

Supplemental Digital Content is available in the text.
Keywords: angiogenesis modulating agents, mice, myc, semaphorins, thrombospondins, yolk sac
Abstract
Objective—
Molecular pathways governing blood vessel patterning are vital to vertebrate development. Because of their ability to counteract proangiogenic factors, antiangiogenic secreted Sema3 (class 3 semaphorins) control embryonic vascular morphogenesis. However, if and how Sema3 may play a role in the control of extraembryonic vascular development is presently unknown.
Approach and Results—
By characterizing genetically modified mice, here, we show that surprisingly Sema3F acts instead as a selective extraembryonic, but not intraembryonic proangiogenic cue. Both in vivo and in vitro, in visceral yolk sac epithelial cells, Sema3F signals to inhibit the phosphorylation-dependent degradation of Myc, a transcription factor that drives the expression of proangiogenic genes, such as the microRNA cluster 17/92. In Sema3f-null yolk sacs, the transcription of Myc-regulated microRNA 17/92 cluster members is impaired, and the synthesis of Myc and microRNA 17/92 foremost antiangiogenic target Thbs1 (thrombospondin 1) is increased, whereas Vegf (vascular endothelial growth factor) signaling is inhibited in yolk sac endothelial cells. Consistently, exogenous recombinant Sema3F inhibits the phosphorylation-dependent degradation of Myc and the synthesis of Thbs1 in mouse F9 teratocarcinoma stem cells that were in vitro differentiated in visceral yolk sac epithelial cells. Sema3f−/− mice placentas are also highly anemic and abnormally vascularized.
Conclusions—
Sema3F functions as an unconventional Sema3 that promotes extraembryonic angiogenesis by inhibiting the Myc-regulated synthesis of Thbs1 in visceral yolk sac epithelial cells.
Highlights.
Sema3 (class 3 semaphorin)-F is a novel extraembryonic proangiogenic factor.
Sema3F inhibits the degradative phosphorylation of Myc on Thr58 in visceral yolk sac epithelial cells, both in vivo and in vitro.
In the mouse yolk sac, Sema3F fosters the transcription of members of the Myc-dependent proangiogenic miR-17/92 cluster.
Sema3F inhibits the synthesis of the Myc target and antiangiogenic protein thrombospondin 1 in visceral yolk sac epithelial cells, both in vivo and in vitro.
Sema3F promotes the phosphorylation of vascular endothelial growth factor receptor 2 in yolk sac vascular endothelial cells.
Effective delivery of oxygen and nutrients is mandatory for proper development of mammalian embryos.1 Although initially supported by molecular diffusion only, around day 8 of gestation (E8), the mouse embryo starts relying in full on blood circulation to further develop. By this time, heart starts beating and extraembryonic yolk sac blood islands, that is, mesoderm-derived clusters of hematopoietic cells surrounded by a layer of endothelial cells (EnCs), coalesce to form a vascular network filled with large nucleated primitive erythroblasts.2 This allows vital molecules to diffuse from the maternal blood, which is contained in giant trophoblast cell-lined uterine sinuses, into visceral yolk sac blood vessels that directly connect to the embryonic vasculature. Between E12.5 and E14.5, the mature chorioallantoic placenta progressively replaces the yolk sac as main nutrient exchange organ supporting the development of the mouse embryo.3
Large embryonic blood vessels and the vasculature of several embryonic organs and extraembryonic yolk sac initially arise by vasculogenesis, namely, the differentiation of angioblasts into EnCs that self-assemble in a homogeneously-sized primitive vascular plexus.1 The latter is then remodeled by angiogenesis into a hierarchically sized mature vascular tree that facilitates and optimizes the distribution of blood.1 In this context, EnC mechanosensitive adhesion receptors translate blood fluid shear traction forces into biochemical signals that allow vascular remodeling and maturation.4 Although it is well established that extraembryonic EnCs do not contribute to intraembryonic blood vessels, the molecular mechanisms that differentially regulate yolk sac and embryonic vascularization are poorly defined.1
Sema3 (class 3 semaphorins) are secreted guidance cues that in the developing embryo signal through the cytosolic GAP (GTPase-activating protein) domain of Plexin receptors5 to inhibit R-Ras, M-Ras, and Rap1, 3 small GTPases that are crucial positive regulators of integrin-mediated cell adhesion in different cell types, EnC included.6,7 The chemorepulsive activity of Sema3A8–10 and Sema3E11 was previously reported to control the vascular patterning in chick, zebrafish, and mouse embryos.12 In addition, thanks to their ability to inhibit angiogenesis, both Sema3A and Sema3E exert an effective anticancer activity.13,14 Similarly, Sema3F was also found to display powerful antiangiogenic and antitumor effects13,14; however, if and how Sema3F may play a role in embryonic vascular development is unknown. Surprisingly, at odds with the antiangiogenic function of all other Sema3 proteins, here we unveil how Sema3F drives a Myc-dependent proangiogenic program that is selectively restricted to the extraembryonic vasculature. In addition to shedding light on the molecular mechanisms that selectively drive extraembryonic but not embryonic blood vessel formation, our findings also suggest how an aberrant expression of Sema3F might be involved in placenta vascular malformations.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
Targeted Disruption of Mouse Sema3f Gene Results in Partially Penetrant Embryonic Lethality but Does Not Control Embryonic Vascular Development
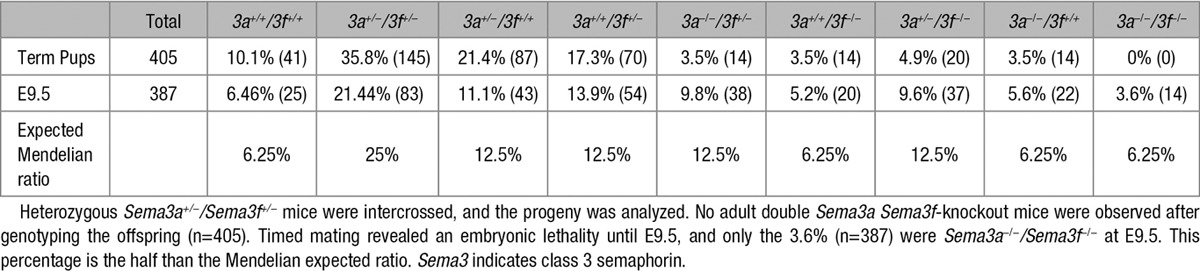
To understand whether Sema3F may control both embryonic and postnatal mouse development, we characterized and compared Sema3f- and, for control purposes, Sema3a-null mice.8 Heterozygous Sema3a and Sema3f mice were phenotypically normal and fertile. As expected,8 the percentage of born Sema3a-null mice was also much lower than the Mendelian ratio (5.7% Sema3a−/−, 39.1% Sema3a+/+, 55.2% Sema3a+/−; n=460). Similarly, Walz et al15 already described that Sema3f−/− mice were born significantly less (14%) than that predicted by Mendelian ratio. As shown in Table 1, we confirmed that the frequency of born Sema3f-knockout pups is indeed lower than expected (5.7% Sema3f−/−, 29.4% Sema3f+/+, 64.9% Sema3f+/−; n=465). The higher embryonic lethality we observed in our Sema3f−/− mice compared with that reported by Walz et al15 might be likely because of differences in the percentage of C57Bl/6 and 129P2/OlaHsd genetic backgrounds between the 2 colonies (see Materials and Methods and Figure I in the online-only Data Supplement). Thus, similarly to Sema3a−/− mice, most of Sema3f-null mice also die in utero.
Table 1.
Genotypes of the Offspring Generated From Sema3f and Sema3a Heterozygous Intercrosses on a C57BL/6 Background
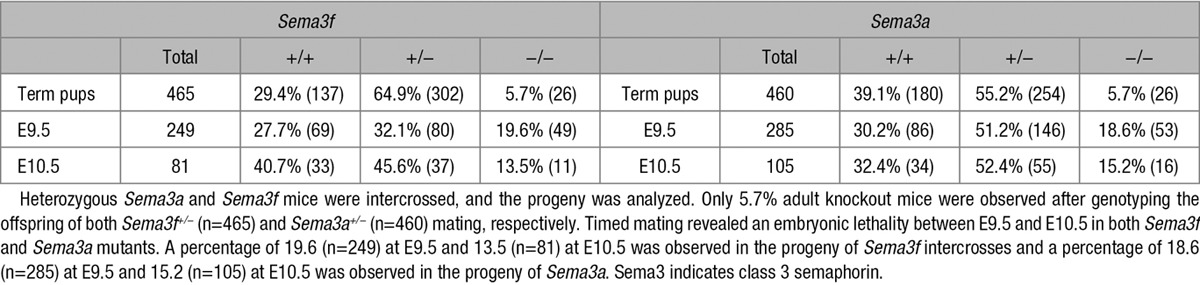
To assess the timing of embryolethality, we genotyped embryos collected at different developmental stages. Between E9.5 and E10.5, a clear significant decrease in Sema3f- and Sema3a-null embryos emerged. In particular, the Sema3f-null embryos were 19.6% at E9.5 (n=249) and 13.5% at E10.5 (n=81), whereas those of Sema3a-null embryos were 18.6% at E9.5 (n=285) and 15.2% at E10.5 (n=105). No gross phenotypic differences were present in the few outliving Sema3f- or Sema3a-knockout mice. However, as previously reported,16 if matched with wild-type mice of the same litter, 3-week-old Sema3a−/− mice were smaller in size, their weight was halved, and they had difficulty maintaining an upright posture.
