

Abstract
Background
Enterohemorrhagic Escherichia coli (EHEC) O145 are among the top non-O157 serogroups associated with severe human disease worldwide. Two serotypes, O145:H25 and O145:H28 have been isolated from human patients but little information is available regarding the virulence repertoire, origin and evolutionary relatedness of O145:H25. Hence, we sequenced the complete genome of two O145:H25 strains associated with hemolytic uremic syndrome (HUS) and compared the genomes with those of previously sequenced O145:H28 and other EHEC strains.
Results
The genomes of the two O145:H25 strains were 5.3 Mbp in size; slightly smaller than those of O145:H28 and other EHEC strains. Both strains contained three nearly identical plasmids and several prophages and integrative elements, many of which differed significantly in size, gene content and organization as compared to those present in O145:H28 and other EHECs. Furthermore, notable variations were observed in several fimbrial gene cluster and intimin types possessed by O145:H25 and O145:H28 indicating potential adaptation to distinct areas of host colonization. Comparative genomics further revealed that O145:H25 are genetically more similar to other non-O157 EHEC strains than to O145:H28.
Conclusion
Phylogenetic analysis accompanied by comparative genomics revealed that O145:H25 and O145:H28 evolved from two separate clonal lineages and that horizontal gene transfer and gene loss played a major role in the divergence of these EHEC serotypes. The data provide further evidence that ruminants might be a possible reservoir for O145:H25 but that they might be impaired in their ability to establish a persistent colonization as compared to other EHEC strains.
Electronic supplementary material
The online version of this article (doi:10.1186/s12866-017-1094-3) contains supplementary material, which is available to authorized users.
Keywords: EHEC O145:H25, Next generation sequencing, Comparative genomics, Phylogenetics, Bacterial adaptation & colonization
Background
Shiga toxin-producing Escherichia coli (STEC) are a genetically and phenotypically extremely diverse group of E. coli strains characterized by the production of one or more Shiga toxins (Stx1 and Stx2). Over 250 different STEC serotypes exist of which at least 100 have been linked to human diarrhea [1, 2]. Certain STEC strains are capable of causing more severe human diseases such as hemorrhagic colitis (HC) and the life-threatening hemolytic uremic syndrome (HUS); these STEC are commonly referred to as enterohemorrhagic E. coli (EHEC). Besides the expression of Stx1 and/or Stx2, classical EHEC strains carry the locus of enterocyte effacement (LEE) responsible for the formation of attaching and effacing (A/E) lesions on epithelial cells, and possess a large virulence plasmid encoding enterohemolysin (EhxA) [3–5]. While EHEC O157:H7 is considered to be the most frequent cause of severe disease, non-O157 EHEC and STEC (LEE-negative STEC e.g. O104 and O113) strains are increasingly recognized as the cause of similar illnesses worldwide [1, 2, 6–9]. In fact, it is estimated that at least 50% of all STEC infections in the US are caused by non-O157 STEC/EHEC strains, many belonging to serogroups O26, O45, O103, O111, O121 and O145 also known as the “big six” [1, 9]. As a result, in addition to O157 all non-intact beef products in the US are required to be tested for the presence of these six serogroups [10].
EHEC O145 has emerged as one of the major EHEC serogroups involved in severe human disease worldwide [7, 8, 11–13]. Most clinical O145 isolates described in the literature encompass motile and nonmotile strains of serotype O145:H28 (n > 150) [8, 11, 13–16], while EHEC O145:H25 appear to be less frequently isolated (10 reported cases) [11, 13, 17, 18]. However, the majority of the documented EHEC O145:H25 isolates are associated with HUS (n = 8), emphasizing the importance of this particular serotype [11, 13, 17, 18]. But little is known about the genetic characteristics of O145:H25 or how they have evolved or were transmitted. In fact, the source of EHEC O145:H25 infections appears to be unknown, as O145:H25 have thus far only been isolated from humans. In contrast, ruminants, especially cattle have been shown to be natural reservoirs of O145:H28 and other EHEC strains [14, 16, 19–22] and the consumption of foods contaminated with ruminant feces such as meat and milk products, fresh produce or drinking water have been identified as important transmission routes for human EHEC infections [3, 23]. It is not clear whether EHEC O145:H25 might be incapable of colonizing the bovine gut or have just failed to be isolated based on the sampling and detection methods applied.
A large number of mobile genetic elements (MGEs) have been identified in O145:H28 and other EHEC strains indicating that horizontal gene transfer (HGT) played a major role in the diversification and evolution of these pathogens [14, 24, 25]. In fact, most known and putative virulence factors are located on MGEs, such as prophages, plasmids and pathogenicity islands (PAI) and the independent acquisition of virulence factors by various serotypes contributes to the emergence of new EHEC/STEC strains [3, 14, 24–28]. Interestingly, in contrast to other non-O157 EHEC strains, EHEC O145:H28 appear to have evolved from a common ancestor with O157:H7 [14, 24]. Information regarding the evolutionary relatedness of O145:H25 and O145:H28 are sparse, but restriction fragment length polymorphisms (RFLP) analysis of the flagellar antigen-associated fliC genes of H25 and H28 indicated that EHEC O145:H25 and O145:H28 may belong to two different clonal lineages [13]. However, complete genome sequencing data of O145:H25 that could support this hypothesis are currently not available. Hence, in an attempt to determine the evolutionary relatedness of EHEC O145:H25 and O145:H28 as well as the genetic diversity and virulence repertoire, we sequenced the whole genome of two HUS-associated O145:H25 strains. Furthermore, in order to gain insight on the colonizing properties of O145:H25 we investigated the presence of various colonization-contributing factors (CCF) such as fimbrial and afimbrial adhesins and compared the obtained CCF profile with those found in O145:H28 and other EHEC strains.
Results and Discussion
General genomic features of EHEC O145:H25 strains CFSAN004176 and CFSAN004177
We sequenced the whole genomes of two EHEC O145:H25 strains that were isolated from HUS patients in the USA in 2003 and 2004. The chromosomes of strains CFSAN004176 and CFSAN004177 are 5193,734 bp and 5191,331 bp in size and consist of 5179 and 5193 coding DNA sequences (CDS), respectively. The chromosomal backbones of the two EHEC O145:H25 strains are nearly identical (Fig. 1); differences in chromosome size are primarily the result of variations in size and gene content of acquired MGEs (details discussed in the next sections). Furthermore, both strains belong to the same sequence type (ST) 7061, indicating their high relatedness. The two strains harbor three, nearly identical plasmids, one generally termed as pEHEC of 52,297 bp in size (pCFSAN004176_EHEC, pCFSAN004177_EHEC), one 34,714 bp plasmid (pCFSAN004176_sfp, pCFSAN004177_sfp) and one 95,721 bp/96,228 bp (CFSAN004176_PP/CFSAN004177_PP) prophage plasmid similar to Enterobacteria phage P1 (GenBank: NC_005856.1). Similar to other EHEC strains their chromosomes were found to be interrupted by various MGEs [14, 24, 25] (Fig. 1). In particular, CFSAN004176 and CFSAN004177 both carry 14 prophages, 9 integrative elements (IE) and 101 and 102 insertion sequences (IS), respectively (Table 1).
Fig. 1.
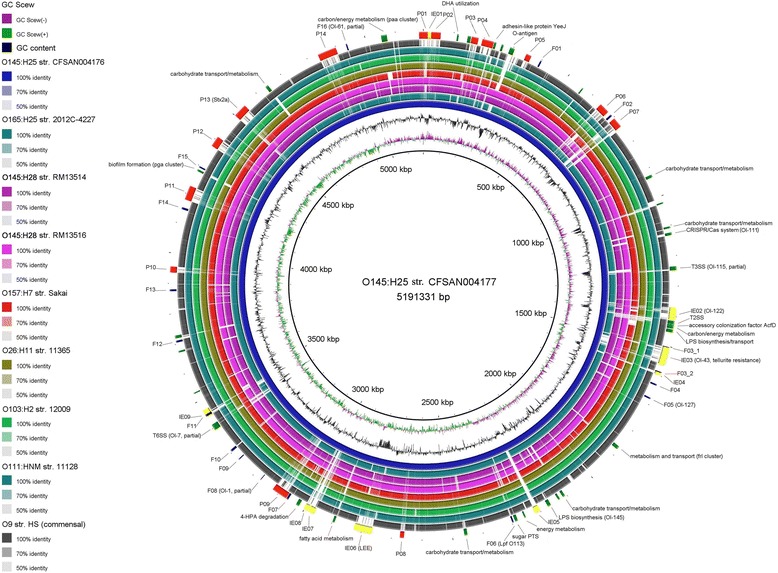
Circular map of EHEC O145:H25 strain CFSAN004177 in comparison to other EHEC strains. BLASTN comparison of the eight fully sequenced EHEC chromosomes including commensal strain HS against the chromosome of EHEC O145:H25 (CFSAN004177). Color key codes for the nine E. coli strains are shown on the left with nucleotide sequence identity as compared to the reference strain. The outermost ring displays prophages (red), IEs (yellow), fimbrial gene cluster (navy) and other genetic features (green) identified in the reference strain. The circular map was generated using BLAST Ring Image Generator (BRIG) with default settings
Table 1.
