were sustained for up to 16 months of follow-up. Thus, inhibition of BRAF activation with the targeted agent vemurafenib is extremely promising. On the cell signaling level, mutations in BRAF result in the activation of RAS-RAF-MEK-ERK pathway resulting in accelerated cell proliferation and survival. Recently, additional discovery of NRAS mutation in ECD strongly suggests that the entire signal transduction pathway plays a pathogenic role in ECD, thus raising interest in dual BRAF/MEK inhibition.
Lessons we have learned from this case include that baseline FDG PET scan in a patient with a suspected diagnosis of IgG4-related disease is helpful to assess the extent of organ involvement. Bone involvement is not typical of IgG4-related disease and the absence of response to steroids weighs even more strongly against that possibility. CNS involvement in IgG4-related disease is uncommon and usually manifests as pachymeningitis [10], destructive bone skull lesions [11, 12], and hypophysitis [13], without arterial involvement. However, to our knowledge, this is the first case of ECD reported to have cerebral arteries involvement. ECD can be misdiagnosed as probable IgG4-related disease without proper molecular testing. Therefore, since the disease is so rare and protean in its presentation, the integration of all clinical, radiological, and laboratory findings with the histopathological findings is paramount to raising suspicion for and arriving at the correct diagnosis of ECD.
Conclusion
Due to its rarity and multisystem involvement, ECD may be easily confused with other entities. IgG4-related disease is prime among them, particularly if a patient with ECD presents with retroperitoneal fibrosis. Standard interferon alpha therapy has poor tolerability. In BRAF mutation-bearing forms of ECD, no other drug to date has been shown to be as active as vemurafenib, including based on prospective studies [8]. FDG PET/CT scan is an appropriate and helpful modality to monitor response to the agent. Patients should be closely followed for a number of possible side effects of vemurafenib, particularly its renal and cutaneous toxicities.
Statement of Ethics
Written consent of the patient in presence of a witness was obtained for publication of this case report.
Disclosure Statement
None of the authors has or had financial or nonfinancial competing interests.
Author Contributions
All the authors substantially contributed to the diagnostic workup, treatment, and follow-up and participated in the preparation of the manuscript. Specifically, A.N. drafted the manuscript, K.E. participated in the discussion and conclusion of the manuscript, G.C. participated in the case report presentation section and images interpretations, S.P. participated in the images selection and interpretation, T.P.-H. participated in the pathology images selection and interpretation, and H.K. and G.G. participated in the revision of the manuscript and are the treating physicians.
Acknowledgements
We would like to acknowledge Dr. John Stone and Dr. Cory Perugino from the Boston Massachusetts Hospital for their expert opinion and substantial help for the diagnosis of this difficult case.
References
- 1.Swerdlow SH, Campo E, Harris NL, et al., editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. (4th ed) Lyon: International Agency for Research of Cancer; 2008. pp. 252–253. [Google Scholar]
- 2.Campochiaro C, Tomelleri A, Cavalli G, Berti A, Dagna L. Erdheim-Chester disease. Eur J Intern Med. 2015;26:223–229. doi: 10.1016/j.ejim.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 3.Stone JH, Brito-Zerón P, Bosch X, Ramos-Casals M. Diagnostic approach to the complexity of IgG4-related disease. Mayo Clin Proc. 2015;90:927–939. doi: 10.1016/j.mayocp.2015.03.020. [DOI] [PubMed] [Google Scholar]
- 4.Deshpande V, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25:1181–1192. doi: 10.1038/modpathol.2012.72. [DOI] [PubMed] [Google Scholar]
- 5.Khosroshahi A, Stone JH. Treatment approaches to IgG4-related systemic disease. Curr Opin Rheumatol. 2011;23:67–71. doi: 10.1097/BOR.0b013e328341a240. [DOI] [PubMed] [Google Scholar]
- 6.Khosroshahi A, Bloch DB, Deshpande V, Stone JH. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum. 2010;62:1755–1762. doi: 10.1002/art.27435. [DOI] [PubMed] [Google Scholar]
- 7.Arnaud L, et al. CNS involvement and treatment with interferon-α are independent prognostic factors in Erdheim-Chester disease: a multicenter survival analysis of 53 patients. Blood. 2011;117:2778–2782. doi: 10.1182/blood-2010-06-294108. [DOI] [PubMed] [Google Scholar]
- 8.Haroche J, et al. Cardiovascular involvement, an overlooked feature of Erdheim-Chester disease: report of 6 new cases and a literature review. Medicine (Baltimore) 2004;83:371–392. doi: 10.1097/01.md.0000145368.17934.91. [DOI] [PubMed] [Google Scholar]
- 9.Haroche J, et al. Reproducible and sustained efficacy of targeted therapy with vemurafenib in patients with BRAF(V600E)-mutated Erdheim-Chester disease. J Clin Oncol. 2015;33:411–418. doi: 10.1200/JCO.2014.57.1950. [DOI] [PubMed] [Google Scholar]
- 10.Soussan M, Medjoul A, Badelon I, Guyot A, Martin A, Abad S. IgG4-related diffuse perineural disease. Neurology. 2014;83:1877–1878. doi: 10.1212/WNL.0000000000000979. [DOI] [PubMed] [Google Scholar]
- 11.Cain RB, Colby TV, Balan V, Patel NP, Lal D. Perplexing lesions of the sinonasal cavity and skull base: IgG4-related and similar inflammatory diseases. Otolaryngol Head Neck Surg. 2014;151:496–502. doi: 10.1177/0194599814533648. [DOI] [PubMed] [Google Scholar]
- 12.Alt JA, Whitaker GT, Allan RW, Vaysberg M. Locally destructive skull base lesion: IgG4-related sclerosing disease. Allergy Rhinol (Providence) 2012;3:e41–e45. doi: 10.2500/ar.2012.3.0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caputo C, Bazargan A, McKelvie PA, Sutherland T, Su CS, Inder WJ. Hypophysitis due to IgG4-related disease responding to treatment with azathioprine: an alternative to corticosteroid therapy. Pituitary. 2014;17:251–256. doi: 10.1007/s11102-013-0498-9. [DOI] [PubMed] [Google Scholar]