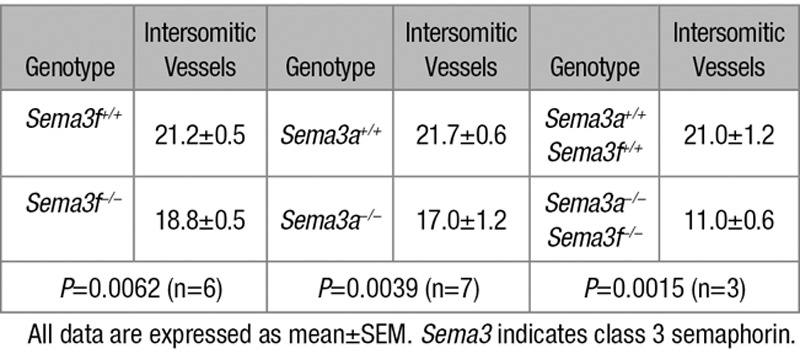
Sema3A signaling was previously reported to control vascular morphogenesis in zebrafish,9 chick,10 and mouse embryos.8,17–19 We hence investigated whether, similar to Sema3A, Sema3F may also regulate mouse embryonic angiogenesis. We examined by fluorescence confocal microscopy the vasculature of whole-mount endomucin-stained wild type (Figure 1A through 1D), Sema3f-null (Figure 1F through 1I), and Sema3a-null (Figure 1K through 1N) embryos at E9.5. Compared with control littermates (Figure 1E), Sema3f- knockout embryos (Figure 1J) displayed retarded growth (E9.5 wild-type embryos: 21.2±0.5 somites; E9.5 Sema3f−/− embryos: 18.8±0.5 somites; Table 2) but not major vascular defects (Figure 1A through 1D and 1F through 1I). In agreement with previous studies8 and differently from Sema3f-null embryos (Figure 1J), Sema3a-knockout embryos (Figure 1O) displayed an even stronger growth retardation (E9.5 Sema3a−/− embryos: 17±1.2 somites, n=7; Figure 1O) that was associated with angiogenic remodeling defects (Figure 1K through 1N). The cephalic plexus of wild-type (Figure 1B and 1C) and Sema3f−/− (Figure 1G and 1H), but not Sema3a−/− E9.5 embryos (Figure 1L and 1M), was stereotypically reshaped into a pattern wherein cephalic veins were discernible. Moreover, in E9.5 wild-type (Figure 1D) and Sema3f-null (Figure 1I) but not Sema3a-null (Figure 1N) embryos, the dorsal longitudinal anastomotical vessel started to be remodeled into a mature perineural capillary plexus.
Figure 1.
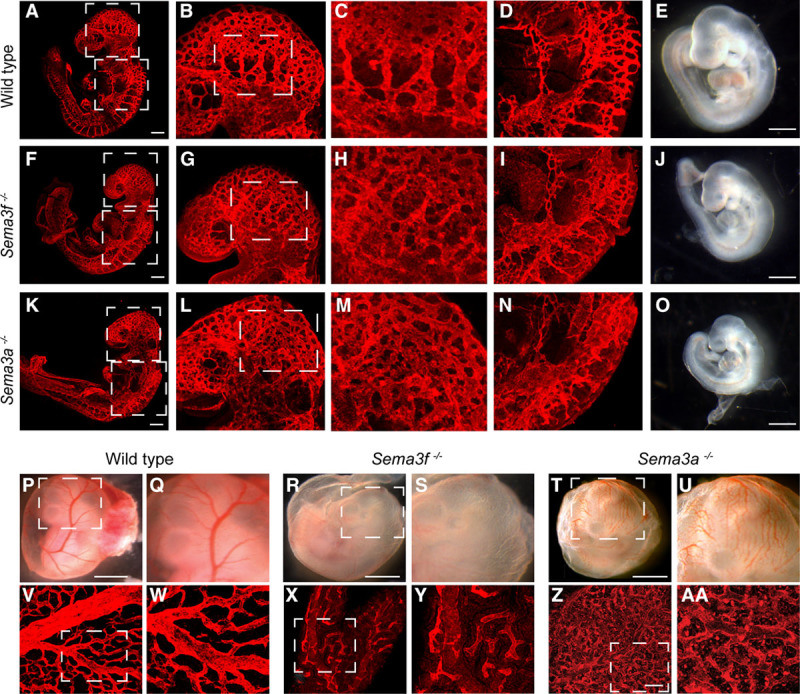
Sema3 (class 3 semaphorin)-A is necessary for embryonic angiogenic remodeling, whereas Sema3F is required for yolk sac blood vessel formation. Whole-mount endomucin stained E9.5 wild-type (A through D), Sema3f−/− (F through I), and Sema3a−/− (K through N) embryos. The cephalic plexus of wild-type (B and C) and Sema3f−/− (G and H) but not Sema3a−/− (L and M) embryos is remodeled to give rise to easily distinguishable cephalic veins. The cephalic perineural vascular plexus develops through remodeling of the dorsal longitudinal anastomotical vessel in wild-type (D) and Sema3f−/− (I) but not in Sema3a−/− embryos (N). E, J, and O, Stereomicroscopic analyses reveals how, when compared with age-matched wild-type embryos (E), the development of both Sema3f−/− (J) and Sema3a−/− (O) E9.5 embryos is delayed. Developmental delay is more severe in Sema3a−/− (O) than that in Sema3f−/− (J) embryos. B, G, and L, Magnifications of the top boxed areas in (A), (F), and (K), respectively. C, H, and M, Magnifications of the boxed areas in (B), (G), and (L), respectively. D, I, and N, Magnifications of the bottom boxed areas in (A), (F), and (K), respectively. Scale bars, 300 μm (A, F, and K) and 100 μm (E, J, and O). Stereomicroscopy (P through U) and endomucin-staining confocal microscopy (V through AA) analyses of E10.5 wild-type (P, Q, V, and W), Sema3f−/− (R, S, X, and Y) and Sema3a−/− (T, U, Z, and AA) yolk sacs. When compared with wild-type yolk sacs (P, Q, V, and W), a dramatic decrease in blood vessels is observed in Sema3f−/− yolk sacs (R, S, X, and Y), while a poorly remodeled primary capillary plexus and the persistence of some blood islands characterize Sema3a−/− (T, U, Z, and AA) yolk sacs. Q, S, U, W, Y, and AA, Magnifications of (P), (R), (T), (V), (X), and (Z), respectively. Scale bars, 100 μm (P, R, and T) and 150 μm (V, X, and Z).
Table 2.
Number of Intersomitic Vessels Evaluated in Sema3f, Sema3a, and Sema3a/Sema3f-Knockout Embryos (E9.5)
Sema3F Drives Extraembryonic Angiogenesis
In quest of explanations for the growth retardation associated with an essentially normal vascular development of Sema3f- knockout embryos, we reasoned that it may be due to defective angiogenesis in extraembryonic organs, namely, yolk sac and placenta. Accordingly, we observed that, although yolk sacs of E10.5 wild-type embryos contained a dense hierarchically ordered vascular network (Figure 1P, 1Q, 1V, and 1W), Sema3f-null yolk sacs unexpectedly displayed a dramatic reduction in blood vessel number and organization into a mature vascular tree (Figure 1R, 1S, 1X, and 1Y). Sema3f −/− mice placentas were also highly anemic and abnormally vascularized (Results section; Discussion section, and Figure II in the online-only Data Supplement). A not remodeled primary vascular plexus and occasional scattered blood islands were instead present in Sema3a-null yolk sacs (Figure 1T, 1U, 1Z, and 1AA).