General genomic characteristics of EHEC O145:H25 in comparison with O145:H28 and other EHEC genomes
| Strains | O145:H25 | O145:H28 | O26:H11 | O103:H2 | O111:HNM | O157:H7 | O165:H25 | ||
|---|---|---|---|---|---|---|---|---|---|
| CFSAN004176 | CFSAN004177 | RM13514 | RM13516 | 11,365 | 12,009 | 11,128 | Sakai | 2012C-4227 | |
| Chromosome | |||||||||
| Size (kbp) | 5194 | 5191 | 5586 | 5402 | 5697 | 5449 | 5371 | 5498 | 5203 |
| GC (%) | 50.5 | 50.5 | 50.7 | 50.7 | 50.7 | 50.7 | 50.6 | 50.5 | 50.7 |
| CDSs | 5179 | 5193 | 5613 | 5325 | 5780 | 5457 | 5409 | 5204 | 5032 |
| rRNA operons | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 |
| tRNA’s | 99 | 96 | 104 | 98 | 101 | 98 | 106 | 105 | 106 |
| Prophages (PP) | 14 | 14 | 20 | 12 | 21 | 15 | 17 | 18 | 20 |
| IEs | 9 | 9 | 7 | 7 | 9 | 6 | 7 | 6 | 3 |
| IS elements | 101 | 102 | 70 | 61 | 84 | 76 | 84 | 60 | 100 |
| STa | 7061 | 7061 | 32 | 6130 | 119 | 21 | 17 | 16 | 11 |
| Plasmids | |||||||||
| size (kbp) | 96/52/35 | 96/52/35 | 87/65 | 98/59 | 85/63/6/4 | 76 | 205/98/78/8/7 | 93/3 | 98/75 |
| GC (%) | 48/47/49 | 48/47/49 | 48/53 | 50/42 | 48/53/46/44 | 49 | 47/48/50/50/50 | 48/43 | 48/48 |
| CDSs | 117/58/46 | 120/58/46 | 94/69 | 115/73 | 98/93/10/3 | 67 | 222/121/72/10/10 | 85/3 | 111/81 |
| IS elements | 20 | 19 | 29 | 14 | 22 | 9 | 28 | 10 | 12 |
| Total genome size (kbp) | 5377 | 5374 | 5738 | 5559 | 5855 | 5525 | 5767 | 5594 | 5376 |
pEHEC-like plasmids are indicated in bold, prophage plasmids are underlined
aFor MLST allelic profile see Additional file 4: Table S1
Whole genome-based phylogenetic analysis among 69 fully sequenced E. coli strains
To determine the evolutionary relatedness of EHEC O145:H25 and O145:H28 among 67 fully sequenced E. coli strains (Additional file 1: Table S1), we applied core genome multi locus sequence typing (cgMLST) followed by genome-wide SNP-based analysis of 1371 orthologous genes (Additional file 2: Dataset S1 and S2). The genome-wide SNP-based tree displayed an overall similar phylogeny of EHEC strains as previously reported [14] (Fig. 2). Furthermore, the phylogenetic analysis demonstrates that EHEC O145:H25 and O145:H28 in fact have evolved from two different clonal lineages as has been previously suspected [13]. Interestingly, while O145:H25 share a common evolutionary lineage with EHEC O26:H11 (str. 11,365), O103:H2 (str. 12,009) and O111:HNM (str. 11,128), O145:H25 appear to be most closely related to O165:H25 (str. 2012C-4227) (Fig. 2) (in the following sections referred to as O26, O103, O111 and O165). Indeed, a common evolution of O145:H25 and O165:H25 was recently assumed [29].
Fig. 2.
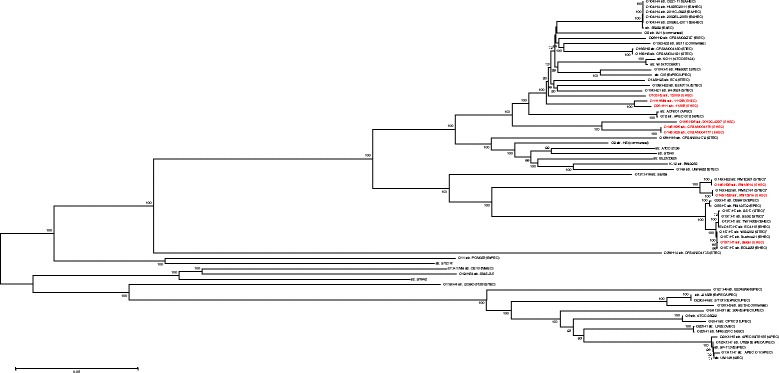
Genome-wide phylogenetic analysis of 69 fully sequenced E. coli strains. Whole genome wide phylogenetic tree based on SNP’s identified in 1371 core genes shared among all 69 E. coli strains (Additional file 2: Dataset S1 and S2). The phylogenetic analysis involved a total dataset of 86,350 SNP positions with all ambiguous positions being removed for each sequence pair. The SNP-based tree was constructed in MEGA 6 [71] using the Neighbour-joining algorithm [72] with the evolutionary history and evolutionary distance inferred using the Minimum Evolution method [73] and the Kimura 2-parameter model [74], respectively. Bootstrap values are shown on the nodes of each branch. Scale bar indicates the number of substitutions per site. EHEC strains used in this study are marked in red. *Carry EHEC virulence factors (stx, eae and ehxA) but were not clinical isolates associated with disease
Comparative genomics and in silico MLST analysis of O145:H25 and other EHEC strains
Based on the phylogenetic analysis and the apparently close evolutionary relationship of EHEC O145:H25 with EHEC O26, O103, O111 and O165 and EHEC O145:H28 with O157 Sakai (Fig. 2) these strains were included in the comparative genomic analysis. In order to gain insight on the genomic diversity between O145:H25 and O145:H28 as well as the other EHEC strains their genome sequences were analyzed and compared among each other. A BLASTN comparison of the nine fully sequenced EHEC strains against the chromosome of O145:H25 strain CFSAN004177 revealed a well conserved chromosomal backbone, as has been demonstrated for other EHEC strains (Fig. 1) [14, 24, 25]. However, significant differences were identified in the number and size of acquired MGEs resulting in the slightly smaller genome sizes of O145:H25 as compared to O145:H28 and other EHEC strains analyzed herein (except O165) (Table 1).
Expectedly, genomic dissimilarities were particularly observed in areas of integrated prophages and IEs (MGEs other than IS and prophages, e.g. LEE) (details discussed in next sections). However, we identified a number of non-homologous regions that did not correlate with the insertion of MGEs (here defined by the presence of an integrase gene) (Fig. 1). In particular, these regions included genes associated with LPS biosynthesis, type II secretion system (T2SS), type VI secretion system (T6SS), CRISPR (clustered regularly interspersed palindromic repeats) loci including the associated cas genes, and several clusters involved in metabolism and fimbrial biosynthesis (Fig. 1). Interestingly, some regions, particularly those involved in metabolism and fimbriae, were present or partially present in EHEC strains belonging to different lineages (Fig. 1, Additional file 3: Table S1). Furthermore, the flanking regions of these cluster were found to be identical in the EHEC genomes analyzed and no integrase or transposase genes were identified that could indicate recent HGT. This observation suggests that these clusters might have been integral parts of ancestral E. coli strains, that were retained in some EHEC lineages while being lost in others. Overall, however the majority of the identified clusters in O145:H25 was found to be absent in distantly related O145:H28 and O157 while being largely preserved in more closely related strains O26, O103, O111 and O165 including commensal E. coli strain HS (Fig. 1, Additional file 3: Table S1). Similarly, most of the O157-specific genomic O islands (OI) [25] were conserved in the closely related O145:H28 [14] while being absent in O145:H25 and other non-O157 EHEC strains (Additional file 3: Table S2). Hence, the data presented indicates a distinct evolutionary path for O145:H25 and O145:H28 and that in addition to HGT, gene loss appeared to have played a significant role in the diversification of these EHEC strains.
