

Abstract
Aims: The susceptibility of CA1 over CA3 to damage from cerebral ischemia may be related to the differences in reactive oxygen species (ROS) production/removal between the two hippocampal subfields. We aimed to measure CA1/CA3 differences in net ROS production in real time in the first 30 min of reperfusion in pyramidal cells. We aimed to determine the underlying cause of the differential vulnerability of CA1 and CA3.
Results: Real-time determinations of mitochondrial H2O2 and, independently, glutathione (GSH) redox status from roGFP-based probes in individual pyramidal cells in organotypic hippocampal cultures during oxygen–glucose deprivation (OGD)–reperfusion (RP) demonstrate a significantly more oxidizing environment during RP in CA1 than CA3 mitochondria. Protein levels (immunohistochemistry and Western blots), roGFP2-based probe measurements during controlled mitochondrial production of ROS, and thioredoxin reductase (TrxR) inhibition by auranofin are consistent with a more effective mitochondrial thioredoxin (Trx) system in CA3. Inhibition of TrxR eliminates the differences in redox status and cell death between the regions. Overexpression of cytosolic Trx1 does not influence mitochondrial H2O2 production.
Innovation: Real-time changes of mitochondrial H2O2 and GSH in tissue cultures during early RP, and also during controlled production of superoxide and peroxide, reveal significant differences between CA1 and CA3. The mitochondrial Trx system is responsible for the observed differences during RP as well as for delayed cell death 18 h afterward.
Conclusion: Greater mitochondrial Trx efficacy in CA3 pyramidal cells results in less vulnerability to ischemia/reperfusion because of the less oxidizing environment in CA3 mitochondria during RP. Antioxid. Redox Signal. 27, 534–549.
Keywords: : mitochondria, ROS, thioredoxin, glutathione, OGD-RP, hippocampal pyramidal cell
Introduction
A case of selective ischemic damage of the hippocampal CA1 region was documented in 1962 (9). Later observations of the brains from patients who had suffered transient global ischemia confirmed that CA1 is more vulnerable than CA3 (6, 19, 52, 73). The selective vulnerability of CA1 pyramidal cells also has been found in animal models of ischemic stroke (54, 55) and in organotypic hippocampal slice cultures (OHSCs) (23) after oxygen–glucose deprivation (OGD)–reperfusion (RP), an in vitro ischemia model (3, 53). Understanding the mechanism underlying the selective ischemic vulnerability of CA1 is of great interest for clarification of the pathophysiology of memory loss after global ischemia in man and thus for possible pharmacological interventions.
Innovation.
We hypothesized that differences in how hippocampal pyramidal neurons manage reactive oxygen species (ROS) in the first minutes of reperfusion (RP) following oxygen–glucose deprivation (OGD) were responsible for the differences in their susceptibility to damage from OGD-RP. Based on observations of real-time redox changes in mitochondria during OGD-RP and during controlled generation of ROS in single pyramidal cells within organotypic hippocampal slice cultures, we conclude that the mitochondrial thioredoxin system is responsible for more effective ROS management by CA3. The novel measurements lead to the unexpected conclusion that this is responsible for relatively lower susceptibility of CA3 to damage from ischemia.
One pivotal factor involved in the selective vulnerability of CA1 in OHSCs is oxidative stress caused by reactive oxygen species (ROS), which leads ultimately to necrosis and apoptosis (41). Longer-term effects of excessive ROS include the expression of pro-oxidant enzymes and cytokines, provoking an inflammatory response leading to additional ROS production and neuronal death (46). Notably, the increase of ROS is greater in CA1 than in CA3 under ischemia (13, 72), and hippocampal damage following focal or global ischemia can be alleviated if increased production of ROS is suppressed (12), but the reasons for the differential ROS levels in CA1 and CA3 are not known.
Wang et al. attempted to decipher the mechanism leading to differential vulnerabilities to excessive ROS in CA1 and CA3 by focusing on changes in gene expression following oxidative stress (67). They concluded that CA1 normally has higher ROS levels as well as many ROS-related transcripts, both pro- and antioxidant, at higher levels than in CA3. These observations are important, but they do not address the early stages of ROS creation that may occur in less than an hour. These early events are critical because they may launch the affected cell into a path to necrosis or apoptosis (15, 40, 41). Recently, Stanika et al. reported that N-methyl-D-aspartate (NMDA) increased mitochondrial Ca2+ in CA1 more than in CA3 in OHSCs (63). Elevated mitochondrial Ca2+ leads to ROS formation (61).
Of course, cells have natural systems to minimize damage from various ROS, including the glutathione (GSH) and thioredoxin (Trx) systems (20, 29, 47, 57). It is the balance of ROS production and removal that ultimately dictates whether ROS levels achieve a toxic magnitude. Thus, knowing the changes in ROS levels and their effect on the redox status of the most abundant antioxidant system, GSH, over time is important. Reversible green fluorescent protein (GFP)-based probes make this possible (25, 44). These provide the reversibility and selectivity absent from the more widely used small-molecule probes (4). We are unaware of attempts to monitor the real-time changes of ROS production and ROS-defeating systems to reveal the influence of transient ischemia–reperfusion (IR, <1 h) (16) on CA1 and CA3 pyramidal cells using reversible fluorescent probes. The ability to do so can lead to new understanding of why CA1 and CA3 have different susceptibilities to IR.
One intriguing question relates to the relative roles of the GSH and Trx systems in reducing ROS formed during IR. There is disagreement about the relative contributions of the GSH and Trx systems to the reduction of endogenous peroxide in isolated rat brain mitochondria and exogenous peroxide in OHSCs (45). It is well known that the concentration of GSH is much greater than that of other thiols, including protein thiols in cells, thus it may be inferred that the GSH system dominates ROS management. Indeed, in reviewing the roles of these two systems, Patenaude et al. emphasized the role of Trx in signal transduction and regulation of apoptosis in contrast to GSH, which takes care of the maintenance of an appropriate redox status (50). On the other hand, a detailed kinetic analysis determined that the flux of reducing equivalents used to reduce exogenous hydrogen peroxide through the cytoplasmic Trx/peroxiredoxin (Prx) system, while less than that for the cytoplasmic GSH/glutathione peroxidase (Gpx) system, is quite significant (2). Cellular compartmentalization of ROS-related functions makes it necessary to assess ROS management at the subcellular level. At this point, there is no clear understanding of the relative roles of the two systems in ROS removal.
Our results from the real-time, roGFP-based probe measurements during OGD (20 min)-RP (30 min) show that changes in H2O2 and GSH redox status during OGD-RP are confined to the mitochondria. During OGD-RP and also during controlled production of superoxide and, separately, hydrogen peroxide, we see significant differences between CA1 and CA3 pyramidal cells. CA1 suffers larger changes in the GSH redox status and higher peroxide levels than CA3. Auranofin (AF) (45), a thioredoxin reductase (TrxR) inhibitor, abolishes the differences seen during OGD-RP, during controlled ROS production, and in cell death. Moreover, upregulation of cytosolic Trx has no effect on mitochondrial ROS. These suggest that the CA3 region suffers less damage because it maintains a greater reducing capability than CA1 due to a larger contribution from the mitochondrial Trx system in CA3 in the early stage of IR. To our knowledge, this is the first study to show significant involvement of the mitochondrial Trx system into the differential resilience of CA1 and CA3 pyramidal cells to injury from IR.
Results
Real-time redox changes in H2O2 and GSH systems of individual pyramidal cells during OGD-RP
The roGFP2-based H2O2 (26) and GSH (25) probes provide ratiometric fluorescence measurements of the degree of oxidation (OxD), which is the ratio [disulfide sensor form]/[total sensor] where total refers to the sum of the concentrations of the oxidized and reduced sensor forms. We measured the degrees of oxidation of the H2O2 probe (OxDP) and of the GSH probe (OxDG) in mitochondria and cytosol of pyramidal cells in hippocampal regions CA1 and CA3, respectively (Fig. 1B and Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/ars). There were no significant changes in cytoplasmic OxDP or OxDG during OGD-RP. Mitochondrial OxDP (mit-OxDP) and mit-OxDG are not significantly different in CA1 and CA3 pyramidal cells during the basal period and OGD, but they differ during RP (Fig. 1E, F). In the last 15 min of RP, mit-OxDP increases slightly more and is higher in CA1 than in CA3 (Fig. 1E and Supplementary Table S1). During RP, mit-OxDG in CA1 increases sharply in the first 8 min, then stabilizes at a much higher level than in CA3 (Fig. 1F and Supplementary Table S1). While the small difference between the mitochondrial H2O2 system in CA1 and CA3 during RP is significant, the difference in mit-OxDG in the two Cornu Ammonis (CA) subfields during RP is striking, indicating that CA1 and CA3 manage ROS differently and the GSH system is involved in that difference.
FIG. 1.