The mouse yolk sac consists of mesoderm-derived blood islands and vessels that are positioned between endoderm-derived visceral yolk sac (VYS) epithelial cell (EpC) layer and parietal yolk sac (PYS) EpC layer.20 VYS EpCs control nutrient and gas exchanges and the development of blood islands and vessels; PYS EpCs instead protect the embryo.20 To understand which cell types synthesize Sema3F and express the major Sema3F coreceptor Nrp2, we performed confocal microscopy analyses on E9.5 yolk sacs that were double stained with either anti-Sema3F or anti-Nrp2 and antibodies recognizing vascular EnC or VYS EpC or PYS EpC markers, respectively, represented by endomucin, cytokeratin 8/18 (CK8/18), and Snail1 proteins.21 Sema3F protein was present in VYS EpCs and in part also in vascular EnCs of Sema3f+/+ but not in Sema3f−/− yolk sacs (Figure 2A and 2C). PYS EpCs essentially do not produce Sema3F (Figure 2A and 2C). We next analyzed the expression of Sema3F in E9.5 embryos. As illustrated in Figure IIIC and IIID in the online-only Data Supplement, Sema3F was mainly expressed in vascular EnCs of both wild-type and Sema3a−/−, but not in Sema3f−/−, embryos. Similarly, Sema3A was expressed in endomucin+ vessels of Sema3f−/− yolk sacks (Figure IIIA and IIIB in the online-only Data Supplement).
Figure 2.
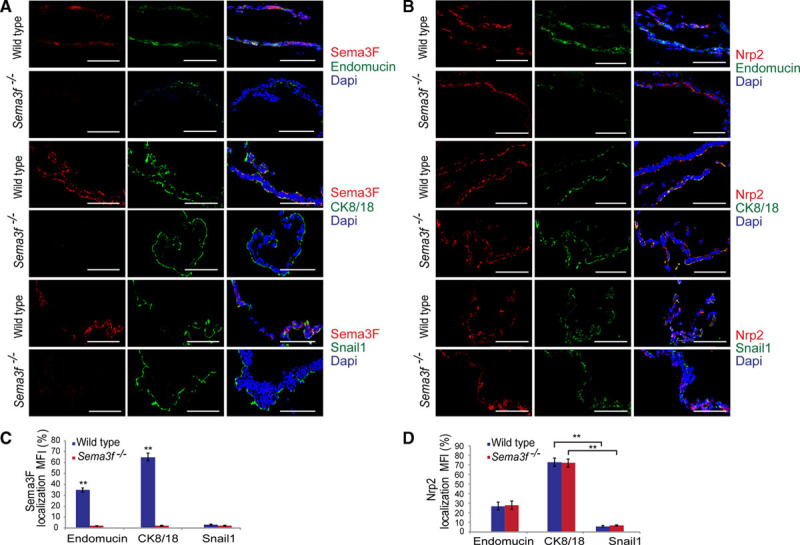
Sema3 (class 3 semaphorin)-F and Nrp2 are mainly expressed by the CK8/18 (cytokeratin 8/18)+ epithelial cells of the yolk sac. Frozen sections of yolk sacs were decorated for Sema3F (red) in costaining alternatively with endomucin, CK8/18, or Snail1 antibodies (green) and Dapi (4′,6-diamidino-2-phenylindole; blue). A, Sema3F largely colocalized with the CK8/18 visceral epithelial cells marker. As expected, no Sema3F expression was detected in Sema3f−/− samples. B, Confocal analysis showed that in both Sema3f+/+ and Sema3f−/− mice, Nrp2 is greater expressed by the visceral yolk sac as assessed by Nrp2-CK8/18 costaining. Confocal analysis was performed on tissue sections from 6 mice per group; scale bars, 100 μm. C and D, Graphs show the percentage of Sema3F and Nrp2 colocalization with endomucin, CK8/18, and Snail1 in both Sema3f+/+ and Sema3f−/− yolk sacs (**P<0.01, Student t test).
We also observed that Nrp2, the major Sema3F coreceptor, is expressed in VYS EpCs and vascular EnCs but not in PYS EpCs (Figure 2B and 2D). Thus, in the yolk sac, Nrp2-expressing EnCs and VYS EpCs may be stimulated by paracrine/autocrine Sema3F.
Sema3F Signaling Inhibits the Degradation of Myc Transcription Factor in Visceral Yolk Sac Epithelial Cells
Our data indicate that, at odds with its well-characterized antiangiogenic effects reported in the adult animal,13 Sema3F plays a proangiogenic role in extraembryonic tissues. Because it is well documented that in EnCs Sema3F elicits canonical antiangiogenic signals,22 we considered the option that Sema3F might exert its proangiogenic activity at least in part by controlling gene transcription in extraembryonic tissues, for example, by acting on Nrp2-expressing VYS EpCs. In this regard, the basic helix-loop-helix transcription factor Myc is a well-established regulator of vascular development,23 and in Caenorhabditis elegans, a Myc-like network was found to cooperate with semaphorin signaling in the control of cell migration.24 Hence, we first assessed whether the loss of Sema3f was associated with the modulation of yolk sac Myc protein expression. Confocal immunofluorescence analysis on E9.5 wild-type and Sema3f−/− yolk sacs revealed how Myc protein was decreased by 0.73-fold in Sema3f-knockout animals (Figure 3A and 3B).
Figure 3.
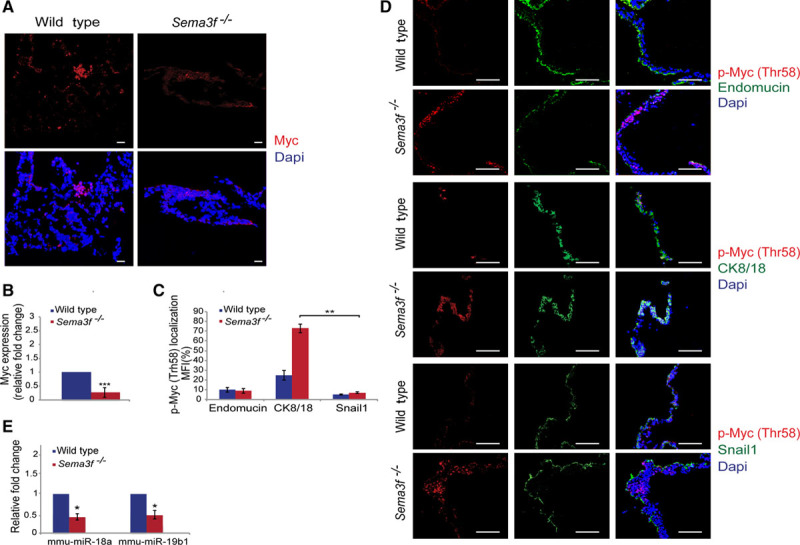
Sema3 (class 3 semaphorin)-F activates a Myc- and miR-18a/miR-19b–dependent proangiogenic pathway in the mouse yolk sac. A and B, Quantification of immunofluorescence analysis of E9.5 yolk sacs using Myc antibody (red) counterstained with Dapi (4′,6-diamidino-2-phenylindole; blue) showed that Myc expression decreases by 0.73-fold in Sema3f−/− yolk sacs. C and D, Myc phosphorylation at Thr58 significantly increased in E9.5 Sema3f−/− yolk sacs compared with the wild type. Furthermore, a double staining with anti-p-Myc (Thr58) and either anti-endomucin or anti–cytokeratin 8/18 (CK8/18), or anti-Snail1 revealed that p-Myc (Thr58) is mainly expressed in CK8/18+ visceral yolk sac epithelial cells, whereas only few amounts of Thbs1 were associated with endomucin+ vascular endothelial cells. E, Real-time polymerase chain reaction revealed how, compared with wild-type yolk sacs, proangiogenic miR-18a and miR-19b diminished in Sema3f−/− yolk sacs. (***P<0.0001; **P<0.01; *P<0.05, Student t test). Scale bars, 100 μm.
Myc protein stability is regulated by phosphorylation and ubiquitin-dependent degradation. In particular, GSK3 (glycogen synthase kinase 3)-mediated phosphorylation of Myc at Thr58 gives rise to a binding site that is directly recognized by the E3 ubiquitin ligase Fbw7(F-box and WD repeat domain-containing 7), resulting in ubiquitination followed by proteasomal degradation of Myc.25 Consistent with previous in vitro data,25 we found that in E9.5 Sema3f−/− yolk sacs the reduction in Myc protein amount was associated with a selective increase of Myc Thr58 phosphorylation in VYS EpCs, but neither in PYS EpCs nor in EnCs (Figure 3C and 3D). Altogether, our findings support a model in which in the mouse yolk sac paracrine/autocrine Sema3F signals to inhibit the (GSK3-dependent) phosphorylation on Thr58 and ensuing degradation of the proangiogenic Myc transcription factor in VYS EpCs. Hence, paracrine/autocrine Sema3F signals inhibit the phosphorylation on Thr58 and ensuing degradation of Myc in VYS EpCs.
