

ABSTRACT
Purpose of Review: Although increasingly recognized, atypical parkinsonian syndromes remain challenging to diagnose and are underrecognized due to overlap with other parkinsonisms. This article provides a diagnostic approach to atypical parkinsonian syndromes, including progressive supranuclear palsy (PSP), multiple system atrophy (MSA), corticobasal degeneration (CBD), and dementia with Lewy bodies. The goal of this review is to aid the clinician in recognizing key clinical and pathologic features and to raise awareness of recent advances in diagnostics and treatment.
Recent Findings: Diagnostic criteria for atypical parkinsonian syndromes are evolving to encompass increasingly recognized heterogeneity in the presentation of these disorders and information gleamed from clinicopathologic correlations. PSP and CBD in particular now share similar pathologic clinical features and include a number of phenotypic variants. Pathologic diagnoses are increasingly used in clinical practice, and there is frequent reference now by clinicians to tauopathies, including PSP and CBD, and the synucleinopathies, which include MSA and dementia with Lewy bodies (as well as Parkinson disease). Research into biomarkers, including both tissue and imaging modalities and genetics, has the potential to increase disease recognition and make earlier diagnosis and treatment possible. Although novel therapeutics are being studied for atypical parkinsonian syndromes such as PSP, no new breakthrough interventions have emerged for the treatment of PSP, CBD, and MSA. Current therapeutic management for these disorders frequently uses a multidisciplinary team approach.
Summary: The approach to atypical parkinsonian syndromes requires recognition of a constellation of overlapping but distinct clinical features that help with identifying and distinguishing them from Parkinson disease and other similar disorders.
INTRODUCTION
Parkinsonism is defined as a hypokinetic syndrome and is characterized by the presence of resting tremor, muscular rigidity, bradykinesia or akinesia, and postural instability. While many secondary or acquired causes of parkinsonism exist, the most common primary or neurodegenerative cause of parkinsonism is Parkinson disease (PD). A smaller but significant number of patients present with a parkinsonian syndrome that has atypical features such as early dementia, frequent falls, ocular dysmotility, prominent dysautonomia, or ataxia. These syndromes typically involve multisystem degeneration and are referred to as atypical parkinsonian syndromes. They commonly include progressive supranuclear palsy (PSP), multiple system atrophy (MSA), corticobasal degeneration (CBD), and dementia with Lewy bodies (DLB), as well as other rarer causes. It is critical to distinguish these disorders from classic PD as disease progression and subsequent functional decline is often more rapid than in PD. In these syndromes, treatment with standard PD therapies frequently lacks efficacy and is fraught with complications. Patients often have complex care needs that necessitate a multidisciplinary approach. This Continuum article focuses on the diagnostic approach to atypical parkinsonian syndromes and aims to help the clinician recognize key clinical and pathologic features as well as recent advances in diagnostics and treatment.
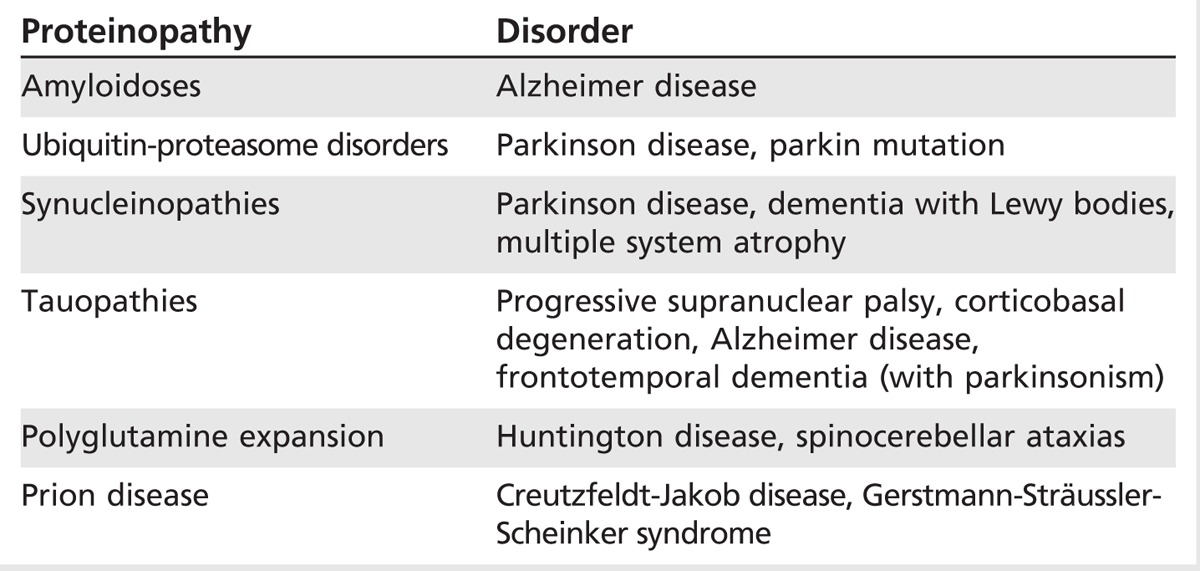
The considerable overlap of signs and symptoms for atypical parkinsonian syndromes with PD, secondary parkinsonisms, and heredodegenerative disorders makes clinical diagnosis challenging (Figure 5-1). These issues may lead to underrecognition, delay in diagnosis, and even misdiagnosis. Table 5-11 includes a list of primary causes of atypical parkinsonisms. Despite the diversity of these conditions, there has been an evolution of our understanding of the pathophysiology of these disorders. Increasingly, clinicopathologic terms are being used to describe atypical parkinsonian syndromes in the clinic because of the diagnostic uncertainty and overlap of symptoms (Figure 5-2). One commonality among neurodegenerative disorders is the presence of abnormal proteinaceous deposits in pathologic brain tissue that have been linked to disease mechanisms, giving rise to the term neuroproteinopathy. Table 5-2 includes examples of various proteinopathies and diseases linked to proteins that accumulate in intracellular inclusions or extracellular plaques (eg, α-synuclein, ubiquitin, tau, and β-amyloid).
Figure 5-1.
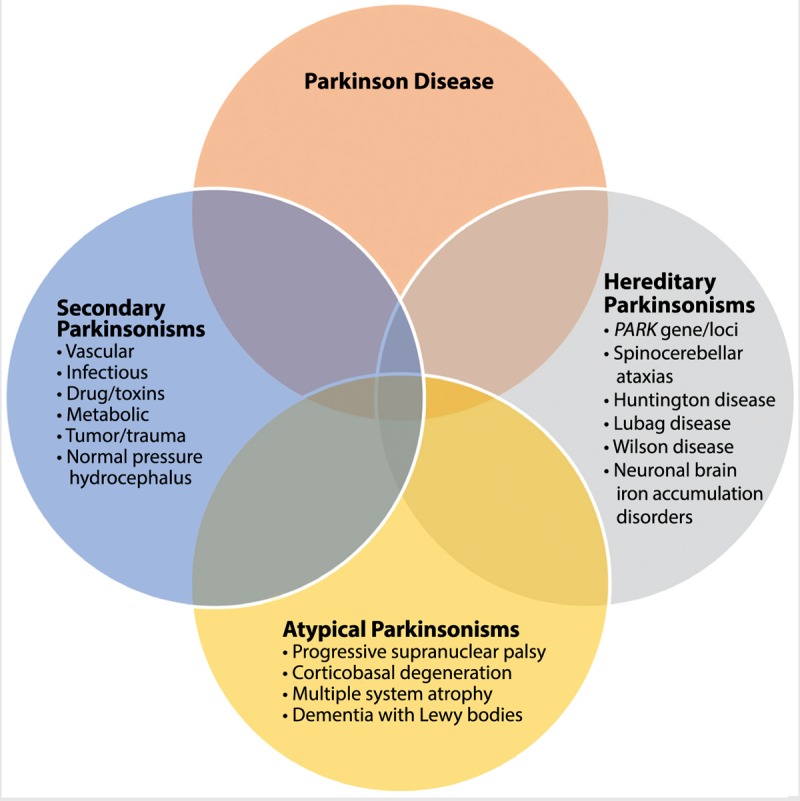
Overlap of parkinsonian syndromes. Atypical parkinsonisms have common features with Parkinson disease, secondary parkinsonisms, and heredodegenerative disorders with parkinsonism.
Table 5-1.
Primary Causes of Atypical Parkinsonisma

Figure 5-2.
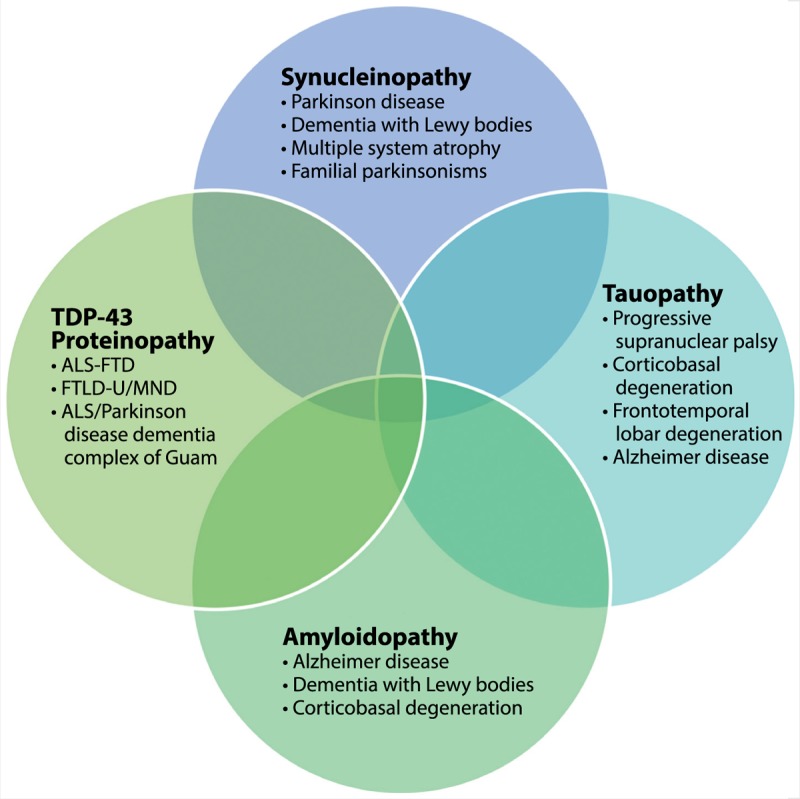
Clinicopathologic overlap of neurodegenerative proteinopathies. Atypical parkinsonian syndromes share abnormal accumulation of proteins such as α-synuclein, tau, amyloid, and TDP-43.
ALS = amyotrophic lateral sclerosis; FTD = frontotemporal dementia; FTLD-U = frontotemporal lobar degeneration with ubiquitin; MND = motor neuron disease; TDP-43 = TAR DNA binding protein 43.
Table 5-2.
Classification of Atypical Parkinsonism as Proteinopathies
Despite recent research and diagnostic advances, the diagnosis of atypical parkinsonian syndromes still relies primarily on clinical evaluation. Ancillary tests such as brain imaging can be supportive, but there are no identified, reliable, and specific biomarkers that have been established as diagnostic. However, certain features or “red flags” have been identified that help distinguish atypical parkinsonian syndromes from PD.2 These include rapid disease progression, early gait instability and falls, absence or paucity of tremor, autonomic failure, and poor or absent response to levodopa, including pain/dysesthesia. Additional features may include oculomotor abnormalities, pyramidal tract or cerebellar signs (ataxia), prominent dysautonomia, severe dysarthria or dysphonia, laryngeal stridor, myoclonus, alien limb, apraxia, and early dementia (Table 5-3). This article focuses on some of the most common atypical parkinsonian syndromes and their identifying features.
