

Abstract
The chemokine receptor CXCR5 is primarily expressed on B cells and Tfh cells and facilitates their migration towards B cell follicles. In the present study we investigated the role of the CXCL13/CXCR5 axis in the pathogenesis of rheumatoid arthritis (RA) and specifically addressed the impact of CXCR5-mediated T and B cell migration in this disease. Employing collagen-induced arthritis (CIA) we identify CXCR5 as an absolutely essential factor for the induction of inflammatory autoimmune arthritis. Cxcr5-deficient mice and mice selectively lacking Cxcr5 on T cells were completely resistant to CIA, showed impaired germinal center responses and failed to mount an IgG1 antibody response to collagen II. Selective ablation of CXCR5 expression in B cells also led to suppression of CIA owing to diminished GC responses in secondary lymphoid organs (SLO) and impaired anti-collagen II antibody production. Chimeric mice harboring Cxcr5-proficient and Cxcr5-deficient immune cells revealed SLO and not the synovial tissue as the compartment where CXCR5-mediated cell migration induces autoimmune inflammation in arthritis. Thus our data demonstrate that CXCR5-mediated co-localization of Tfh cells and B cells in SLOs is absolutely essential for the induction of RA and identify CXCR5 and Tfh cells as promising therapeutic targets for the treatment of RA.
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory autoimmune disease mainly affecting peripheral diarthrodial joints with a prevalence ranging from 0.4 to 1.3%1. The hallmark of RA is synovial inflammation and hyperplasia that can lead to progressive cartilage and bone erosion, resulting in severe pain and disability. The immunopathogenesis of RA is still not completely elucidated, however it is well established that both innate and adaptive immune system components are crucially involved in RA development and chronicity2. A key factor in RA pathophysiology is the production of autoantibodies3. Autoantibodies in RA lead to immune complex formation in the joints, complement activation and subsequent recruitment and activation of inflammatory leukocytes2. Several autoantibody systems have been identified in RA. Rheumatoid factor (RF) and anti-citrullinated protein/peptide antibodies (ACPAs) can be detected in approximately 50–80% of RA patients and can appear many years prior to the development of arthritic symptoms4–7. The presence of RF or ACPAs in serum of RA patients correlates with a higher disease severity and manifestation of systemic extra articular symptoms8, 9.
Robust high affinity autoantibody production is usually preceded by activation, somatic diversification and affinity maturation of autoreactive B cells. These processes take place in germinal centers (GC) where activated B cells interact with follicular dendritic cells (FDCs) and T follicular helper cells (Tfh) and upon successful selection differentiate into either memory B cells or antibody secreting plasmablasts10. The localization of both B cells and Tfh cells in lymphoid organs is orchestrated by signaling mediated through chemokine receptors such as CCR7, CXCR4, and CXCR511. Expression of CXCR5 allows cells to follow gradients of the chemokine CXCL13 to reach follicular areas. CXCL13 is constitutively produced in B cell follicles and ectopic lymphoid tissue by FDCs and macrophages12, 13. Studies on Cxcr5 −/− and Cxcl13 −/− mice have revealed essential roles of this chemokine axis in lymphoid organ organogenesis and immune cell migration14–16. The splenic architecture is altered in Cxcr5 −/− mice. B cells are not organized into follicles but form a rim around the T cell zone which is located in the center of the follicle but not polarized as in WT mice15. Adoptive transfers of B cells from Cxcr5 −/− mice demonstrated a requirement for CXCR5 expression on B cells for successful migration into splenic follicular areas but not for migration into LN follicles15. Whereas GCs supporting B cell affinity maturation can be induced in Cxcr5 −/− mice they are aberrantly localized in the splenic PALS15, 17. These findings suggest that the CXCL13/CXCR5 axis could also be involved in autoimmunity and especially in diseases with ample autoantibody production such as RA. Indeed, CXCL13 levels are increased in sera, synovial fluid and synovial tissue of RA patients and have been proposed to be a reliable marker of synovial inflammation18–20. CXCL13 neutralization by anti-CXCL13 mAbs was shown to ameliorate the severity of collagen-induced arthritis (CIA) in mice but anti-collagen II (CII) Ab levels and GC response remained unaffected21, 22. Additionally, recent studies show an increased frequency of circulating CXCR5+ T cells in peripheral blood of RA patients that correlate with serum ACPA levels and disease severity23, 24.
In the present study we investigated the role of CXCR5-mediated cell migration in the induction of RA. By using a model of collagen-induced arthritis (CIA) enabling high disease incidence in the relatively resistant C57BL/6 genetic background we demonstrate that Cxcr5 −/− mice were overall resistant to CIA induction, showed no signs of synovial inflammation and exhibited profoundly decreased levels of anti-collagen II antibodies. We show that CXCR5-mediated cell migration was redundant for the formation of the inflammatory infiltrate in arthritic paws but indispensable for robust autoimmune GC responses in secondary lymphoid organs (SLOs) during CIA. Mice with a selective Cxcr5 deficiency in B cells (B-CXCR5−/−) exhibited a highly defective GC- and anti-CII antibody response and profoundly ameliorated CIA incidence and severity. Mice with a selective Cxcr5 deficiency in T cells (T-CXCR5−/−) were characterized by hampered GC formation, very weak antibody response to CII upon CIA induction and decreased serum levels of several pro-inflammatory cytokines. Most importantly, T-CXCR5−/− mice did not develop arthritic paws throughout the observation period. Thus our data suggest that the CXCR5-mediated migration of Tfh cells in B-cell follicles is essential for the induction of RA and that CXCR5 and Tfh cells represent promising therapeutic targets in RA.
Results
Cxcr5-deficient mice are resistant to the development of CIA
In order to address the role of CXCR5 signaling in the pathogenesis of RA we used a CIA mouse model in Cxcr5 −/− mice on a C57BL/6 genetic background. Cxcr5 −/− mice were completely resistant to CIA showing no arthritic symptoms throughout the monitoring period (Fig. 1A–C). Histopathologic analysis of joints in paws of Cxcr5 −/− mice lacked signs of cellular infiltration, synovial hyperplasia, and joint destruction and joint spaces were well preserved (Fig. 1D). Because the antibody response to CII is an essential pathogenic driver in CIA25, 26, serum anti-CII antibody levels were determined. In sera of Cxcr5 −/− mice the specific antibody response to both chicken and murine CII was severely suppressed and especially in case of IgG1 antibodies almost absent (Fig. 1E). Taken together these data show that functional CXCR5 signaling is essential for the development of robust anti-CII Ab responses and the development of CIA.
Figure 1.