Sema3F Promotes the Transcription of Myc-Regulated AngiomiRs in the Mouse Yolk Sac
Myc may drive yolk sac blood vessel development downstream of Sema3F signaling by promoting the transcription of different proangiogenic genes.23,26 In particular, it was reported that in cultured embryonic stem cells, Myc loss impairs Vegfa (vascular endothelial growth factor A) gene expression.23 However, even if Myc protein abundance was dramatically decreased (Figure 3A and 3B), we did not detect any significant reduction of Vegfa mRNA in Sema3f-null yolk sacs, as evaluated by real-time quantitative polymerase chain reaction (Figure IVA in the online-only Data Supplement). These findings indicate that in the mouse yolk sac, Sema3F should induce the transcription of additional Myc-dependent proangiogenic genes other than Vegfa.
Specific miRNA genes, dubbed angiomiRs, are master regulators of developmental angiogenesis and transcription of the key proangiogenic miR-17/92 cluster26 lies under the control of Myc.27 Thus, we considered the possibility that Myc protein downmodulation in Sema3f−/− yolk sacs may impair miR-17/92 gene cluster transcription. By real-time quantitative polymerase chain reaction, we analyzed the transcription of genes belonging to miR-17/92 cluster in E9.5 wild-type and Sema3f −/− yolk sacs. Among miR-17/92 gene cluster members, miR-18a and miR-19b-1 emerged as the most modulated miRNAs in Sema3f −/− yolk sacs. Compared with wild type, miR-18a and miR-19b-1 levels, respectively, decreased by 0.5- and 0.8-fold in Sema3f −/− yolk sacs (Figure 3E). Hence, Sema3F signaling drives the Myc-mediated transcription of the proangiogenic miR 17/92 cluster members miR-18a and miR-19b-1.
Sema3F Inhibits the Expression of the Antiangiogenic Factor Thbs1 (Thrombospondin 1) in Mouse Visceral Yolk Sac Epithelial Cells
Thbs1 (thrombospondin 1) is an effective angiogenesis inhibitor28 whose expression is inhibited by Myc protein23 and Myc-induced miR-17/92 cluster miRNAs,29 in particular, miR-18a and miR-19b-1.30–32 To assess whether Thbs1 expression was modulated in E9.5 Sema3f −/− yolk sacs compared with their wild-type counterpart, we first analyzed Thbs1 gene transcription by real-time quantitative polymerase chain reaction (Figure IVB in the online-only Data Supplement). Compared with wild-type yolk sacs, Thbs1 mRNA levels increased by 1.5-fold in Sema3f −/− yolk sacs. Next, we assessed Thbs1 protein expression by confocal immunofluorescence microscopy. Quantitave analysis revealed that, in comparison to wild-type yolk sacs, Thbs1-positive areas increased by 2.5-fold in Sema3f −/− yolk sacs (Figure 4A and 4B).
Figure 4.
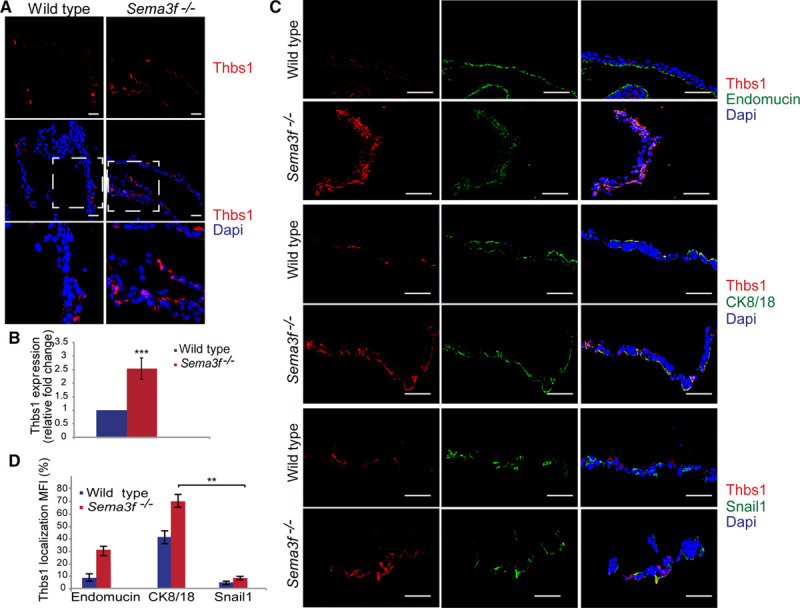
Sema3 (class 3 semaphorin)-F signaling inhibits Thbs1 (thrombospondin 1) protein expression. A and B, Quantification of immunofluorescence analyses of E9.5 yolk sacs showed that Thbs1 expression increases by 2.5-fold in Sema3f−/− yolk sacs. Scale bars, 300 μm (**P<0.001; ***P<0.0001, Student t test). C, Confocal analysis showed that Thbs1 protein expression is enriched in CK8/18 (cytokeratin 8/18)+ visceral yolk sac epithelial cells compared with the other cell types. Scale bars, 100 μm. D, Bar graph shows the percentage of Thbs1 colocalization with endomucin, CK8/18, and Snail1 in both wild-type and Sema3f−/− yolk sacs (**P<0.001, Student t test).
To understand which cell type(s) synthesize Thbs1, we analyzed by confocal microscopy E9.5 yolk sacs that were double stained with anti-Thbs1 and either antiendomucin or anti-CK8/18, or anti-Snail1. We detected Thbs1 protein mainly in CK8/18+ VYS EpCs, whereas only a few amount of Thbs1 was associated with endomucin+ vascular EnCs (Figure 4C and 4D). Of note, we did not observe any difference in the expression of Thbs2 in the different cell types of Sema3f−/−, compared with wild-type yolk sacs (Figure V in the online-only Data Supplement).
It has been clearly documented how Thbs1 effectively impairs VEGFA function mostly by inhibiting VEGF-R2 (VEGF receptor 2 phosphorylation).28,33,34 Therefore, we investigated whether endothelial VEGF signaling may be affected in Sema3f-knockout yolk sacs and uncovered that indeed tyrosine phosphorylation VEGF-R2 was reduced dramatically in vascular EnCs of E9.5 Sema3f −/− yolk sacs (Figure 5). Hence, in Sema3f-null yolk sacs, VYS EpCs synthesize high amount of Thbs1 protein that in turn effectively impairs VEGF-R2 activation in vascular EnCs.
Figure 5.
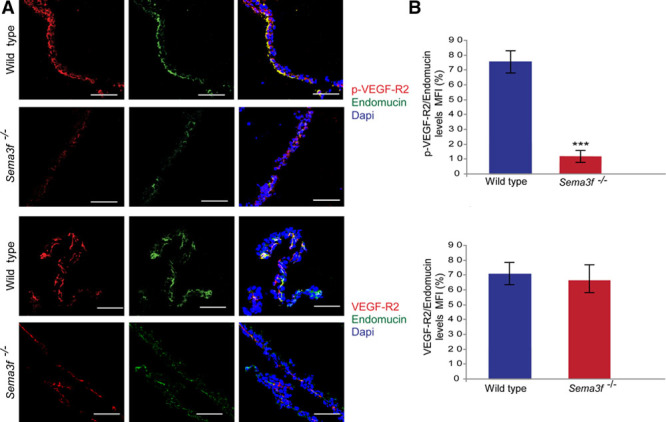
Sema3f-knockout impairs VEGF-R2 (vascular endothelial growth factor receptor R2) phosphorylation in the yolk sac. A, Confocal microscopy analysis showed that VEGF-R2 phosphorylation is strongly reduced in Sema3f−/− yolk sacs compared with wild-type controls, whereas VEGF-R2 expression is not affected. Scale bars, 100 μm. B, Percentage of p-VEGF-R2 and total VEGF-R2 relative levels in endomucin+ endothelial cell. (***P<0.0001, Student t test). Sema3 indicates class 3 semaphorin.
In In Vitro Differentiated VYS EpCs, Exogenous Recombinant Sema3F Inhibits the Myc-Dependent Expression of the Antiangiogenic Factor Thbs1
To directly assess whether Sema3F may stimulate Myc protein accumulation and the decrease of the antiangiogenic factor Thbs1, we set up an in vitro system to create differentiated CK8/18+ VYS Eps that may then be treated with recombinant Sema3F (rSema3F). To this aim, we exploited the well-characterized in vitro model of F9 testicular teratocarcinoma stem cells that, when cultured as aggregates in suspension and stimulated with retinoic acid, differentiate in VYS Eps.35,36 Fluorescent confocal microscopy confirmed that, similar to what observed in the mouse yolk sac (Figure 2B and 2D), in vitro differentiated CK8/18+ VYS EpCs expressed Sema3F receptor Nrp2 as well (Figure 6A). Of note, quantitative analysis of immunofluorescent staining revealed how exogenously added rSema3F effectively inhibits the degradative GSK-dependent phosphorylation of Myc on Thr58 (Figure 6B and 6D) and increases total Myc protein levels (Figure 6B and 6D), while significantly decreasing Thbs1 (Figure 6C and 6D), whose expression is known to be inhibited by Myc.23 Thus, direct in vitro stimulation of cultured VYS Eps with rSema3F promotes the accumulation of Myc and impairs the expression of the antiangiogenic factor Thbs1.