Mit-OxDP and OxDG show significant differences during OGD-RP in pyramidal cells (stratum pyramidale). OxDG, but not OxDP, is higher during RP in CA1 than CA3. (A) Image of OHSC with outline of CA regions. (B) (Top) Representative images of Grx1-roGFP2 expressed in mitochondria or cytoplasm in pyramidal cells from CA1. They are imaged with excitation at 405 and 488 nm. roGFP2-Orp1 has similar fluorescence parameters as Grx1-roGFP2 (bottom). Four chimeric protein probes are expressed in CA1 and CA3 (Supplementary Fig. S1): Mito-Grx1-roGFP2 and Cyto-Grx1-roGFP2 (GSH probe targeting mitochondria or cytoplasm) and Mito-roGFP2-Orp1 and Cyto-roGFP2-Orp1 (H2O2 probe targeting mitochondria or cytoplasm). (C) (Top) Orp1 in H2O2 probe and Grx1 in GSH probe regulate the oxidation and reduction of their functional fluorescent subunits, roGFP2. Orp1 is oxidized by H2O2 and reduced by Trx with rates that are first order in H2O2 and Trx, respectively. Grx1 is oxidized by oxidized GSH (GSSG) and reduced by GSH with rates that are first order for GSH and GSSG, respectively. k1, k2, k1′, and k2′ are the rate constants (bottom). OxDS is the fraction: oxidized sensor/total sensor. (D) (Top) OxDP and (bottom) OxDG represent the OxDS of H2O2 and GSH probes, respectively. The probes are calibrated with oxidant (H2O2) and reductant (DTT). Each trace is representative of 40 OHSCs (mean ± SEM). (E) OxDP and (F) OxDG in pyramidal cells showing mean ± SEM (n = 6; OHSCs) for each trace. Data from mitochondria and cytoplasm are shown in the top and bottom, respectively. Statistical analysis was applied with t-test with bootstrap and one-way ANOVA test (n.s., no significant difference, p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001, see Experimental Methods section in Supplementary Data for more details). Here and in figures below, the horizontal lines with symbols represent comparisons using ANOVA. The vertical lines with symbols represent t-test results. Student's t-tests were performed on the data from the 5-min period before the visible transient caused by solution exchange. Although there is little change in the cytoplasmic probes, the mitochondrial probes change significantly during OGD-RP. In (E), there is no significant difference between CA1 and CA3 in mit-OxDP in OGD-RP except during the latter period of RP, R2. In (F), noticeable differences are found in mit-OxDG between CA1 and CA3 during RP. CA, Cornu Ammonis; GSH, glutathione; OGD-RP, oxygen–glucose deprivation–reperfusion; OHSC, organotypic hippocampal slice cultures; OxDG, oxidation degree of the GSH probe; OxDP, oxidation of the H2O2 probe; Trx, thioredoxin.
NADPH is the source of reducing equivalents for removing H2O2 (2). NAD(P)H [two-photon imaging (Supplementary Fig. S3A)] in the non-nuclear portion of the pyramidal cell layer during OGD-RP responds differently in CA1 and CA3 (Supplementary Fig. S3B). NAD(P)H reaches a steady state after 10 min before OGD. During OGD, the fluorescence signal increases in CA1 and CA3 (Supplementary Table S2). During RP, NAD(P)H fluorescence slowly decays in CA3, while in CA1, NAD(P)H increases significantly and oscillates. These observations suggest the existence of differences in ROS management in the two regions' pyramidal cells.
Controlled generation of superoxide and H2O2 in mitochondria
The differences in the response of mitochondrial H2O2 and mitochondrial GSH between CA1 and CA3 could be due to differences in ROS production rates, removal rates, or both. Thus, we investigated the probe responses with controlled generation of superoxide and peroxide (Fig. 2A). We demonstrate below that complex I concentrations are not statistically different in CA1 and CA3 both by Western blot (WB) and by immunohistochemistry. Thus, the free radical-forming compound, menadione (MD), acting at complex I (18, 68) would produce the same flux of mitochondrial superoxide in the two regions. Superoxide forms peroxide without catalysis or via superoxide dismutase (SOD) (62). The artificial manganese superoxide dismutase mimic, MnTnBuOE-2-PyP5+ [MnSODm, currently in clinical trials (NCT02655601)], is an excellent catalyst for dismutation (7, 8, 56). Both MD and MnSODm reversibly partition between the extracellular space and mitochondria. MD will produce superoxide, some of which will be converted to hydrogen peroxide naturally. Exposure of OHSCs to both MD and MnSODm will increase peroxide production. We expect that there is a concentration above which further addition of MnSODm has no effect on peroxide production because the existing SOD2 and the added MnSODm together maximally convert superoxide to peroxide. With maximal conversion of superoxide to peroxide, OxDP reflects the balance of superoxide production and peroxide removal.
FIG. 2.

Difference in ROS management between pyramidal cells in CA1 and CA3 is evidenced by artificially stimulating the generation and removal of ROS. (A) (1) Superoxide is generated by inhibiting complex I with MD; (2) superoxide dismutation can be accelerated with MnSODm. MnSODm at 20 μM was found to be sufficient to fully convert superoxide into H2O2 (see Supplementary Fig. S4 for more details). (3) H2O2 is removed by the GSH and Trx systems. In (B–E), there are five stages (labeled with I to V) in the treatment. Each trace is represented as mean ± SEM, n = 6 OHSCs. (B) OxDP of the H2O2 probe is monitored in CA1 and CA3 during 5 min of basal conditions (I), 15 min; 10 μM MD (II), 15 min; WO (III); followed by 15 min 10 μM MD +20 μM MnSODm (IV); and finally15 min WO (V). ACSF is used for basal and WO. One-way ANOVA test results of CA1 and CA3 are indicated by black and red horizontal lines, respectively. Student's t-test results are indicated by vertical lines. (C) OxDP is monitored in CA1 and CA3 during the treatment as above, but with higher MD concentration. (D) OxDG of GSH probe is monitored in CA1 and CA3 when OHSC is treated in the same condition as in (B). (E) OxDG is monitored in CA1 and CA3 treated in the same conditions as in (C). See Figure 1 legend for indicators of statistical significance. ACSF, artificial cerebrospinal fluid; MD, menadione; ROS, reactive oxygen species; WO, washout.
OxDP increases when increasing [MnSODm] from 10 to 20 μM, but there were no significant changes when increasing [MnSODm] from 20 to 30 μM (Supplementary Fig. S4A, B). Thus, we used 20 μM MnSODm in the following experiments to maximally convert superoxide to peroxide. MnSODm itself did not alter OxDP or OxDG (Supplementary Fig. S4D). We exposed cultures to the following solutions/times: 5 min basal/15 min MD/15 min washout (WO)/15 min MD+MnSODm/15 min WO while monitoring mit-OxDP and mit-OxDG (Fig. 2B–E).
Consider first mit-OxDP. Figures 2B (IV) and C (IV) show responses from MD with MnSODm that maximally convert O2− to peroxide as explained above. The indistinguishable OxDP in Figure 2B and C (IV) is consistent with O2− production rates being similar in CA1 and CA3 on exposure to equal concentrations of MD. On exposure to 10 μM MD alone, Figure 2B (II), mit-OxDP shows no response, while at 20 μM, Figure 2C (II), the response in CA1 is greater than CA3 (See also Supplementary Table S3). Clearly, the mitochondrial flux of O2− is well handled in both regions at the lower concentration. At the higher concentration, it is not, so excess peroxide formation results. The excess is lower in CA3 than CA1. As the peroxide generation rates are the same, the difference between CA1 and CA3 seen in Figure 2C (II) is ascribed to differences in peroxide reduction capacity.
Figures 2D and E show mit-OxDG for the same treatments. First, note that the mit-OxDG picture is broadly similar to the mit-OxDP picture. The exception is in Figure 2D (IV) where mit-OxDPs are the same, but mit-OxDG is lower in CA3 than CA1. We infer that the oxidation status of the mitochondrial GSH system changes less in CA3 than in CA1 in the face of the same hydrogen peroxide generation rate. The comparisons of mit-OxDP and OxDG in CA1 and CA3 in Figure 2C and E (II) are consistent with this conclusion. The overall tentative conclusion is that under conditions where superoxide production rate is constant, peroxide production is greater in CA1 than CA3 and the GSH system is at the same time more oxidized in CA1.