Table 5-3.
Red Flags for Differentiating Atypical Parkinsonism From Parkinson Disease

PROGRESSIVE SUPRANUCLEAR PALSY
PSP was described over 50 years ago by Drs Steele, Richardson, and Olszewski.3 It is the most common form of atypical parkinsonism, comprising about 5% to 6% of those patients presenting with parkinsonism. The estimated prevalence and annual incidence of PSP is about 5 per 100,000 in individuals between the ages of 50 and 99 years, but is likely higher due to misdiagnosis and underrecognition. The average age of onset is typically in the sixties (average age of 63 to 66 years), and the mean survival from diagnosis is reported between 5 to 8 years. Hallmarks of the disease include prominent, early postural instability, unexplained falls, vertical supranuclear palsy, and progressive dementia.
Gait instability and early falls are key features of PSP and distinguish it from other parkinsonian syndromes. Relative to other parkinsonian syndromes, falls occur early, within the first year or two, and often lead to significant injury and fractures.4 Gait in PSP is characteristically stiff, broad based, with knees extended and arms abducted. It is often described as clumsy like a “drunken sailor” or “dancing bear,” and includes large lateral deviations and step asymmetry. When turning, persons with PSP tend to pivot rather than turn en bloc as is more typical in PD. The cause of falls is often multifactorial and includes axial rigidity, bradykinesia, loss of postural reflexes, freezing, a visual-vestibular component, and decreased insight. “Rocket sign” occurs in patients with PSP who have lost insight into their postural instability and “rocket” out of their chair without assistance, resulting in a high risk for falling. Retropulsion and unchecked sitting (falling into their chair) are also common.
A key feature of PSP includes inability to perform volitional saccades and progressive supranuclear ophthalmoparesis. Although limitation of upgaze is often described as a sign of PSP, it is nonspecific and can be seen in other neurodegenerative disorders as well as in aging. Limitation of downgaze is most sensitive, and the syndrome frequently progresses to include upgaze and lateral gaze palsies. Slowed saccades and reduced optokinetic nystagmus—vertical more affected than horizontal—are also frequently observed and are early predictors of progression to supranuclear gaze palsy. Another common finding is square-wave jerks, characterized by rapid back and forth mini saccadic intrusions during fixation. Complete gaze palsy and involuntary ocular fixation (as well as lack of eye blink) contribute to the classic “Mona Lisa” stare or stone face. Ability to overcome gaze limitation with vestibular or cervical-ocular reflex maneuvers defines supranuclear palsy, but can be difficult to assess in patients with PSP who have significant axial rigidity. Visual concerns are also common. Blurred vision occurs due to a combination of factors including decreased blink and tear production, drying of the cornea, and ocular irritation. Diplopia is common not only in PSP but also in other parkinsonisms and is most frequently due to convergence insufficiency. Reading is affected and may be helped by prisms in select cases. Despite visual aids, patients often continue to experience blurred vision and difficulty scanning text due to progressive gaze palsy. In addition, patients may also experience photosensitivity (wearing sunglasses inside), decreased eye blink, blepharospasm, and eye-opening apraxia, leading to eyebrow furrowing (procerus contraction), and vertical wrinkling of the forehead, referred to as procerus sign.5
Additional symptoms include a characteristic facial appearance from the rigid bradykinesia and dystonia in facial musculature. The hypertonic facial muscles produce facial folds and a worried, astonished expression. Bulbar features include a progressive dysarthria that is often spastic or hypernasal, hypokinetic, and monotonous. Speech can be slow and can include stuttering, echolalia, and occasional involuntary vocalizations. An apraxia of phonation has also been reported. Most concerning, however, is progressive dysphagia that can lead to aspiration, pneumonia, and early death. As with other parkinsonisms, mood disturbance is frequently present and characterized by apathy, depression, or both.6 Disinhibition, dysphoria, anxiety, and irritability are also possible. Emotional lability (also called emotional incontinence), referred to as pseudobulbar affect, also can occur and cause significant distress for both the patient and caregivers. PSP has the highest rate of pseudobulbar laughing or crying across all parkinsonian syndromes.7 Cognitive decline and dementia occur with disease progression. A frontal subcortical dementia is typical, with slowed processing, or bradyphrenia, reduced verbal fluency, and executive dysfunction. In PSP there is often perseveration of automatic behaviors, as exemplified by the three-clap test or applause sign. (The clinician should demonstrate to the patient three claps and ask him or her to copy. The applause sign is present if the patient claps more than three times and continues [perseverates]). Grasping, imitative behaviors may also be present.
Diagnosis, Heterogeneity, and Progressive Supranuclear Palsy Variants
In 1996 the National Institute of Neurological Disorders and Stroke (NINDS) and the Society for PSP (SPSP) international workshop proposed criteria for the diagnosis of classic PSP (Richardson syndrome).8 The criteria for possible or probable PSP included a progressive disorder with onset after the age of 40 with postural instability, significant falls, slowing of vertical saccades, or vertical gaze palsy. Definite PSP added the requirement of pathologic evidence. Supportive findings included symmetric rigidity, diminished response to levodopa, and early cognitive impairment. Factors excluding the diagnosis of PSP were encephalitis, focal brain lesion, hallucinations, dysautonomia, and alien limb syndrome. Cerebellar features were also previously included as exclusionary, but a recent description of a cerebellar variant of PSP has called this exclusion into question.9
The diagnosis of PSP is further complicated by its heterogeneous presentation, resulting in increasing recognition of clinical variants or phenotypes.10 In classic presentations, the diagnosis of Richardson syndrome is relatively straightforward. However, at least five phenotypic variants have recently been described: PSP-parkinsonism, PSP–pure akinesia with gait freezing, PSP–corticobasal syndrome (PSP-CBS) (or primary nonfluent aphasia), PSP–behavioral variant of frontotemporal dementia (FTD), and two other possible PSP variants with features that overlap with either primary lateral sclerosis (PLS) or cerebellar ataxia. Table 5-4 lists the clinical and pathologic features that best differentiate these PSP variants. Although each PSP variant may present differently, they are united by later onset of the more typical PSP features of postural instability, significant falls, and supranuclear gaze palsy. Additionally, although the distribution of tau pathology may differ, each variant shares pathognomonic PSP-related tau pathology. The definition of PSP is continually undergoing revision, and new criteria are likely in the near future. In 2014, because of the overlap in features, Respondek and colleagues10 proposed a more comprehensive approach to describing the phenotypic spectrum of PSP based on the predominant clinical feature seen within the first 2 years of presentation. Predominance types were defined including classic Richardson syndrome, postural instability predominant, oculomotor predominant, Parkinsonism, CBS, FTD, and those unclassified. Validation of these predominance types and atypical PSP variants is ongoing.
Table 5-4.
Progressive Supranuclear Palsy Syndromes
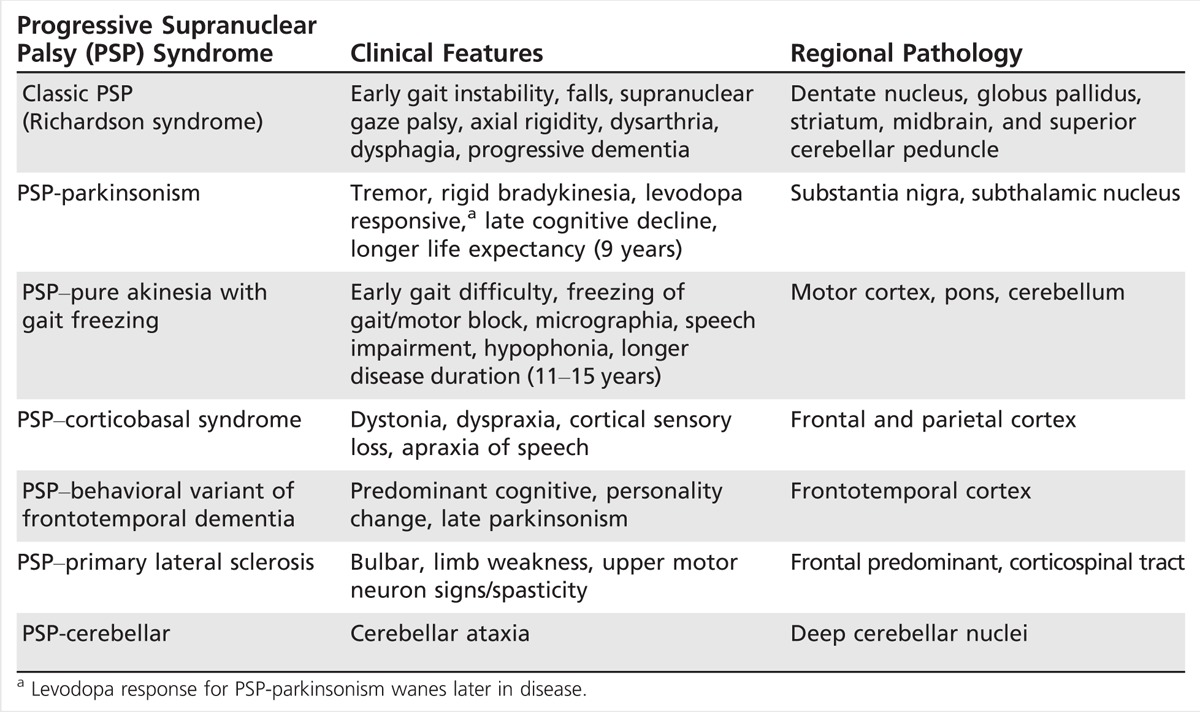
Progressive supranuclear palsy–parkinsonism. PSP-parkinsonism is the most common variant of PSP with features of tremor, early asymmetric bradykinesia, and axial rigidity that mimic PD.11 As illustrated in Case 5-1, a striking feature is relatively normal eye movements early on, although slowed saccades and reduced optokinetic nystagmus may be present, suggesting atypical disease. Patients are also frequently levodopa responsive, and that response can be sustained for years until the later onset of PSP symptoms are manifest. Life expectancy in PSP-parkinsonism is also typically longer than in Richardson syndrome, averaging 9 or more years from diagnosis.
Pure akinesia with gait freezing. In pure akinesia with gait freezing, patients characteristically present with primary freezing of gait, usually with initiation, and frequent falls.12 Micrographia, masked face, and apraxia of eyelids may also be present. However, tremor, rigidity, and ophthalmoplegia are not typical. In 2007, diagnostic criteria were developed that require gradual onset of freezing of gait or speech, without tremor, a lasting response to levodopa, or imaging suggestive of Binswanger disease. In the first 5 years, there should be no evidence of supranuclear palsy, dementia, or limb rigidity.12
Progressive supranuclear palsy–corticobasal syndrome. PSP-CBS is one of the rarest presentations of PSP. In contrast to classic PSP, early gait difficulty and postural instability are not typical.13 The clinical features of PSP-CBS include asymmetrical dyspraxia, alien limb phenomenon, and dystonia. Rigidity and bradykinesia are common and do not respond to levodopa. These patients additionally have difficulty with saccadic eye movements, most often to the side affected by apraxia, as opposed to the typical vertical gaze palsy.