Figure 6.
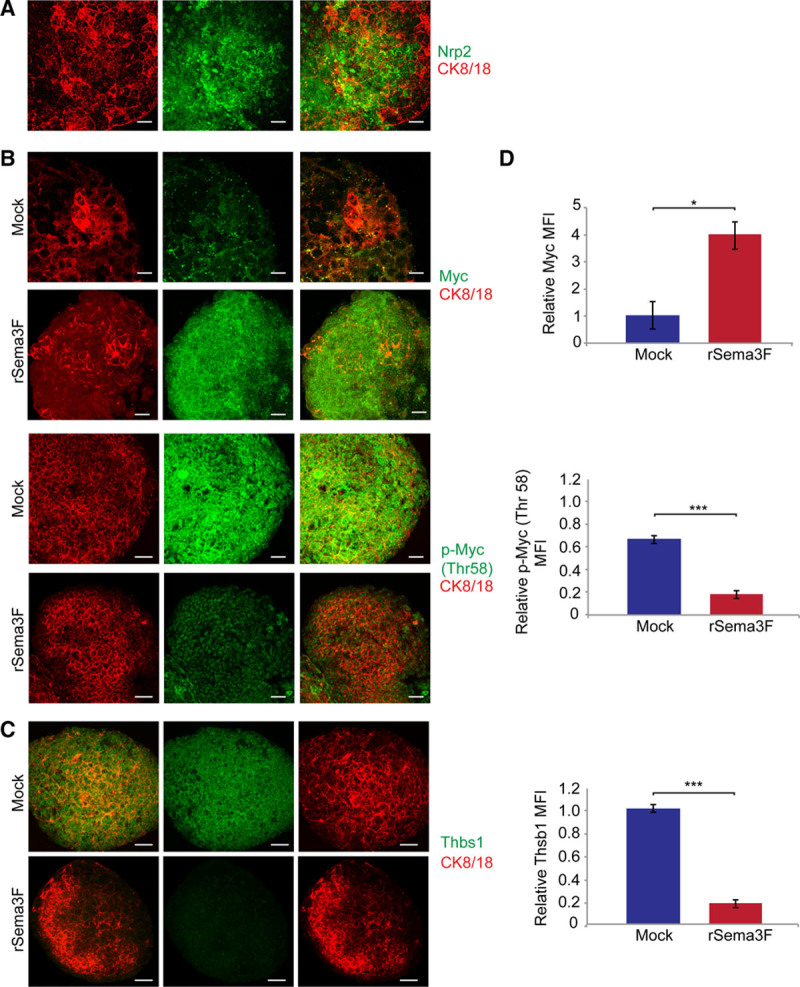
Exogenous recombinant Sema3 (class 3 semaphorin)-F impairs Myc degradation and inhibits Thbs1 (thrombospondin 1) expression in in vitro differentiated visceral yolk sac (VYS) epithelial cells (EpCs). A, Mouse F9 teratocarcinoma stem cells differentiated in VYS EpCs express Sema3F receptor Nrp2, as revealed by immunofluorescence staining. B, Stimulation with exogenous rSema3F increases Myc expression and reduces Myc Thr58 phosphorylation in in vitro differentiated VYS EpCs derived from mouse F9 teratocarcinoma stem cells. C, rSema3F stimulation also decreases the expression of antiangiogenic Thbs1 protein in VYS EpCs derived from mouse F9 teratocarcinoma stem cells. D, The relative fluorescence intensity of Myc, Thr58 phosphorylated Myc, and Thbs1 was measured in VYS EpCs derived from mouse F9 teratocarcinoma stem cells stimulated or not with rSema3F. Data are mean±SEM. n=15 cell aggregates per condition pooled from 3 independent experiments. (***P<0.0001; *P<0.05, Student t test). Scale bars, 25 μm.
Sema3a Sema3f Double Knockdown Results in Early Mouse Embryonic Lethality and Severely Impairs Both Embryonic and Extraembryonic Angiogenesis
To understand whether Sema3A and Sema3F may cooperate in regulating embryonic development, we first mated Sema3a+/− and Sema3f+/− mice to create double Sema3a+/− Sema3f+/− mice that in F1 progeny were born in a normal Mendelian ratio. Then, to generate double Sema3a/Sema3f-knockout mice, Sema3a+/− Sema3f+/− animals were mated. Among 405 weaned mice, no double Sema3a−/− Sema3f −/− mice were born. These data indicate that all double Sema3a Sema3f-knockout mice died in utero. To assess the time of embryo lethality, double Sema3a−/− Sema3f −/− embryos were genotyped at different gestation times, observing a large loss of double Sema3a−/− Sema3f −/− embryos around E9.5 (Table 3).
Table 3.
Genotypes of Progeny from Sema3a+/−/Sema3f+/− Intercrosses
Compared with wild-type animals (Figure 7D), Sema3a−/− Sema3f −/− embryos were significantly smaller (Figure 7H), and their growth delay at E9.5 was more important than that observed in Sema3a−/− embryos (Figure 1O). In fact, on average, at E9.5, Sema3a−/− Sema3f −/− embryos displayed 11 somites compared with the 21 somites of wild-type littermates (Table 2). Confocal immunofluorescence microscopy on whole-mount endomucin-stained Sema3a−/− Sema3f −/− embryos (Figure 7E through 7G) unveiled how vascular abnormalities were considerably more severe than those observed in Sema3a-null mice (Figure 1K through 1N). In particular, compared with wild-type embryos (Figure 7A through 7C), both cephalic vascular plexus (Figure 7E and 7F) and intersomitic blood vessels (Figure 7E and 7G) were poorly formed and, when formed, they were not remodeled. Short noninterconnected sprouts enclosed wide avascular spaces. Next, we evaluated the impact of Sema3a Sema3f-double knockout on yolk sac vascularization. Because of the difficulties in recovering viable double Sema3a−/− Sema3f−/− mutant embryos at E10.5, we decided to analyze the vasculature of E9.5 yolk sacs, which, differently from wild-type yolk sacs (Figure 7I), appeared extremely pale and essentially avascular (Figure 7J). Thus, abnormalities of Sema3a−/− Sema3f−/− yolk sac blood vessels (Figure 7J) were considerably more serious than those observed in either Sema3f−/− (Figure 1R, 1S, 1X, and 1Y) or Sema3a−/− (Figure 1T, 1U, 1Z, and 1AA) yolk sacs.
Figure 7.
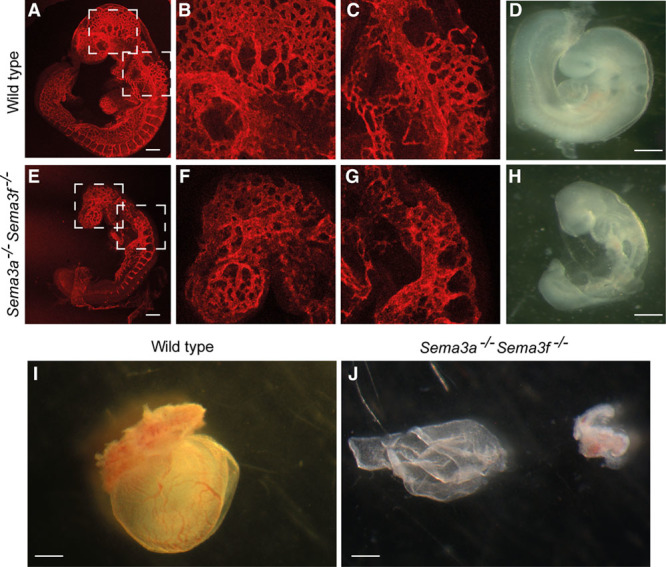
Sema3 (class 3 semaphorin)-F and Sema3A cooperate to control developmental angiogenesis. Whole-mount endomucin-stained E9.5 wild-type (A, B, and C) and Sema3a−/− Sema3f−/−-double knockout (E, F, and G) embryos. Remodeling of the cephalic plexus into veins occurs in wild type (A and B) but not in Sema3a−/− Sema3f−/− (E and F) embryos. Similarly, the dorsal longitudinal anastomotical vessel is remodeled in the cephalic perineural vascular plexus in wild type (A and C) but not in Sema3a−/− Sema3f−/− (E and G) embryos. Stereomicroscopic analyses revealed how, when compared with age-matched wild-type embryos (D), the development of Sema3a−/− Sema3f−/− (H) E9.5 embryos is severely delayed. Stereomicroscopy analysis (I and J) of E10.5 wild-type (I) and Sema3a−/− Sema3f−/− (J) yolk sacs. Sema3a−/− Sema3f−/− yolk sacs (J) are essentially avascular. B and F, Magnifications of the top boxed areas in (A) and (E), respectively. (C) and (G) are magnifications of bottom boxed areas in (A) and (E), respectively. Scale bars, 300 μm (A and E) and 100 μm (D, H, I, and J).