In silico MLST based on the seven E. coli housekeeping genes (adk, fumC, gyrB, icd, purA and recA) revealed that both EHEC O145:H25 strains belong to a new ST, 7061 (Table 1). Furthermore, except for fumC and purA that were identical between O145:H25 and O165, no similarities to the allelic profile of any of the other EHEC strains including O145:H28 were found (Additional file 4: Table S1).
Plasmids
As reported previously, the EHEC strains differed significantly in the number, size, and gene contents of plasmids [14, 24]. Specifically, while the two O145:H25 strains were found to carry three plasmids, both O145:H28 strains carried two whereas the other EHEC strains analyzed herein possessed one to five plasmids (Table 1). None of the EHEC serotypes carried the same set of plasmids and in contrast to O26, O111, and O145:H28 (RM13514), no antibiotic resistance genes were identified in any of the three plasmids present in O145:H25 [14, 24]. We previously determined that the pEHEC plasmids of O145:H25 are much smaller than other pEHEC plasmids and that they lacked several pEHEC-characteristic genes (ecf-cluster, espP, toxB, katP and stcE) many of which were present in various combinations in O145:H28 and other EHEC strains (Table 2) [17]. The presence of several IS elements/transposase genes (Additional file 4: Table S4) and the vast genetic diversity among these pEHEC plasmids indicate a distinct evolutionary path.
Table 2.
Identified plasmid and chromosome-encoded virulence genes and other genetic features of O145:H25 and other EHEC genomes
| Strains | O145:H25 | O145:H28 | O26:H11 | O103:H2 | O111:HNM | O157:H7 | O165:H25 | ||
|---|---|---|---|---|---|---|---|---|---|
| CFSAN004176 | CFSAN004177 | RM13514 | RM13516 | 11,365 | 12,009 | 11,128 | Sakai | 2012C-4227 | |
| chromosome-encoded | |||||||||
| aidA-I | - | - | 2 (1) | 3 (3) | 2 | 2 (1) | 1 | 1 | - |
| astA | 1 | 1 | 1 | 1 | 1 | - | 1 | 1 | 1 |
| bor (iss) | 1 | 1 | 1 | - | 2 | 1 | 1 | 2 | 1 |
| eae type | β | β | γ | γ | β | ε | θ | γ | ε |
| efa1 | 2 (1) | 2 | (+/−) | 1 | 1 | 2 | 1 | (+/−) | 1 |
| ehaA | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| espI | 1 | 1 | 1 | - | - | 1 | - | - | 1 |
| gad | 1 | 1 | 1 | - | 1 | 1 | 1 | 1 | 1 |
| iee | (+/−) | (+/−) | 1 | 1 | 1 | 1 | 1 | 1 | - |
| iha | - | - | 1 | 1 | 2 | - | 1 | 1 | - |
| lpf cluster | 1 | 1 | 1 (1) | 1 | 2 | 1 | 2 | 2 (1) | 1 |
| pagC | - | - | - | 1 | - | 2 | 1 | 1 | - |
| sod (Cu/Zn) | 1 | 1 | 2 | 2 | - | 2 | 2 | 2 | 1 |
| stx genes | stx 2a | stx 2a | stx 2a | stx 2a | stx 1a | stx 1a + stx 2a | stx 1a + stx 2a | stx 1a + stx 2a | stx 1a + stx 2a |
| tir | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| plasmid-encoded | |||||||||
| cba | 1 | 1 | - | - | - | - | - | - | - |
| cma | 1 | 1 | - | - | - | - | - | - | - |
| ecf | - | - | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| ehxA subtype | E | E | C | C | C | F | C | B | C |
| espI | 1 | 1 | - | - | - | - | - | - | - |
| espP | - | - | 1 | - | - | - | (+/−) | 1 | 1 |
| katP | - | - | - | - | 1 | - | - | 1 | 1 |
| sfp | 1 | 1 | - | - | - | - | - | - | 1 |
| sta1 | 1 | 1 | - | - | - | - | - | - | - |
| stcE | - | - | - | 1 | - | 1 | - | 1 | - |
| toxB | - | - | 1 | - | 1 | - | - | 1 | - |
present (≥ 90% identity, ≥ 80% gene coverage); −, absent; (+/−), partial (≥ 90% identity, 10–80% coverage);
numbers in parentheses indicate pseudogene(s)
Of the remaining two plasmids identified in O145:H25, one was a prophage plasmid similar to Enterobacteria phage P1 (GenBank: NC_005856.1) (99% nucleotide sequence identity with 80% sequence coverage) also found to be present in O165 and O111 (Table 1). The third plasmid present in both EHEC O145:H25 carried the sfp fimbrial gene cluster that was present on the pEHEC plasmids of O165 and the sorbitol-fermenting O157:HNM [17, 30, 31].
Prophages and IS elements
MGEs are important factors in EHEC genome diversity and plasticity and play a major role in virulence evolution by means of HGT among and within bacterial species [14, 24, 25]. In fact, the most important virulence factors of all disease-associated STEC and EHEC strains, the Shiga toxin-encoding genes stx 1 and stx 2, are located on mobile bacteriophages. Overall, we identified 14 prophages in both O145:H25 strains, of which lambda-like prophages are predominant (Additional file 4: Table S2). Similarly, O145:H28 and other EHEC strains were found to carry 12 to 20 predominantly lambda-like prophages (Table 1) [14, 24]. Furthermore, the majority of the O145:H25 prophages were located near common integration sites and similar to previous findings were found to carry several non-LEE encoded effector (Additional file 4: Table S2 and Table S5) [14, 24]. Like O145:H28, both O145:H25 strains carry a prophage encoding the highly HUS-associated Stx2a variant. However, while the two O145:H25 strains carry a closely related approximately 45 kbp lambda-like Stx2 a prophage located adjacent to the yecE locus, O145:H28 (RM13514) was found to carry an approximately 60 kbp Podoviridae family Stx2 a prophage inserted at argW [14]. Phylogenetic analysis suggests that the lambda-like Stx2a prophages of O145:H25, O145:H28 (RM13516) and O111 are closely related to each other while being distantly related to those found in O145:H28 (RM13514) and the other EHEC strains (Fig. 3), indicating their lineage-independent acquisition. Interestingly, CFSAN004176 contains a second “Stx2 a-like” prophage (CFSAN004176_P01) with several nonsense mutations within the stx 2A subunit and CFSAN004177 carries a closely related prophage (CFSAN004177_P01) at the same insertion site (yciE) without the stx 2A and stx 2B genes (Fig. 3). Both “Stx2-like” prophages appear to be defective as they are lacking genes primarily encoding DNA packaging-, phage tail- and lysis-associated proteins (Fig. 3).
Fig. 3.
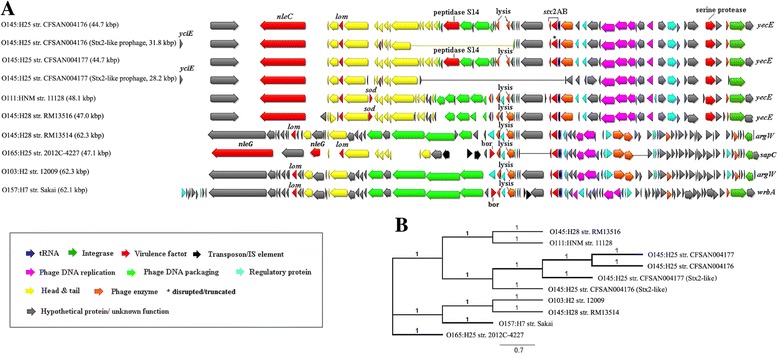
Genetic organization and phylogeny of the Stx2a prophages identified in O145:H25 and other EHEC strains. a Integration sites are displayed on either sites and genes are colored based on functional characteristics as described in the figure legend. Approximate prophage sizes are indicated in parentheses. Nucleotide sequences were aligned in Geneious 9.1.5 using MAFFT with default settings. b Phylogenetic analysis of the Stx2a prophages. The maximum-likelihood tree was constructed in Geneious 9.1.5 using RAxML and the GTR + GAMMA + Invariable sites model and 500 bootstrap replicates. The scale bar indicates the number of substitutions per base; branch support values 1 representing 100
IS elements are widely distributed among bacteria and play an important role in genome evolution and diversification [14, 24, 32]. They have further been proposed to contribute to improved bacterial fitness and adaptation to environmental changes due to their capability to modulate gene expression and induce insertional gene inactivation [33]. Overall, we identified 101 and 102 IS elements in the chromosomes of O145:H25, a considerably higher IS copy number as compared to O145:H28 and other EHEC strains (excluding O165) (Table 1). The majority of IS elements was found to be located on prophages, plasmids and IEs, supporting their possible implication in the fixation of MGEs in the bacterial genome [32]. Interestingly, we observed significant differences in the prevalence of certain IS elements among EHEC strains. For example, while IS629 was found to be the predominant IS among all EHEC strains with copy numbers ranging from 9 to 37 [14, 24] we identified only 3, non-intact IS629 in both O145:H25 strains. In contrast, O145:H25 carry over 50 copies of IS600 while the majority of the other EHEC strains carries less than 10 copies or zero (Additional file 4: Table S3). Moreover, most IS600 copies (45) appear to be active, as indicated by the presence of complete terminal repeats on both sides, suggesting that the evolution and diversification of O145:H25 was primarily driven by IS600-mediated mutations and not by IS629 as has been shown for O157 and suggested for other EHEC strains [14, 24, 32].