Progressive supranuclear palsy–frontotemporal dementia. PSP-FTD is a rare variant of PSP. These patients may present with behavioral and cognitive dysfunction either as the only feature or as the predominant feature of disease. Other typical symptoms of PSP follow later in the course, including falls, supranuclear gaze palsy, and levodopa-unresponsive parkinsonism.14 Because of the predominant behavioral and cognitive issues, these patients may be misdiagnosed with FTD or another primary dementia.15
Progressive supranuclear palsy–primary lateral sclerosis. Several reports have suggested possible overlap of a PSP syndrome with PLS.16,17 Indeed, there are common features that overlap. PLS is similarly progressive and typically manifests bulbar symptoms, spastic gait, and sometimes ocular dysmotility, although usually not supranuclear palsy. Purported PSP-PLS cases are reported to have a tauopathy mainly in motor areas linked by the corticospinal tract.18
Cerebellar variant of progressive supranuclear palsy. Recently, a number of reports have described a novel variant of PSP with features of cerebellar ataxia either as the presenting or predominant symptom and tau pathology in the deep cerebellar nuclei.9,19 This cerebellar subtype of PSP appears rare, but may be more prevalent in Asian populations given recent reports.
Case 5-1
A 65-year-old man presented with a 5-year history of reported Parkinson disease. He was diagnosed 2 years ago, but had noticed onset of a mild right hand resting tremor 3 years earlier and progressive slowing and stiffness of his right arm and gait. After his initial diagnosis, he was started on carbidopa/levodopa 25 mg/100 mg with initial positive response. However, gait instability progressed with onset of falls, slurred speech, and mild dysphagia. His wife also noticed a progressive change in his facial expression. He seemed to stare more and rarely initiated conversation.
On examination (Supplemental Digital Content 5-1, http://links.lww.com/CONT/A178), he was fully oriented. His speech was hypophonic and dysarthric, but language and cognition were preserved. His facial expression was masked, and he tended to squint. Vertical saccades (upgaze and downgaze) were reduced, and horizontal saccades were slowed. Off levodopa he had moderate bradykinesia of hand movements, finger tapping, and rigidity in the limbs, on the right more than the left. Mild left hand dystonic posturing was also noted. He had marked neck rigidity, and he found it difficult to achieve full range of motion. He stood without assistance, and his gait was stooped, slow, and his stance was narrowed and unstable with a tendency to hold onto the examiner or walls. Pull test revealed minimal to no compensation. He would have fallen if not caught. Brain MRI was notable for age-related cerebral atrophy and mild prominence of basal cisterns. Further increase in carbidopa/levodopa dose was unhelpful. A year later, he continued to slow in all of his movements, his dysarthria worsened, and his vertical gaze palsy became more apparent. A diagnosis of progressive supranuclear palsy–parkinsonism was suspected.
Comment. This case illustrates several features suggestive of probable progressive supranuclear palsy–parkinsonism. Early on, the patient’s features were typical of Parkinson disease, including tremor, asymmetric rigid bradykinesia, and response to levodopa. Later, he developed oculomotor abnormalities, bulbar features (eg, dysarthria, mild dysphagia), increased postural instability, and waning levodopa response. There is an increased awareness of levodopa-responsive cases of progressive supranuclear palsy, especially early in the disease course.
Pathology
The hallmark of PSP is abnormal deposition of tau and associated pathology (Figure 5-3). The tufted astrocyte is pathognomonic, but other features include coiled bodies, neuropil threads, pretangles, and neurofibrillary tangles.20 Tau pathology generally spares the cortex and involves the basal ganglia, dentate, pontine, and oculomotor nuclei. There is associated gliosis and degeneration that is marked by midbrain atrophy, loss of pigmented cells in the substantia nigra, and atrophy of the subthalamic nucleus, superior cerebellar and middle cerebellar peduncles, dentate nucleus, and frontal cortex.
Figure 5-3.
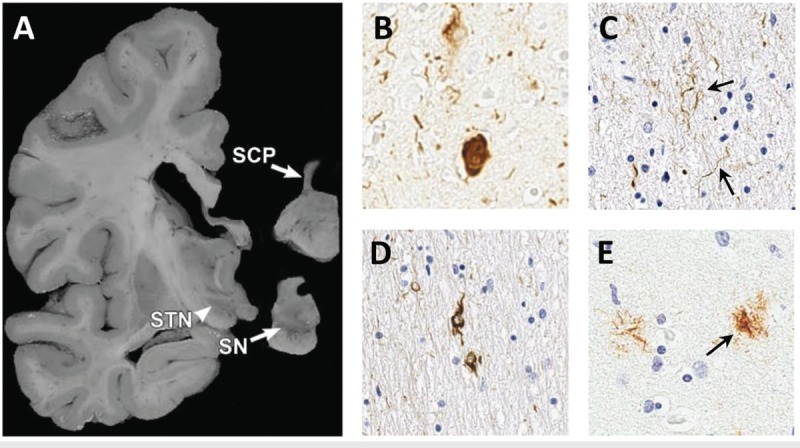
Typical neuropathologic findings in progressive supranuclear palsy. The macroscopic photo shows atrophy of the superior cerebellar peduncle (SCP) and midbrain structures including the subthalamic nucleus (STN) and loss of pigmented cells in the substantia nigra (SN) (A).The photomicrographs show classic pathologic findings including neurofibrillary tangles (B), neuropil threads (C, arrows), coiled bodies (D), and tufted astrocyte (E, arrow) stained with PHF1 antibody to human tau.
Panel A is reprinted with permission from Dickson DW, et al, Curr Opin Neurol.18 © 2010 Lippincott Williams & Wilkins, Inc. journals.lww.com/co-neurology/Abstract/2010/08000/Neuropathology_of_variants_of_progressive.9.aspx.
The tauopathies (PSP, CBS, Alzheimer disease, and FTD) are characterized by abnormal tau hyperphosphorylation and deposition. Tau is encoded by MAPT and normally functions to stabilize microtubules. The protein has six isoforms based on splice variants, including three or four microtubule binding repeat (R) domains. In PSP the 4R:3R tau ratio in brain is increased (lower in PSP-parkinsonism) compared to controls. By contrast, the opposite is observed in FTD. Although MAPT is not mutated in sporadic PSP, it is closely linked to parkinsonisms including PSP. Mutation in MAPT is most commonly associated with FTD with parkinsonism linked to chromosome 17 (FTDP-17). A recent genome-wide association study was performed and identified three additional putative genes associated with PSP: STX6 (encodes syntaxin 6), EIF2AK3 (PERK, which involves the endoplasmic reticulum unfolded response pathway), and MOBP (myelin-associated oligodendrocytic basic protein, which is concentrated in pathologic regions).21
Diagnostics
To date there is no available blood, CSF, or single imaging marker for PSP. Brain imaging such as MRI, however, remains most helpful. In PSP, atrophy of the midbrain and superior cerebellar peduncles can help distinguish PSP from other neurodegenerative disorders. Measurement of suprapontine anteroposterior diameter with MRI has been used for analyzing midbrain atrophy, but remains controversial. Another proposed method of assessing midbrain atrophy is by measurement of the midsagittal area of the midbrain tegmentum and ratio to area of the pons, which is significantly reduced in PSP compared to that in PD, MSA-parkinsonism, and healthy controls.22 As a result, the brainstem is often beaked and takes on the appearance of a hummingbird or penguin body (Figure 5-4). The width of the superior versus the middle cerebral peduncle has also been used.23 Diffusion-tensor imaging (DTI) shows both gray and white matter reduction, with predilection for the anterior and medial thalamic nuclei.24 Consistent with that found pathologically, positron emission tomography (PET)/single-photon emission computed tomography (SPECT) scans of patients with PSP reveal hypometabolism of bilateral frontal cortex, caudate heads, thalami, cingulate gyri, and midbrain.25 Recent tau PET ligands, such as 18F-T807/8 and 18F-THK523, that have been developed for Alzheimer disease also show promise for detecting tau in PSP (and other tau disorders), but will require further validation.26
Figure 5-4.
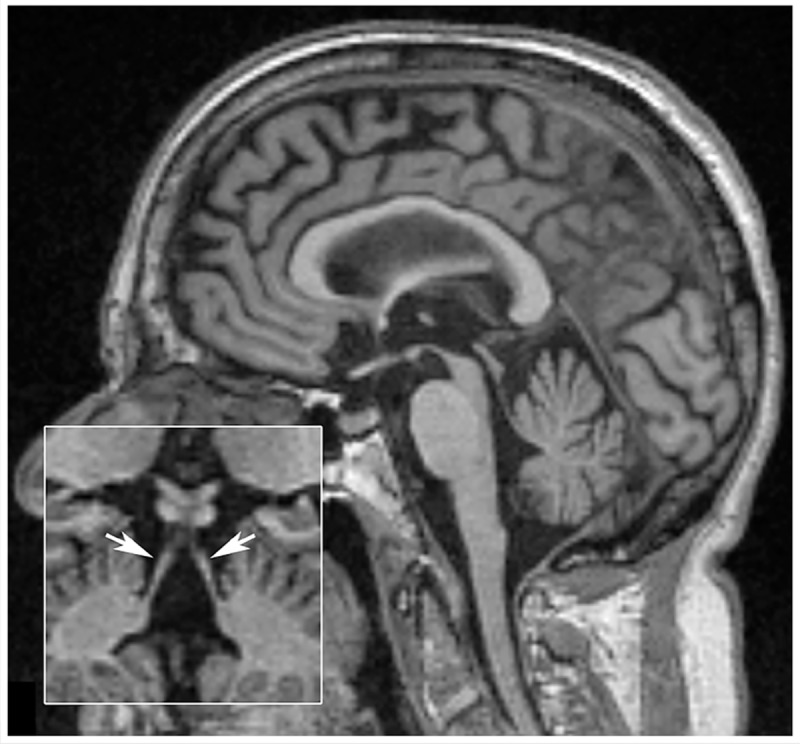
Sagittal T1-weighted MRI of a patient with progressive supranuclear palsy–Richardson syndrome. Marked midbrain atrophy is present, suggesting the appearance of a hummingbird or penguin sign. Inset shows atrophy of the superior cerebellar peduncles (arrows).
Therapeutic Strategies
Currently there is no specific treatment or cure for PSP. Treatment is focused primarily on symptomatic improvement and, ideally, should involve a multidisciplinary team approach including physical and occupational therapy, speech pathology, neuropsychology, psychiatry, social work, and palliative care. Physical therapy is critical for gait and postural instability, fall prevention, and to develop an exercise program to maintain mobility. Occupational therapy can help patients (and caregivers) with activities of daily living. Speech therapy is important early on to treat dysarthria. More advanced patients may need cognitive therapy for speech apraxia or to discuss alternative communication modes if markedly dysarthric or anarthric. Dysphagia and aspiration are the leading causes of morbidity and mortality in PSP. The author recommends early and regular swallowing evaluations (6-month intervals) to assess risk and to modify the diet. Often in later stages of the disease, the question of a feeding tube arises. Discussion with the patient, family/caregiver, and speech pathologist regarding the benefits and risks of a feeding tube is recommended. A percutaneous gastrostomy tube can help maintain nutrition, hydration, and medication administration, but does not reduce the risk of aspiration. The decision regarding gastrostomy should be carefully approached and discussed as it raises ethical, cultural, and personal considerations regarding quality of life. A social worker or case manager is often critical to help family and caregivers plan for care needs. Palliative care also should be considered, and frank discussion of advance directives should be pursued.