Discussion
The formation of properly patterned intraembryonic and extraembryonic blood vessel networks is crucial to development of vertebrate organisms.1 By counteracting the activity of proangiogenic factors such as VEGFA, antiangiogenic Sema3 proteins, eg, Sema3A and Sema3E, play key roles in shaping blood vascular patterns of developing embryos.12 Here, we unveil how, as a notable exception, Sema3F selectively exerts an effective proangiogenic activity during extraembryonic, but not intraembryonic, vascular development. The defective vascularization of Sema3f-null yolk sacs phenocopies the defects of Nrp1−/− Nrp2−/− embryos that have nearly avascular yolk sacs.37 In addition, we observed that Sema3f−/− placentas are also anemic (Results section and Discussion section in the online-only Data Supplement). Sema3F signaling mainly relies on Nrp2 coreceptor.6 However, in the absence of Nrp2, Sema3F can also signal via Nrp138 and dissociation constants for Sema3F binding to Nrp1 and Nrp2 are 1.1 and 0.09 nmol/L, respectively.39 Altogether, our and previous data suggests that both Nrp1 and Nrp2 may be required to allow Sema3F-promoting extraembryonic vascular development.
In Sema3f−/− yolk sacs, we observed a significant downregulation of Myc, a transcription factor that regulates the expression of several gene sets, such as those promoting developmental23 and tumor29 angiogenesis. Myc protein levels are regulated by GSK3 that, by phosphorylating Myc on Thr58, generates a phosphodegron that enables Fbw7 ubiquitin ligase binding, Myc ubiquitylation, and proteasomal degradation.25 Notably, as previously reported in cultured cells,25 we also found that in Sema3f−/− yolk sacs, the drop in Myc protein abundance was combined with an increased Myc Thr58 phosphorylation in Nrp2-expressing VYS EpCs. Fittingly, we also observed that the treatment of in vitro differentiated Nrp2+ VYS EpCs with exogenous Sema3F decreases Myc Thr58 phosphorylation, thus promoting Myc accumulation. In EnCs, Sema3F inhibits the phosphatidylinositol 3 kinase-dependent activation of Akt22 that is known to phosphorylate and inactivate GSK3.40 It seems that instead in VYS EpCs autocrine/paracrine Sema3F, by either activating a GSK3 kinase (such as Akt, p90RSK, p70S6K, and PKA) or inhibiting a GSK3-phosphatase (eg, PP2A), impairs the GSK3-dependent phosphorylation of Myc on Thr58 and its ensuing degradation. Further work is needed to establish the diverse molecular mechanisms by which Sema3F may activate or inhibit GSK3 function in different cell types.
MiRNAs are central regulators of cardiovascular development, and Myc promotes the transcription of the miR-17/92 cluster,26,27 whose members miR-18a and miR-19b-1 target the angiogenesis inhibitor Thbs1 hence promoting blood vessel formation.30,31 We revealed how lack of Sema3F results in reduced miR-18a and miR-19b-1 gene transcription, increased Thbs1 protein in VYS EpCs, and dramatically reduced VEGF-R2 tyrosine phosphorylation in vascular EnCs of E9.5 Sema3f−/− yolk sacs. Consistently, we also observed that exogenous Sema3F strongly inhibits the expression of Thbs1 protein in in vitro differentiated Nrp2+ VYS EpCs.
Our findings unveil a reciprocal cellular cross-talk that supports extraembryonic angiogenesis in the yolk sac (depicted in Figure VI in the online-only Data Supplement). In VYS EpCs, Sema3F, which is produced by both VYS EpCs and EnCs, signals to counteract the GSK3-driven degradative phosphorylation of Myc on Thr58, thus fostering the Myc-dependent transcription of proangiogenic miR-17/92 cluster members that in turn inhibit the synthesis of the antiangiogenic protein Thbs1 in VYS EpCs themselves, finally allowing EnCs to be elicited by VEGFA, which is also secreted by VYS EpCs and required for yolk sac vascularization.41 In sum, we provide evidence that the extraembryonic proangiogenic activity of Sema3F relies on the Myc-dependent inhibition of the antiangiogenic protein Thbs1, which in turn is an effective inhibitor of VEGF signaling.33 Indeed, the Thbs1 receptor CD36 has been shown to physically interact with VEGF-R2 and hamper its ligand-induced phosphorylation and biological activity on EnCs.28,33 Mechanistically, it was reported that Thbs1 binding to CD36 recruits the SHP-1(Src homology 2 domain–containing protein tyrosine phosphatase-1) to the CD36–VEGF-R2 complex, where SHP-1 dephosphorylates and inactivates VEGF-R2 proangiogenic signaling.34
In light of the fact that another Nrp2-binding semaphorin, that is, SEMA3B, was already suggested to be involved in human preeclampsia,42 our finding that Sema3f knockdown severely impairs placentas development and vascularization suggests that an abnormal expression of Sema3F might be implicated in placenta vascular malformations and malignant tumors (Results section, Discussion section, and Figure II in the online-only Data Supplement).
As previously reported in zebrafish,9 chick,10 and mouse embryos,8,17–19 here we further substantiate how, differently from Sema3F, Sema3A is a crucial driver of embryonic vascular morphogenesis. Our findings that ≈80% of Sema3a-knockout embryos die in utero and display a strong growth retardation compared with wild-type embryos reconcile previous apparently contradicting observations that, based on age and stage matching between wild-type and Sema3a-null embryos, concluded that Sema3A does not influence embryonic angiogenesis.43 Furthermore, we establish how Sema3A similarly controls the remodeling of the yolk sac vasculature.
We found that in Sema3a-knockout mice, embryonic and extraembryonic blood vessels develop but display severe defects remodeling into an optimized hierarchically branched mature vascular tree. On the contrary, in Sema3f-knockout mice, embryonic blood vessels are effectively remodeled into a mature and functional tree, whereas vascularization of extraembryonic tissues is strongly reduced and poorly branched. It seems that, compared with Sema3f−/− yolk sacs, the simultaneous knockdown of Sema3a and Sema3f gene further impairs the vascularization of the yolk sac that, until E12.5, is the primary means of nutrient, gas, and waste exchange for the mouse embryo.3 Therefore, our findings suggest that the further dramatic impairment of embryonic vascular remodeling observed in Sema3a/Sema3f-double knockout embryos, compared with Sema3a−/− embryos, is conceivably because of the severe disruption of the vasculature of the yolk sac, which constitutes the main nutrient and gas exchange organ between mother and embryo. Our data are in agreement with the fact that the yolk sac constitutes the first main nutrient and gas exchange organ between mother and embryo and with previous observations that mutations causing severe disruption yolk sac blood vessel formation are lethal and result in severe defects in embryo development.44–51 Finally, the severe disruption of the embryonic vasculature observed in Sema3a/Sema3f-double knockout phenocopy the previously described vascular phenotype of double targeted Nrp1−/− Nrp2−/− embryos.37
Acknowledgments
E. Giraudo, and G. Serini conceived the project; E. Giraudo, G. Serini, F. Bussolino, and F. Maione designed the experiments; D. Regano, F. Maione, A. Visintin, F. Clapero, and D. Valdembri performed the experiments; E. Giraudo, G. Serini, D. Regano, F. Maione, A. Visintin, F. Clapero, and D. Valdembri analyzed the data; E. Giraudo, G. Serini, D. Regano, F. Maione, A. Visintin, F. Clapero, and D. Valdembri interpreted the results; D. Regano, F. Maione, F. Clapero, D. Valdembri, G. Serini, and E. Giraudo wrote the paper; and all authors read and approved the article.