The traces of OxDP and OxDG in Figure 2 appear to follow first-order kinetics [Eq. (S14)]. We derived equations for the change in OxD with time based on first-order reactions for oxidation and reduction of each probe [Eqs. (S5–S12)]. Four kinetic quantities can be determined from the curves for more insight about OxD curves, namely the rates of probe oxidation 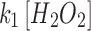 and reduction
and reduction 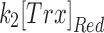 for the peroxide probe and the analogous rates
for the peroxide probe and the analogous rates 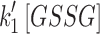 and
and 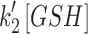 for the GSH probe (Supplementary Table S4). To aid in this analysis, we determined the concentration of total reduced thiols as a surrogate for GSH in the pyramidal cell layer. The [GSH] in mitochondria and whole cell is statistically indistinguishable in CA1 and CA3 pyramidal cells (Fig. 3). The lumped rate constants,
for the GSH probe (Supplementary Table S4). To aid in this analysis, we determined the concentration of total reduced thiols as a surrogate for GSH in the pyramidal cell layer. The [GSH] in mitochondria and whole cell is statistically indistinguishable in CA1 and CA3 pyramidal cells (Fig. 3). The lumped rate constants, 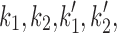 contain many parameters and concentrations, but comparisons are nonetheless revealing. Comparisons of these rates are plotted in Supplementary Figure S9A and B. The rates derived from the peroxide probe for the data in Figure 2B (IV) and C (IV) (MD and MnSODm) show no differences when comparing CA1 with CA3 as expected given the earlier evidence for equal rates of production of peroxide. However, for the data with 20 μM MD alone, the rates are significantly different with CA1
contain many parameters and concentrations, but comparisons are nonetheless revealing. Comparisons of these rates are plotted in Supplementary Figure S9A and B. The rates derived from the peroxide probe for the data in Figure 2B (IV) and C (IV) (MD and MnSODm) show no differences when comparing CA1 with CA3 as expected given the earlier evidence for equal rates of production of peroxide. However, for the data with 20 μM MD alone, the rates are significantly different with CA1 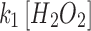 exceeding that of CA3 and CA3
exceeding that of CA3 and CA3 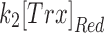 exceeding that of CA1. The rates related to the GSH probe show analogous CA1/CA3 differences with 20 μM MD alone, but in addition, show differences with 10 μM MD and 20 μM MnSODm consistent with the significantly higher [GSSG] in CA1 expected from a higher mit-OxDG with no significant difference in total reduced thiols and mit-OxDP.
exceeding that of CA1. The rates related to the GSH probe show analogous CA1/CA3 differences with 20 μM MD alone, but in addition, show differences with 10 μM MD and 20 μM MnSODm consistent with the significantly higher [GSSG] in CA1 expected from a higher mit-OxDG with no significant difference in total reduced thiols and mit-OxDP.
FIG. 3.

Similar reduced thiol concentration in mitochondria or whole cell between CA1 and CA3 pyramidal cells. (A) Fluorescence imaging begins before exposure of cultures to the fluorogenic reagent thiol probe IV. The reaction occurs in seconds. Nonfluorescent thiols turn fluorescent after exposure to 100 μM thiol probe IV (top). The increasing fluorescence rising to a steady value is indicative of the reaction occurring during the rise and then ceasing when the thiols are all reacted. Representative images from CA1 (bottom) are displayed: (left) BF image, (right) fluorescence image (ex 405 nm, em 440–480 nm). Imaging protocol is optimized as shown in Supplementary Figure S2. (B) Threshold was set to the fluorescence image in (A) to define the areas of pyramidal cells (gray), above threshold (green), and below threshold (blue). CA1 and CA3 pyramidal cells reach a constant fluorescence after about 8 min, indicating that the reaction is complete. (C) A mask of mitochondria is created from a pyramidal cell expressing MitoGFP in CA1 (left) and applied to count the thiol fluorescence in mitochondria (right). (D) The CA1/CA3 ratio of reduced thiols in mitochondria and, separately, whole cell is ∼1.0 (n = 17 OHSCs). BF, bright-field.
WBs and immunofluorescence imaging demonstrate differences in CA1 and CA3
If the differences in mitochondrial ROS management in pyramidal cells are not due either to differences in ROS production rate or to total thiol content, then it is likely that there are differences in the effectiveness of major antioxidant systems, namely the GSH and Trx systems. Therefore, we determined the relative content of complex I, SOD2, and mitochondrial Trx (Trx2) in CA1 and CA3 by WB and immunofluorescence (IF), and the results from WB and IF are consistent (Fig. 4). The consistency is important. WBs on OHSCs require laser capture microdissection (LCM) on 150 cultures to obtain a mean and SEM for n = 3 measurements. IF can be accomplished with many fewer cultures. Thus, we also measured levels of (mitochondrial) TrxR2, Prx3, Prx5, and Grx2 and (cytoplasmic) Trx1, TrxR1, Prx1, and Prx2 with IF. In addition, we measured levels of two proteins, Gpx1 and glutathione reductase (GR), which are found in both mitochondria and cytoplasm.
FIG. 4.

Expression of Trx2, Trx1, and SOD2 in pyramidal cells is significantly higher in CA3 than in CA1. (A, B) Comparison of the intensities of ND4L, SOD2, and Trx2 in CA1 and CA3 in WBs. (A) The pyramidal cell layers of CA1 and CA3 visualized by H&E staining were collected separately by LCM. For each individual WB, 50 LCM-generated bits of tissue were collected. Bottom, WB bands of ND4L, SOD2, and Trx2 from CA1 and CA3 subfields (see uncropped blots in Supplementary Fig. S10). (B) Expression of each protein from CA1 and CA3 is compared quantitatively (n = 3 repeats). BIR is used to refer the targeted protein to internal standard, α-tubulin. (C–E) Comparison of the contents of 13 proteins in CA1 and CA3 with IF. (C, D) Representative fluorescence images of subfield CA1 from immunostained OHSCs. The pyramidal cell layer is indicated by white dash lines. (C) Seven mitochondrial proteins are labeled with green fluorescent antibody shown in green. (D) First four proteins are mainly located in cytosol. GPx1 and GR exist both in mitochondria and cytosol. They are all labeled with red fluorescent antibody shown in red. (E) FR of the targeted protein (mitochondrial proteins referenced to MitoTracker, cytosolic proteins referenced to α-tubulin) is quantitatively analyzed and compared between CA1 and CA3 (n = 10 OHSCs, see Supplementary Figure S5 and Experimental Methods section in Supplementary Data for more details). Data of ND4L, SOD2, and Trx2 are consistent with that acquired with WB in (B). See Figure 1 legend for indicators of statistical significance. BIR, band intensity ratio; FR, fluorescence ratio; Gpx, glutathione peroxidase; GR, glutathione reductase; Grx, glutaredoxin; IF, immunofluorescence; LCM, laser capture microdissection; ND4L, NADH dehydrogenase subunit 4L; Prx, peroxiredoxin; SOD, superoxide dismutase; Trx, thioredoxin; TrxR, thioredoxin reductase; WB, Western blot.
Pyramidal cells in CA1 and CA3 have statistically indistinguishable amounts of complex I. This is consistent with our observation that similar amounts of superoxide are generated in CA1 and CA3 with a given MD concentration. Interestingly, the amount of SOD2 in CA3 is about 1.5 times greater than that in CA1, which indicates that H2O2 generation from superoxide is more efficient in CA3. This adds support to the conclusion made above that the CA3 relative robustness is not due to less ROS production compared with CA1. Some differences in the GSH system noted in Figure 4E are modest. We see slightly greater Gpx1 ( × 1.3), but slightly lower GR ( × 0.8), in CA3. These differences would tend to counteract each other in determining the efficiency of peroxide removal by GSH.
More differences are seen in Trx/TrxR/Prx system. In the cytoplasm, we see modestly, but significantly, higher Trx1 in CA3. In mitochondria, we see somewhat lower TrxR2 and Prx5 in CA3. By far, the biggest difference is that the concentration of Trx2 in CA3 is about 2.3 times greater than in CA1. This is consistent with the idea that the mitochondrial GSH system is less oxidized in CA3 with similar OxDP due to the parallel contribution of reducing capacity of the mitochondrial Trx system. The contribution of differences in the Trx system to the differences between CA1 and CA3 in ROS management during OGD-RP can be established by inhibiting TrxR from participating in the regeneration of reduced Trx and monitoring the consequential influence on OxDP and OxDG.
Inhibition of TrxRs, but not overexpression of cytoplasmic Trx, leads to differential changes in CA1 versus CA3 mitochondrial OxDs
Mit-OxDs were monitored with controlled production of peroxide using 10 μM MD +20 μM MnSODm following exposure of cultures to the irreversible TrxR (1 and 2) inhibitor AF at a concentration of 1 μM (45, 59) 1 h (17) before treatment as shown in Figure 5A. Figure 5B and C shows that escalations of mit-OxDP due to AF: ΔOxDP in CA3 (0.32 ± 0.02) are significantly greater than in CA1 (0.24 ± 0.04). More dramatic changes in mit-OxDG are found in CA3 in comparison with CA1. In fact, in CA1 pyramidal cells, the change in steady-state OxDG caused by AF is not statistically significant, whereas it is highly significant in CA3 (ΔOxDP in CA1 vs. CA3, 0.056 ± 0.034 vs. 0.40 ± 0.07). These data support the idea that CA3 pyramidal cells are affected to a larger extent by TrxR inhibition than CA1 pyramidal cells. More intriguingly, AF eliminates the CA1/CA3 differences of mit-OxDP and mit-OxDG caused by controlled generation of mitochondrial peroxide (Fig. 5C). The OxDP and OxDG (Fig. 5B, C) kinetics can be determined as above. The resulting rates, 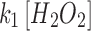 ,
, 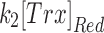 ,
, 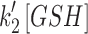 , and
, and 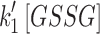 , provide additional information (shown in Supplementary Fig. S9C and D and Supplementary Table S5) in support of the inferences reached from comparisons of the (near) steady-state OxDs.
, provide additional information (shown in Supplementary Fig. S9C and D and Supplementary Table S5) in support of the inferences reached from comparisons of the (near) steady-state OxDs.
FIG. 5.