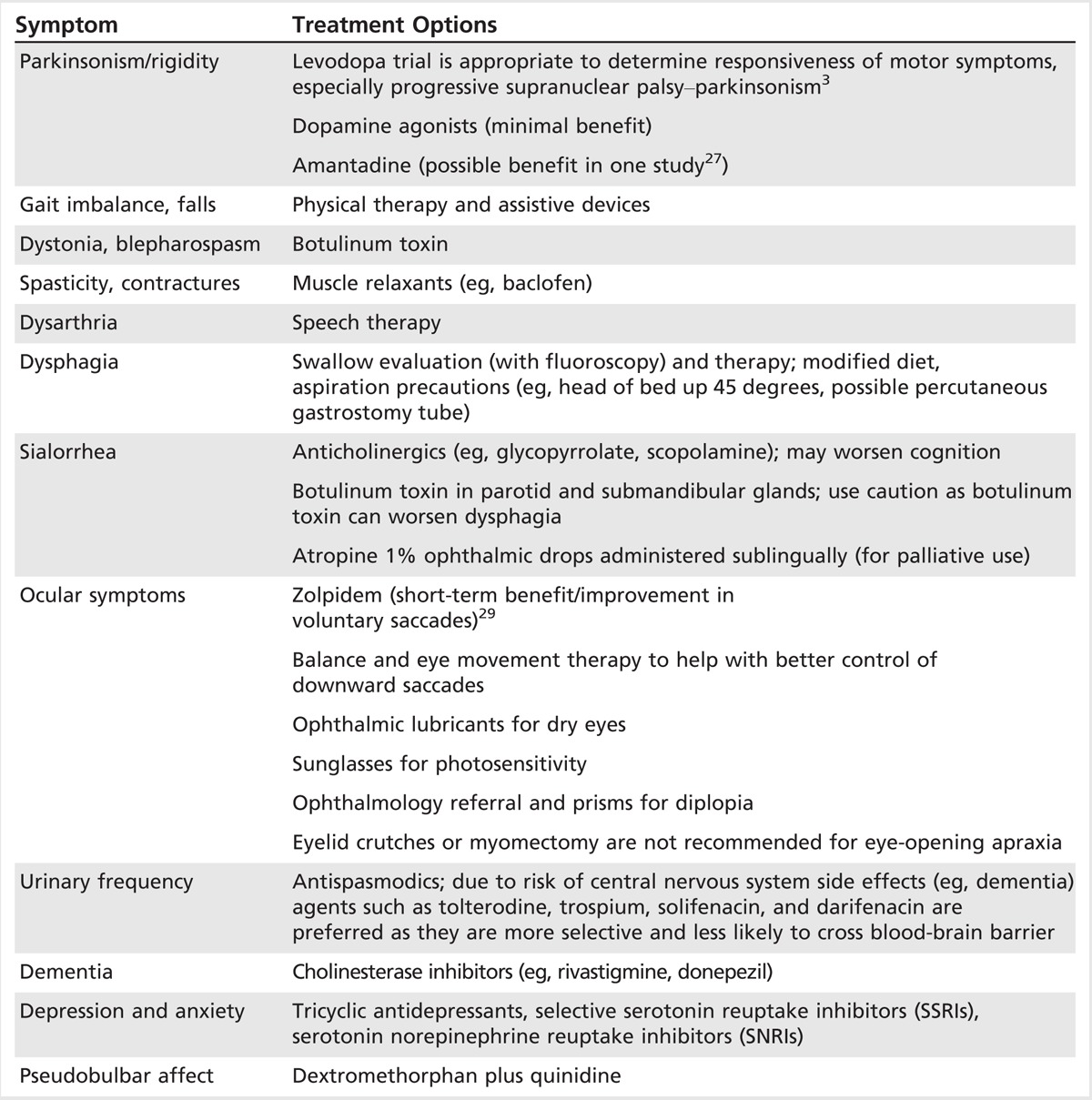
Although no one medication treats all the symptoms of PSP, specific treatments may be helpful. For parkinsonism, a levodopa trial up to 1200 mg/d (up to 300 mg per dose if it can be tolerated) for 1 month is recommended to determine responsiveness. PSP-parkinsonism in particular is often levodopa responsive initially and contrasts with that observed in classic Richardson syndrome and other variants. Dopamine agonists, however, often provide minimal benefit. Amantadine has been reported to have benefit for gait and dysphagia, but studies to support this treatment have been limited.27 Anticholinergics should be avoided due to the potential for confusion. Coenzyme Q10 is a well-tolerated supplement and has recently been shown in a small randomized clinical trial to provide modest benefit in PSP, including slight improvement in cognitive function.28 Painful dystonic posturing of the neck and limbs, blepharospasm, and eye-opening apraxia may be treated with botulinum toxin. For cognitive decline and dementia, cholinesterase inhibitors such as rivastigmine may provide modest benefit. Mood disturbance including depression, anxiety, and irritability should be identified and treated with antidepressants. For apathy, activating antidepressants such as bupropion, venlafaxine, sertraline, and fluoxetine are preferred over other selective serotonin reuptake inhibitors (SSRIs), which may worsen apathy. If pseudobulbar affect is marked, dextromethorphan-quinidine, a tricyclic antidepressant, or an SSRI may be helpful. Treatments for other symptoms are summarized in Table 5-5.
Table 5-5.
Overlapping Symptomatic Treatment Options for Progressive Supranuclear Palsy and Corticobasal Degeneration
CORTICOBASAL DEGENERATION
CBD is an atypical parkinsonian syndrome with predominant involvement of the cortex and basal ganglia that presents with varied phenotypes. The classic presentation with asymmetric rigidity, dystonia, and ideomotor apraxia is now referred to as CBS, but CBD is increasingly also recognized to present with features that may overlap with FTD, primary progressive aphasia, Alzheimer disease, posterior cortical atrophy, and PSP. Typically, marked asymmetry of involvement is the most striking feature and helps differentiate CBD from other degenerative disorders. The most common presenting feature is asymmetric hand clumsiness followed by early bradykinesia, a frontal syndrome, tremor, and rigidity.30 The mean onset of disease occurs in the sixth decade, and prognosis is generally poor with a mean survival of about 7 years from diagnosis. Typical features include marked asymmetry, focal rigidity, coarse rest/action tremor, limb dystonia (followed by contractures), alien limb phenomenon, hand, limb, gait, or speech apraxia, myoclonus, cortical sensory loss, language deficits, frontal/cortical dementia, oculomotor dysfunction (gaze palsy, impaired convergence), bulbar impairment, postural instability, gait difficulty, hyperreflexia, and extensor plantar response. Poor levodopa response tends to occur, but high-dose trials are warranted early in the disease as in PSP (levodopa dose of up to 1200 mg/d for 2 to 3 months as tolerated) and sometimes provide partial benefit. Case 5-2 illustrates a typical case of CBS.
Ideomotor apraxia, myoclonus, asymmetric rigid-bradykinetic syndrome, and later-onset gait/balance disturbance have been reported as the best predictors for CBS diagnosis.31 Ideomotor apraxia is defined by an inability to perform a skilled motor task despite having intact language, motor, and sensory function. Examples include inability to imitate gestures or mime a certain task (eg, use a screwdriver or cut with a pair of scissors). This type of apraxia can be difficult to distinguish from limb-kinetic apraxia, which is frequently seen in parkinsonisms, but is independent of modality (imitation versus miming). Peculiar to CBS, alien limb phenomenon appears as abnormal grasping, posturing, or spontaneous levitation of an arm or leg, but can also include pursuit or avoidance of a tactile stimulus in the opposite or contralateral limb. Dementia in CBS is actually a late feature with typically preserved semantic memory.32 Neuropsychiatric testing often shows a fronto-striatal-parietal predominance with deficits in attention, concentration, verbal fluency, language, praxis, and executive and visuospatial function.33 Cortical findings such as aphasia, limb apraxia, and graphesthesia depend on the hemisphere predominantly affected.
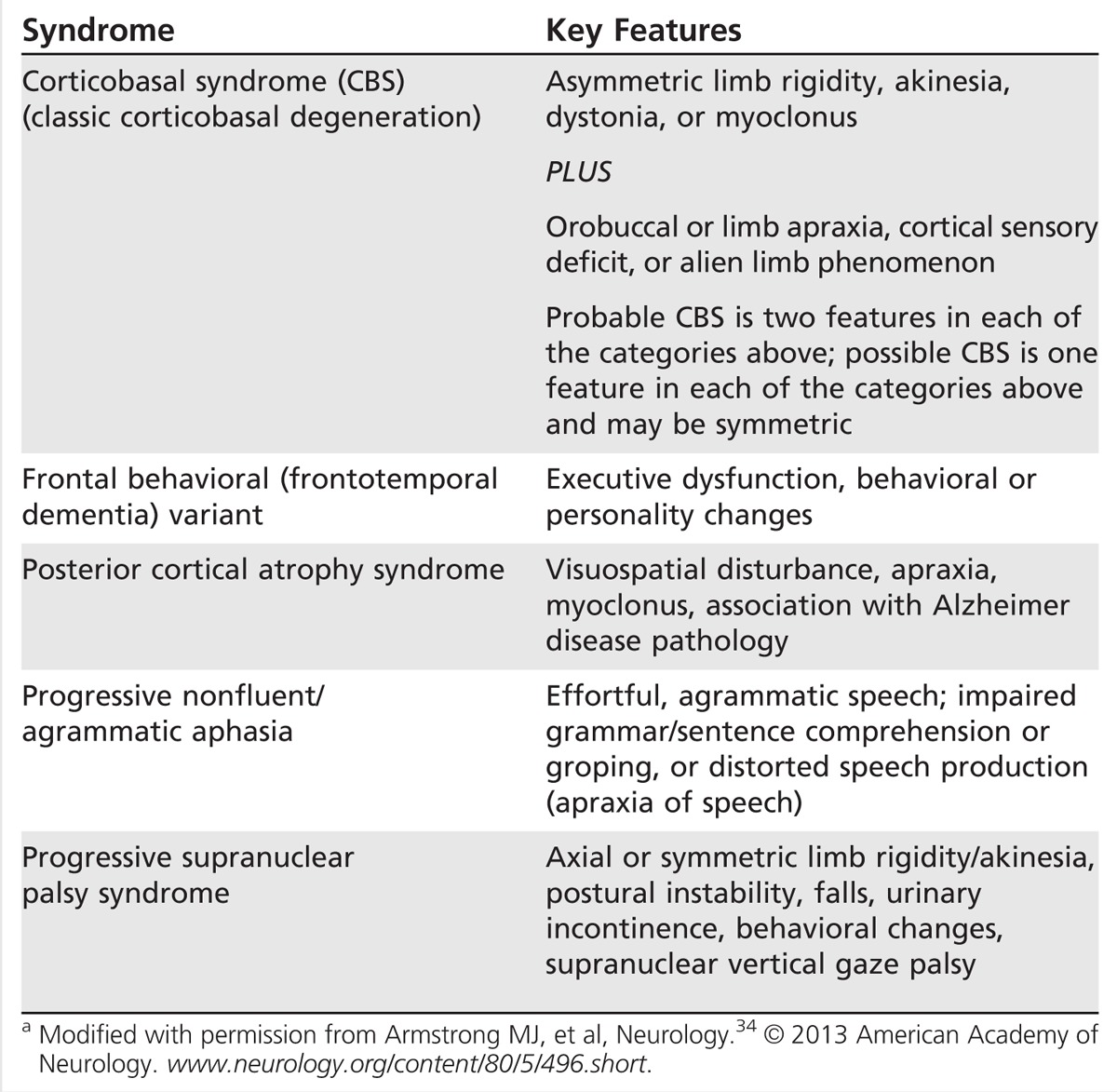
Given the varied presentations and evolving clinicopathologic understanding of CBD, clinical criteria for diagnosis have recently been revisited. Armstrong and colleagues34 proposed new diagnostic criteria for four CBD phenotypes including CBS, frontal behavioral-spatial syndrome, nonfluent/agrammatic primary progressive aphasia, and a PSP syndrome. Probable and possible criteria are described and include insidious onset, gradual progression of more than 1 year, age of onset of 50 years or more, with or without a similar family history or known tau mutations (Table 5-6).