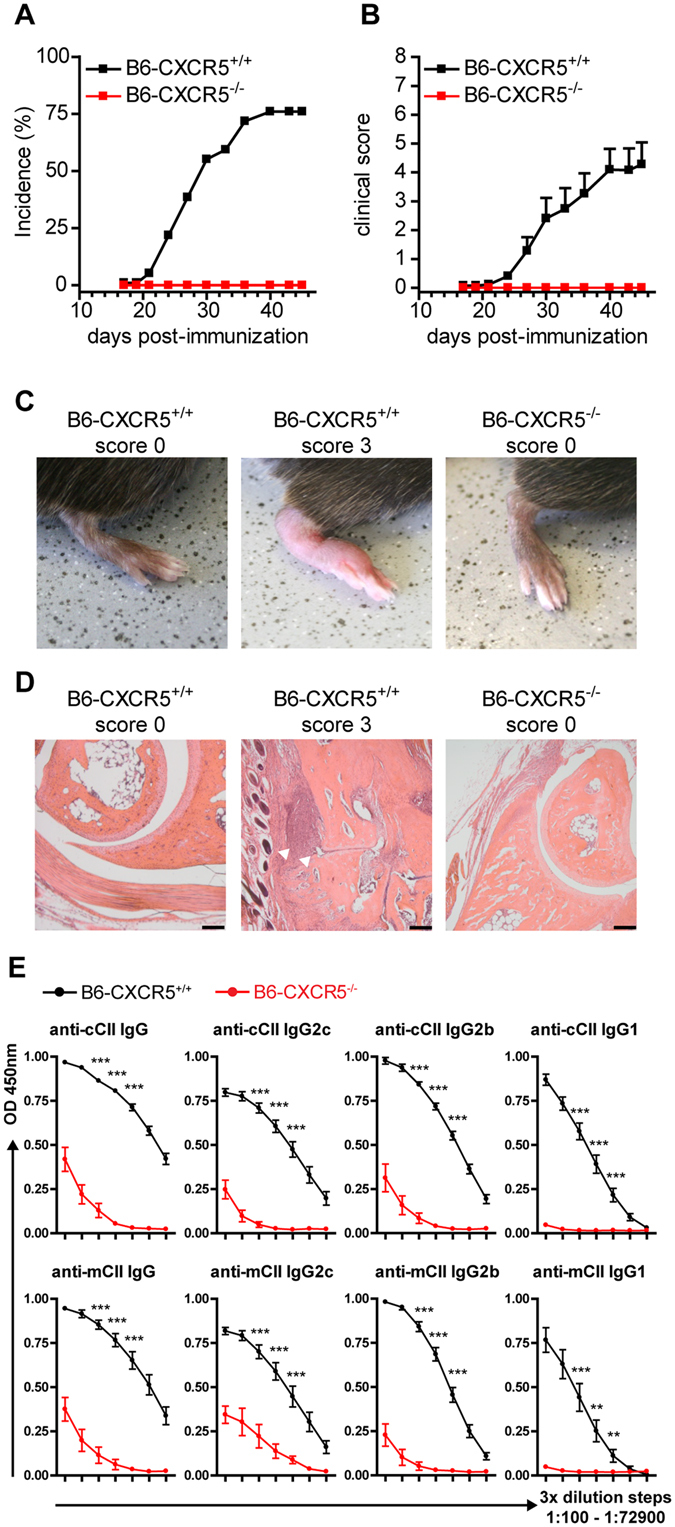
Cxcr5 −/− mice are resistant to CIA induction. Incidence (A) and clinical score (B) of collagen-induced arthritis in WT and Cxcr5 −/− mice on the B6 background. Results are shown from ≥12 mice per group. Data in (B) are presented as mean ± SEM. Representative images of clinical signs of arthritis (C) and histopathology of hind paws (D) are shown for healthy and arthritic WT mice and Cxcr5 −/− mice. White arrowheads point to areas with inflammatory infiltration. (E) Sera were collected at day 45 post-CIA induction from WT and Cxcr5 −/− mice and total IgG, IgG1 and IgG2c antibody levels of anti-chicken CII (cCII) Abs (top row) or anti-murine CII (mCII) Abs (bottom row) were measured by ELISA in serially diluted serum samples (3-fold dilution steps, 1:100–1:72900). Results are shown from 8–9 mice per group. Data are presented as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
Cxcr5 deficiency does not prevent leukocyte migration to arthritic paws
The absence of inflammatory cellular infiltrates in the paws of Cxcr5 −/− mice in this CIA model prompted us to investigate the role of CXCR5 in the migration of leukocytes into arthritic joints. The frequency of CXCR5+ cells in arthritic paws of WT mice was approximately 1% of all CD45+ cells (Fig. 2B). Approximately 65% of all CXCR5 - expressing cells in the inflamed paws were B cells (B220+) and some 30% were myeloid cells (CD11b+; Fig. 2C). In order to delineate if Cxcr5 deficiency affects the migration and/or retention of leukocytes in arthritic paws and thereby the composition of the inflammatory infiltrate we generated bone marrow (BM) chimeras reconstituted with a mixture of Cxcr5 −/− and WT BM cells (Fig. 3A). Upon development of CIA in the chimeric mice, the cellular composition of the inflammatory infiltrate in affected paws was analyzed by flow cytometry. The origin of cells, either donor WT, donor Cxcr5 −/−, or recipient, was identified by the use of congenic markers (Fig. 3C). Similar frequencies of cells in the inflammatory infiltrate in arthritic paws were derived from WT or Cxcr5 −/− donor mice for monocytes (CD11b+ Ly6chi Ly6G−), neutrophils (CD11b+ Ly6G+), DCs (CD11b+ Ly6c− Ly6G− CD11c+) and B cells (CD11b− B220+; Fig. 3D) but not for T cells, reflecting the diminished reconstitution of Cxcr5 −/− T cells in the recipients. The same analysis was performed on cells isolated from peripheral blood, joint-draining lymph nodes (JDLN) and spleen of arthritic WT/Cxcr5 −/− mixed chimeras. Neutrophils, monocytes, DCs, and follicular B cells generated from Cxcr5 −/− BM were found in these organs at frequencies similar to cells of WT BM origin (Fig. 3D). However in spleen and JDLN the frequency of Cxcr5 −/− GC B cells (B220+ Fas+) was significantly reduced compared to GC B cells of WT origin. Furthermore, significantly lower frequencies of Cxcr5 −/− T cells compared to WT T cells were found in JDLN, SPL and peripheral blood (Fig. 3D). These results suggest an essential role for CXCR5 signaling in the generation of the germinal center response in secondary lymphoid organs (SLOs) during CIA but not in the local cellular inflammatory response in arthritic paws.
Figure 2.

Detection of CXCR5 - expressing cells in murine arthritic paws. (A) Representative plots showing identification and characterization of CXCR5+ cells in the inflammatory infiltrate of arthritic paws. Cells were pre-gated as DAPI− as shown in suppl. Figure 1. (B) Frequency of CXCR5 - expressing cells in arthritic paws of WT mice. (C) Frequency of B cells (CD11b− B220+) and myeloid cells (CD11b+) in the CXCR5+ population in arthritic paws. Results are shown from 8 mice.
Figure 3.

Cxcr5 deficiency does not influence the composition of the inflammatory infiltrate in arthritic paws. (A) Generation of mixed Cxcr5 −/−/WT chimeric mice. BM from Cxcr5 −/− donor mice (CD45.2+) was mixed at a ratio of 3:1 with BM from WT donor mice (CD45.1+CD45.2+) and injected in lethally irradiated WT recipient mice (CD45.1+). (B) Reconstitution of total leukocytes, B and T cells in peripheral blood of mixed Cxcr5 −/−/WT chimeric mice before CIA induction. The left bar indicates the color code of cells of given origin. (C) Gating strategy for the identification of WT and Cxcr5 −/− neutrophils in arthritic paws of mixed Cxcr5 −/−/WT chimeric mice. The origin of cells, either donor WT, donor Cxcr5 −/−, or recipient WT, was identified by the expression of CD45.1 and CD45.2. (D) Frequencies of donor WT (black bars), donor Cxcr5 −/− (white bars) or recipient WT (grey bars) cells for the cell types indicated in arthritic paws, peripheral blood, spleen and JDLN of arthritic mixed Cxcr5 −/−/WT chimeric mice. Data are presented as mean ± SEM from 7 mice. Frequencies of donor WT and donor Cxcr5 −/− cells were compared for statistical significance. Ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
Mice with B cell - specific Cxcr5 deficiency show severely impaired CIA
CXCR5 is expressed mainly on B cells and Tfh cells11. In order to delineate the role of CXCR5 signaling in B cells in CIA we generated mixed BM chimeras with a B cell-specific Cxcr5 deficiency (B-CXCR5−/−; Fig. 4A). In B-CXCR5−/− mice no CXCR5-expressing B cells could be identified in splenic follicles by using a novel anti-murine CXCR5 mAb (clone 6C3) enabling faithful identification of CXCR5 - expressing cells in organ sections by IF microscopy (Fig. 4B). B cells in spleens of B-CXCR5−/− mice failed to form follicles but were aberrantly located outside the marginal sinus (Fig. 4C). B cell-specific Cxcr5 deficiency severely lowered the incidence and score of CIA. (Fig. 4D,E). Evaluation of the anti-CII antibody response revealed significantly reduced levels of anti-chicken CII as well as anti-murine CII IgG, IgG2c, IgG1 and IgG2b antibodies but unaltered levels of anti-murine CII IgM in sera of B-CXCR5−/− mice (Fig. 4F). Evaluation of the GC response in spleen and JDLN of B-CXCR5−/− mice revealed substantially reduced GC numbers especially in the spleen (Fig. 4G–I). These results suggest an essential role of B cell-expressed CXCR5 in the formation of the GC response and anti-CII antibody production in CIA.