The mitochondrial H2O2 and GSH systems experience larger changes in OxD in CA3 than in CA1 when the Trx system is inhibited by AF. The tissue is pretreated with or without 1 μM AF 1 h before imaging. Tissue is superfused with ASCF in basal condition for ∼8 min, then with 10 μM MD +20 μM MnSODm for 15 min. (A) H2O2 is consumed by the GSH and Trx systems via several redox enzymes, Gpx, Prx, and Orp. roGFP2-Orp1, the H2O2 probe used here, has the subunit Orp (underlined in A), which is reduced by Trx. AF inhibits the Trx system by irreversibly inhibiting TrxR. (B) AF causes an increase in OxDP in CA1 (left) and in CA3 (right). The change in OxDP is greater in CA3 than in CA1. (C) AF affects OxDG slightly in CA1, whereas in CA3, the effect is noticeable. In (B, C), each trace is represented as mean ± SEM (n = 6 OHSCs). ΔOxD is the difference between the steady-state (the last 5 min of MD+MnSODm) values of OxD (AF) and OxD (no AF). See Figure 1 legend for indicators of statistical significance. AF, auranofin; Gpx, glutathione peroxidase; Orp, oxidant receptor protein; TrxR, thioredoxin reductase; Prx, peroxiredoxin.
AF also selectively alters mitochondrial OxDs observed during OGD-RP (Fig. 6A, B; Supplementary Table S6). In CA3 mitochondria, AF causes a significant increase of mit-OxDP during RP. Remarkably, the inhibition of TrxR has no effect on OxDP in CA1 mitochondria and it has only a very modest effect on OxDG. Compared with CA1, the rise of OxDG in CA3 caused by AF is large and more significant.
FIG. 6.

Inhibition of the Trx system by AF causes pyramidal cells in CA3 to perform similarly to those in CA1. OxD profiles of CA1 (left) and CA3 (middle) during 10/20/30 min basal/OGD/RP are represented as mean ± SEM, n = 6 OHSCs. The steady OxDS of each period is acquired from the time frame (5 min) indicated by the horizontal line shown in right. The profiles of mit-OxDP and OxDG, and cytosolic OxDP and OxDG are displayed from (A) to (D). Statistical significance of the effect of AF in CA1 (or CA3) is indicated above the OxD traces. Statistical significance of CA1/CA3 differences (with or without AF) is indicated above the bar graphs on the right. Inhibition of the Trx system by AF increases OxDs in both mitochondria and cytosol and removes the difference of mit-OxDs between CA1 and CA3. See Figure 1 legend for indicators of statistical significance. n.s. is not remarked.
We have focused our attention on mitochondrial events because there are no observable changes in cytoplasmic OxDG or OxDP during OGD-RP. However, it is important not to overlook potential contributions from cytoplasmic events. Thus, we also measured the profiles of OxDs in cytosol during OGD-RP in the presence of AF (Fig. 6C, D). AF causes cytosolic OxDs to increase during RP, but CA1 and CA3 cytosolic OxDs are not significantly different with or without AF. In other words, inhibition of TrxR has a large effect, but the effect is the same in CA1 and CA3. However, we cannot ignore the possibility that the cytosolic Trx system could influence mitochondrial OxDs. If there is a difference between CA1 and CA3 in their ability to reduce cytosolic hydrogen peroxide, this may result in a difference in the peroxide concentration gradient across the mitochondrial membranes, leading to a difference in mitochondrial peroxide due to the difference in cytosolic consumption of peroxide.
While there is no statistically significant difference in cytosolic TrxR or Prxs between CA1 and CA3 (Fig. 4E), we noted above that CA3 has higher Trx1 than CA1. We thus tested the hypothesis that a higher Trx1 concentration will significantly decrease mitochondrial OxDs. We introduced human Trx1 (39) into single pyramidal cells in CA1 and CA3 in OHSCs (Supplementary Fig. S7). We observed a significant, two- to three-fold, increase in total (human+rat) Trx1 resulting from the transfection (Supplementary Fig. S7). However, the mit-OxDs are not altered significantly in either CA1 or CA3 under either the controlled production protocol (MD/MnSODm) or OGD-RP (Supplementary Fig. S8). The foregoing observations suggest that the cytosolic Trx systems in CA1 and CA3 are critical for maintaining low cytosolic ROS levels following OGD during RP. However, while there is a statistically significant and greater Trx1 level in CA3 compared with CA1, we find that there is no significant effect of an increased Trx1 level on mitochondrial OxDs. It is thus difficult to ascribe the differential susceptibility of CA1/CA3 to cytosolic ROS consumption in the short-term (<1 h) timescale.
To sum up, in all of the experiments carried out with AF, the differences of mit-OxDs in CA1 and CA3 pyramidal cells are abolished. This strongly suggests that the Trx/Prx system is responsible for differences observed in short-term (<1 h) ROS management. As there is no statistically significant effect on mit-OxDs caused by overexpressing Trx1 in cytoplasm, we suggest that the mitochondrial Trx system is responsible for the different mit-OxDs. This led us to wonder if these short-term events are correlated with later cell death.
Cell death measured in propidium iodide assay and its correlation with OxDS
Cell death after 18 h resulting from controlled ROS production conditions (Fig. 2) was assessed using propidium iodide (PI) fluorescence intensities (Fig. 7B). No significant cell death resulted from exposure to 10 μM MD or 20 μM MnSODm, consistent with there being no change of mit-OxDP and OxDG. Exposure to 20 μM MD +20 μM MnSODm is significantly and similarly lethal in CA1 and CA3, consistent with the extreme mit-OxDs described in Figure 2. However, there are significant differences in cell death in CA1 and CA3 when treated either with 20 μM MD alone or 10 μM MD +20 μM MnSODm (Fig. 7B) that correlate with the OxDs. Figure 7C shows that 1 μM AF causes less than 10% cell death in CA1 and CA3. In the absence of AF, CA1 has more cell death than CA3 after OGD-RP or 10 μM MD +20 μM MnSODm. When OHSCs were pretreated with AF, then treated with OGD-RP or 10 μM MD +20 μM MnSODm, cell death increased both in CA1 and CA3, and importantly, the difference in cell death between the two CA areas disappeared.
FIG. 7.

Cell death in OHSCs is correlated with OxDs. (A–C) Fluorescence images of OHSCs stained by PI after different treatments. (A) Live control is OHSC without any treatment (assigned 0% cell death). Dead control is OHSC treated with methanol (assigned 100% cell death). The calculation of cell death percentage is described in the Experimental Methods section in Supplementary Data. (B, C) Differential mortality of pyramidal cells in areas CA1 and CA3 is found after the treatment with OGD-RP and 10 μM MD +20 μM MnSODm. Data are represented as mean ± SEM (n = 10 OHSCs) and analyzed with one-way ANOVA test. See Figure 1 legend for indicators of statistical significance. (D) Correlation tests were run on data acquired in treatments included in (B, C). Pearson's correlation coefficient, R, is notated at each subplot and highlighted in red when p < 0.05, indicating that the correlation is significant. Cell death percentage is positively correlated with OxDP and OxDG. PI, propidium iodide.
We correlated the PI data (Fig. 7D) with mit-OxD data from both protocols, OGD-RP and controlled ROS production, with and without AF. The correlations are significant. This correlation is consistent with the idea that mit-OxDs in the first minutes after OGD are relevant to hypoxia/ischemia injury.
Discussion
Our goal is to decipher a decades-old observation that CA1 pyramidal cells are less resilient under hypoxia/ischemia than those of CA3 (73). Significant attention has been given to the longer-term effects of hypoxia/ischemia, such as delayed cell death (14). In this study, we have focused on the short-term events during and following OGD. We have specifically designed our investigations to minimize the likelihood that transcription/translation are responsible for the observed effects. We improved the OGD-RP procedure to switch solutions rapidly (70) so that ROS changes could be measured within a few minutes. We restricted our attention to pyramidal cells, but did not isolate them, thus allowing the natural interaction between glia and neurons to occur.
We find that there is no significant change in cytoplasmic OxDP or OxDG during OGD-RP. The lack of significant ROS production during OGD-RP in cytoplasm on this timescale has been noted (1), consistent with the fact that antioxidants are more abundant in cytoplasm than mitochondria (42). At longer timescales, NADPH oxidase activity, which is cytoplasmic, and superoxide production are higher in CA1 than CA3 after 10-min global cerebral ischemia and a 1-h (72) or a 1-day (13) RP.
We turn our attention to mitochondrial ROS. Mit-OxDP and mit-OxDG decrease during OGD and increase during RP in both CA1 and CA3. During a 30-min RP, mit-OxDG in CA1 is higher than in CA3 (Fig. 1). Mit-OxDG in CA1 is higher than in CA3 during controlled mitochondrial production of H2O2 with 10 μM MD +20 μM MnSODm, while the steady-state mit-OxDP is similar in the two regions (Fig. 2B, D). Thus, the CA3 GSH system is less oxidized than CA1 under similar conditions of oxidative stress. With even higher controlled mitochondrial superoxide production (20 μM MD), CA1 mit-OxDP is greater compared with CA3 (Fig. 2C, E). CA3 mitochondria seem to maintain a less oxidized environment whether the flux of ROS is predominantly peroxide (the former controlled production example) or a mixture of peroxide and superoxide (the latter example).