Table 5-6.
Corticobasal Syndrome Phenotypesa
Case 5-2
An 80-year-old right-handed man with history of hypertension, prostatic hypertrophy, and deep venous thrombosis presented with symptoms of progressive gait difficulty, falls, increasing fatigue, tremulousness, and left hand dysfunction that he had experienced over the past 2 years. The patient’s wife reported that his gait and movement had slowed several years prior, but had worsened further in the setting of deep venous thrombosis and leg edema. Recently, he had begun dragging his left leg more. The patient noted increasing stiffness in his left arm and tended to hold it flexed at his side. He felt that his symptoms were beginning to spread to his right side and reported symptoms of micrographia. Additional symptoms included hoarse, slowed speech, a decrease in facial expression, and mild difficulty swallowing. If he ate too quickly he would hiccup, and swallowing pills had become difficult. The patient denied changes in cognition, memory, or mood. He had previously been evaluated by a local neurologist, diagnosed with parkinsonism, and treated with a dopamine agonist with some subjective symptomatic benefit. The addition of carbidopa/levodopa did not provide additional benefit.
On examination (Supplemental Digital Content 5-2, http://links.lww.com/CONT/A204), the patient was fully oriented but displayed mild disorganized thought processes, and his cognition and language were otherwise intact. The patient’s speech was characterized by mild hypokinetic dysarthria. He had slight facial hypomimia, reduced vertical saccades, and slowed optokinetic nystagmus. His left upper extremity tone was rigid, and he tended to hold the arm flexed at his side, but without tremor. Ideomotor apraxia and reduced graphesthesia were noted in his left hand, but no neglect. The patient’s gait was slow and stiff with the left arm held flexed, and his left leg dragged. Postural reflex was reduced on pull testing. Review of his brain MRI suggested possible asymmetric cerebral atrophy, right greater than left, and “beaking” of his midbrain.
After his initial presentation, the patient’s symptoms rapidly progressed with increasing left-sided rigidity, dystonic posturing, alien limb phenomenon, limb-kinetic apraxia, and cortical sensory deficits. He had increasing symptoms of vertical gaze palsy, gait difficulty, falls, and he required a wheelchair and assistance with activities of daily living. His speech remained mildly dysarthric, but was characterized by some grandiosity and nonsensical statements. He remained on a dopamine agonist (due to subjective benefit over levodopa) and received botulinum toxin injections for left upper extremity dystonia and mild antecollis.
Comment. This case demonstrates progressive asymmetric limb dysfunction, rigid bradykinesia, dystonia, apraxia, and cortical sensory deficits consistent with probable corticobasal syndrome. Later progression of cognitive deficits, axial rigidity, falls, and supranuclear palsy raise the possibility of a corticobasal–progressive supranuclear palsy syndrome.
Pathology
Although classically defined by as a distinct clinicopathologic entity, in recent studies CBD has increasingly been shown to present with other distinct pathologies. Clinical diagnosis of CBD correlates with CBD pathology in only 25% to 56% of cases.34 Pathologically, CBD is characterized by symmetrical cerebral atrophy, which is typically present despite the asymmetric clinical presentation usually seen in CBD. Neurodegeneration is widespread and, in addition to the cortex, includes the basal ganglia, thalamus, substantia nigra, subthalamic nucleus, and red nucleus. Pathologic diagnosis is characterized by widespread but topographic deposition of 4R-predominant, hyperphosphorylated tau in neurons and glia, astrocytic plaques, and corticobasal inclusions.35 In subcortical nuclei, such as the substantia nigra, intranuclear inclusions are reminiscent of globose neurofibrillary tangles, but those in cortex are granular, crescent shaped, or globular.36 Ballooned neurons with diffuse cytoplasmic immunoreactivity to αβ-crystallin and ubiquitin are common, but not specific to CBD. “Coiled bodies” are frequent in oligodendroglia, but, unlike PSP, tufted astrocytes are absent.
Diagnostics
Neuroimaging is most useful in CBD. Asymmetric frontoparietal atrophy can be identified on CT/MRI and can sometimes be helpful in differentiating CBD from other parkinsonian syndromes.37 Fludeoxyglucose positron emission tomography (FDG-PET) similarly can sometimes reveal asymmetric cortical metabolism. Uptake on levodopa–positron emission tomography (DOPA-PET) likewise is reduced in the striatum and highly asymmetric in the cortex. [123I]β-CIT (iodine-123-2β-carbomethoxy-3β-[4-iodophenyl]tropane) SPECT shows a similar pattern of reduced asymmetric striatal binding, but is nonspecific to CBD (similar to dopamine transporter SPECT [DAT-SPECT]). Both tau and β-amyloid ligand are also being explored for use in distinguishing CBD from other related disorders.38
Therapeutic Strategies
Effective pharmacologic therapies for CBD are still lacking, and treatment remains mainly supportive. Similar to PSP, rehabilitative services and multidisciplinary approaches are critical to maintain function as long as possible despite progressive decline. Regular exercise and activity early in the disease are most beneficial and may delay disability. Despite the limitations in treatment, a levodopa dose trial up to 800 mg/d to 1200 mg/d should be attempted; however, dopaminergic therapy rarely provides improvement and may induce dyskinesia or intolerance. Clonazepam or levetiracetam may improve myoclonus, and muscle relaxants (eg, baclofen) and botulinum toxin can help painful rigidity and dystonic contractures that limit function. Experimental therapeutics have focused on tau pathology (see previous section on PSP therapeutics).
MULTIPLE SYSTEM ATROPHY
MSA is characterized by variable presentations of parkinsonism, cerebellar and pyramidal signs, and autonomic dysfunction. It was first described in the 1960s under the subheadings of Shy-Drager syndrome, olivopontocerebellar atrophy, and striatonigral degeneration, depending on the predominant presenting symptoms.39 However, the discovery of oligodendroglial cytoplasmic inclusions and associated multisystem neurodegeneration in patients with MSA, regardless of the clinical phenotype, suggested a common pathology and has led to the terminology used today. Two clinical phenotypes are generally distinguished by predominant parkinsonism (MSA–parkinsonian type [MSA-P]) or predominant cerebellar ataxia (MSA–cerebellar type [MSA-C]) (Table 5-7). Median age of onset for MSA is 58 years of age, which is younger than that of PSP and CBD. No MSA cases have been identified younger than age 30, whereas for PSP the cutoff age is 50 years. Disease progression is faster than in PD and mean survival is approximately 6 to 9 years, consistent with more widespread neurodegeneration.
Table 5-7.
Multiple System Atrophy Phenotypes
Clinical Features
In the Western hemisphere, MSA-P is more common than MSA-C. Patients present with features of bradykinesia, rigidity, and tremor and manifest a more symmetric appearance than in PD. The pill-rolling type of tremor typically seen in PD is uncommon in patients with MSA-P. By contrast, tremor in MSA-P is often higher frequency, lower amplitude, and sometimes has a jerky, stimulus-sensitive, myoclonic component.40 Postural instability is a later feature in MSA-P compared to that in PSP. Speech is often characterized by a mixed spastic, hypokinetic dysarthria or dysphonia as opposed to that observed in PSP. Dysphagia is not uncommon, earlier, and more marked than observed in PD. Some patients also develop respiratory or laryngeal stridor. Other suggestive features include hyperreflexia, Babinski signs, dystonia, anterocollis, and early striatal deformities (Table 5-8). MSA-P is often poorly responsive to dopaminergic therapy. Patients who do respond may do so transiently, and therapy can be limited by exacerbation of orthostatic symptoms. Levodopa-induced dyskinesia is also possible; the development of early orofacial dystonia is a red flag for MSA-P. There are, however, select cases of MSA-P that respond well to levodopa for many years. Case 5-3 illustrates many of the features of MSA-P.
Table 5-8.
Differentiating Multiple System Atrophy–Parkinsonian Type and Multiple System Atrophy–Cerebellar Type from Idiopathic Parkinson Disease

In MSA-C, patients present with cerebellar signs such as gait and balance impairment, limb ataxia, and staccato speech or dysarthria. Oculomotor disturbances (nystagmus, jerky pursuits, and hypometric/hypermetric saccades) may be present. The gait ataxia in MSA-C may be indistinguishable from other sporadic, adult-onset cerebellar ataxias. Other clues in the history, such as the presence of parkinsonism, dysautonomia, or rapid progression, should point toward the diagnosis of MSA-C.40 Notably, the predominant motor features of the disease can change with disease progression so that patients who present with cerebellar features may eventually also develop parkinsonism.
Features common to both subtypes of MSA include sleep disturbance, autonomic failure, and respiratory dysfunction, which can precede motor signs by several months to years.41 Orthostatic hypotension is a frequent, debilitating symptom, often complicated by treatment. Urogenital dysfunction, such as incomplete bladder emptying or urinary incontinence, is common in women, whereas erectile dysfunction is common in men. Urogenital dysfunction tends to occur early in MSA compared to PD, in which symptoms present only at more advanced disease stages. Other MSA features include Pisa syndrome, a form of axial dystonia that manifests as lateral bending of the trunk; camptocormia, or abnormal forward flexion of the trunk; and respiratory insufficiency.42 These red flags may help to distinguish MSA from PD (Table 5-8). Although dementia historically has not been included as a feature of MSA, there is increasing recognition of cognitive dysfunction and estimates of dementia in 14% to 16% of patients with MSA.43
Case 5-3
A 66-year-old man presented with a 4-year history of progressive bilateral upper extremity and lower facial tremors, rare myoclonus, bradykinesia, and speech and gait difficulty with recent falls and freezing. Over the past couple of years he had been treated with carbidopa/levodopa and required a high dose (up to 500 mg every 4 hours for effect). However, on presentation off medication, he reported little change in symptoms except mild decrease in tremor. Additional symptoms included dysphagia, reduced appetite, urinary frequency and urgency, erectile dysfunction, constipation, dream enactment, depression, and anxiety.
On examination (Supplemental Digital Content 5-3, http://links.lww.com/CONT/A179) while seated in a wheelchair, the patient had abnormal posture with mild right head tilt, hypokinetic dysarthric speech with mild tremulousness, and jaw-opening dystonia. Facial hypomimia and reduced/slowed volitional saccades vertically were noted. Jerky tremor was apparent only with sustained posture and intention. He had moderate axial and bilateral limb rigidity and bradykinesia, and his gait was slow and marked by hesitation and freezing. On pull testing he took several steps and was caught by the examiner. Brain MRI demonstrated moderate cerebral atrophy and reduced T2 signal in the posterior putamen bilaterally and slit hyperintensity.
Comment. This case illustrates features of multiple system atrophy–parkinsonism including rapid progression, dysarthria, dystonia, abnormal posture, atypical tremor, axial and limb-rigid bradykinesia that is reasonably symmetric, early gait and postural instability, rapid eye movement (REM) sleep behavior disorder, and dysautonomia with urinary frequency and urgency, erectile dysfunction, and probable gastrointestinal dysmotility. Note also the presence of a wheelchair (the “wheelchair sign”), which can be a red flag for atypical parkinsonism and indicate significant, early postural instability. Symptoms were partially responsive to high-dose levodopa (500 mg 5 times daily). REM sleep behavior disorder was treated with clonazepam.