IS3 family members, such as IS629 and IS600, are believed to transpose in a replicative manner. While IS proliferation is generally an important factor in genome evolution as they can generate beneficial mutations that promote bacterial adaptation [34, 35], uncontrolled IS proliferation can be detrimental to the host. Thus, various genetic strategies have evolved to maintain stable IS copy numbers [33, 36, 37], one of which is the insertion sequence excision enhancer (IEE) that promotes the excision and deletion of IS3 family members [38]. While O145:H28, O157 Sakai, O26, O111 and O103 all carry the iee gene, in both O145:H25 strains iee is disrupted by IS600 while being completely absent in O165 (Fig. 4). This may explain the rather high copy number present in both O145:H25 strains and O165. However, in the presence of high copy numbers, negative regulators that inhibit or attenuate IS transpositions have been shown to be overexpressed [33, 36]. By implication, lowering the copy number (i.e. due to IEE-mediated IS deletion) increases IS transposition activity. Hence, the absence of iee accompanied by high IS copy numbers in O145:H25 and O165 suggests that IS transposition might be continuously depressed in those strains while being promoted in O145:H28 and the other EHEC strains. Furthermore, lower IS activity associated with decreased ability to change suggests that EHEC O145:H25 and O165 may not be able to adapt as rapidly to environmental changes as O145:H28 and other EHEC strains, which in turn might be one explanation why these serotypes are less commonly isolated [2, 13, 16]. Indeed, highly pathogenic and clinically relevant EHEC/STEC strains were found to carry iee and IS3 family members while less common EHEC/STEC strains were devoid of at least one of these elements [39].
Fig. 4.
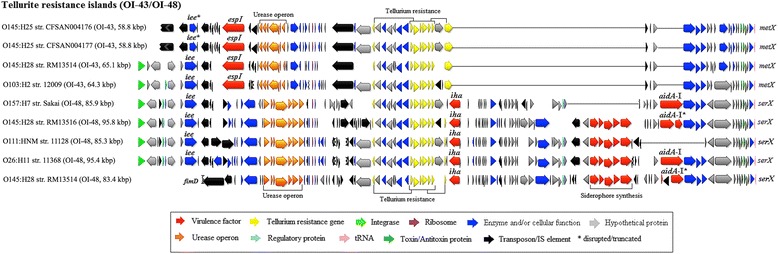
Genetic organization and content of the tellurite resistance islands OI-43/OI-48 identified in the EHEC strains. Approximate sizes of the OIs are indicated in parentheses. tRNA integration sites are displayed on the right and genes are colored based on functional characteristics as described in the figure legend. Nucleotide sequences were aligned in Geneious 9.1.5 using MAFFT with default settings
Genomic islands and Integrative elements
Genomic islands occupy large chromosomal regions (> 10 kbp) and often carry genes providing selective advantages to the bacteria that carry them [25]. Based on their gene content, genomic islands are referred to metabolic, fitness and pathogenicity islands, the latter of which are absent in nonpathogenic bacteria. Several genomic islands have been identified in EDL933 [25], of which OI-28 (RTX-toxin), OI-35 (energy metabolism), OI-47 (adhesin and fatty-acid biosynthesis) and OI-138 (fatty acid biosynthesis) appear to separate the non-O157 EHEC from the O157 lineages (including EPEC O55 and EHEC O145:H28) as they are absent in O145:H25 and all other non-O157 EHEC strains while being present in O145:H28 (OI-47 is partially conserved in RM13514) (Additional file 3: Table S2). Other large OIs such as OI-115 (T3SS and Salmonella/Shigella-associated host-cell invasion genes) and OI-7 (T6SS) are partially conserved in O145:H25 and other EHEC strains while being absent in O145:H28 (lacks OI-7) and O165 (lacks both OIs) indicating potential evolutionary regression (Additional file 3: Table S2). The remaining large OIs, namely the PAIs OI-43 and OI-48 (tellurite resistance and urease gene cluster), OI-122 (toxins), and OI-148 (LEE) are present in all EHEC strains (O165 lacks OI-43/48) albeit with significant variations in size and gene contents (Figs. 4 and 5). In particular, the LEE islands of O145:H25 are approximately 58 kbp in size, are integrated at tRNA pheU and carry the intimin subtype beta. In contrast, the LEE islands of O145:H28 are about 10 kbp smaller, are integrated at the tRNA selC locus and carry the intimin subtype gamma, analogous to O157 [14]. Furthermore, while the LEE accessory regions (LEE-AR) of O145:H25 were found to contain the OI-122 encoding the virulence associated adhesin Efa1 and three T3SS effector proteins [3, 27, 40] the LEE-AR of O145:H28, O157, and O165 are devoid of any virulence factors but carry OI-122 outside the LEE (Fig. 5). Others proposed that the LEE and OI-122 were originally acquired as one large mosaic PAI and that in some strains, such as O157, the LEE and OI-122 separated later on [27]. However, both O145:H25 and other EHEC strains carry a second OI-122, suggesting that these two PAIs may indeed have separate origins [24, 28]. In fact, while the LEE core regions are well conserved among various EHEC strains the LEE-ARs are highly heterogeneous or unoccupied [14, 24] (Fig. 4). Thus, it appears likely that insertions (e.g. OI-122) were introduced into a preexisting LEE core and later on passed horizontally among different serotypes as exemplified by O145:H25 and O26 that carry a nearly identical LEE-OI-122 PAI (99% sequence identity). Indeed, phylogenetic analysis indicates that the LEEs of O145:H25 and O26 are highly related whereas the LEEs of O145:H28 are more closely related to the LEE in O157 (Fig. 5).
Fig. 5.
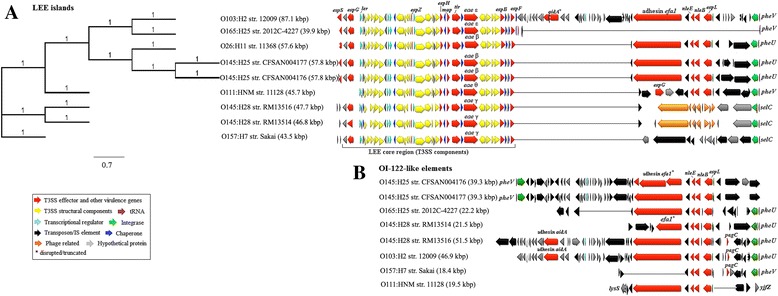
Genetic organization of the LEE islands and OI-122 identified in O145:H25 and other EHEC strains. tRNA integration sites are displayed on either sites and genes are colored based on functional characteristics as defined in the figure legend. Approximate sizes of the LEE and OI-122 are indicated in parentheses. Nucleotide sequences of the LEE and OI-122 were aligned in Geneious 9.1.5 using MAFFT with default settings. a Phylogeny and gene organization of the identified LEE islands. The maximum-likelihood tree was constructed in Geneious 9.1.5 using RAxML and the GTR + GAMMA + Invariable sites model and 500 bootstrap replicates. The scale bar indicates the number of substitutions per base; branch support values 1 representing 100. b Gene organization of OI-122 identified in the EHEC strains in comparison to the LEE-AR displayed above
Of the two identical tellurite resistance islands (OI-43 and OI-48) identified in EDL933 [25] both O145:H25 strains were found to carry OI-43 that is highly similar to those found in O145:H28 (RM13514) and O103. However, RM13514 carries a second, much larger tellurite resistance island (OI-48) similar to those present in O145:H28 (RM13516), O26, O111 and O157 Sakai [14] (Fig. 4). In contrast to EDL933, significant differences were observed in the gene contents of these two islands. Specifically, while OI-43 encodes the putative virulence-associated serine protease EspI [41], OI-48 was found to carry genes encoding two adherence-associated proteins, Iha and AIDA-I, and a siderophore cluster involved in iron acquisition [42–44]. The presence of several IS elements within both tellurite resistance islands suggests that IS-mediated deletions/insertions played a role in the evolution of these PAIs (Fig. 4) and because closely related O165 was devoid of OI-43/48, these PAIs were acquired in a lineage-independent manner.