The steady-state levels of ROS and OxDs arise, in the absence of exogenous agents, from natural production and removal rates. Production rate differences between CA1 and CA3 are unlikely to explain the differences observed in OxDs and cell death because their complex I concentrations are not significantly different (Fig. 4). We find indistinguishable mit-OxDP in CA1 and CA3 under controlled production of peroxide (Supplementary Fig. S4) consistent with similar complex I concentrations. It is unlikely given the same superoxide production rate that H2O2 production rate is higher in CA1 than CA3 in the absence of MnSODm because it has less SOD2 than CA3 [Fig. 4 and Ref. (43)]. We infer that the differences seen in the response to OGD-RP are due to ROS removal rates being different in CA1 and CA3.
It has been noted that certain pro-oxidant and antioxidant transcriptional levels are higher in CA1 than CA3 perhaps because CA1 incorporates more ROS signaling pathways (66). However, transcriptional patterns cannot predict the steady-state levels of ROS following IR. As for differences in ROS removal, we found that the total reduced thiol content, primarily GSH, is similar in CA1 and CA3 (Fig. 3D). Immunohistochemistry shows that Gpx1, which is found both in cytoplasm and mitochondria, is statistically significantly, although modestly, higher in CA3 than CA1. One might infer that the GSH system was more active in peroxide removal in CA3 than CA1 because of the higher Gpx1. However, the mitochondrial GSH system is less oxidized in CA3 compared with CA1 during RP or controlled generation of ROS. Why?
We found a significantly higher Trx2 and Trx1 protein content in CA3 than CA1 (Fig. 4) and, at the same time, somewhat lower level of the TrxR2, but no difference in reduced thiols, an approximate measure of GSH (Fig. 3). In addition, we find that Trx2 and Trx1 levels are significantly higher in CA3 versus CA1 in older rats (Supplementary Fig. S6). The relative levels have reversed, however: the CA3/CA1 ratio of Trx1 is greater compared with Trx2 in the older rat (Supplementary Fig. S6), which is the opposite of the situation for the younger animals. There is one report confirming higher Trx2 in CA3 in older rats (47), while another finds no difference (5). Perhaps the Trx system is more active in CA3 than in CA1.
According to Mitozo et al. (45), the GSH system is more important than the Trx system in decomposing a relatively high concentration (100 μM) of exogenous H2O2 applied to acute hippocampal slices. On the other hand, Kudin et al. noted that the Trx system outperforms the GSH system in endogenous H2O2 removal in isolated rat hippocampal mitochondria. Drechsel and Patel found the same result, namely that the Trx system removes more endogenous peroxide than the GSH system in isolated neuronal mitochondria. Trx exists at lower concentrations than GSH in general, but the kinetics in the Trx system are faster than the GSH system. The reaction of Trx and H2O2 occurs through Prx, while the reaction of GSH and H2O2 is catalyzed by Gpx (2). Oxidized GSH and Trx are reduced by NADPH via GSH reductase and TrxR, respectively (2). Considering estimates of the rates of these processes in Jurkat cell cytoplasm, Adimora et al. reached the conclusion that the cytoplasmic GSH system was responsible for removal of about 36% more exogenous peroxide than the cytoplasmic Trx system. This is a considerably more significant contribution from the Trx system than might be expected from estimates of the concentrations of GSH and reduced Trx. Thus, assuming that the mitochondrial rate constants do not differ dramatically from those in cytoplasm, the mitochondrial Trx system may manage smaller increases in ROS rapidly, while the GSH system becomes more important when ROS production is higher.
AF inhibits TrxRs irreversibly (17, 59). It has a dramatic impact on the mit-OxDs during RP in CA3, but not CA1 (Fig. 6A, B). In fact, it does not appear that the CA1 mitochondrial Trx system plays any role in suppressing ROS during RP. The inhibition of TrxRs makes the response of CA3 mitochondria to OGD-RP and controlled mitochondrial ROS production resemble that of CA1. It seems clear that the Trx system is responsible for the CA1/CA3 difference in mitochondrial ROS management.
As it inhibits both TrxR1 and TrxR2, the AF effect alone cannot tell us whether the cytoplasmic or mitochondrial Trx system is responsible for the differences in ROS management seen during OGD-RP. However, it is very clear based on our evidence that OGD (20 min)-RP (30 min) has a dramatic effect on mitochondrial ROS, but no effect at all on cytoplasmic ROS. Either cytoplasmic ROS levels remain low or cytoplasmic ROS are effectively eliminated in both regions. Obviously, the Trx system in cytoplasm is important as AF exposure causes cytoplasmic ROS to increase dramatically during RP in both regions (Fig. 6C, D). In contrast to similar findings related to cytoplasmic OxDs, mit-OxDs in CA1 and CA3 respond to AF very differently (Fig. 6A, B). CA1 OxDP is unmoved with exposure to AF, and the change in OxDG is modest. In CA3, AF leads to significant increases in OxDs during RP. In addition, while acknowledging the adage that correlation does not imply causation, it is noteworthy that mitochondrial changes in ROS from OGD-RP and MD/MnSODm, both with and without AF, correlate with cell death very well (Fig. 7). We suggest that the mitochondrial Trx system is more active in CA3, which renders CA3 pyramidal cells more resilient to OGD-RP.
It is certainly plausible that cytoplasmic ROS consumption could suppress observed increases in mitochondrial OxDs by acting as a sink for peroxide diffusing into the cytoplasm. It has been proposed that Trx1 may migrate to the mitochondrial intermembrane space (48), which would be an ideal location for a mitochondrial ROS sink. In addition, we found that Trx1 is statistically significantly greater in CA3 than CA1. This excess could result in a more effective cytoplasmic sink in CA3 than CA1. It is noteworthy that a transgenic mouse overexpressing hTrx1 improves survival rates early in life (51). Thus, Trx1 is important. However, increasing Trx1 content (Supplementary Fig. S7) results in no attenuation of mit-OxDs under either of our ROS-producing protocols, namely controlled production or OGD-RP (Supplementary Fig. S8). This finding is consistent with You et al. who found in HeLa cells that mitochondrial ROS (measured with MitoSox) did not decrease following overexpression of hTrx1 (71). Our finding suggests that at least for short periods of excess mitochondrial ROS generation, Trx1 does not influence the mitochondrial oxidation status noticeably.
Another question is whether the two main antioxidant systems can be considered as separate entities. The Trx and GSH systems are not independent. Several disulfide exchange reactions among the Trx and GSH systems exist (27, 28, 34, 38, 60), although they are not at equilibrium (22). They share a source of reducing equivalents. In addition, the GSH system enzyme, glutaredoxin (Grx), can influence the activity of the Trx system by its deglutathionylation capability (11, 37). A higher Grx activity can, in principle, increase Trx system activity if Trx glutathionylation is suppressing its activity. Figure 4E shows that mitochondrial Grx levels are not significantly different in CA1 and CA3, thus it is not likely that differential control of Trx glutathionylation status is responsible for the differences we observe in mit-OxDs.
The Trx systems, Trx2 in particular, are important in cellular health in general, including following IR in brain and other organs (27). Upregulation of Trx2 is neuroprotective in rat neurons (33) and gerbil hippocampus (69). It works by reducing apoptosis after global ischemia (32) possibly through the apoptosis signal-regulating kinase one-dependent pathway in mouse myocytes (30). On the other hand, Trx2 could be proapoptotic by denitrosylation of procaspase-3 following cerebral IR (64). Clearly, more work is needed to understand this ambiguity.
We are not cognizant of other work indicating that the mitochondrial Trx system controls mitochondrial oxidation status in the short-term events following OGD and, by implication, hypoxia/ischemia. Our work shows that the mitochondrial Trx system, perhaps the relative abundance of Trx2, appears to be the determining factor in the differences in oxidative stress and cell death following OGD-RP or MD/MnSODm in CA1 and CA3. This is supported by the observation that AF eliminates the differences of mit-OxDP, OxDG, and cell death in CA1 versus CA3. This conclusion is supported by the overarching concept of redox compartmentalization (17, 22).
While we have specifically designed the OxD experiments to observe 30 min of RP, many workers have noted changes in translational or transcriptional activity as a result of an insult or exposure to exogenous ROS. Papadia et al. found that NMDA exposure can increase Trx2 expression. Thioredoxin-interacting protein (Txnip) activity was recorded 16 h after NMDA exposure, and mRNA was measured 4 h after NMDA exposure. The timescale of these observations is well beyond ours. We note also that it is likely that Trx2 control is not through regulation of its synthesis directly, but rather through the control of its natural inhibitor, Txnip. Trx2 mRNA has a lifetime of greater than 35 h, not typical for proteins whose control is transcriptional regulation (35). On the other hand, Txnip control is more rapid. Exposure to bicuculline decreases Txnip mRNA at 60 min, but not at 30 min (49). While fast, this timescale is longer than our experiments. We suggest that new protein synthesis is not a significant factor in our observations.
It is interesting to speculate on why the CA3 and CA1 regions differ in their need to manage small rapid changes in mitochondrial ROS. Gamma oscillations (30–80 Hz) involving pyramidal cells and fast-spiking interneurons play a central role in the mnemonic functions of CA3 (10). As a result, CA3 has the highest levels of O2 consumption and mitochondrial performance within the hippocampal formation (31, 36). ROS production in normoxic conditions is proportional to metabolic activity (65), so during γ-oscillations, ROS production is likely to be high. It is thus conceivable that pyramidal cells in CA3 have evolved a mechanism involving Trx2 to maintain the ROS levels at nontoxic levels during the rapid intense metabolism that accompanies gamma oscillations.