Figure 4.

Severely ameliorated CIA in mice with B cell - specific Cxcr5 deficiency. (A) Schematic representation of the generation of B-CXCR5−/− and B-CXCR5+/+ mixed chimeric mice. B-CXCR5−/− and B-CXCR5+/+ mice were generated by reconstitution of lethally irradiated WT recipient mice with BM from Tcra −/− Cxcr5 −/− and µMT donor mice and BM from TCRa −/− Cxcr5 +/+ and µMT donor mice respectively. (B) Absence of CXCR5 expression on B cells in splenic follicles of B-CXCR5−/− mice. Spleen sections from B-CXCR5−/− and B-CXCR5+/+ mice were evaluated for CXCR5- expressing cells using a novel in-house generated anti-murine-CXCR5 mAb (clone 6C3) enabling faithful CXCR5 staining in organ sections (bar 100 µm). (C) B cells in spleen of B-CXCR5−/− mice are aberrantly localized outside the marginal zone adjacent to the red pulp. The marginal zone (white arrowheads) was designated by a rim of CD169+ macrophages and the red pulp by F4/80+ macrophages. Representative pictures from 9–10 mice per group are shown. Bar, 100 µm. Incidence (D) and mean clinical score (E) of collagen induced arthritis in B-CXCR5−/− and B-CXCR5+/+ mixed chimeric mice. Upon a reconstitution period of 10 weeks CIA was induced in B-CXCR5−/− and B-CXCR5+/+ mice and mice were monitored for signs of arthritis until day 45 post-immunization. Data are mean ± SEM from 9–10 mice per group. (F) Sera were collected from B-CXCR5−/− and B-CXCR5+/+ mice upon CIA induction and levels of anti-chicken-CII Abs (top row) and anti-murine-CII Abs (bottom row) were measured by ELISA in serially diluted serum samples (3-fold dilution steps, 1:100–1:72900). Data are shown from 9–10 mice per group. (G) Composite micrographs of medial longitudinal splenic sections from B-CXCR5−/− and B-CXCR5+/+ mice upon CIA induction (day 45). GCs were identified by PNA staining (white arrowheads). Bar, 500 µm. (H) GCs, identified by PNA staining as shown in (G) were counted in composite micrographs of longitudinal medial splenic sections and in sections of JDLN (I) in B-CXCR5−/− and B-CXCR5+/+ mice at day 45 upon CIA induction. In (I) the number of GCs per analyzed JDLN is shown. Data are presented as mean ± SD, n = 9–10 per group. Ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
Mice with T cell - specific Cxcr5 deficiency are resistant to CIA
Apart from B cells, CXCR5 expression can be identified in mice on CD4+ Tfh cells. The frequency of Tfh cells (CD4+ CXCR5+ PD-1+) in CIA was approximately 5.0% in spleen and 2.0% in JDLN (Fig. 5B). Tfh cells were also detected on sections of JDLN of arthritic mice being in close interaction with PNA+ GC B cells (Fig. 5C). In order to assess the role of CXCR5 expression on T cells in CIA we used mice with T cell-specific Cxcr5 deficiency (T-CXCR5−/−; Fig. 6A). Interestingly, the incidence of CIA was 0% in T-CXCR5−/− mice throughout the monitoring period (Fig. 6B). Furthermore, the anti-chicken CII as well as the anti-murine CII antibody response were significantly reduced in T-CXCR5−/− compared to T-CXCR5+/+ mice for all IgG isotypes analyzed, but not for IgM antibodies (Fig. 6D). Of note, the anti CII IgG1 antibody response was almost absent in T-CXCR5−/− mice. Evaluation of the GC response by immunofluorescence microscopy (Fig. 6E) revealed a slight but not significant reduction in the number of GCs in JDLNs and spleens of T-CXCR5−/− mice (Fig. 6F,G). Assuming that these slight differences in GC numbers might probably not account for the significant reduction of the anti-CII Ab responses observed in T-CXCR5−/− mice in CIA, we measured the size of splenic GCs and evaluated GC morphology in JDLN sections. In spleens of T-CXCR5+/+ mice significantly bigger GCs formed compared to spleens of T-CXCR5−/− mice (Fig. 6E and H). In JDLN of T-CXCR5+/+ mice most GCs exhibited polarization with distinct FDC (CD21/CD35+) -rich (light zone) and GC B cell (Bcl-6+) -rich (dark zone) areas. In contrast, only 25% of the GCs in JDLN of T-CXCR5−/− mice showed such polarization (Fig. 6I,J). Despite CXCR5 deficiency we were able to detect T cells in GCs of T-CXCR5−/− mice (Fig. 6I) however their numbers were strongly reduced compared to T-CXCR5+/+ mice (Fig. 6K). The complete resistance to CIA induction in T-CXCR5−/− mice prompted us to investigate the systemic cytokine response. In T-CXCR5−/− mice serum levels of IL-1α and CCL2 were significantly lower compared to T-CXCR5+/+ mice (Fig. 6L). Serum levels of IL-1β, TNF-α, IL-5 and IL-12 were non-significantly reduced while other pro-inflammatory cytokines such as IL-6 and IL-17 were not differentially expressed between T-CXCR5+/+ and T-CXCR5−/− mice (Fig. 6L). Taken together these data suggest that CXCR5 expression in T cells is essential for the induction of CIA and has a significant impact on the anti-CII antibody and inflammatory cytokine response during CIA.
Figure 5.

Detection of Tfh cells in SLOs of arthritic BL6 mice. (A) Tfh cells were gated as CD4+ B220− PD-1+ CXCR5+ cells. (B) Frequency of Tfh cells gated as shown in (A) in spleens and JDLN of arthritic BL6 mice (red bars) at day 45 post CII/CFA immunization and non-immunized mice (grey bars). Representative data are presented as mean ± SD (n = 5 mice per group) from more than 2 experiments. ***P < 0.001; (C) Tfh cells (CD4+ CXCR5+, purple color, white arrowheads) were identified by immunofluorescence microscopy in splenic sections of arthritic WT mice (left image) close to GC B cells (PNA+) using a novel in-house generated anti-murine-CXCR5 mAb (clone 6C3) (Bar 20 µm). The specificity of the Tfh cell staining was established in splenic sections of Cxcr5 −/− mice upon CIA induction (middle image) and in splenic sections of WT arthritic mice with control staining (secondary antibody only, right image). DAPI+ CD4− PNA− cells are follicular B cells. Representative data are shown from more than 6 mice analyzed.