Pathology
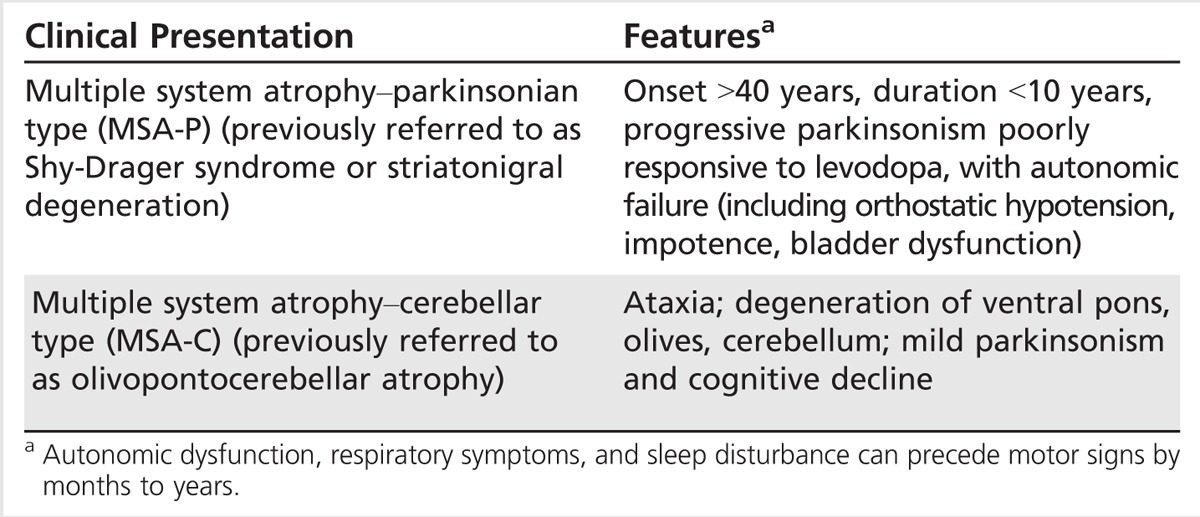
MSA is characterized by oligodendroglial cytoplasmic inclusions and multisystem neurodegeneration, including neuronal loss and gliosis involving the putamen, substantia nigra, pons, inferior olivary nucleus, cerebellum, and intermediolateral cell column of the thoracic and sacral spinal cord.44 The degree of involvement (nigrostriatal system, pons, cerebellum) determines the predominant presentation or subtype of MSA. In patients with autonomic features, the dorsal motor nucleus of the vagus, locus coeruleus, and ventrolateral medulla are affected. Oligodendroglial cytoplasmic inclusions are argyrophilic, sickle-shaped, or oval cytoplasmic aggregates that are enriched with α-synuclein and correlate with the severity of neuronal loss and disease duration (Figure 5-5).45
Figure 5-5.
Glial cytoplasmic inclusions in multiple system atrophy. Oligodendroglial cytoplasmic inclusions here are stained with antibody to human α-synuclein.
Diagnostics
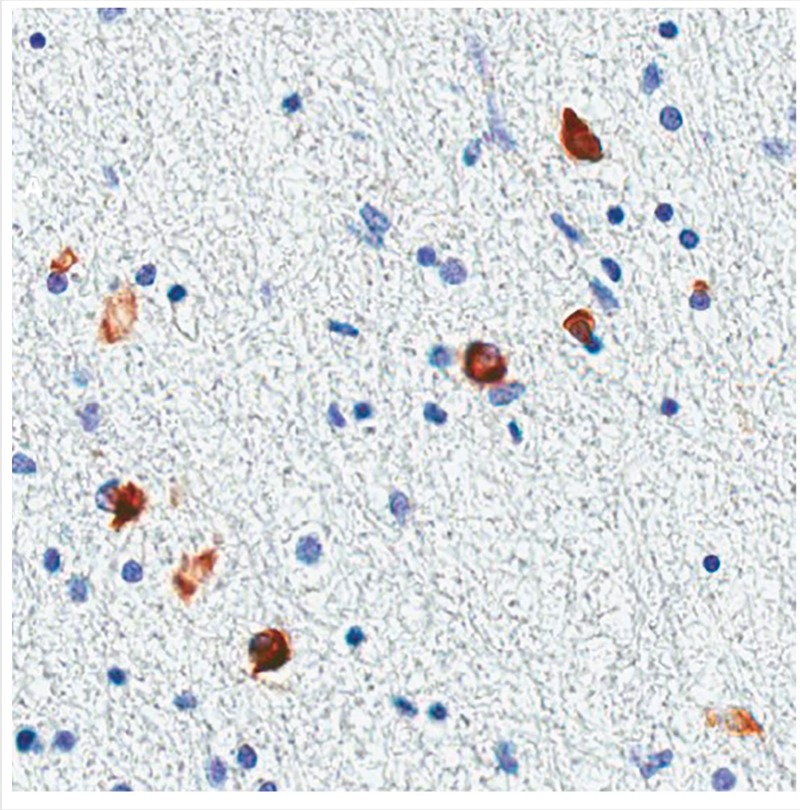
Although the diagnosis of MSA is primarily based on clinical criteria, several diagnostic studies may be helpful. So far no blood test exists to distinguish MSA from other parkinsonian syndromes. Biomarkers such as α-synuclein are being explored, but remain controversial. Neuroimaging such as MRI is often helpful. MRI findings supportive of the diagnosis include bilateral T2 hypointensity in the posterolateral putamen, representing iron deposition, and slit hyperintensity in the lateral margin of the putamen (Figure 5-6). Olivopontocerebellar atrophy is consistent with MSA-C. Pontine atrophy and gliosis may be apparent on T2-weighted images in a hot cross bun–like pattern. Additionally, PET scans often show decreased striatal and frontal metabolism. DAT (125I-ioflupane) SPECT also typically shows asymmetric reduced striatal binding.46 Autonomic testing also can be helpful and include tilt-table testing, 24-hour ambulatory blood pressure and heart rate monitoring, and baroreceptor sensitivity (ie, the baroreflex mechanism or ability to regulate blood pressure by controlling heart rate, contractility, and peripheral resistance) for orthostatic hypotension. In the office, testing should include supine blood pressure with heart rate, then standing blood pressure and heart rate after 3 minutes. A drop in systolic blood pressure of more than 20 mm Hg or drop in diastolic blood pressure of more than 10 mm Hg with minimal rise in heart rate is diagnostic. Other tests may include sweat testing (eg, quantitative sudomotor axon reflex test [QSART]) and gastric emptying study (for gastroparesis), and urodyamics for urinary dysfunction. If a sleep disturbance is suspected, polysomnogram should be performed to identify sleep apnea, periodic limb movements, and rapid eye movement (REM) sleep behavior disorder.
Figure 5-6.
Axial T2-weighted MRI of a patient with multiple system atrophy–parkinsonian type demonstrating slitlike hyperintensity (A, arrows) at the rim of the right putamen, fluid-attenuated inversion recovery (FLAIR) hypointensity of posterior putamen (B, arrows), and typical hot cross bun sign representing atrophy and gliosis of the pons (C).
Therapeutic Strategies
Treatment in MSA focuses mainly on supportive therapy and should involve allied health care and rehabilitation services because of the multisystem nature of disease. Some 30% to 60% of patients with MSA initially respond to dopaminergic therapy. In the absence of other treatments, a trial of up to 1000 mg/d to 1200 mg/d of levodopa (300 mg per dose if tolerated) for a period of 3 months is supported and is the mainstay of initial therapy for parkinsonism in MSA. Dopamine agonists, because of autonomic and other issues, are generally inferior to levodopa for MSA and have not been studied carefully in this setting. The challenge is to balance the benefits of therapy with the development of motor complications that may appear earlier in MSA as compared to classic PD. These MSA features include dyskinesia, frequently involving the jaw and face, and orthostatic hypotension. Alternative therapies such as deep brain stimulation are not recommended due to poor response.
Autonomic symptoms such as orthostatic hypotension are treated first with conservative measures including oral hydration, increased salt intake, and compression stockings or abdominal binder. If still symptomatic, pharmacologic therapy with fludrocortisone or desmopressin, which increase blood volume, may be helpful but are contraindicated in patients with heart failure. Midodrine, an α1 agonist, can also be used but has the potential to cause supine hypertension. Recently, the US Food and Drug Administration (FDA) approved the use of droxidopa for neurogenic orthostatic hypotension. Droxidopa is a synthetic amino acid precursor for norepinephrine and epinephrine. Pyridostigmine is an alternative that provides a mild boost in blood pressure without supine hypertension but most of the data supportive of its use come from patients with neuropathy. For neurogenic bladder, antispasmodics or botulinum toxin injections are often helpful. Alpha-blockers for benign prostatic hypertrophy, however, have the potential to cause hypotension. Some patients require intermittent self-catheterization or placement of a suprapubic catheter.
PARKINSON DEMENTIAS: DEMENTIA WITH LEWY BODIES
DLB (also known as [diffuse] Lewy body dementia or disease) is an early-onset, rapidly progressive dementia that is part of the spectrum of PD. It is the second most common form of neurodegenerative dementia, after Alzheimer disease, and has similar features to other dementias, including PD. The clinical criteria for DLB in addition to early dementia include: (1) parkinsonism that is coincident with or follows dementia onset; (2) fluctuating cognition, awareness, or alertness; and (3) recurrent visual hallucinations.47 Additional features include gait instability, falls, syncope or transient loss of consciousness, delusions/paranoia, depression, REM sleep behavior disorder, and neuroleptic sensitivity. A combination of dementia and psychosis in general is considered a poor prognostic predictor in this population.
Pathology
Diffuse Lewy bodies are the hallmark in DLB as well as in PD dementia (Figure 5-7). Incidental Lewy bodies, however, are also found at autopsy in individuals with no clinical signs of parkinsonism, but may indicate preclinical disease.48 The degree of Lewy body involvement of the cortex correlates with the severity of dementia.49 Spreading Lewy body pathology, or synucleinopathy, from brainstem to neocortex is described by Braak and colleagues50 and is thought to contribute to progressive cognitive impairment. Coincident Alzheimer disease pathology is also frequently found in cases and contributes to dementia.
Figure 5-7.
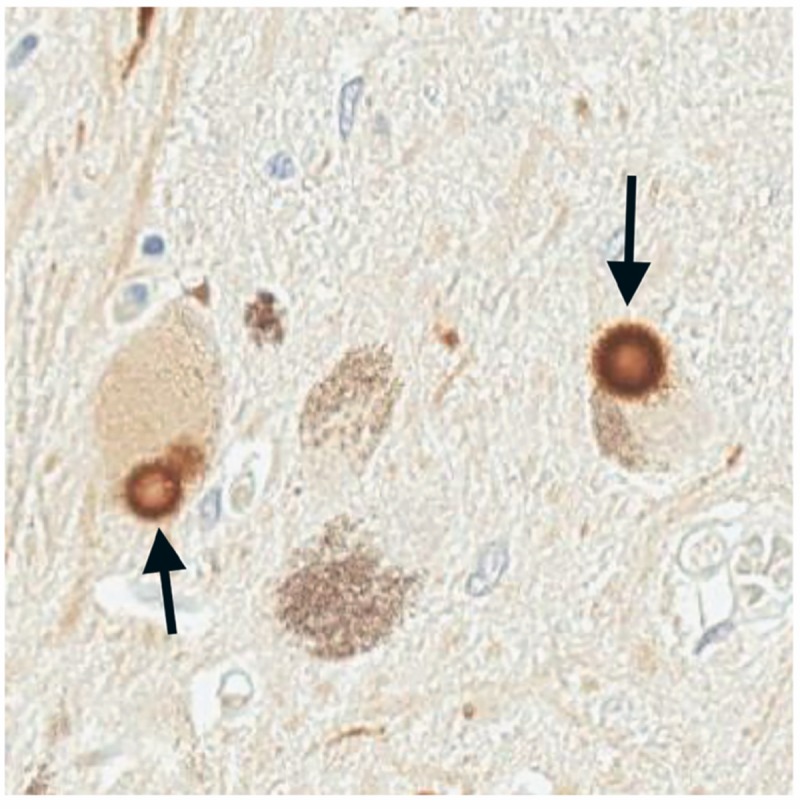
Typical Lewy bodies are found in the pigmented cells of the substantial nigra in both Parkinson disease and dementia with Lewy bodies. Here Lewy bodies (arrows) are stained with an antibody specific to the pathologic phosphorylated form (serine 129) of α-synuclein found enriched in Lewy bodies. They have a characteristic round appearance with lighter core and dark halo.