In summary, during OGD-RP, changes in cytoplasmic OxDs are not detectable. Changes in mit-OxDs are significant and different in CA1 and CA3 pyramidal cells during the 30-min RP. The rates of production of superoxide plus peroxide in the mitochondria of CA1 and CA3 are indistinguishable. As there are no changes in cytoplasmic OxDs during OGD-RP, and equivalent ROS production at complex I, we focused on differences in ROS removal in CA1 and CA3 mitochondria. Differential cell death after OGD-RP is highly correlated with the differential mit-OxDP and mit-OxDG. These differences are entirely eliminated by inhibition of the Trx system. Trx1 is confirmed not to influence mit-OxDP. We suggest that differences in Trx2 levels and mitochondrial Trx system may be responsible for the differences seen in OxDs during RP and in cell death.
Materials and Methods
Animals
All experiments on animals were approved by the IACUC of the University of Pittsburgh. All experiments were carried on the two CA subfields, CA1 and CA3, within OHSCs collected from postnatal 7-day-old Sprague-Dawley rats.
Materials
Tryptone and yeast extract were from BD bioscience, and 1.6 μm gold nanoparticles and other accessories used for biolistic transfection (70) were purchased from Bio-Rad Laboratories, Inc. Thiol probe IV, Opti-MEM, and HBSS were obtained from Invitrogen. MnSODm, MnTnBuOE-2-PyP5+, was synthesized and purified according to (7, 8, 56). RIPA buffer and SignalFire™ Elite ECL Reagent (No. 12757) were obtained from Cell Signaling Technology. All other chemical reagents were purchased from Sigma Aldrich and used as received. Various redox probes based on roGFP2 construct (Mito-Grx1-roGFP2, Cyto-Grx1-roGFP2, Mito-roGFP2-Orp1, Cyto-roGFP2-Orp1) were generous gifts from Tobias P. Dick (25, 26). pCMV-tdTomato (Clontech) and pCS2+ (Addgene) were kindly provided by Zachary P. Wills (University of Pittsburgh). pMito-tdTomato was constructed in the Deiters laboratory (University of Pittsburgh) (70). The antibodies and dye used in WBs and IF are listed in Supplementary Data.
Measurement of OxDP and OxDG during OGD-RP with/without AF
All fluorescence images in this work were acquired on a Leica TCS SP5II broadband confocal microscope. We followed the protocol established in our previous work (70). OHSCs were collected from a postnatal 7-day-old rat and cultured 3 days before transfection with plasmids for H2O2 or GSH probes. The expression of probes was usually apparent in 2 days. The 5- to 7-day-old cultures were then treated with OGD-RP conditions. OxDP and OxDG were determined from the ratiometric fluorescence signal of the probe from single cells in OHSCs (70). AF is an irreversible inhibitor and it was applied 1 h before reagent treatment if used.
Thiol staining and NADPH imaging
The total reduced thiol pool can be determined quantitatively via fluorescence imaging (exi/emi: 405/440–480 nm) after derivatization with thiol probe IV (EMD Millipore). OHSCs with single cells expressing tdTomato were incubated with 100 μM thiol probe IV and monitored until the signal reached a steady value. MitoGFP (exi/emi: 488/500–530 nm) as a mitochondrial indicator was introduced by gene gun and expressed in single pyramidal cell for creating a mitochondrial mask. Two-photon imaging was applied to image NAD(P)H (Ex: 740 nm, Em: 580–630 nm) on the confocal microscope equipped with a Mai Tai Ti:Sapphire laser (Spectra-Physics, CA) and nondescanned detector. tdTomato (Ex: 561 nm, Em: 580–600 nm)-expressing cells were used as the target for autofocusing. During a single imaging process, images were taken alternately of CA1 and CA3 in the same OHSC using the function “Mark & Find and Tile Scan” in the confocal microscope (see more details in Supplementary Fig. S2).
MD/MnSODm/AF experiment
OHSCs expressing H2O2 or GSH probe were treated with 5 min basal/15 min MD, MnSODm, or both in artificial cerebrospinal fluid (ACSF)/15 min WO, and imaged. OxDP and OxDG were calculated from the fluorescence ratiometric imaging data. AF is an irreversible inhibitor, thus it was applied 1 h before reagent treatment.
Overexpression of hTrx1
Recombinant human Trx1 (hTrx1) was overexpressed in OHSCs by gene gun (39). The transfection efficacy is confirmed by IF described below. Details are described in Supplementary Data.
LCM/WB and IF experiment
To restrict the WB measurements to (mostly) pyramidal cells, we used LCM by following Espina's protocol (21). About 10 μg of total protein could be extracted from the CA subfields of 50 frozen sections by following the protocol from Cell Signaling Technology for WB. This amount of material was used for one lane. NADH dehydrogenase subunit 4L (ND4L, a subunit of complex I), SOD2, and Trx2 were targeted with their specific antibodies. The band intensity of each protein was referred to that of the internal standard, α-tubulin [Eq. (S1)]. Thirteen proteins were measured with IF (24). Fluorescence from immunolabeled proteins selectively located in mitochondria was referenced to MitoTracker, while the others were referenced to α-tubulin. Look for more details in Supplementary Figure S5, Equations (S2) and (S3), and descriptions in Supplementary Data. In IF, only pyramidal cell layers from CA1 and CA3 in OHSCs were imaged and fluorescence microscopy intensity of each protein was referred to that of the internal standard [Eqs. (S2–S3)]. The values obtained from CA3 were normalized to those from CA1 in each culture.
PI staining
PI was used to quantify the existence of dead cells (58). Methanol-treated OHSCs were used as dead (100%) control, while untreated OHSCs were used as live (0%) control. The cell death percentage of reagent-treated OHSCs was calculated as Equation (S4).
Image processing and statistical analyses
Images were processed in ImageJ (http://imagej.nih.gov/ij/), then the numerical data were processed in MATLAB (version R2014b; The MathWorks, Inc.) and OrginPro (version 9.0; OriginLab Corp.). Linear and nonlinear regression statistical analyses were operated in MATLAB or R. The parameters, 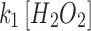 ,
, 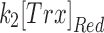 , and
, and 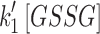 and
and 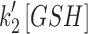 , were derived from OxDP and OxDG
versus time traces, which exhibited first-order exponential rise and fall through nonlinear regression (see details in Supplementary Data).
, were derived from OxDP and OxDG
versus time traces, which exhibited first-order exponential rise and fall through nonlinear regression (see details in Supplementary Data).
Supplementary Material
Abbreviations Used
- AF
auranofin
- CA
cornu ammonis
- GFP
green fluorescent protein
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- Gpx
glutathione peroxidase
- GR
glutathione reductase
- Grx
glutaredoxin
- IF
immunofluorescence
- IR
ischemia–reperfusion
- LCM
laser capture microdissection
- MD
menadione
- MnSODm
manganese superoxide dismutase mimic
- ND4L
NADH dehydrogenase subunit 4 L
- NMDA
N-methyl-D-aspartate
- OGD-RP
oxygen–glucose deprivation–reperfusion
- OHSCs
organotypic hippocampal slice cultures
- Orp
oxidant receptor protein
- OxD
degree of oxidation of probe
- OxDG
oxidation of GSH probe
- OxDP
oxidation of the H2O2 probe
- PI
propidium iodide
- Prx
peroxiredoxin
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- SOD2
mitochondrial (Mn) superoxide dismutase
- Trx
thioredoxin
- Trx2
mitochondrial Trx
- TrxR
thioredoxin reductase
- Txnip
thioredoxin-interacting protein
- WB
Western blot
- WO
washout
Acknowledgments
This work is supported by NIH Grants R01 GM066018 (S.G.W., G.B.) and GM044842 (S.G.W., M.S.). The authors thank Tom Harper (Department of Biological sciences, University of Pittsburgh) for advice on imaging; Prof. Alexander Deiters (Department of Chemistry, University of Pittsburgh) for help on the WB experiment; Prof. Mona F. Melhem (Veterans' Affairs Medical Center in Pittsburgh) and Krishna Prasadan (Children's Hospital of Pittsburgh) for LCM. The authors are thankful for the thoughtful and productive discussion with Prof. Satish Lyengar (Department of Statistics, University of Pittsburgh) about the statistical analysis of linear and nonlinear curves. His student, Bowen Yi, generously assisted with coding in the software R.
Author Disclosure Statement
I.B.H. is a consultant with BioMimetix JVLLC and holds equities in BioMimetix JVLLC. I.B.H. and Duke University have patent rights and have licensed technologies to BioMimetix JVLLC. B.Y., G.B., M.S., and S.G.W. declare that no competing financial interests exist.