Figure 6.
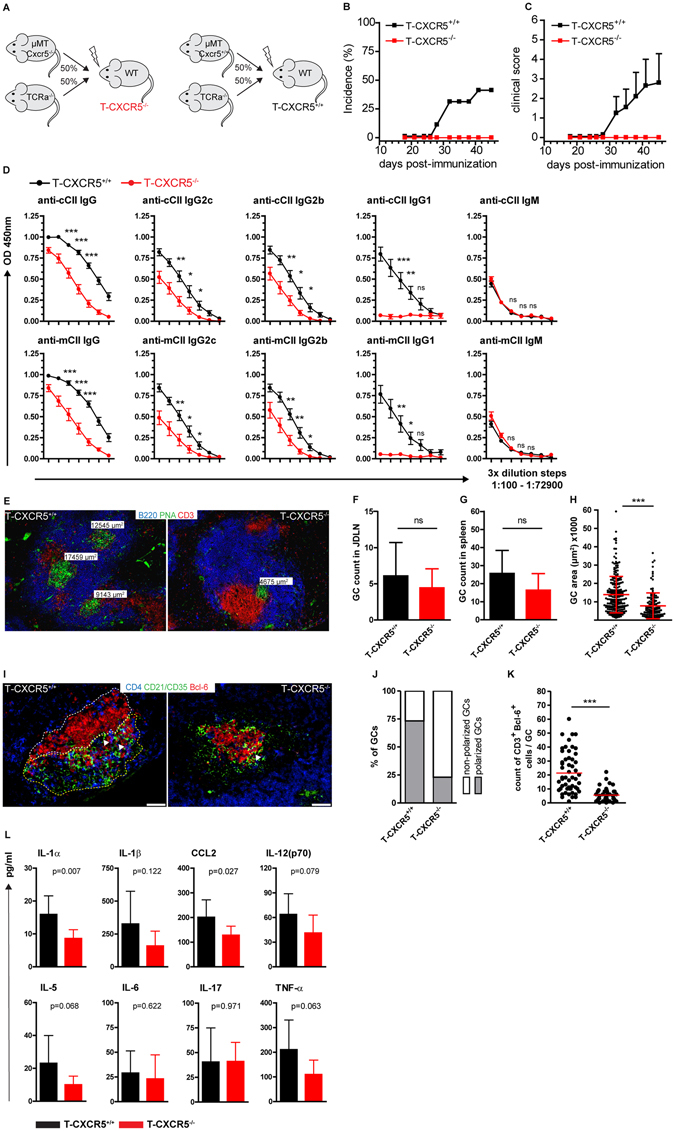
Mice with T cell - specific Cxcr5 deficiency are resistant to the development of CIA. (A) Schematic representation of the generation of T-CXCR5−/− and T-CXCR5+/+ mixed chimeric mice. T-CXCR5−/− and T-CXCR5+/+ mice were generated by reconstitution of lethally irradiated WT recipient mice with BM from µMT Cxcr5 −/− and Tcra −/− donor mice and BM from µMT Cxcr5 +/+ and Tcra −/− donor mice respectively. (B) Incidence and (C) mean clinical score of CIA in T-CXCR5−/− and T-CXCR5+/+ mice. (D) Levels of anti-chicken-CII (top row) and anti-murine-CII (bottom row) Abs were measured by ELISA in serial serum dilutions. (B–D) Data are mean ± SEM from 8–9 mice per group. (E) Spleen sections were stained with anti-B220, PNA and anti-CD3 and the area of GCs (PNA+) in µm2 is indicated. Counts of GCs, identified as shown in (E) per JDLN (F) and in longitudinal medial sections of spleens (G). Data are mean ± SD from 8–9 mice per group. (H) The area of GCs (PNA+ regions) was measured in composite micrographs of longitudinal medial splenic sections. Dots represent individual GCs; red bars mean ± SD derived from 8–9 mice per group; ***P < 0.0001; (I) JDLN sections were stained with anti-CD4, anti-CD21/CD35 and anti-Bcl-6. The GC dark zone is demarcated by white and the GC light zone by yellow dotted lines. White arrowheads point to Tfh cells (CD4+ Bcl-6+). Bar 50 µm. (J) The frequency of GCs with distinct FDC-rich zone and Bcl-6+ GC B cell rich zone (polarized GCs) vs GCs without zone distinction (non-polarized GCs) was assessed in sections shown in (I),n ≥ 40 GCs from at least 8 mice per group. (K) Sections of JDLN were stained with anti-CD3, PNA and anti-Bcl-6. Number of intra-GC Tfh cells assessed by counting CD3+ Bcl-6+ cells in Bcl-6+ PNA+ regions. Data are shown from more than 44 GC analyzed from at least 8 mice per group. ***P < 0.001; Data shown in (E–K)) were obtained from organs harvested at day 45 post CIA induction; (L) Selective reduction of pro-inflammatory cytokines in T-CXCR5−/− mice in CIA. Serum cytokine concentrations were measured in a luminex based assay. Data are presented as mean ± SD from ≥8 mice per group.
Discussion
The present study reveals a fundamental role for CXCR5 in the pathogenesis of CIA. We provide genetic evidence that expression of this chemokine receptor on B cells as well as Tfh cells plays an essential role for GC formation and function especially during secondary immune responses. So far only scarce mechanistic data has been reported regarding the role of CXCR5 in RA including upregulated CXCR5 expression in human RA synovium27. We were able to detect CXCR5-expressing cells in murine arthritic paws in CIA albeit at low frequency. The majority of these cells were B cells and only few CD11b+ myeloid cells showed CXCR5 expression. CXCR5 expression on myeloid cells has been previously reported for migratory skin-derived DCs, a subset of marginal zone DCs, DCs in mesenteric LN of Heligmosomoides polygyrus-infected mice and macrophages in rheumatoid synovium27–30. The very few activated T cells we were able to identify by flow cytometry in arthritic paws were CXCR5− (data not shown), similar to most of the activated PD-1+ CD4+ T cells recently described in RA synovial samples31. These CXCR5− ‘peripheral helper’ T cells were shown to localize outside lymphoid aggregates adjacent to B cells and express B cell-help associated factors31. Further, we show that leukocytes can migrate to and be retained in the arthritic inflammatory infiltrate independent of CXCR5 and that its expression does not affect the composition of the cellular infiltrate. In contrast, in JDLN and spleens of arthritic mice, CXCR5 expression seems to be essential for the generation of a robust GC B cell response.
Experiments described in this study revealed an outstanding role for the expression of CXCR5 on Tfh cells for the development of CIA. Chimeric mice with a T cell - specific Cxcr5 deficiency were completely resistant to CIA induction, presented a moderately reduced GC response and showed a severely defective anti-CII antibody production. Furthermore, we could show that during the course of CIA a robust Tfh cell pool is formed in SLOs that probably sustains high anti-CII Ab production. The literature reports conflicting results regarding the role of CXCR5 on T cells in RA models depending on the adoptive transfer of transgenic CD4+ T cells carrying a TCR specific for glucose-6-phoshate isomerase (GPI). While one study indicates that CXCR5 expression on T cells is essential for robust disease induction32, another group provides evidence for a redundant role for CXCR5 expression on T cells in RA induction and anti-GPI IgG1 production33. The reasons for these differences are unclear but it is known that the different genetic backgrounds used in these studies have a major effect on RA initiation and progression. In the present study we used the well characterized CIA model in mice on a clean C57BL/6 genetic background and without the potential bias of out-competition of adoptively transferred CD4 T cells by the endogenous CD4 T cell populations. Using this model we provide strong evidence for an indispensable role of CXCR5 expression on T cells in autoimmune Ab responses and arthritis development. T cell migration into follicular areas was not completely abrogated in this model in the absence of CXCR5 as we were able to detect by IF microscopy occasional Cxcr5 −/− T cells in GCs and in extra GC follicular areas (Fig. 6E,I and K). We assume that other chemokine receptors may compensate for Cxcr5 deficiency and guide T cells to follicular areas or that CCR7 downregulation is sufficient for some Cxcr5 −/− activated T cells to enter follicular areas as previously suggested16, 34.