Noteworthy, besides the identified CRISPR/Cas system involved in phage immunity, both O145:H25 strains acquired what appear to be two distinct restriction-modification (RM) systems (identified by the presence of endonuclease and methylase genes) associated with phage-resistance (IE05 and IE07) (Fig. 1, Additional file 4: Table S2) [45]. Both of these IEs were absent in all EHEC genomes analyzed herein, in fact a BLASTN comparison against the NCBI database revealed that IE05 appears to be unique to O145 while IE07 was also present in O104:H7. Furthermore, IE05 was found to carry a potentially third phage-resistant system associated with abortive infection (Abi). In contrast to RM and CRISPR that prevent phage infections, Abi systems are triggered post phage infection and were shown to lead to bacterial cell death. However, whether the presence of these phage-resistant islands is associated with the acquisition of fewer prophages in O145:H25 as compared to the other EHEC genomes, as well as, the rarity of this particular serotype remains to be determined as many phages have been shown to be capable of evading phage-resistance systems [45].
Adhesins and other colonization-contributing factors
Bacterial adhesion and their ability to persist in a host depends on a variety of different colonization-contributing factors (CCF). The LEE-encoded adhesin intimin (encoded by eae) and its translocated receptor Tir as well as several non-LEE-encoded effectors have been shown to play a key role for the intimate attachment of EHEC strains in the human and bovine gastrointestinal tract (GIT) [3, 26, 40]. However, several other genetic features are involved in this complex process including the plasmid-encoded EspP [46], ToxB [47], Ecf cluster [48] and StcE [49] as well as a number of fimbrial (i.e. Sfp and Lpf) [31, 40, 50, 51] and afimbrial adhesins (i.e. Iha, Efa1, AIDA-I and EhaA) [42, 52–54]. Similar to other EHEC serotypes, EHEC O145:H25 was found to carry eae, numerous non-LEE-encoded effectors (Additional file 4: Table S5), efa1, ehaA, and lpf but lacked aidA-I, iha, espP, stcE and toxB, many of which were equally absent in the other EHEC serotypes especially in O165 (Table 2). Furthermore, resistance genes such as bor [55], sod [56], gad [57], katP [58] and pagC [59] associated with serum survival and resistance towards oxidative stress were less prevalent in O145:H25 as compared to the other EHEC strains. (Table 2). More importantly, in contrast to all other EHEC strains analyzed herein, O145:H25 lacked the ecf-cluster associated with bacterial persistence in the bovine GIT which might explain why O145:H25 have thus far failed to be detected in any ruminant species. In fact, while ecf-positive strains were shown to persist in the bovine gut for up to 3 months ecf-deletion mutants were not recovered beyond seven days [48].
In addition, the acquisition of nutrients as well as other resources such as iron are equally important for the successful colonization of any given environment [44, 60]. Iron has been shown to be essential for bacterial growth and many EHEC strains including O145:H28, O26 and O157 were found to have acquired additional iron-acquisition systems such as siderophores or those located in OI-140 [14, 24, 25] (Fig. 4, Additional file 3: Table S2). In contrast, EHEC O145:H25 was devoid of any of these systems, indicating that they might be impaired in their ability to acquire iron in a stable microbiota, particularly during early stages of the colonization process. Thus, while O145:H25 generally appear to be able of colonizing the bovine gut, the absence of the ecf-cluster as well as additional iron acquisition systems suggests that they might be impaired in their ability to establish a persistent colonization.
Fimbriae are believed to mediate initial attachment of the bacteria to the epithelial cells and thus have been proposed to influence the tissue tropism observed in O157 and other EHEC strains [51, 61, 62]. Overall, we identified 15 apparently intact fimbrial gene cluster in O145:H25 a similar range as compared to other EHEC strains (Table 3). Interestingly, while many fimbrial gene cluster were shared among all EHEC strains others appear to be lineage-specific. In particular, whereas clusters F03, F04, F10 and F13 were present in all EHEC strains of the non-O157 lineages, in O145:H28 and O157 these clusters were either absent or they carried non-identical homologs instead (Table 3, Additional file 3: Table S3). Similarly, cluster F08 (OI-1) was present in closely related O157 and O145:H28 while being only partially conserved in the other EHEC strains. Furthermore, all EHEC strains were found to carry lpf-clusters however, while the majority of the non-O157 EHECs carried the lpf-cluster initially identified in O113 [50], O157 carried lpf OI-141 and lpf OI-154 while O145:H28 carried lpf OI-141. The observed differences among the fimbrial gene cluster present in the various EHEC serotypes may indicate adaptation to distinct ecological niches. In fact, it was previously shown that O157 primarily colonizes the lowest portion of the small intestine whereas other EHEC strains such as O26 and O111 were found to interact primarily with the large intestinal mucosa [63–66]. The observed tissue tropism was further proposed to be influenced by the intimin type. Hence, since the fimbrial profile and intimin type of O145:H25 are more similar to that of O26 and O111, and O145:H28 are more similar to O157 (Tables 2 and 3), O145:H25 and O145:H28 likely adapted to different ecological niches within the mammalian host.
Table 3.
Fimbrial gene clusters present in EHEC O145:H25 and other E. coli strains
| Fimbrial cluster | O145:H25 | O145:H28 | O26:H11 | O103:H2 | O111:HNM | O157:H7 | O165:H25 | commensal | ||
|---|---|---|---|---|---|---|---|---|---|---|
| CFSAN004176 | CFSAN004177 | RM13514 | RM13516 | 11,365 | 12,009 | 11,128 | Sakai | 2012C-4227 | HS | |
| F01 | + | + | + | + | + | + | + | H | + | + |
| F02 | + | + | Ha | Ha | Hb | + | Hb | Hc | Hb | Hb |
| F03 | (+) | (+) | − | − | + | + | (+) | − | (+) | + |
| F04 | + | + | − | − | (+) | + | + | − | + | + |
| F05 (OI-127) | + | + | − | − | + | + | + | (+) | + | + |
| F06 (Lpf_O113/OI-154) | + | + | − | − | + | − | + | H | + | − |
| F07 | + | + | + | + | + | + | + | + | + | + |
| F08 (OI-1) | (+/−) | (+/−) | (+) | (+) | (+/−) | (+/−) | (+/−) | (+) | (+/−) | (+/−) |
| F09 | + | + | + | + | + | + | + | + | + | + |
| F10 | + | + | Hd | Hd | + | + | + | Hd | (+) | (+/−) |
| F11 | + | (+) | + | + | + | + | − | + | − | (+/−) |
| F12 | + | + | + | + | + | + | + | + | + | + |
| F13 | + | + | He | He | + | (+) | + | He | + | + |
| F14 | + | + | + | + | + | (+) | + | (+) | (+) | + |
| F15 | + | + | + | (+) | + | + | + | + | (+) | (+) |
| F16 (OI-61) | (+/−) | (+/−) | + | (+/−) | + | (+/−) | + | + | (+/−) | (+/−) |
| F17 (OI-141) | − | − | (+) | + | Hf | Hf | Hf | (+) | − | − |
| F18 (OI-47) | − | − | (+/−) | (+/−) | − | − | − | + | − | − |
| F19 (sfp cluster) | + | + | − | − | − | − | − | − | + | − |
| Totalg | 15 (2) | 15 (2) | 13 (1) | 12 (2) | 16 (1) | 14 (2) | 15 (1) | 16 | 14 (2) | 11 (4) |
+, present (> 90% identity, > 90% gene coverage); −, absent; (+/−), partial; (+), contains pseudogene(s); H homolog (< 90% sequence identity); (see also Additional file 3: Table S3)
a, b, c, d, e, fsame letters indicate same homolog
gnumbers in parentheses indicate the number of partially conserved clusters
Conclusion
Phylogenetic analysis supported by a comprehensive comparative genomic scrutiny revealed that EHEC O145:H25 and O145:H28 evolved from two different clonal lineages. The lineage-independent acquisition of various MGEs as well as gene loss contributed to the divergence of these pathogens also reflected in variations seen in their virulence repertoire. Furthermore, based on the identified CCF profile, ruminants such as cattle could be a natural reservoir for EHEC O145:H25 as has been shown for other EHEC strains. However, the absence of several genetic features such as the ecf-cluster and additional iron-acquisition systems present in other EHEC strains suggests that EHEC O145:H25 might be impaired in their ability to establish a persistent colonization. This might explain why these strains have not been detected in any ruminant species. In addition, marked variations in fimbrial gene clusters and intimin subtypes between EHEC O145:H25 and O145:H28 suggests a potential adaptation to different colonization sites within the mammalian host’s GIT.