References
- 1.Abramov AY, Scorziello A, and Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci 27: 1129–1138, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adimora NJ, Jones DP, and Kemp ML. A model of redox kinetics implicates the thiol proteome in cellular hydrogen peroxide responses. Antioxid Redox Signal 13: 731–743, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahlgren H, Henjum K, Ottersen O, and Rundén-Pran E. Validation of organotypical hippocampal slice cultures as an ex vivo model of brain ischemia: different roles of NMDA receptors in cell death signalling after exposure to NMDA or oxygen and glucose deprivation. Cell Tissue Res 345: 329–341, 2011 [DOI] [PubMed] [Google Scholar]
- 4.Albrecht Simone C, Barata Ana G, Großhans J, Teleman Aurelio A, and Dick Tobias P. In vivo mapping of hydrogen peroxide and oxidized glutathione reveals chemical and regional specificity of redox homeostasis. Cell Metab 14: 819–829, 2011 [DOI] [PubMed] [Google Scholar]
- 5.Aon-Bertolino ML, Romero JI, Galeano P, Holubiec M, Badorrey MS, Saraceno GE, Hanschmann E-M, Lillig CH, and Capani F. Thioredoxin and glutaredoxin system proteins—immunolocalization in the rat central nervous system. Biochim Biophys Acta 1810: 93–110, 2011 [DOI] [PubMed] [Google Scholar]
- 6.Bartsch T, Dohring J, Reuter S, Finke C, Rohr A, Brauer H, Deuschl G, and Jansen O. Selective neuronal vulnerability of human hippocampal CA1 neurons: lesion evolution, temporal course, and pattern of hippocampal damage in diffusion-weighted MR imaging. J Cereb Blood Flow Metab 35: 1836–1845, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Batinic-Haberle I, Tovmasyan A, Roberts ERH, Vujaskovic Z, Leong KW, and Spasojevic I. SOD therapeutics: latest insights into their structure-activity relationships and impact on the cellular redox-based signaling pathways. Antioxid Redox Signal 20: 2372–2415, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Batinić-Haberle I, Tovmasyan A, and Spasojević I. Mn porphyrin-based redox-active therapeutics. In: Redox-Active Therapeutics, edited by Batinić-Haberle I, Rebouças JS, and Spasojević I. Cham: Springer International Publishing, 2016, pp. 165–212 [Google Scholar]
- 9.Brierley JB, and Cooper JE. Cerebral complications of hypothensive anaesthesia in a healthy adult. J Neurol Neurosurg Psychiatry 25: 24–30, 1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buzsáki G, and Wang X-J. Mechanisms of gamma oscillations. Annu Rev Neurosci 35: 203–225, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Casagrande S, Bonetto V, Fratelli M, Gianazza E, Eberini I, Massignan T, Salmona M, Chang G, Holmgren A, and Ghezzi P. Glutathionylation of human thioredoxin: A possible crosstalk between the glutathione and thioredoxin systems. Proc Natl Acad Sci U S A 99: 9745–9749, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab 21: 2–14, 2001 [DOI] [PubMed] [Google Scholar]
- 13.Chan PH, Kawase M, Murakami K, Chen SF, Li Y, Calagui B, Reola L, Carlson E, and Epstein CJ. Overexpression of SOD1 in transgenic rats protucts vulnerable neurons against ischemic damage after global cerebral ischemia and reperfusion. J Neurosci 18: 8292–8299, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen H, Yoshioka H, Kim GS, Jung JE, Okami N, Sakata H, Maier CM, Narasimhan P, Goeders CE, and Chan PH. Oxidative stress in ischemic brain damage: mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid Redox Signal 14: 1505–1517, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Circu ML, and Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med 48: 749–762, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collino M, Aragno M, Mastrocola R, Benetti E, Gallicchio M, Dianzani C, Danni O, Thiemermann C, and Fantozzi R. Oxidative stress and inflammatory response evoked by transient cerebral ischemia/reperfusion: Effects of the PPAR-α agonist WY14643. Free Radic Biol Med 41: 579–589, 2006 [DOI] [PubMed] [Google Scholar]
- 17.Cox AG, Brown KK, Arner ESJ, and Hampton MB. The thioredoxin reductase inhibitor auranofin triggers apoptosis through a Bax/Bak-dependent process that involves peroxiredoxin 3 oxidation. Biochem Pharmacol 76: 1097–1109, 2008 [DOI] [PubMed] [Google Scholar]
- 18.Criddle DN, Gillies S, Baumgartner-Wilson HK, Jaffar M, Chinje EC, Passmore S, Chvanov M, Barrow S, Gerasimenko OV, Tepikin AV, Sutton R, and Petersen OH. Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. J Biol Chem 281: 40485–40492, 2006 [DOI] [PubMed] [Google Scholar]
- 19.Cummings JL, Tomiyasu U, Read S, and Benson DF. Amnesia with hippocampal lesions cardiopulmonary arrest. Neurology 34: 679, 1984 [DOI] [PubMed] [Google Scholar]
- 20.Dringen R, Pawlowski PG, and Hirrlinger J. Peroxide detoxification by brain cells. J Neurosci Res 79: 157–165, 2005 [DOI] [PubMed] [Google Scholar]
- 21.Espina V, Wulfkuhle JD, Calvert VS, VanMeter A, Zhou W, Coukos G, Geho DH, Petricoin EF, and Liotta LA. Laser-capture microdissection. Nat Protoc 1: 586–603, 2006 [DOI] [PubMed] [Google Scholar]
- 22.Go Y-M, and Jones DP. Redox compartmentalization in eukaryotic cells. Biochim Biophys Acta 1780: 1273–1290, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gogolla N, Galimberti I, DePaola V, and Caroni P. Preparation of organotypic hippocampal slice cultures for long-term live imaging. Nat Protoc 1: 1165–1171, 2006 [DOI] [PubMed] [Google Scholar]
- 24.Gogolla N, Galimberti I, DePaola V, and Caroni P. Staining protocol for organotypic hippocampal slice cultures. Nat Protoc 1: 2452–2456, 2006 [DOI] [PubMed] [Google Scholar]
- 25.Gutscher M, Pauleau A-L, Marty L, Brach T, Wabnitz GH, Samstag Y, Meyer AJ, and Dick TP. Real-time imaging of the intracellular glutathione redox potential. Nat Methods 5: 553–559, 2008 [DOI] [PubMed] [Google Scholar]
- 26.Gutscher M, Sobotta MC, Wabnitz GH, Ballikaya S, Meyer AJ, Samstag Y, and Dick TP. Proximity-based protein thiol oxidation by H2O2-scavenging peroxidases. J Biol Chem 284: 31532–31540, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanschmann E-M, Godoy JR, Berndt C, Hudemann C, and Lillig CH. Thioredoxins, glutaredoxins, and peroxiredoxins—molecular mechanisms and health significance: from cofactors to antioxidants to redox signaling. Antioxid Redox Signal 19: 1539–1605, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanschmann E-M, Lönn ME, Schütte LD, Funke M, Godoy JR, Eitner S, Hudemann C, and Lillig CH. Both thioredoxin 2 and glutaredoxin 2 contribute to the reduction of the mitochondrial 2-Cys peroxiredoxin Prx3. J Biol Chem 285: 40699–40705, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holmgren A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid Redox Signal 2: 811–820, 2000 [DOI] [PubMed] [Google Scholar]
- 30.Huang Q, Zhou HJ, Zhang H, Huang Y, Hinojosa-Kirschenbaum F, Fan P, Yao L, Belardinelli L, Tellides G, Giordano FJ, Budas GR, and Min W. Thioredoxin-2 inhibits mitochondrial ROS generation and ASK1 activity to maintain cardiac function. Circulation 131: 1082–1097, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huchzermeyer C, Berndt N, Holzhütter H-G, and Kann O. Oxygen consumption rates during three different neuronal activity states in the hippocampal CA3 network. J Cereb Blood Flow Metab 33: 263–271, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hwang IK, Yoo K-Y, Kim DW, Lee CH, Choi JH, Kwon Y-G, Kim Y-M, Choi SY, and Won M-H. Changes in the expression of mitochondrial peroxiredoxin and thioredoxin in neurons and glia and their protective effects in experimental cerebral ischemic damage. Free Radic Biol Med 48: 1242–1251, 2010 [DOI] [PubMed] [Google Scholar]
- 33.Jia J, Zhang X, Hu Y-S, Wu Y, Wang Q-Z, Li N-N, Wu C-Q, Yu H-X, and Guo Q-C. Protective effect of tetraethyl pyrazine against focal cerebral ischemia/reperfusion injury in rats: Therapeutic time window and its mechanism. Thromb Res 123: 727–730, 2009 [DOI] [PubMed] [Google Scholar]
- 34.Johansson C, Lillig CH, and Holmgren A. Human mitochondrial glutaredoxin reduces S-glutathionylated proteins with high affinity accepting electrons from either glutathione or thioredoxin reductase. J Biol Chem 279: 7537–7543, 2004 [DOI] [PubMed] [Google Scholar]
- 35.Jurado J, Prieto-Álamo M-J, Madrid-Rísquez J, and Pueyo C. Absolute gene expression patterns of thioredoxin and glutaredoxin redox systems in mouse. J Biol Chem 278: 45546–45554, 2003 [DOI] [PubMed] [Google Scholar]