An anti-CII IgG1Ab response was virtually absent in Cxcr5 −/−, T-CXCR5 −/− and B-CXCR5−/− mice whereas the anti-CII IgG2 response was reduced but still readily detectable in all these strains. Thus we propose that CIA is highly dependent on an anti-CII IgG1 response, robust GC formation and on Tfh cells, while the IgG2 Ab response is less affected by the absence of CXCR5 and impaired GC formation. Using a murine influenza immunization modeI, it was recently shown that IgG2 responses are mediated by TH1 cells35. Interestingly, the TH1 response in T-CXCR5−/− and B-CXCR5−/− mice is most probably unaffected as TH1 cytokines such as IFN-γ and IL-12 were not significantly deregulated in the serum of these mice upon CIA induction in the present study (not shown).
The role of B cells and autoantibodies in RA has gained much attention in recent years in particular due to the therapeutic success of B cell depletion by Rituximab and other drugs36, 37. It is widely believed that B cells participate in RA not only by autoantibody production but also through Ag presentation and co-stimulation of autoreactive T cells and through production of pro-inflammatory cytokines38. However B cell depletion represents a rather crude approach leading to long term diminution of the overall B cell compartment. Thus there is a clear need for more fine-tuned approaches targeting autoreactive B cells and autoreactive antibody responses. Belonging to the family of seven transmembrane-spanning receptors chemokine receptors have been considered attractive therapeutic targets for interfering with unwanted immune cell trafficking in a variety of immunological disorders including autoimmune diseases. However, most trials targeting chemokine receptors reported largely disappointing results so far39–41. Whereas some effects of chemokine receptor inhibitors have been found in tumor settings, HIV infection and animal studies using models of autoimmunity, no promising clinical data on the efficacy of chemokine receptor inhibitors in RA have been reported41, 42 but no studies have been described so far targeting CXCR5. This is striking since several studies implicate an important role for CXCR5+ cells and the CXCR5/CXCL13 axis in the pathogenesis and progress of several autoimmune conditions including RA. CXCR5+ T cells were detected in inflammatory infiltrates in spinal cords of mice with experimental autoimmune encephalomyelitis (EAE) and EAE was attenuated in CXCL13−/− mice43. CXCL13 was detected within the perivascular infiltrates in actively demyelinating lesions and was elevated in the cerebrospinal fluid (CSF) of MS patients44. Meningeal B cell follicles were associated with severe pathology and disease progress in secondary progressive MS patients and all CSF B cells in samples from MS patients and approximately 20% of CSF CD4+ T cells expressed CXCR544, 45. CXCR5 and CXCL13 expression were also readily detected in gut associated lymphoid tissue (GALT) and lymphoid aggregates in gut samples of ulcerative colitis patients. CXCR5+ CD4+ T cells producing IL-21, a cytokine that promotes mucosal inflammation and B cell activation, were demonstrated in colon samples from IBD patients46, 47. In salivary glands and blood of primary Sjögren syndrome (pSS) patients CD4+ CXCR5+ T cells were shown to be significantly increased48, 49. Expansion of circulating CXCR5+ Tfh cells (cTfh) with a B cell helper phenotype in the blood has been described besides pSS patients in a variety of autoimmune conditions such as MS, autoimmune thyroiditis, SLE and especially RA50–55. The expansion of cTfh cells was associated in most cases with high autoantibody titers and correlated with high disease activity. Especially in RA patients the frequency of cTfh cells showed a positive correlation with anti-CCP Ab levels23 and 28-joint count disease activity score (DAS28)56.
These studies together with results of the present work suggest that targeting CXCR5 would be an ideal candidate for the treatment of RA for several reasons: i) genetic deficiency for Cxcr5 completely prevents CIA, a well-established model of RA sharing several immunological patho-mechanisms and genetic susceptibility features with human RA. The CIA model would be well suited in order to verify the efficacy of CXCR5 targeting, being dependent both on T cells and B cells, mainly driven by anti-CII autoantibodies and due to its widespread use to develop current RA therapeutics such as TNFα inhibitors in pre-clinical trials57. ii) as CXCL13 is the sole ligand for CXCR5, receptor antagonists have to be only optimized for competing one natural ligand. iii) since only few cell types, primarily B cells and Tfh cells, express CXCR5, targeting of this chemokine receptor would leave large parts of the cellular components of the innate and adaptive immune system unaffected. We propose that targeting CXCR5 in RA would disrupt co-localization of autoreactive B cells and Tfh cells, inhibiting thereby autoreactive GC responses and resulting autoimmune Ab production making the development of CXCR5 inhibitors a promising future approach in RA therapy and probably the therapy of other autoantibody driven autoimmune conditions.
Materials and methods
Mice
Mice used were bred at the Central Animal Facility of Hannover Medical School. Cxcr5 −/− mice (B6.129S2(Cg)-Cxcr5 tm1Lipp/J) on a BL6 genetic background, B cell deficient µMT (B6.129S2-Ighm tm1Cgn/J)-, αβ T-cell receptor deficient Tcra −/−(B6.129S2-Tcra tm1Mom/J), B6 CD45.1(B6.SJL-Ptprc a Pepc b/BoyJ)- and B6 CD45.1/CD45.2 heterozygous mice have been described before16, 58, 59. µMT Cxcr5 −/− and Tcra −/− Cxcr5 −/− mice were generated by crossing Cxcr5 −/− mice to µMT and TCRa−/− mice respectively and were used as bone marrow (BM) cell donors for the generation of mixed BM chimeras as described below. All animals were maintained under SPF conditions. All animal experiments have been performed in accordance with institutional guidelines and approved by the Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit.
Collagen-induced arthritis and evaluation of arthritis
CIA was induced on a H-2b genetic background as previously described60.10- to 14-week-old male mice were used in all experiments. Mice were immunized intradermally at the base of the tail with 100 µg chicken collagen II in dilute acetic acid (MD Bioproducts, Switzerland) mixed with an equal volume of CFA (Difco), 100 µl total injection volume per mouse. 21 days later mice received an identical boost immunization. Mice were monitored and scored several times a week for clinical signs of arthritis as follows: 0 = normal, 1 = mild swelling and erythema affecting single paw joints, 2 = pronounced swelling and erythema affecting one or more paw joints, 3 = severe joint swelling and erythema and ankylosis / joint deformity. The maximum clinical score per mouse was 12.