Methods
Bacterial Strains
EHEC O145:H25 strains CFSAN004176 and CFSAN004177 were isolated from patients in the USA that developed HUS in 2003 and 2004, respectively. Both strains were obtained from the Centers for Disease Control and Prevention (Atlanta, GA).
Genome sequencing and assembly
Bacteria were grown in tryptic soy broth as previously described [17]. Bacterial DNA was extracted from 2 mL of bacterial cell culture using the automated QIAcube extraction system (Qiagen Inc., Valencia, CA) according to the manufacturer’s instructions for Gram-negative bacteria.
Sequencing was carried out using single molecule real time (SMRT) DNA sequencing on the Pacific Bioscience RS II Sequencer (PacBio, Menlo Park, CA) as previously described [17]. Briefly, template DNA was sheared to ≥10-kbp using g-TUBEs (Covaris, Inc., Woburn, MA). Whole genome libraries were prepared according to the PacBio 10-kbp insert library protocol using DNA Template Kit 1.0 and afterwards size-selected using the BluePippin size-selection system (Sage Science, Inc., Beverly, MA) according to the manufacturer’s instruction. Libraries were sequenced using P4/C2 Chemistry Kits on five SMRT cells with a 180-min collection protocol. The obtained sequence reads were analyzed by SMRT Analysis 2.3.0 (PacBio, Menlo Park, CA) and de novo assembled using the PacBio hierarchical genome assembly process 3 (HGAP3.0)/Quiver software package.
Gap closure, genome annotation and accession numbers
The resulting assemblies for each strain were confirmed by optical maps generated with 30 fold coverage on the Argus Mapping Station (OpGen, Gaithersburg, MD) as previously described [67]. Briefly, high molecular weight DNA was immobilized onto derivatized glass surfaces, digested with BamHI restriction enzyme (OpGen), and fluorescently stained with YOYO-1 dye (OpGen). The fragments were then size measured using an automated fluorescent microscope (OpGen). Each sequence contig was aligned to the optical map using MapSolver Software version 3.1 (OpGen) in order to identify contig order, overlapping contigs, potential mis-assemblies and inversions [68]. Overlapping contigs were combined by removing the overlapping, identical sequence of one contig. The resulting, combined contig was verified by mapping all PacBio raw sequencing reads back to this contig using SMRT Analysis. Finally, contig closure was determined by creating dot plots for each contig utilizing the Gepard software as previously described [17].
Genome sequences were annotated by the NCBI Prokaryotic Genome Automatic Annotation Pipeline (PGAAP) (http://www.ncbi.nlm.nih.gov/genome/annotation_prok/) and are available at GenBank under the following accession numbers: CFSAN004176 chromosome (NZ_CP014583), pCFSAN004176_EHEC (NZ_CP012493), pCFSAN004176_sfp (NZ_CP012492), pCFSAN004176_PP (NZ_CP012491), CFSAN004177 chromosome (CP014670), pCFSAN004177_EHEC (CP012495), pCFSAN004177_sfp (CP012496), pCFSAN004177_PP (CP012494).
Comparative genomics and phylogenetic analysis
In order to identify genomic dissimilarities among O145:H25 and other EHEC strains BLAST Ring Image Generator (BRIG) [69] was used for comparison and visualization purposes. EHEC O145:H25 strain CFSAN004177 served as a reference genome and the nucleotide sequences of the following EHEC chromosomes were BLASTed against the reference strain with default settings: O145:H25 strain CFSAN004176, O165:H25 strain 2012C-4227 (GenBank: CP013029.1), O145:H28 strain RM13514 (GenBank: NZ_CP006027.1), O145:H28 strain RM13516 (GenBank: NZ_CP006262.1), O157:H7 strain Sakai (GenBank: NC_002695.1), O26:H11 strain 11,365 (GenBank: NC_013361.1), O103:H2 strain 12,009 (GenBank: NC_013353.1), and O111:HNM strain 11,128 (GenBank: NC_013364.1). Non-homologous regions as identified by the BRIG image were further manually inspected using CLC Genomics Workbench 7.5 (CLC bio, Boston, MA).
The phylogenetic relatedness of the newly sequenced O145:H25 EHEC strains among 67 fully sequenced E. coli strains (Additional file 1: Table S1) was determined by cgMLST analysis using Ridom SeqSphere + 2.4.0 as previously described [70]. First a cgMLST scheme was defined by using the Ridom software’s target definer tool with default settings and E. coli O157:H7 Sakai (GenBank: NC_002695.1) as reference genome (5498,450 bases, 5204 genes). To establish a list of core and accessory genome genes the following six fully sequenced genomes were used for comparison with the reference genome: E. coli O157:H7 strain EC4115, E. coli O157:H7 strain EDL933, E. coli O157:H7 strain TW14359, E. coli O55:H7 strain CB9615, E. coli O55:H7 strain RM12579, and E. coli O157:H7 strain Xuzhou21 (Additional file 1: Table S1). Multiple gene copies identified in any of the seven genomes were removed from the analysis as failed genes (553 targets). Then a task template was created containing both core and accessory genes for future analysis. Each individual locus of the identified core (3860 targets) and accessory genes (791 targets) was assigned allele number 1. Afterwards, each individual E. coli genome was queried against the created task template and all identified loci that were 100% identical to the reference and query genomes were assigned allele number 1; otherwise a new allele number was called for that locus. The number of shared loci among the 69 genomes was determined to be 1371 loci (using Sakai genome as reference). The cgMLST performed a gene-by-gene analysis of each individual genome for the 69 strains, and SNP’s found within the identified 1371 shared alleles were used to determine their genetic distance (Additional file 2: Dataset S1 and S2). The phylogenetic analysis involved 69 nucleotide sequences and a total dataset of 86,350 SNP’s positions with all ambiguous positions being removed for each sequence pair. The SNP-based phylogenetic tree was constructed in MEGA 6 [71] using the Neighbour-joining algorithm [72] with the evolutionary history and evolutionary distance inferred using the Minimum Evolution method [73] and the Kimura 2-parameter model [74], respectively.
In silico MLST analysis, serotyping and determination of virulence and antimicrobial resistance genes
In silico MLST analysis was performed based on the seven housekeeping genes (adk, fumC, gyrB, icd, mdh, purA and recA) previously described for E. coli [75]. Allelic variants of those seven gene loci were identified using Ridom SeqSphere + 2.4.0 (Ridom GmbH, Münster, Germany) and alleles numbers and STs were assigned according to the E. coli MLST database (http://mlst.warwick.ac.uk/mlst/dbs/Ecoli). Briefly, each genome sequence was screened against all in the MLST database available alleles for the seven loci. Once a 100% sequence match was identified an allele number was assigned to that loci. STs were only assigned if the obtained allele combination for the seven loci was already described in the MLST database otherwise a new ST was called.
Serotypes were confirmed in silico using Ridom software by screening the obtained genome sequences for the presence of O-type (wzx and wzy) and H-type genes (fliC) as previously described [76, 77]. Genome sequences were further screened for all virulence [78] and antimicrobial resistance genes [79] deposited in the Center for Genomic Epidemiology database (http://www.genomicepidemiology.org/) using Ridom for performing batch screening of the genomes analyzed as described elsewhere [76]. Briefly, all the genes were divided into classes or groups by homology in fasta format (e.g. all astA alleles were in a single fasta file), and used as a task template. Afterwards a project was created using all these task templates, and each WGS was screened for the presence of each gene class (virulence or antimicrobial gene). Ninety-five virulence genes and 14 antimicrobial classes were analyzed by this method. The threshold used for screening for these genes using the Ridom software was > 90% identity and > 80% aligning or gene coverage in order to be called as present. When a pseudogene is present, Ridom visualize it as a red gene, when present and complete is visualized as green, and when absent the name appears as gray out.