Sources of Funding
This work was supported by grants from Italian Association for Cancer Research (AIRC-IG grant number: 15645 to E. Giraudo; grant numbers 13016 and 16702 to G. Serini; grant number 10133 to F. Bussolino; and AIRC 5x1000 grant number 12182 to F. Bussolino); FPRC-ONLUS grant MIUR 2010 Vaschetto - 5 per mille 2010 MIUR (to E. Giraudo and G. Serini); FPRC 5xmille 2014 Ministero Salute (to E. Giraudo and G. Serini); Swiss National Science Foundation, Sinergia grant (number CRSII3 160742/1 to E. Giraudo); Fondi di Ricerca Locale (ex 60% 2015, 2016), University of Turin (to E. Giraudo and G. Serini); Fondo Investimenti per la Ricerca di Base RBAP11BYNP (Newton to F. Bussolino); European Community- FP7, contract 318035 (Biloba to F. Bussolino). Telethon Italy (GGP09175 to G. Serini); and Associazione Augusto per la Vita (to G. Serini). D. Regano was supported by FPRC-ONLUS grant MIUR 2010 Vaschetto-Chiodo Fellowship; and F. Maione was supported by postdoctoral fellowships 2014 granted by Fondazione Umberto Veronesi.
Disclosures
None.
Supplementary Material
Nonstandard Abbreviations and Acronyms
- CK8/18
- cytokeratin 8/18
- EnCs
- endothelial cells
- EpCs
- epithelial cells
- Fbw7
- F-box and WD repeat domain-containing 7
- GAP
- GTPase-activating protein
- PYS
- parietal yolk sac
- rSEMA3F
- recombinant semaphorin 3F
- SNAIL1
- snail family transcriptional repressor 1
- Thbs1
- thrombospondin 1
- VYS
- visceral yolk sac
These authors contributed equally to this article as first authors.
These authors contributed equally to this article as senior authors.
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.117.308226/-/DC1.
References
- 1.Herbert SP, Stainier DY. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat Rev Mol Cell Biol. 2011;12:551–564. doi: 10.1038/nrm3176. doi: 10.1038/nrm3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirschi KK. Hemogenic endothelium during development and beyond. Blood. 2012;119:4823–4827. doi: 10.1182/blood-2011-12-353466. doi: 10.1182/blood-2011-12-353466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Watson ED, Cross JC. Development of structures and transport functions in the mouse placenta. Physiology (Bethesda) 2005;20:180–193. doi: 10.1152/physiol.00001.2005. [DOI] [PubMed] [Google Scholar]
- 4.Hahn C, Schwartz MA. Mechanotransduction in vascular physiology and atherogenesis. Nat Rev Mol Cell Biol. 2009;10:53–62. doi: 10.1038/nrm2596. doi: 10.1038/nrm2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Worzfeld T, Swiercz JM, Sentürk A, Genz B, Korostylev A, Deng S, Xia J, Hoshino M, Epstein JA, Chan AM, Vollmar B, Acker-Palmer A, Kuner R, Offermanns S. Genetic dissection of plexin signaling in vivo. Proc Natl Acad Sci USA. 2014;111:2194–2199. doi: 10.1073/pnas.1308418111. doi: 10.1073/pnas.1308418111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Worzfeld T, Offermanns S. Semaphorins and plexins as therapeutic targets. Nat Rev Drug Discov. 2014;13:603–621. doi: 10.1038/nrd4337. doi: 10.1038/nrd4337. [DOI] [PubMed] [Google Scholar]
- 7.Serini G, Napione L, Bussolino F. Integrins team up with tyrosine kinase receptors and plexins to control angiogenesis. Curr Opin Hematol. 2008;15:235–242. doi: 10.1097/MOH.0b013e3282fa745b. doi: 10.1097/MOH.0b013e3282fa745b. [DOI] [PubMed] [Google Scholar]
- 8.Serini G, Valdembri D, Zanivan S, Morterra G, Burkhardt C, Caccavari F, Zammataro L, Primo L, Tamagnone L, Logan M, Tessier-Lavigne M, Taniguchi M, Püschel AW, Bussolino F. Class 3 semaphorins control vascular morphogenesis by inhibiting integrin function. Nature. 2003;424:391–397. doi: 10.1038/nature01784. doi: 10.1038/nature01784. [DOI] [PubMed] [Google Scholar]
- 9.Torres-Vázquez J, Gitler AD, Fraser SD, Berk JD, Pham VN, Fishman MC, Childs S, Epstein JA, Weinstein BM. Semaphorin-plexin signaling guides patterning of the developing vasculature. Dev Cell. 2004;7:117–123. doi: 10.1016/j.devcel.2004.06.008. doi: 10.1016/j.devcel.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 10.Bates D, Taylor GI, Minichiello J, Farlie P, Cichowitz A, Watson N, Klagsbrun M, Mamluk R, Newgreen DF. Neurovascular congruence results from a shared patterning mechanism that utilizes semaphorin3A and neuropilin-1. Dev Biol. 2003;255:77–98. doi: 10.1016/s0012-1606(02)00045-3. [DOI] [PubMed] [Google Scholar]
- 11.Gu C, Yoshida Y, Livet J, Reimert DV, Mann F, Merte J, Henderson CE, Jessell TM, Kolodkin AL, Ginty DD. Semaphorin 3E and plexin-D1 control vascular pattern independently of neuropilins. Science. 2005;307:265–268. doi: 10.1126/science.1105416. doi: 10.1126/science.1105416. [DOI] [PubMed] [Google Scholar]
- 12.Wälchli T, Wacker A, Frei K, Regli L, Schwab ME, Hoerstrup SP, Gerhardt H, Engelhardt B. Wiring the vascular network with neural cues: a CNS perspective. Neuron. 2015;87:271–296. doi: 10.1016/j.neuron.2015.06.038. doi: 10.1016/j.neuron.2015.06.038. [DOI] [PubMed] [Google Scholar]
- 13.Tamagnone L. Emerging role of semaphorins as major regulatory signals and potential therapeutic targets in cancer. Cancer Cell. 2012;22:145–152. doi: 10.1016/j.ccr.2012.06.031. doi: 10.1016/j.ccr.2012.06.031. [DOI] [PubMed] [Google Scholar]
- 14.Serini G, Bussolino F, Maione F, Giraudo E. Class 3 semaphorins: physiological vascular normalizing agents for anti-cancer therapy. J Intern Med. 2013;273:138–155. doi: 10.1111/joim.12017. doi: 10.1111/joim.12017. [DOI] [PubMed] [Google Scholar]
- 15.Walz A, Feinstein P, Khan M, Mombaerts P. Axonal wiring of guanylate cyclase-D-expressing olfactory neurons is dependent on neuropilin 2 and semaphorin 3F. Development. 2007;134:4063–4072. doi: 10.1242/dev.008722. doi: 10.1242/dev.008722. [DOI] [PubMed] [Google Scholar]
- 16.Taniguchi M, Yuasa S, Fujisawa H, Naruse I, Saga S, Mishina M, Yagi T. Disruption of semaphorin III/D gene causes severe abnormality in peripheral nerve projection. Neuron. 1997;19:519–530. doi: 10.1016/s0896-6273(00)80368-2. [DOI] [PubMed] [Google Scholar]
- 17.Reidy KJ, Villegas G, Teichman J, Veron D, Shen W, Jimenez J, Thomas D, Tufro A. Semaphorin3a regulates endothelial cell number and podocyte differentiation during glomerular development. Development. 2009;136:3979–3989. doi: 10.1242/dev.037267. doi: 10.1242/dev.037267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joza S, Wang J, Tseu I, Ackerley C, Post M. Fetal, but not postnatal, deletion of semaphorin-neuropilin-1 signaling affects murine alveolar development. Am J Respir Cell Mol Biol. 2013;49:627–636. doi: 10.1165/rcmb.2012-0407OC. doi: 10.1165/rcmb.2012-0407OC. [DOI] [PubMed] [Google Scholar]
- 19.Joza S, Wang J, Fox E, Hillman V, Ackerley C, Post M. Loss of semaphorin-neuropilin-1 signaling causes dysmorphic vascularization reminiscent of alveolar capillary dysplasia. Am J Pathol. 2012;181:2003–2017. doi: 10.1016/j.ajpath.2012.08.037. doi: 10.1016/j.ajpath.2012.08.037. [DOI] [PubMed] [Google Scholar]
- 20.Bielinska M, Narita N, Wilson DB. Distinct roles for visceral endoderm during embryonic mouse development. Int J Dev Biol. 1999;43:183–205. [PubMed] [Google Scholar]
- 21.Yagi S, Tagawa Y, Shiojiri N. Transdifferentiation of mouse visceral yolk sac cells into parietal yolk sac cells in vitro. Biochem Biophys Res Commun. 2016;470:917–923. doi: 10.1016/j.bbrc.2016.01.149. doi: 10.1016/j.bbrc.2016.01.149. [DOI] [PubMed] [Google Scholar]