Flow cytometry
The isolation of cells from arthritic paws was performed as previously described61. Cell suspensions from lymph nodes, spleen, blood and paws were analyzed on a LSR II (BD Biosciences) upon staining with the following mAbs: anti-CD44-eFluor 450 (IM7), anti-CD4 PerCp (RM4-5), anti-PD-1 PE-Cy7 (J43), anti-CD45.2 APC-eFluor 780 (104), anti-CD11b PE-Cy7 (M1/70), anti-Ly-6C PerCp-Cy5.5 (HK1.4), anti-Ly-6G PE (1A8), anti-CD11c PE-Cy7 (N418), anti-CD45.1 APC (A20), anti-CD3e PE-Cy7 (145-2C11), anti-CD62L APC-Cy7 (Mel-14), anti-Fas PE (15A7) (all from eBioscience), anti-CD19 Alexa 488 (6D5) (Biolegend), anti-B220 Pacific Orange (RA3-6B2) (ThermoFisher Scientific), anti-I-Ab FITC (AF6-120.1) (BD Biosciences) and anti-CD11b biotinylated (MAC-1) grown in our laboratories. Staining of biotinylated Abs was followed by incubation with Pacific Orange labeled streptavidin (ThermoFisher Scientific). Expression of CXCR5 was assessed using anti-CXCR5 biotinylated (clone 2G8, BD Biosciences), 30 min at room temperature, followed by APC-eFluor 780 or Cy5 labeled streptavidin (eBioscience). Fluorescence-minus-one (FMO) or isotype control samples were used to assess background fluorescence and unspecific Ab binding. Data were analyzed with FACSDiva (BD Biosciences) and FlowJo Software (Treestar).
Histological analysis
Paws were fixed in 10% (vol/vol) neutral buffered formalin, decalcified in EDTA and embedded in paraffin blocks. Paw sections (5 µm) were stained with H&E for microscopic evaluation. The degree of inflammatory infiltration, synovial hyperplasia and cartilage/bone erosion was assessed in a blinded fashion. For immunohistological analysis of SLOs, spleen and joint draining lymph nodes were embedded in OCT and frozen on dry ice. 8 µm thick sections were prepared on a cryostat (CM3050; Leica) and fixed for 10 min in ice-cold acetone. Cryosections were rehydrated in Tris-buffered saline with 0.05% Tween 20, blocked with 5% mouse or rat serum and stained with the following antibodies at room temperature: anti-CD3 Cy3 (17A2), anti-CD3 Cy5 (17A2), anti-CD4 Cy5 (GK1.5), anti-CXCR5 (6C3), anti-B220 Cy5 (RA3-3A1) (all grown and labeled in our laboratories), anti-F4/80 PE (BM8) (ThermoFisher Scientific), anti-CD169 FITC (MOMA-1) (AbD Serotec), anti-CD21/CD35 FITC (7G6) (BD Biosciences) and anti-Bcl-6 (7D1) (Santa-Cruz) followed by anti-rat IgG Cy3 (Jackson Immunoresearch). Germinal centers were identified using PNA biotinylated (Sigma-Aldrich) followed by Streptavidin Alexa 488 (ThermoFisher Scientific). Images were acquired using a motorized epifluorescence microscope (BX61; UPlanSApo lenses: 10x/0.4, 20x/0.75 and 40x/0.9) with a fluorescence camera (F-View II) and cellSens software (Olympus). For analysis of the germinal center response longitudinal sections from the middle of the spleen were used, composite images of 10x magnification were produced and the number and area of germinal centers determined.
Cytokine measurement
Levels of cytokines and chemokines in mouse sera were measured in a Luminex-based cytokine array according to manufacturer’s instructions (Bio-Rad Laboratories, Hercules, USA).
ELISA
Levels of anti-chicken collagen II and anti-murine collagen II antibodies were assessed in serial dilutions of sera by ELISA. 96-well immunosorbent plates (Nunc, Germany) were coated with 2 µg/ml chicken- or murine- collagen II (MD Bioproducts, Switzerland and Chondrex, Inc. USA, respectively) and anti-collagen II antibodies in mouse sera were detected using HRP conjugated anti-mouse-IgM (ThermoFisher Scientific), biotinylated anti-mouse -IgG (Jackson Immunoresearch), -IgG1, -IgG2b (BD Biosciences)and -IgG2c antibodies (Bethyl Laboratories). Upon incubation with peroxidase-conjugated streptavidin (Jackson Immunoresearch) followed by tetramethylbenzidine (TMB) substrate (Sigma Aldrich) optical density (OD) was measured at 450 nm in an automated microplate reader (Tecan).
Bone-marrow chimeras
Recipient C57BL/6 male mice were irradiated lethally with a single dose of 9 Gy and reconstituted by i.v. injection of 10 × 106 bone marrow cells isolated from male donor mice. Reconstitution of the lymphoid compartment was assessed 7–8 weeks later by flow cytometric analysis of peripheral blood. 9–10 weeks upon reconstitution collagen-induced arthritis was induced as described above. Mice with a B cell-specific Cxcr5 deficiency, designated B-CXCR5−/− mice were generated by reconstitution of lethally irradiated male WT recipients with 50% B cell-deficient µMT BM cells and 50% Tcra −/− Cxcr5 −/− BM cells. In these recipients T cells were generated from the µMT BM cells that cannot give rise to B cells and Cxcr5 −/− B cells from the TCRα −/− Cxcr5 −/− BM cells. Other hematopoietic lineages were contributed by both groups of BM cells. Chimeric mice harboring Cxcr5-proficient B cells (B-CXCR5+/+) were generated similarly by reconstitution with 50% µMT BM cells and 50% Tcrα −/− BM cells. Mice with a T cell specific Cxcr5 deficiency, designated T-CXCR5−/− mice were generated by reconstitution of lethally irradiated male WT recipient mice with BM cells from B cell-deficient Cxcr5-deficient (µMT Cxcr5 −/−) mice and αβ TCR-deficient (Tcrα −/−) mice mixed at a ratio of 1:1. In this setup Cxcr5 −/− T cells are derived from µMT Cxcr5 −/− BM cells and B cells from the Tcrα −/− BM cells. Other hematopoietic lineages were generated by both groups of BM cells. For the generation of T-CXCR5+/+ control mice BM cells from µMT Cxcr5 +/+ and Tcrα −/− mice were mixed at a 1:1 ratio and injected in lethally irradiated male WT recipient mice.
Generation of anti-murine-CXCR5 antibodies
Wistar rats were immunised subcutaneously (s.c.) and intraperitoneally (i.p.) with a mixture of membrane fractions isolated from murine Cxcr5 transfected RBL cells in 500 µl PBS, 5 nmol CpG2006 (TIB MOLBIOL, Berlin, Germany), and 500 µl incomplete Freund’s adjuvant. 6 weeks later, a boost without Freund’s adjuvant was given i.p. and s.c. 3 days before fusion. Fusion of the myeloma cell line P3 × 63-Ag8.653 with the rat immune spleen cells was performed using polyethylene glycol 1500 according to standard procedures62. Supernatants were tested by ELISA for IgG production and analyzed by flow cytometry for specificity using lymphocyte suspensions isolated from WT and Cxcr5 −/− mice. Hybridoma cells from supernatants that reacted specifically were subcloned at least twice by limiting dilution. Experiments in this study were performed with clone 6C3 (IgG2a/k).
Statistics
Statistical analysis was performed with Prism 4 (Graph-Pad Software, Inc.). Statistical significance between groups was determined using unpaired two-tailed Student’s t test. *P < 0.05; **P < 0.01; ***P < 0.001.
Data Availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Electronic supplementary material
Acknowledgements
We thank G. Bernhardt for critically reading the manuscript. We thank Jasmin Bölter, Andrew Flatley and Kerstin Daemen for excellent technical assistance and Mathias Herberg and Svetlana Piter for excellent animal care. This work was supported by Deutsche Forschungsgemeinschaft (DFG) grant KFO 250-FO 334/2-1 to R. Förster.