- 36.Kann O, Huchzermeyer C, Kovács R, Wirtz S, and Schuelke M. Gamma oscillations in the hippocampus require high complex I gene expression and strong functional performance of mitochondria. Brain 134: 345–358, 2011 [DOI] [PubMed] [Google Scholar]
- 37.Lillig CH, Berndt C, Holmgren A. Glutaredoxin systems. Biochim Biophys Acta 1780: 1304–1317, 2008 [DOI] [PubMed] [Google Scholar]
- 38.Lillig CH, and Holmgren A. Thioredoxin and related molecules—from biology to health and eisease. Antioxid Redox Signal 9: 25–47, 2006 [DOI] [PubMed] [Google Scholar]
- 39.Liu H, Nishitoh H, Ichijo H, and Kyriakis JM. Activation of apoptosis signal-regulating kinase 1 (ASK1) by tumor necrosis factor receptor-associated factor 2 requires prior dissociation of the ASK1 inhibitor thioredoxin. Mol Cell Biol 20: 2198–2208, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mailloux RJ, and Harper M-E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic Biol Med 51: 1106–1115, 2011 [DOI] [PubMed] [Google Scholar]
- 41.Manzanero S, Santro T, and Arumugam TV. Neuronal oxidative stress in acute ischemic stroke: Sources and contribution to cell injury. Neurochem Int 62: 712–718, 2013 [DOI] [PubMed] [Google Scholar]
- 42.Marí M, Morales A, Colell A, García-Ruiz C, Kaplowitz N, and Fernández-Checa JC. Mitochondrial glutathione: Features, regulation and role in disease. Biochim Biophys Acta 1830: 3317–3328, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mattiasson G, Friberg H, Hansson M, Elmér E, and Wieloch T. Flow cytometric analysis of mitochondria from CA1 and CA3 regions of rat hippocampus reveals differences in permeability transition pore activation. J Neurochem 87: 532–544, 2003 [DOI] [PubMed] [Google Scholar]
- 44.Meyer AJ, and Dick TP. Fluorescent protein-based redox probes. Antioxid Redox Signal 13: 621–650, 2010 [DOI] [PubMed] [Google Scholar]
- 45.Mitozo PA, de Souza LF, Loch-Neckel G, Flesch S, Maris AF, Figueiredo CP, dos Santos ARS, Farina M, and Dafre AL. A study of the relative importance of the peroxiredoxin-, catalase-, and glutathione-dependent systems in neural peroxide metabolism. Free Radic Biol Med 51: 69–77, 2011 [DOI] [PubMed] [Google Scholar]
- 46.Morgan MJ, and Liu Z-g. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res 21: 103–115, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murphy MP. Mitochondrial thiols in antioxidant protection and redox signaling: distinct roles for glutathionylation and other thiol modifications. Antioxid Redox Signal 16: 476–495, 2011 [DOI] [PubMed] [Google Scholar]
- 48.Nakao LS, Everley RA, Marino SM, Lo SM, de Souza LE, Gygi SP, and Gladyshev VN. Mechanism-based Proteomic Screening Identifies Targets of Thioredoxin-like Proteins. J Biol Chem 290: 5685–5695, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Papadia S, Soriano FX, Leveille F, Martel M-A, Dakin KA, Hansen HH, Kaindl A, Sifringer M, Fowler J, Stefovska V, McKenzie G, Craigon M, Corriveau R, Ghazal P, Horsburgh K, Yankner BA, Wyllie DJA, Ikonomidou C, and Hardingham GE. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci 11: 476–487, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patenaude A, Murthy MRV, and Mirault ME. Emerging roles of thioredoxin cycle enzymes in the central nervous system. Cell Mol Life Sci 62: 1063–1080, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perez VI, Cortez LA, Lew CM, Rodriguez M, Webb CR, Van Remmen H, Chaudhuri A, Qi W, Lee S, Bokov A, Fok W, Jones D, Richardson A, Yodoi J, Zhang Y, Tominaga K, Hubbard GB, and Ikeno Y. Thioredoxin 1 overexpression extends mainly the earlier part of life span in mice. J Gerontol Ser A 66A: 1286–1299, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petito CK, Feldmann E, Pulsinelli WA, and Plum F. Delayed hippocampal damage in humans following cardiorespiratory arrest. Neurology 37: 1281, 1987 [DOI] [PubMed] [Google Scholar]
- 53.Pringle AK, Iannotti F, Wilde GJC, Chad JE, Seeley PJ, and Sundstrom LE. Neuroprotection by both NMDA and non-NMDA receptor antagonists in in vitro ischemia. Brain Res 755: 36–46, 1997 [DOI] [PubMed] [Google Scholar]
- 54.Pulsinelli WA, and Brierley JB. A new model of bilateral hemispheric ischemia in the unanesthetized rat. Stroke 10: 267–272, 1979 [DOI] [PubMed] [Google Scholar]
- 55.Pulsinelli WA, Brierley JB, and Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol 11: 491–498, 1982 [DOI] [PubMed] [Google Scholar]
- 56.Rajic Z, Tovmasyan A, Spasojevic I, Sheng H, Lu M, Li AM, Gralla EB, Warner DS, Benov L, and Batinic-Haberle I. A new SOD mimic, Mn(III) ortho N-butoxyethylpyridylporphyrin, combines superb potency and lipophilicity with low toxicity. Free Radic Biol Med 52: 1828–1834, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rhee SG, Yang K-S, Kang SW, Woo HA, and Chang T-S. Controlled elimination of intracellular H2O2: regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxid Redox Signal 7: 619–626, 2005 [DOI] [PubMed] [Google Scholar]
- 58.Riccardi C, and Nicoletti I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc 1: 1458–1461, 2006 [DOI] [PubMed] [Google Scholar]
- 59.Rigobello MP, Folda A, Baldoin MC, Scutari G, and Bindoli A. Effect of auranofin on the mitochondrial generation of hydrogen peroxide. Role of thioredoxin reductase. Free Radic Res 39: 687–695, 2005 [DOI] [PubMed] [Google Scholar]
- 60.Rouhier N, Gelhaye E, and Jacquot JP. Glutaredoxin-dependent peroxiredoxin from poplar: protein-protein interaction and catalytic mechanism. J Biol Chem 277: 13609–13614, 2002 [DOI] [PubMed] [Google Scholar]
- 61.Schäfer MKE, Pfeiffer A, Jaeckel M, Pouya A, Dolga AM, and Methner A. Regulators of mitochondrial Ca2+ homeostasis in cerebral ischemia. Cell Tissue Res 357: 395–405, 2014 [DOI] [PubMed] [Google Scholar]
- 62.Sheng Y, Abreu IA, Cabelli DE, Maroney MJ, Miller A-F, Teixeira M, and Valentine JS. Superoxide dismutases and superoxide reductases. Chem Rev 114: 3854–3918, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stanika RI, Winters CA, Pivovarova NB, and Andrews SB. Differential NMDA receptor-dependent calcium loading and mitochondrial dysfunction in CA1 vs. CA3 hippocampal neurons. Neurobiol Dis 37: 403–411, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sun N, Hao JR, Li XY, Yin XH, Zong YY, Zhang GY, and Gao C. GluR6-FasL-Trx2 mediates denitrosylation and activation of procaspase-3 in cerebral ischemia/reperfusion in rats. Cell Death Dis 4: e771, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol 552: 335–344, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang X, and Michaelis EK. Selective neuronal vulnerability to oxidative stress in the brain. Front Aging Neurosci 2: 12, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang X, Pal R, Chen X-w, Limpeanchob N, Kumar KN, and Michaelis EK. High intrinsic oxidative stress may underlie selective vulnerability of the hippocampal CA1 region. Mol Brain Res 140: 120–126, 2005 [DOI] [PubMed] [Google Scholar]
- 68.Wyatt KM, Skene C, Veitch K, Hue L, and McCormack JG. The antianginal agent ranolazine is a weak inhibitor of the respiratory Complex I, but with greater potency in broken or uncoupled than in coupled mitochondria. Biochem Pharmacol 50: 1599–1606, 1995 [DOI] [PubMed] [Google Scholar]
- 69.Yan BC, Park JH, Ahn JH, Lee YJ, Lee TH, Lee CH, Cho JH, Kim MJ, Kim TY, Kang I-J, and Won M-H. Comparison of the immunoreactivity of Trx2/Prx3 redox system in the hippocampal CA1 region between the young and adult gerbil induced by transient cerebral ischemia. Neurochem Res 37: 1019–1030, 2012 [DOI] [PubMed] [Google Scholar]
- 70.Yin B, Barrionuevo G, and Weber SG. Optimized real-time monitoring of glutathione redox status in single pyramidal neurons in organotypic hippocampal slices during oxygen-glucose deprivation and reperfusion. ACS Chem Neurosci 6: 1838–1848, 2015 [DOI] [PubMed] [Google Scholar]
- 71.You BR, and Park WH. Suberoylanilide hydroxamic acid-induced HeLa cell death is closely correlated with oxidative stress and thioredoxin 1 levels. Int J Oncol 44: 1745–1755, 2014 [DOI] [PubMed] [Google Scholar]
- 72.Zhang Q-G, Raz L, Wang R, Han D, De Sevilla L, Yang F, Vadlamudi RK, and Brann DW. Estrogen attenuates ischemic oxidative damage via an estrogen receptor α-mediated inhibition of NADPH oxidase activation. J Neurosci 29: 13823–13836, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zola-Morgan S, Squire L, and Amaral D. Human amnesia and the medial temporal region: enduring memory impairment following a bilateral lesion limited to field CA1 of the hippocampus. J Neurosci 6: 2950–2967, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.