Diagnostics
Disease-specific biomarkers for DLB/PD dementia and related parkinsonian-dementia syndromes are generally lacking, and diagnosis remains primarily clinical. Imaging studies have been promising for development of potential biomarkers to distinguish DLB.51 Brain MRI generally shows diffuse cerebral atrophy with relative preservation of the occipital and mesial temporal lobes when compared to Alzheimer disease. SPECT studies focused on examining occipital hypoperfusion have reported some specificity/sensitivity in discriminating DLB from Alzheimer disease. FDG-PET has also been used to examine occipital (and parietal) lobe changes in DLB. There is emerging evidence for the cingulate island sign, or preservation of FDG-PET metabolism in the posterior cingulate relative to the cuneus and precuneus, that correlates with DLB versus Alzheimer pathology.52 Dopaminergic imaging (ie, DAT-SPECT) likewise may be useful if there is question regarding the diagnosis of DLB, but is not specific. Pittsburg compound B (PiB) scanning also can be helpful to assess amyloid burden, given the association with DLB more than in PD dementia.53 In Alzheimer disease, CSF total and phosphorylated tau levels are increased and amyloid-β level is reduced. By contrast, CSF tau (and possibly also α-synuclein) is significantly lower in DLB than in Alzheimer disease.54
Therapeutic Strategies
As with other atypical parkinsonian syndromes, treatment of DLB is symptomatic and involves coordination of caregivers and allied health personnel. Treatment of dementia-related psychosis in parkinsonisms often requires balancing use of dopaminergic medications, such as levodopa, and use of antipsychotics. Levodopa is usually beneficial for parkinsonism but in some cases can exacerbate psychosis and hallucinations. Conversely, reduction in levodopa dose, particularly at night, may benefit hallucinations, agitation, and psychosis. Dopaminergic agonists are typically not used or are weaned, and nondopaminergic agents such as monoamine oxidase inhibitors (MAOIs), amantadine, and anticholinergics are typically avoided due to their potential to worsen cognition and psychosis. If not improved with medication changes, hallucinations and psychosis are often treated with atypical antipsychotics. Of note, use of antipsychotics for dementia-related psychosis carries risk of increased mortality (mainly cardiovascular or infectious) and should be used judiciously. Although quetiapine is often the drug chosen by many clinicians and may be helpful in mild cases, studies have not supported its efficacy, particularly for control of hallucinations. In contrast, clozapine has proven efficacy but requires frequent routine blood monitoring for the risk of agranulocytosis.55 Other potential side effects of clozapine include orthostatic hypotension and depression. Recently pimavanserin, a selective 5-hydroxytryptamine, serotonin receptor 2 (5-HT2) inverse agonist, has been studied in phase 3 trials and holds promise for controlling psychosis without exacerbating parkinsonism.56
Treatment of cognitive impairment begins with formal neuropsychiatric assessment and cognitive therapy. Often, comorbid depression, apathy, and anxiety are found and should be treated. For cognitive dysfunction, cholinesterase inhibitors such as rivastigmine and donepezil have been used, and some evidence supports improvements in cognition.57 Memantine also may provide mild benefit.58
CONCLUSION
The approach to atypical parkinsonian syndromes requires careful attention to details of the patient’s history, such as mode of presentation and disease progression, as well as identification of signs and symptoms that may provide clues to diagnosis. Growing recognition of the clinical heterogeneity, overlap, and varied phenotypes of these syndromes may further aid in diagnosis and will be particularly important as novel therapies emerge. Our increasing understanding of the clinicopathologic correlates for these syndromes will aid in the development of diagnostic biomarkers and will likely bolster efforts aimed at the development of disease-modifying therapeutics.
VIDEO LEGENDS
Supplemental Digital Content 5-1
Progressive supranuclear palsy–parkinsonism. Video shows the 65-year-old man in Case 5-1 demonstrating hypophonic and dysarthric speech with preserved language and cognition. His facial expression is masked, and he tends to squint. Vertical saccades (upgaze and downgaze) are reduced, and horizontal saccades are slowed. Off levodopa he demonstrates moderate bradykinesia of hand movements, finger tapping, and rigidity in the limbs, on the right more than the left. His gait is stooped and slow, and his stance is narrowed and unstable with a tendency to hold onto the examiner or walls. Pull test reveals minimal to no compensation. He would have fallen if not caught.
http://links.lww.com/CONT/A178
© 2016 American Academy of Neurology.
Supplemental Digital Content 5-2
Corticobasal degeneration. Video shows the 80-year-old man in Case 5-2 demonstrating asymmetric limb dysfunction, rigidity, bradykinesia, dystonia, apraxia, and cortical sensory deficits consistent with probable corticobasal degeneration.
http://links.lww.com/CONT/A204
© 2016 American Academy of Neurology.
Supplemental Digital Content 5-3
Multiple system atrophy–parkinsonism. Video shows the 66-year-old man in Case 5-3 demonstrating hypokinetic dysarthric speech, jaw-opening dystonia, atypical tremor, symmetric rigid bradykinesia, and early postural instability consistent with multiple system atrophy–parkinsonism.
http://links.lww.com/CONT/A179
© 2016 American Academy of Neurology.
KEY POINTS
Key features of progressive supranuclear palsy include early gait instability, unexplained falls, supranuclear gaze palsy, axial rigidity, dysarthric speech, and dementia.
The rocket sign occurs in patients with progressive supranuclear palsy who have lost insight into their postural instability and “rocket” out of their chair without assistance, resulting in a high risk for falling.
Early signs of supranuclear gaze palsy in patients with progressive supranuclear palsy include slowed vertical saccades and reduced optokinetic nystagmus. Square-wave jerks, or minute saccadic eye movements, may also be present, representing fixation instability.
To assess for the applause sign, a clinician can demonstrate three claps to the patient and ask him or her to copy. The applause sign is present if the patient claps more than three times and continues (perseverates).
Atrophy of the midbrain and superior cerebellar peduncles correlates with disease progression in progressive supranuclear palsy.
The approach to treatment of patients with progressive supranuclear palsy should include a multidisciplinary team and involve physical, occupational, and speech therapy; psychiatry; neuropsychology; social work; and palliative care.
Levodopa therapy should be tried in most progressive supranuclear palsy cases, with a levodopa dose of up to 1200 mg/d in divided doses as tolerated. Partial response is possible in early progressive supranuclear palsy, particularly in progressive supranuclear palsy–parkinsonism.
The most common presenting features for corticobasal degeneration are asymmetric hand clumsiness or apraxia followed by early bradykinesia, frontal syndrome, tremor, and rigidity.
Ideomotor apraxia is defined by an inability to perform a skilled motor task despite having intact language, motor, and sensory function. Examples include inability to imitate gestures or mime a certain task (eg, use a screwdriver or cut with a pair of scissors). This type of apraxia can be difficult to distinguish from limb-kinetic apraxia, which is frequently seen in parkinsonisms, but is independent of modality (imitation versus miming).
Multiple system atrophy–parkinsonian type may be differentiated from Parkinson disease by its more symmetrical appearance, atypical tremor, dystonia (antecollis), early dysarthria/dysphonia, gait and postural instability, dysautonomia, and rapid progression.
Multiple system atrophy–cerebellar type is one of the most common causes of sporadic, adult-onset ataxia and is distinguished by parkinsonism, dysautonomia, and rapid progression.
Glial cytoplasmic inclusions that are enriched with α-synuclein are a pathologic hallmark of multiple system atrophy.
Pharmacologic treatment of orthostatic hypotension may include enhancing blood volume with fludrocortisone or desmopressin or adding drugs that increase vascular resistance such as midodrine, droxidopa, or pyridostigmine.
Dementia with Lewy bodies is characterized by rapid-onset dementia, parkinsonism (coincident or following cognitive decline), mental status fluctuations, and hallucinations.