Author Contributions
G.L.M., A.B. and M.F. performed experiments; G.L.M., C.S.F., R.Fe. and R.Fö. analyzed experiments; G.L.M. and R.Fö. designed experiments and wrote the paper; The manuscript was approved by all authors.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-08935-6
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Georgios L. Moschovakis, Email: Moschovakis.Leandros@mh-hannover.de
Reinhold Förster, Email: Foerster.Reinhold@mh-hannover.de.
References
- 1.Sacks JJ, Luo YH, Helmick CG. Prevalence of specific types of arthritis and other rheumatic conditions in the ambulatory health care system in the United States, 2001-2005. Arthritis Care Res (Hoboken) 2010;62:460–4. doi: 10.1002/acr.20041. [DOI] [PubMed] [Google Scholar]
- 2.Klareskog L, Catrina AI, Paget S. Rheumatoid arthritis. Lancet. 2009;373:659–72. doi: 10.1016/S0140-6736(09)60008-8. [DOI] [PubMed] [Google Scholar]
- 3.Conigliaro P, et al. Autoantibodies in inflammatory arthritis. Autoimmun Rev. 2016;15:673–83. doi: 10.1016/j.autrev.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Majka DS, et al. Duration of preclinical rheumatoid arthritis-related autoantibody positivity increases in subjects with older age at time of disease diagnosis. Ann Rheum Dis. 2008;67:801–7. doi: 10.1136/ard.2007.076679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rantapaa-Dahlqvist S, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48:2741–9. doi: 10.1002/art.11223. [DOI] [PubMed] [Google Scholar]
- 6.Nielen MM, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. 2004;50:380–6. doi: 10.1002/art.20018. [DOI] [PubMed] [Google Scholar]
- 7.Steiner G, Smolen J. Autoantibodies in rheumatoid arthritis and their clinical significance. Arthritis Res 4. 2002;Suppl 2:S1–5. doi: 10.1186/ar551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bas S, et al. Diagnostic tests for rheumatoid arthritis: comparison of anti-cyclic citrullinated peptide antibodies, anti-keratin antibodies and IgM rheumatoid factors. Rheumatology (Oxford) 2002;41:809–14. doi: 10.1093/rheumatology/41.7.809. [DOI] [PubMed] [Google Scholar]
- 9.Vencovsky J, et al. Autoantibodies can be prognostic markers of an erosive disease in early rheumatoid arthritis. Ann Rheum Dis. 2003;62:427–30. doi: 10.1136/ard.62.5.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. 2012;30:429–57. doi: 10.1146/annurev-immunol-020711-075032. [DOI] [PubMed] [Google Scholar]
- 11.Schulz O, Hammerschmidt SI, Moschovakis GL, Forster R. Chemokines and Chemokine Receptors in Lymphoid Tissue Dynamics. Annu Rev Immunol. 2016;34:203–42. doi: 10.1146/annurev-immunol-041015-055649. [DOI] [PubMed] [Google Scholar]
- 12.Carlsen HS, Baekkevold ES, Morton HC, Haraldsen G, Brandtzaeg P. Monocyte-like and mature macrophages produce CXCL13 (B cell-attracting chemokine 1) in inflammatory lesions with lymphoid neogenesis. Blood. 2004;104:3021–7. doi: 10.1182/blood-2004-02-0701. [DOI] [PubMed] [Google Scholar]
- 13.Manzo A, et al. Systematic microanatomical analysis of CXCL13 and CCL21 in situ production and progressive lymphoid organization in rheumatoid synovitis. Eur J Immunol. 2005;35:1347–59. doi: 10.1002/eji.200425830. [DOI] [PubMed] [Google Scholar]
- 14.Ansel KM, et al. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature. 2000;406:309–14. doi: 10.1038/35018581. [DOI] [PubMed] [Google Scholar]
- 15.Forster R, et al. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell. 1996;87:1037–47. doi: 10.1016/S0092-8674(00)81798-5. [DOI] [PubMed] [Google Scholar]
- 16.Hardtke S, Ohl L, Forster R. Balanced expression of CXCR5 and CCR7 on follicular T helper cells determines their transient positioning to lymph node follicles and is essential for efficient B-cell help. Blood. 2005;106:1924–31. doi: 10.1182/blood-2004-11-4494. [DOI] [PubMed] [Google Scholar]
- 17.Voigt I, et al. CXCR5-deficient mice develop functional germinal centers in the splenic T cell zone. Eur J Immunol. 2000;30:560–7. doi: 10.1002/1521-4141(200002)30:2<560::AID-IMMU560>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 18.Bugatti S, et al. High expression levels of the B cell chemoattractant CXCL13 in rheumatoid synovium are a marker of severe disease. Rheumatology (Oxford) 2014;53:1886–95. doi: 10.1093/rheumatology/keu163. [DOI] [PubMed] [Google Scholar]
- 19.Rosengren S, Wei N, Kalunian KC, Kavanaugh A, Boyle DL. CXCL13: a novel biomarker of B-cell return following rituximab treatment and synovitis in patients with rheumatoid arthritis. Rheumatology (Oxford) 2011;50:603–10. doi: 10.1093/rheumatology/keq337. [DOI] [PubMed] [Google Scholar]
- 20.Bugatti S, et al. Serum levels of CXCL13 are associated with ultrasonographic synovitis and predict power Doppler persistence in early rheumatoid arthritis treated with non-biological disease-modifying anti-rheumatic drugs. Arthritis Res Ther. 2012;14 doi: 10.1186/ar3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finch DK, Ettinger R, Karnell JL, Herbst R, Sleeman MA. Effects of CXCL13 inhibition on lymphoid follicles in models of autoimmune disease. Eur J Clin Invest. 2013;43:501–9. doi: 10.1111/eci.12063. [DOI] [PubMed] [Google Scholar]
- 22.Klimatcheva E, et al. CXCL13 antibody for the treatment of autoimmune disorders. BMC Immunol. 2015;16 doi: 10.1186/s12865-015-0068-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma J, et al. Increased frequency of circulating follicular helper T cells in patients with rheumatoid arthritis. Clin Dev Immunol. 2012;2012 doi: 10.1155/2012/827480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J, et al. High frequencies of activated B cells and T follicular helper cells are correlated with disease activity in patients with new-onset rheumatoid arthritis. Clin Exp Immunol. 2013;174:212–20. doi: 10.1111/cei.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stuart JM, Dixon FJ. Serum transfer of collagen-induced arthritis in mice. J Exp Med. 1983;158:378–92. doi: 10.1084/jem.158.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wooley PH, Luthra HS, Krco CJ, Stuart JM, David CS. Type II collagen-induced arthritis in mice. II. Passive transfer and suppression by intravenous injection of anti-type II collagen antibody or free native type II collagen. Arthritis Rheum. 1984;27:1010–7. doi: 10.1002/art.1780270907. [DOI] [PubMed] [Google Scholar]
- 27.Schmutz C, et al. Chemokine receptors in the rheumatoid synovium: upregulation of CXCR5. Arthritis Res Ther. 2005;7:R217–29. doi: 10.1186/ar1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leon B, et al. Regulation of T(H)2 development by CXCR5+ dendritic cells and lymphotoxin-expressing B cells. Nat Immunol. 2012;13:681–90. doi: 10.1038/ni.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saeki H, Wu MT, Olasz E, Hwang ST. A migratory population of skin-derived dendritic cells expresses CXCR5, responds to B lymphocyte chemoattractant in vitro, and co-localizes to B cell zones in lymph nodes in vivo. Eur J Immunol. 2000;30:2808–14. doi: 10.1002/1521-4141(200010)30:10<2808::AID-IMMU2808>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 30.Yu P, et al. B cells control the migration of a subset of dendritic cells into B cell follicles via CXC chemokine ligand 13 in a lymphotoxin-dependent fashion. J Immunol. 2002;168:5117–23. doi: 10.4049/jimmunol.168.10.5117. [DOI] [PubMed] [Google Scholar]