Identification and comparative analysis of prophages, IEs and IS elements
Prophages and prophage-like elements of the newly sequenced O145:H25 EHEC strains were initially identified using the prophage-predicting PHASTER web server [80]. IEs such as pathogenicity and genomic islands were analyzed using the genomic island prediction web server IslandViewer3 for initial identification [81]. Each of the identified prophages, prophage-like elements and IEs were then manually inspected for accuracy using CLC Genomics Workbench 7.5 by locating nearby integrases and identification of potential integration sites. In order to identify sequence similarity and synteny among the identified prophages and IEs of the newly sequenced O145:H25 strains and other EHEC strains, prophage and IE sequences were aligned, analyzed and visualized using MAFFT Alignment with default settings in Geneious 9.1.5 (Biomatters, Auckland, New Zealand). Phylogenetic analysis was inferred using RAxML with the GTR + GAMMA + Invariable sites model and assessed by 500 bootstrap replicates using Geneious 9.1.5.
IS elements were initially identified using ISfinder [82]. Copy number and presence of each identified IS element were confirmed by BLASTing the nucleotide sequences of each IS element against the genome sequences of the nine EHEC genomes using BLASTN. Nucleotide sequences with at least 90% identity and coverage to the matching IS element were considered a match, coverage between 10% and 90% were considered a partial match. Each IS element was further BLASTed against all identified IS elements of the nine EHEC genomes to identify highly similar sequences among different IS elements. Highly similar IS sequences were BLASTed against the EHEC genomes simultaneously and the BLASTN results compared regarding sequence location and identity in order to avoid multiple matches for different IS elements at the same position. Of note, notably fewer IS were identified in all EHEC strains as compared to previous results, mainly attributable to different search parameters used herein. For example, previously identified IS elements such as IS1414 or IS630 shared less than 90% sequence identity in all EHEC strains and thus were not considered a match. Other copy numbers of certain IS could not be confirmed regardless of which parameters were applied (particularly in both O145:H28 strains).
Availability of supporting data
The genome sequences of CFSAN004176 and CFSAN004177 were deposited in GenBank and are available under the following links:
CFSAN004176 chromosome https://www.ncbi.nlm.nih.gov/nuccore/NZ_CP014583.1;
pCFSAN004176_EHEC https://www.ncbi.nlm.nih.gov/nuccore/NZ_CP012493.1;
pCFSAN004176_sfp https://www.ncbi.nlm.nih.gov/nuccore/NZ_CP012492.1;
pCFSAN004176_PP https://www.ncbi.nlm.nih.gov/nuccore/NZ_CP012491.1;
CFSAN004177 chromosome https://www.ncbi.nlm.nih.gov/nuccore/CP014670.1;
pCFSAN004177_EHEC https://www.ncbi.nlm.nih.gov/nuccore/CP012495.1;
pCFSAN004177_sfp https://www.ncbi.nlm.nih.gov/nuccore/CP012496.1;
pCFSAN004177_PP https://www.ncbi.nlm.nih.gov/nuccore/CP012494.1.
All other supporting data are included in this published article and its supplementary information files.
Additional files
Metadata and accession numbers of the 69 E. coli strains used for phylogenetic analysis. (XLSX 17 kb)
Dataset S1. SNP’s and SNP positions identified in the 1371 gene loci shared among the 69 E. coli genomes. Dataset S2. cgMLST for each of the 1371 identified gene loci shared among the 69 E. coli genomes. (XLSX 23678 kb)
Genomic dissimilarities between O145:H25 and other EHEC genomes. Table S2. Conservation of the EDL933-specific OIs in O145:H25 and the other EHEC strains. Table S3. Fimbrial gene cluster identified in the nine EHEC genomes, including nucleotide sequence identity. (XLSX 65 kb)
In silico MLST analysis of the nine EHEC strains. Table S2. Identified prophages and IEs in O145:H25. Table S3. IS elements present on EHEC chromosomes. Table S4. IS elements present on EHEC plasmids. Table S5. LEE- and non-LEE-encoded effectors present on EHEC chromosomes. (DOCX 59 kb)
Acknowledgements
This project was supported by the appointment of S.C.L. to the Research Fellowship Program for the Center for Food Safety and Applied Nutrition administered by the Oak Ridge Associated Universities through a contract with the FDA. This project is further part of the Ph.D. program (S.C.L.) at the University of Hamburg, Germany under the supervision of Markus Fischer.
Funding
Not applicable.
Availability of data and materials
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
Abbreviations
- CCF
Colonization-contributing factor
- CDS
Coding DNA sequence
- cgMLST
Core genome multi locus sequence typing
- EHEC
Enterohemorrhagic Escherichia coli
- GIT
Gastrointestinal tract
- HC
Hemorrhagic colitis
- HGT
Horizontal gene transfer
- HUS
Hemolytic uremic syndrome
- IE
Integrative element
- IEE
Insertion sequence excision enhancer
- IS
Insertion sequence
- LEE
Locus of enterocyte effacement
- MGE
Mobile genetic element
- OI
O island
- PAI
Pathogenicity island
- SNP
Single nucleotide polymorphism
- STEC
Shiga toxin producing Escherichia coli
Authors’ contributions
SCL carried out the experimental design, data gathering, data analysis, data interpretation and writing of the manuscript. NGE and MLK contributed in data gathering, data analysis and revision of this manuscript. JAK and MF supervised this study, contributed to experimental design and revision of the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12866-017-1094-3) contains supplementary material, which is available to authorized users.
References
- 1.Johnson KE, Thorpe CM, Sears CL. The emerging clinical importance of non-O157 Shiga toxin–producing Escherichia coli. Clin Infect Dis. 2006;43(12):1587–1595. doi: 10.1086/509573. [DOI] [PubMed] [Google Scholar]
- 2.Bettelheim KA. The non-O157 Shiga-toxigenic (verocytotoxigenic) Escherichia coli; Under-rated pathogens. Crit Rev Microbiol. 2007;33(1):67–87. doi: 10.1080/10408410601172172. [DOI] [PubMed] [Google Scholar]
- 3.Caprioli A, Morabito S, Brugère H, Oswald E. Enterohaemorrhagic Escherichia coli: emerging issues on virulence and modes of transmission. Vet Res. 2005;36(3):289–311. doi: 10.1051/vetres:2005002. [DOI] [PubMed] [Google Scholar]
- 4.Levine MM. Escherichia coli that cause diarrhea: enterotoxigenic, enteropathogenic, enteroinvasive, enterohemorrhagic, and enteroadherent. J Infect Dis. 1987;155(3):377–389. doi: 10.1093/infdis/155.3.377. [DOI] [PubMed] [Google Scholar]
- 5.Kaper JB, Nataro JP, Mobley HL. Pathogenic Escherichia coli. Nat Rev Microbiol. 2004;2(2):123–140. doi: 10.1038/nrmicro818. [DOI] [PubMed] [Google Scholar]
- 6.Beutin L, Zimmermann S, Gleier K. Human infections with Shiga toxin-producing Escherichia coli other than serogroup O157 in Germany. Emerg Infect Dis. 1998;4(4):635. doi: 10.3201/eid0404.980415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerber A, Karch H, Allerberger F, Verweyen HM, Zimmerhackl LB. Clinical course and the role of Shiga toxin–producing Escherichia coli infection in the hemolytic-uremic syndrome in pediatric patients, 1997–2000, in Germany and Austria: a prospective study. J Infect Dis. 2002;186(4):493–500. doi: 10.1086/341940. [DOI] [PubMed] [Google Scholar]
- 8.Eklund M, Scheutz F, Siitonen A. Clinical isolates of non-O157 shiga toxin-producing Escherichia coli: serotypes, virulence characteristics, and molecular profiles of strains of the same serotype. J Clin Microbiol. 2001;39(8):2829–2834. doi: 10.1128/JCM.39.8.2829-2834.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brooks JT, Sowers EG, Wells JG, Greene KD, Griffin PM, Hoekstra RM, Strockbine NA. Non-O157 Shiga toxin–producing Escherichia coli infections in the United States, 1983–2002. J Infect Dis. 2005;192(8):1422–1429. doi: 10.1086/466536. [DOI] [PubMed] [Google Scholar]
- 10.U.S. Department of Agriculture Food Safety and Inspection Service Shiga Toxin-producing Escherichia coli in certain raw beef products. Fed Regist. 2012;77:31975–31981. [Google Scholar]