REFERENCES
- 1.Jankovic J, Lang AE. Movement disorders: diagnosis and assessment. In: Bradley WG, Daroff RB, Fenichel GM, Jankovic J, eds. Neurology in clinical practice. 5th edition Philadelphia, PA: Saunders; 2008. [Google Scholar]
- 2.Quinn N. Multiple system atrophy—the nature of the beast. J Neurol Neurosurg Psychiatry 1989;(suppl):78–89. doi:10.1136/jnnp.52.Suppl.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Richardson JC, Steele J, Olszewski J. Supranuclear ophthalmoplegia, pseudobulbar palsy, nuchal dystonia and dementia. A clinical report on eight cases of “heterogenous system degeneration.” Trans Am Neurol Assoc 1963;88:25–29. [PubMed] [Google Scholar]
- 4.Williams DR, Watt HC, Lees AJ. Predictors of falls and fractures in bradykinetic rigid syndromes: a retrospective study. J Neurol Neurosurg Psychiatry 2006;77(4):468–473. doi:10.1136/jnnp.2005.074070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Batla A, Nehru R, Vijay T. Vertical wrinkling of the forehead or procerus sign in progressive supranuclear palsy. J Neurol Sci 2010;298(1-2):148–149. doi:10.1016/j.jns.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 6.Litvan I, Mega MS, Cummings JL, Fairbanks L. Neuropsychiatric aspects of progressive supranuclear palsy. Neurology 1996;47(5):1184–1189. doi:10.1212/WNL.47.5.1184. [DOI] [PubMed] [Google Scholar]
- 7.Strowd RE, Cartwright MS, Okun MS, et al. Pseudobulbar affect: prevalence and quality of life impact in movement disorders. J Neurol 2010;257(8):1382–1387. doi:10.1007/s00415-010-5550-3. [DOI] [PubMed] [Google Scholar]
- 8.Litvan I, Hauw JJ, Bartko JJ, et al. Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. J Neuropathol Exp Neurol 1996;55(1):97–105. [DOI] [PubMed] [Google Scholar]
- 9.Kanazawa M, Shimohata T, Toyoshima Y, et al. Cerebellar involvement in progressive supranuclear palsy: a clinicopathological study. Mov Disord 2009;24(9):1312–1318. doi:10.1002/mds.22583. [DOI] [PubMed] [Google Scholar]
- 10.Respondek G, Stamelou M, Kurz C, et al. The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Mov Disord 2014;29(14):1758–1766. doi:10.1002/mds.26054. [DOI] [PubMed] [Google Scholar]
- 11.Williams DR, de Silva R, Paviour DC, et al. Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson’s syndrome and PSP-parkinsonism. Brain 2005;128(pt 6):1247–1258. doi:10.1093/brain/awh488. [DOI] [PubMed] [Google Scholar]
- 12.Williams DR, Holton JL, Strand K, et al. Pure akinesia with gait freezing: a third clinical phenotype of progressive supranuclear palsy. Mov Disord 2007;22(15):2235–2241. doi:10.1002/mds.21698. [DOI] [PubMed] [Google Scholar]
- 13.Williams DR, Lees AJ. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol 2009;8(3):270–279. doi:10.1016/S1474-4422(09)70042-0. [DOI] [PubMed] [Google Scholar]
- 14.Hassan A, Parisi JE, Josephs KA. Autopsy-proven progressive supranuclear palsy presenting as behavioral variant frontotemporal dementia. Neurocase 2011;18(6):478–488. doi:10.1080/13554794.2011.627345. [DOI] [PubMed] [Google Scholar]
- 15.Donker Kaat L, Boon AJ, Azmani A, et al. Familial aggregation of parkinsonism in progressive supranuclear palsy. Neurology 2009;73(2):98–105. doi:10.1212/WNL.0b013e3181a92bcc. [DOI] [PubMed] [Google Scholar]
- 16.King A, Curran O, Al-Sarraj S. Atypical progressive supranuclear palsy presenting as primary lateral sclerosis. J Neurol Sci 2013;329(1–2):69 doi:10.1016/j.jns.2013.03.015. [DOI] [PubMed] [Google Scholar]
- 17.Nagao S, Yokota O, Nanba R, et al. Progressive supranuclear palsy presenting as primary lateral sclerosis but lacking parkinsonism, gaze palsy, aphasia, or dementia. J Neurol Sci 2012;323(1–2):147–153. doi:10.1016/j.jns.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 18.Dickson DW, Ahmed Z, Algom AA, et al. Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol 2010;23(4):394–400. doi:10.1097/WCO.0b013e32833be924. [DOI] [PubMed] [Google Scholar]
- 19.Jellinger K. Cerebellar involvement in progressive supranuclear palsy. Mov Disord 2010;25(8):1104–1105. doi:10.1002/mds.23045. [DOI] [PubMed] [Google Scholar]
- 20.Stamelou M, de Silva R, Arias- Carrión O, et al. Rational therapeutic approaches to progressive supranuclear palsy. Brain 2010;133(pt 6):1578–1590. doi:10.1093/brain/awq115. [DOI] [PubMed] [Google Scholar]
- 21.Höglinger GU, Melhem NM, Dickson DW, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet 2011;43(7):699–705. doi:10.1038/ng.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oba H, Yagishita A, Terada H, et al. New and reliable MRI diagnosis for progressive supranuclear palsy. Neurology 2005;64(12):2050–2055. doi:10.1212/01.WNL.0000165960.04422.D0. [DOI] [PubMed] [Google Scholar]
- 23.Longoni G, Agosta F, Kostić VS, et al. MRI measurements of brainstem structures in patients with Richardson’s syndrome, progressive supranuclear palsy-parkinsonism, and Parkinson’s disease. Mov Disord 2011;26(2):247–255. doi:10.1002/mds.23293. [DOI] [PubMed] [Google Scholar]
- 24.Knake S, Belke M, Menzler K, et al. In vivo demonstration of microstructural brain pathology in progressive supranuclear palsy: a DTI study using TBSS. Mov Disord 2010;25(9):1232–1238. doi:10.1002/mds.23054. [DOI] [PubMed] [Google Scholar]
- 25.Renard D, Collombier L, Castelnovo G, Labauge P. Teaching NeuroImages: FDG-PET in progressive supranuclear palsy. Neurology 2010;74(14):e60 doi:10.1212/WNL.0b013e3181d7d871. [DOI] [PubMed] [Google Scholar]
- 26.Ariza M, Kolb HC, Moechars D, et al. Tau positron emission tomography (PET) imaging: past, present, and future. J Med Chem 2015;58(11):4365–4382. doi:10.1021/jm5017544. [DOI] [PubMed] [Google Scholar]
- 27.Norris FH, Panitch HS, Denys EH, et al. The treatment of subacute sclerosing panencephalitis with interferon: a case report. J Neurol 1986;233(2):102–107. doi:10.1007/BF00313855. [DOI] [PubMed] [Google Scholar]
- 28.Stamelou M, Reuss A, Pilatus U, et al. Short-term effects of coenzyme Q10 in progressive supranuclear palsy: a randomized, placebo-controlled trial. Mov Disord 2008;23(7):942–949. doi:10.1002/mds.22023. [DOI] [PubMed] [Google Scholar]
- 29.Cotter C, Armytage T, Crimmins D. The use of zolpidem in the treatment of progressive supranuclear palsy. J Clin Neurosci 2010;17(3):385–386. doi:10.1016/j.jocn.2009.05.038. [DOI] [PubMed] [Google Scholar]
- 30.Wenning GK, Litvan I, Jankovic J, et al. Natural history and survival of 14 patients with corticobasal degeneration confirmed at postmortem examination. J Neurol Neurosurg Psychiatry 1998;64(2):184–189. doi:10.1136/jnnp.64.2.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Litvan I, Agid Y, Goetz C, et al. Accuracy of the clinical diagnosis of corticobasal degeneration: a clinicopathologic study. Neurology 1997;48(1):119–125. doi:10.1212/WNL.48.1.119. [DOI] [PubMed] [Google Scholar]
- 32.Graham NL, Bak TH, Hodges JR. Corticobasal degeneration as a cognitive disorder. Mov Disord 2003;18(11):1224–1232. doi:10.1002/mds.10536. [DOI] [PubMed] [Google Scholar]
- 33.Pillon B, Gouider-Khouja N, Deweer B, et al. Neuropsychological pattern of striatonigral degeneration: comparison with Parkinson’s disease and progressive supranuclear palsy. J Neurol Neurosurg Psychiatry 1995;58(2):174–179. doi:10.1136/jnnp.58.2.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013;80(5):496–503. doi:10.1212/WNL.0b013e31827f0fd1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dickson DW, Bergeron C, Chin SS, et al. Office of rare diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol 2002;61(11):935–946. [DOI] [PubMed] [Google Scholar]
- 36.Kouri N, Whitwell JL, Josephs KA, et al. Corticobasal degeneration: a pathologically distinct 4R tauopathy. Nat Rev Neurol 2011;7(5):263–272. doi:10.1038/nrneurol.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whitwell JL, Jack CR, Jr, Boeve BF, et al. Imaging correlates of pathology in corticobasal syndrome. Neurology 2010;75(21):1879–1887. doi:10.1212/WNL.0b013e3181feb2e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murray ME, Kouri N, Lin WL, et al. Clinicopathologic assessment and imaging of tauopathies in neurodegenerative dementias. Alzheimers Res Ther 2014;6(1):1 doi:10.1186/alzrt231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stefanova N, Bücke P, Duerr S, Wenning GK. Multiple system atrophy: an update. Lancet Neurol 2009;8(12):1172–1178. doi:10.1016/S1474-4422(09)70288-1. [DOI] [PubMed] [Google Scholar]
- 40.Köllensperger M, Geser F, Seppi K, et al. Red flags for multiple system atrophy. Mov Disord 2008;23(8):1093–1099. doi:10.1002/mds.21992. [DOI] [PubMed] [Google Scholar]
- 41.Jecmenica-Lukic M, Poewe W, Tolosa E, Wenning GK. Premotor signs and symptoms of multiple system atrophy. Lancet Neurol 2012;11(4):361–368. doi:10.1016/S1474-4422(12)70022-4. [DOI] [PubMed] [Google Scholar]
- 42.Köllensperger M, Geser F, Ndayisaba JP, et al. Presentation, diagnosis, and management of multiple system atrophy in Europe: final analysis of the European multiple system atrophy registry. Mov Disord 2010;25(15):2604–2612. doi:10.1002/mds.23192. [DOI] [PubMed] [Google Scholar]
- 43.Brown RG, Lacomblez L, Landwehrmeyer BG, et al. Cognitive impairment in patients with multiple system atrophy and progressive supranuclear palsy. Brain 2010;133(pt 8):2382–2393. doi:10.1093/brain/awq158. [DOI] [PubMed] [Google Scholar]
- 44.Ahmed Z, Asi YT, Sailer A, et al. The neuropathology, pathophysiology and genetics of multiple system atrophy. Neuropathol Appl Neurobiol 2012;38(1):4–24. doi:10.1111/j.1365-2990.2011.01234.x. [DOI] [PubMed] [Google Scholar]
- 45.Wenning GK, Jellinger KA. The role of alpha-synuclein in the pathogenesis of multiple system atrophy. Acta Neuropathol 2005;109(2):129–140. doi:10.1007/s00401-004-0935-y. [DOI] [PubMed] [Google Scholar]
- 46.Perju-Dumbrava LD, Kovacs GG, Pirker S, et al. Dopamine transporter imaging in autopsy-confirmed Parkinson’s disease and multiple system atrophy. Mov Disord 2012;27(1):65–71. doi:10.1002/mds.24000. [DOI] [PubMed] [Google Scholar]
- 47.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005;65(12):1863–1872. doi:10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 48.DelleDonne A, Klos KJ, Fujishiro H, et al. Incidental Lewy body disease and preclinical Parkinson disease. Arch Neurol 2008;65(8):1074–1080. doi:10.1001/archneur.65.8.1074. [DOI] [PubMed] [Google Scholar]
- 49.Hurtig HI, Trojanowski JQ, Galvin J, et al. Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson’s disease. Neurology 2000;54(10):1916–1921. doi:10.1212/WNL.54.10.1916. [DOI] [PubMed] [Google Scholar]
- 50.Braak H, Rüb U, Jansen Steur EN, et al. Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology 2005;64(8):1404–1410. doi:10.1212/01.WNL.0000158422.41380.82. [DOI] [PubMed] [Google Scholar]
- 51.Sinha N, Firbank M, O’Brien JT. Biomarkers in dementia with Lewy bodies: a review. Int J Geriatr Psychiatry 2012;27(5):443–453. doi:10.1002/gps.2749. [DOI] [PubMed] [Google Scholar]
- 52.Graff-Radford J, Murray ME, Lowe VJ, et al. Dementia with Lewy bodies: basis of cingulate island sign. Neurology 2014;83(9):801–809. doi: 10.1212/WNL.0000000000000734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gomperts SN, Rentz DM, Moran E, et al. Imaging amyloid deposition in Lewy body diseases. Neurology 2008;71(12):903–910. doi:10.1212/01.wnl.0000326146.60732.d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mollenhauer B, Bibl M, Trenkwalder C, et al. Follow-up investigations in cerebrospinal fluid of patients with dementia with Lewy bodies and Alzheimer’s disease. J Neural Transm (Vienna) 2005;112(7):933–948. doi:10.1007/s00702-004-0235-7. [DOI] [PubMed] [Google Scholar]
- 55.Hack N, Fayad SM, Monari EH, et al. An eight-year clinic experience with clozapine use in a Parkinson’s disease clinic setting. PloS One 2014;9(3):e91545 doi:10.1371/journal.pone.0091545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cummings J, Isaacson S, Mills R, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. Lancet 2014;383(9916):533–540. doi:10.1016/S0140-6736(13)62106-6. [DOI] [PubMed] [Google Scholar]
- 57.Rolinski M, Fox C, Maidment I, McShane R. Cholinesterase inhibitors for dementia with Lewy bodies, Parkinson’s disease dementia and cognitive impairment in Parkinson’s disease. Cochrane Database Syst Rev 2012;3:CD006504 doi:10.1002/14651858.CD006504.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emre M, Tsolaki M, Bonuccelli U, et al. Memantine for patients with Parkinson’s disease dementia or dementia with Lewy bodies: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2010;9(10):969–977. doi:10.1016/S1474-4422(10)70194-0. [DOI] [PubMed] [Google Scholar]