- 31.Rao DA, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature. 2017;542:110–114. doi: 10.1038/nature20810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Victoratos P, Kollias G. Induction of autoantibody-mediated spontaneous arthritis critically depends on follicular dendritic cells. Immunity. 2009;30:130–42. doi: 10.1016/j.immuni.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 33.Chevalier N, et al. The Role of Follicular Helper T Cell Molecules and Environmental Influences in Autoantibody Production and Progression to Inflammatory Arthritis in Mice. Arthritis Rheumatol. 2016;68:1026–38. doi: 10.1002/art.39481. [DOI] [PubMed] [Google Scholar]
- 34.Haynes NM, et al. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J Immunol. 2007;179:5099–108. doi: 10.4049/jimmunol.179.8.5099. [DOI] [PubMed] [Google Scholar]
- 35.Miyauchi K, et al. Protective neutralizing influenza antibody response in the absence of T follicular helper cells. Nat Immunol. 2016;17:1447–1458. doi: 10.1038/ni.3563. [DOI] [PubMed] [Google Scholar]
- 36.Cohen SB, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54:2793–806. doi: 10.1002/art.22025. [DOI] [PubMed] [Google Scholar]
- 37.Emery P, et al. Efficacy and safety of different doses and retreatment of rituximab: a randomised, placebo-controlled trial in patients who are biological naive with active rheumatoid arthritis and an inadequate response to methotrexate (Study Evaluating Rituximab’s Efficacy in MTX iNadequate rEsponders (SERENE)) Ann Rheum Dis. 2010;69:1629–35. doi: 10.1136/ard.2009.119933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakken B, et al. B-cells and their targeting in rheumatoid arthritis–current concepts and future perspectives. Autoimmun Rev. 2011;11:28–34. doi: 10.1016/j.autrev.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 39.Lebre MC, et al. Why CCR2 and CCR5 blockade failed and why CCR1 blockade might still be effective in the treatment of rheumatoid arthritis. PLoS One. 2011;6 doi: 10.1371/journal.pone.0021772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schall TJ, Proudfoot AE. Overcoming hurdles in developing successful drugs targeting chemokine receptors. Nat Rev Immunol. 2011;11:355–63. doi: 10.1038/nri2972. [DOI] [PubMed] [Google Scholar]
- 41.Szekanecz Z, Koch AE. Successes and failures of chemokine-pathway targeting in rheumatoid arthritis. Nat Rev Rheumatol. 2016;12:5–13. doi: 10.1038/nrrheum.2015.157. [DOI] [PubMed] [Google Scholar]
- 42.Debnath B, Xu S, Grande F, Garofalo A, Neamati N. Small molecule inhibitors of CXCR4. Theranostics. 2013;3:47–75. doi: 10.7150/thno.5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bagaeva LV, Rao P, Powers JM, Segal BM. CXC chemokine ligand 13 plays a role in experimental autoimmune encephalomyelitis. J Immunol. 2006;176:7676–85. doi: 10.4049/jimmunol.176.12.7676. [DOI] [PubMed] [Google Scholar]
- 44.Krumbholz M, et al. Chemokines in multiple sclerosis: CXCL12 and CXCL13 up-regulation is differentially linked to CNS immune cell recruitment. Brain. 2006;129:200–11. doi: 10.1093/brain/awh680. [DOI] [PubMed] [Google Scholar]
- 45.Magliozzi R, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. 2007;130:1089–104. doi: 10.1093/brain/awm038. [DOI] [PubMed] [Google Scholar]
- 46.Carlsen HS, Baekkevold ES, Johansen FE, Haraldsen G, Brandtzaeg P. B cell attracting chemokine 1 (CXCL13) and its receptor CXCR5 are expressed in normal and aberrant gut associated lymphoid tissue. Gut. 2002;51:364–71. doi: 10.1136/gut.51.3.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sarra M, et al. Interferon-gamma-expressing cells are a major source of interleukin-21 in inflammatory bowel diseases. Inflamm Bowel Dis. 2010;16:1332–9. doi: 10.1002/ibd.21238. [DOI] [PubMed] [Google Scholar]
- 48.Jin L, et al. CD4+ CXCR5+ follicular helper T cells in salivary gland promote B cells maturation in patients with primary Sjogren’s syndrome. Int J Clin Exp Pathol. 2014;7:1988–96. [PMC free article] [PubMed] [Google Scholar]
- 49.Maehara T, et al. Selective localization of T helper subsets in labial salivary glands from primary Sjogren’s syndrome patients. Clin Exp Immunol. 2012;169:89–99. doi: 10.1111/j.1365-2249.2012.04606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Romme Christensen J, et al. Systemic inflammation in progressive multiple sclerosis involves follicular T-helper, Th17- and activated B-cells and correlates with progression. PLoS One. 2013;8 doi: 10.1371/journal.pone.0057820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arroyo-Villa I, et al. Constitutively altered frequencies of circulating follicullar helper T cell counterparts and their subsets in rheumatoid arthritis. Arthritis Res Ther. 2014;16 doi: 10.1186/s13075-014-0500-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Feng X, et al. Inhibition of aberrant circulating Tfh cell proportions by corticosteroids in patients with systemic lupus erythematosus. PLoS One. 2012;7 doi: 10.1371/journal.pone.0051982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fukuyo, S. et al. Abatacept therapy reduces CD28+ CXCR5+ follicular helper-like T cells in patients with rheumatoid arthritis. Clin Exp Rheumatol (2017). [PubMed]
- 54.Simpson N, et al. Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum. 2010;62:234–44. doi: 10.1002/art.25032. [DOI] [PubMed] [Google Scholar]
- 55.Zhu C, et al. Increased frequency of follicular helper T cells in patients with autoimmune thyroid disease. J Clin Endocrinol Metab. 2012;97:943–50. doi: 10.1210/jc.2011-2003. [DOI] [PubMed] [Google Scholar]
- 56.Liu R, et al. A regulatory effect of IL-21 on T follicular helper-like cell and B cell in rheumatoid arthritis. Arthritis Res Ther. 2012;14 doi: 10.1186/ar4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Williams RO. Collagen-induced arthritis in mice: a major role for tumor necrosis factor-alpha. Methods Mol Biol. 2007;361:265–84. doi: 10.1385/1-59745-208-4:265. [DOI] [PubMed] [Google Scholar]
- 58.Prinz I, Klemm U, Kaufmann SH, Steinhoff U. Exacerbated colitis associated with elevated levels of activated CD4+ T cells in TCRalpha chain transgenic mice. Gastroenterology. 2004;126:170–81. doi: 10.1053/j.gastro.2003.10.062. [DOI] [PubMed] [Google Scholar]
- 59.Zietara N, et al. Multicongenic fate mapping quantification of dynamics of thymus colonization. J Exp Med. 2015;212:1589–601. doi: 10.1084/jem.20142143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Inglis JJ, et al. Collagen-induced arthritis in C57BL/6 mice is associated with a robust and sustained T-cell response to type II collagen. Arthritis Res Ther. 2007;9 doi: 10.1186/ar2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Armaka M, Gkretsi V, Kontoyiannis D, Kollias G. A standardized protocol for the isolation and culture of normal and arthritogenic murine synovial fibroblasts. Protocol Exchange. 2009 [Google Scholar]
- 62.Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–7. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.