- 22.Nakayama H, Bruneau S, Kochupurakkal N, Coma S, Briscoe DM, Klagsbrun M. Regulation of mTOR signaling by semaphorin 3F-neuropilin 2 interactions in vitro and in vivo. Sci Rep. 2015;5:11789. doi: 10.1038/srep11789. doi: 10.1038/srep11789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baudino TA, McKay C, Pendeville-Samain H, Nilsson JA, Maclean KH, White EL, Davis AC, Ihle JN, Cleveland JL. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002;16:2530–2543. doi: 10.1101/gad.1024602. doi: 10.1101/gad.1024602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pickett CL, Breen KT, Ayer DE. A C. elegans Myc-like network cooperates with semaphorin and Wnt signaling pathways to control cell migration. Dev Biol. 2007;310:226–239. doi: 10.1016/j.ydbio.2007.07.034. doi: 10.1016/j.ydbio.2007.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu L, Eisenman RN. Regulation of c-Myc protein abundance by a protein phosphatase 2A- glycogen synthase kinase 3β-negative feedback pathway. Genes Cancer. 2012;3:23–36. doi: 10.1177/1947601912448067. doi: 10.1177/1947601912448067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doebele C, Bonauer A, Fischer A, Scholz A, Reiss Y, Urbich C, Hofmann WK, Zeiher AM, Dimmeler S. Members of the microRNA-17-92 cluster exhibit a cell-intrinsic antiangiogenic function in endothelial cells. Blood. 2010;115:4944–4950. doi: 10.1182/blood-2010-01-264812. doi: 10.1182/blood-2010-01-264812. [DOI] [PubMed] [Google Scholar]
- 27.Bui TV, Mendell JT. Myc: maestro of microRNAs. Genes Cancer. 2010;1:568–575. doi: 10.1177/1947601910377491. doi: 10.1177/1947601910377491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lawler PR, Lawler J. Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harb Perspect Med. 2012;2:a006627. doi: 10.1101/cshperspect.a006627. doi: 10.1101/cshperspect.a006627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, Thomas-Tikhonenko A. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006;38:1060–1065. doi: 10.1038/ng1855. doi: 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suárez Y, Fernández-Hernando C, Yu J, Gerber SA, Harrison KD, Pober JS, Iruela-Arispe ML, Merkenschlager M, Sessa WC. Dicer-dependent endothelial microRNAs are necessary for postnatal angiogenesis. Proc Natl Acad Sci USA. 2008;105:14082–14087. doi: 10.1073/pnas.0804597105. doi: 10.1073/pnas.0804597105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohgawara T, Kubota S, Kawaki H, Kondo S, Eguchi T, Kurio N, Aoyama E, Sasaki A, Takigawa M. Regulation of chondrocytic phenotype by micro RNA 18a: involvement of Ccn2/Ctgf as a major target gene. FEBS Lett. 2009;583:1006–1010. doi: 10.1016/j.febslet.2009.02.025. doi: 10.1016/j.febslet.2009.02.025. [DOI] [PubMed] [Google Scholar]
- 32.Zhu X, Yang Y, Han T, Yin G, Gao P, Ni Y, Su X, Liu Y, Yao Y. Suppression of microRNA-18a expression inhibits invasion and promotes apoptosis of human trophoblast cells by targeting the estrogen receptor α gene. Mol Med Rep. 2015;12:2701–2706. doi: 10.3892/mmr.2015.3724. doi: 10.3892/mmr.2015.3724. [DOI] [PubMed] [Google Scholar]
- 33.Zhang X, Kazerounian S, Duquette M, Perruzzi C, Nagy JA, Dvorak HF, Parangi S, Lawler J. Thrombospondin-1 modulates vascular endothelial growth factor activity at the receptor level. FASEB J. 2009;23:3368–3376. doi: 10.1096/fj.09-131649. doi: 10.1096/fj.09-131649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chu LY, Ramakrishnan DP, Silverstein RL. Thrombospondin-1 modulates VEGF signaling via CD36 by recruiting SHP-1 to VEGFR2 complex in microvascular endothelial cells. Blood. 2013;122:1822–1832. doi: 10.1182/blood-2013-01-482315. doi: 10.1182/blood-2013-01-482315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hogan BL, Taylor A, Adamson E. Cell interactions modulate embryonal carcinoma cell differentiation into parietal or visceral endoderm. Nature. 1981;291:235–237. doi: 10.1038/291235a0. [DOI] [PubMed] [Google Scholar]
- 36.Thompson JR, Gudas LJ. Retinoic acid induces parietal endoderm but not primitive endoderm and visceral endoderm differentiation in F9 teratocarcinoma stem cells with a targeted deletion of the Rex-1 (Zfp-42) gene. Mol Cell Endocrinol. 2002;195:119–133. doi: 10.1016/s0303-7207(02)00180-6. [DOI] [PubMed] [Google Scholar]
- 37.Takashima S, Kitakaze M, Asakura M, Asanuma H, Sanada S, Tashiro F, Niwa H, Miyazaki Ji J, Hirota S, Kitamura Y, Kitsukawa T, Fujisawa H, Klagsbrun M, Hori M. Targeting of both mouse neuropilin-1 and neuropilin-2 genes severely impairs developmental yolk sac and embryonic angiogenesis. Proc Natl Acad Sci USA. 2002;99:3657–3662. doi: 10.1073/pnas.022017899. doi: 10.1073/pnas.022017899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nasarre P, Constantin B, Rouhaud L, Harnois T, Raymond G, Drabkin HA, Bourmeyster N, Roche J. Semaphorin SEMA3F and VEGF have opposing effects on cell attachment and spreading. Neoplasia. 2003;5:83–92. doi: 10.1016/s1476-5586(03)80020-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen H, Chédotal A, He Z, Goodman CS, Tessier-Lavigne M. Neuropilin-2, a novel member of the neuropilin family, is a high affinity receptor for the semaphorins Sema E and Sema IV but not Sema III. Neuron. 1997;19:547–559. doi: 10.1016/s0896-6273(00)80371-2. [DOI] [PubMed] [Google Scholar]
- 40.Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010;20:87–90. doi: 10.1016/j.gde.2009.11.002. doi: 10.1016/j.gde.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Damert A, Miquerol L, Gertsenstein M, Risau W, Nagy A. Insufficient VEGFA activity in yolk sac endoderm compromises haematopoietic and endothelial differentiation. Development. 2002;129:1881–1892. doi: 10.1242/dev.129.8.1881. [DOI] [PubMed] [Google Scholar]
- 42.Zhou Y, Gormley MJ, Hunkapiller NM, Kapidzic M, Stolyarov Y, Feng V, Nishida M, Drake PM, Bianco K, Wang F, McMaster MT, Fisher SJ. Reversal of gene dysregulation in cultured cytotrophoblasts reveals possible causes of preeclampsia. J Clin Invest. 2013;123:2862–2872. doi: 10.1172/JCI66966. doi: 10.1172/JCI66966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vieira JM, Schwarz Q, Ruhrberg C. Selective requirements for NRP1 ligands during neurovascular patterning. Development. 2007;134:1833–1843. doi: 10.1242/dev.002402. doi: 10.1242/dev.002402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Millauer B, Wizigmann-Voos S, Schnürch H, Martinez R, Møller NP, Risau W, Ullrich A. High affinity VEGF binding and developmental expression suggest Flk-1 as a major regulator of vasculogenesis and angiogenesis. Cell. 1993;72:835–846. doi: 10.1016/0092-8674(93)90573-9. [DOI] [PubMed] [Google Scholar]
- 45.Dickson MC, Martin JS, Cousins FM, Kulkarni AB, Karlsson S, Akhurst RJ. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development. 1995;121:1845–1854. doi: 10.1242/dev.121.6.1845. [DOI] [PubMed] [Google Scholar]
- 46.Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. doi: 10.1038/376066a0. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- 47.Healy AM, Rayburn HB, Rosenberg RD, Weiler H. Absence of the blood-clotting regulator thrombomodulin causes embryonic lethality in mice before development of a functional cardiovascular system. Proc Natl Acad Sci USA. 1995;92:850–854. doi: 10.1073/pnas.92.3.850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mustonen T, Alitalo K. Endothelial receptor tyrosine kinases involved in angiogenesis. J Cell Biol. 1995;129:895–898. doi: 10.1083/jcb.129.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Winnier G, Blessing M, Labosky PA, Hogan BL. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 1995;9:2105–2116. doi: 10.1101/gad.9.17.2105. [DOI] [PubMed] [Google Scholar]
- 50.Boucher DM, Pedersen RA. Induction and differentiation of extra-embryonic mesoderm in the mouse. Reprod Fertil Dev. 1996;8:765–777. doi: 10.1071/rd9960765. [DOI] [PubMed] [Google Scholar]
- 51.Carmeliet P, Ferreira V, Breier G, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]