- 11.Käppeli U, Hächler H, Giezendanner N, Beutin L, Stephan R. Human infections with non-O157 Shiga toxin-producing Escherichia coli, Switzerland, 2000–2009. Emerg Infect Dis. 2011;17(2):180–185. doi: 10.3201/eid1702.100909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rivas M, Sosa-Estani S, Rangel J, Caletti MG, Vallés P, Roldán CD, Balbi L, de Mollar MCM, Amoedo D, Miliwebsky E. Risk factors for sporadic Shiga toxin–producing Escherichia coli infections in children, Argentina. Emerg Infect Dis. 2008;14 [DOI] [PMC free article] [PubMed]
- 13.Sonntag A-K, Prager R, Bielaszewska M, Zhang W, Fruth A, Tschäpe H, Karch H. Phenotypic and genotypic analyses of enterohemorrhagic Escherichia coli O145 strains from patients in Germany. J Clin Microbiol. 2004;42(3):954–962. doi: 10.1128/JCM.42.3.954-962.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cooper KK, Mandrell RE, Louie JW, Korlach J, Clark TA, Parker CT, Huynh S, Chain PS, Ahmed S, Carter MQ. Comparative genomics of enterohemorrhagic Escherichia coli O145:H28 demonstrates a common evolutionary lineage with Escherichia coli O157:H7. BMC Genomics. 2014;15:1–17. doi: 10.1186/1471-2164-15-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beutin L, Krause G, Zimmermann S, Kaulfuss S, Gleier K. Characterization of Shiga Toxin-Producing Escherichia coli Strains Isolated from Human Patients in Germany over a 3-Year Period. J Clin Microbiol. 2004;42(3):1099–1108. doi: 10.1128/JCM.42.3.1099-1108.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carter MQ, Quinones B, He X, Zhong W, Louie JW, Lee BG, Yambao JC, Mandrell RE, Cooley MB. An environmental Shiga toxin-producing Escherichia coli O145 clonal population exhibits high-level phenotypic variation that includes virulence traits. Appl Environ Microbiol. 2016;82(4):1090–1101. doi: 10.1128/AEM.03172-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lorenz SC, Monday SR, Hoffmann M, Fischer M, Kase JA. Plasmids from Shiga toxin-producing Escherichia coli strains with rare enterohemolysin gene (ehxA) subtypes reveal pathogenicity potential and display a novel evolutionary path. Appl Environ Microbiol. 2016;82(21):6367–6377. doi: 10.1128/AEM.01839-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Toma C, Espinosa EM, Song T, Miliwebsky E, Chinen I, Iyoda S, Iwanaga M, Rivas M. Distribution of putative adhesins in different seropathotypes of Shiga toxin-producing Escherichia coli. J Clin Microbiol. 2004;42(11):4937–4946. doi: 10.1128/JCM.42.11.4937-4946.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krause G, Zimmermann S, Beutin L. Investigation of domestic animals and pets as a reservoir for intimin-(eae) gene positive Escherichia coli types. Vet Microbiol. 2005;106(1):87–95. doi: 10.1016/j.vetmic.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 20.Wieler L, Vieler E, Erpenstein C, Schlapp T, Steinrück H, Bauerfeind R, Byomi A, Baljer G. Shiga toxin-producing Escherichia coli strains from bovines: association of adhesion with carriage of eae and other genes. J Clin Microbiol. 1996;34(12):2980–2984. doi: 10.1128/jcm.34.12.2980-2984.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blanco M, Padola NL, Krüger A, Sanz ME, Blanco JE, González EA, Dahbi G, Mora A, Bernárdez MI, Etcheverría AI. Virulence genes and intimin types of Shiga-toxin-producing Escherichia coli isolated from cattle and beef products in Argentina. Int Microbiol. 2010;7(4):269–276. [PubMed] [Google Scholar]
- 22.Geue L, Segura-Alvarez M, Conraths F, Kuczius T, Bockemühl J, Karch H, Gallien P. A long-term study on the prevalence of shiga toxin-producing Escherichia coli (STEC) on four German cattle farms. Epidemiol Infect. 2002;129(01):173–185. doi: 10.1017/S0950268802007288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erickson MC, Doyle MP. Food as a Vehicle for Transmission of Shiga Toxin–Producing Escherichia coli. J Food Prot. 2007;70(10):2426–2449. doi: 10.4315/0362-028X-70.10.2426. [DOI] [PubMed] [Google Scholar]
- 24.Ogura Y, Ooka T, Iguchi A, Toh H, Asadulghani M, Oshima K, Kodama T, Abe H, Nakayama K, Kurokawa K. Comparative genomics reveal the mechanism of the parallel evolution of O157 and non-O157 enterohemorrhagic Escherichia coli. Proc Natl Acad Sci U S A. 2009;106(42):17939–17944. doi: 10.1073/pnas.0903585106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perna NT, Plunkett G, Burland V, Mau B, Glasner JD, Rose DJ, Mayhew GF, Evans PS, Gregor J, Kirkpatrick HA. Genome sequence of enterohaemorrhagic Escherichia coli O157: H7. Nature. 2001;409(6819):529–533. doi: 10.1038/35054089. [DOI] [PubMed] [Google Scholar]
- 26.Sheng H, Lim JY, Knecht HJ, Li J, Hovde CJ. Role of Escherichia coli O157:H7 virulence factors in colonization at the bovine terminal rectal mucosa. Infect Immun. 2006;74(8):4685–4693. doi: 10.1128/IAI.00406-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morabito S, Tozzoli R, Oswald E, Caprioli A. A mosaic pathogenicity island made up of the locus of enterocyte effacement and a pathogenicity island of Escherichia coli O157: H7 is frequently present in attaching and effacing E. coli. Infect Immun. 2003;71(6):3343–3348. doi: 10.1128/IAI.71.6.3343-3348.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konczy P, Ziebell K, Mascarenhas M, Choi A, Michaud C, Kropinski AM, Whittam TS, Wickham M, Finlay B, Karmali MA. Genomic O island 122, locus for enterocyte effacement, and the evolution of virulent verocytotoxin-producing Escherichia coli. J Bacteriol. 2008;190(17):5832–5840. doi: 10.1128/JB.00480-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beutin L, Delannoy S, Fach P. Sequence Variations in the Flagellar Antigen Genes fliC H25 and fliC H28 of Escherichia coli and Their Use in Identification and Characterization of Enterohemorrhagic E. coli (EHEC) O145: H25 and O145: H28. PLoS One. 2015;10(5):e0126749. doi: 10.1371/journal.pone.0126749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bielaszewska M, Prager R, Vandivinit L, Musken A, Mellmann A, Holt NJ, Tarr PI, Karch H, Zhang W. Detection and characterization of the fimbrial sfp cluster in enterohemorrhagic Escherichia coli O165:H25/NM isolates from humans and cattle. Appl Environ Microbiol. 2009;75(1):64–71. doi: 10.1128/AEM.01815-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Musken A, Bielaszewska M, Greune L, Schweppe CH, Muthing J, Schmidt H, Schmidt MA, Karch H, Zhang W. Anaerobic conditions promote expression of Sfp fimbriae and adherence of sorbitol-fermenting enterohemorrhagic Escherichia coli O157:NM to human intestinal epithelial cells. Appl Environ Microbiol. 2008;74(4):1087–1093. doi: 10.1128/AEM.02496-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ooka T, Ogura Y, Asadulghani M, Ohnishi M, Nakayama K, Terajima J, Watanabe H, Hayashi T. Inference of the impact of insertion sequence (IS) elements on bacterial genome diversification through analysis of small-size structural polymorphisms in Escherichia coli O157 genomes. Genome Res. 2009;19(10):1809–1816. doi: 10.1101/gr.089615.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mahillon J, Chandler M. Insertion sequences. Microbiol Mol Biol Rev. 1998;62(3):725–774. doi: 10.1128/mmbr.62.3.725-774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schneider D, Lenski RE. Dynamics of insertion sequence elements during experimental evolution of bacteria. Res Microbiol. 2004;155(5):319–327. doi: 10.1016/j.resmic.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 35.Wu Y, Aandahl RZ, Tanaka MM. Dynamics of bacterial insertion sequences: can transposition bursts help the elements persist? BMC Evol Biol. 2015;15(1):288. doi: 10.1186/s12862-015-0560-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sekine Y, Ki I, Mizuno T, Ohtsubo E. Inhibition of transpositional recombination by OrfA and OrfB proteins encoded by insertion sequence IS3. Genes Cells. 1997;2(9):547–557. doi: 10.1046/j.1365-2443.1997.1440342.x. [DOI] [PubMed] [Google Scholar]
- 37.Wagner A. Periodic extinctions of transposable elements in bacterial lineages: evidence from intragenomic variation in multiple genomes. Mol Biol Evol. 2006;23(4):723–733. doi: 10.1093/molbev/msj085. [DOI] [PubMed] [Google Scholar]
- 38.Kusumoto M, Ooka T, Nishiya Y, Ogura Y, Saito T, Sekine Y, Iwata T, Akiba M, Hayashi T. Insertion sequence-excision enhancer removes transposable elements from bacterial genomes and induces various genomic deletions. Nat Commun. 2011;2:152. doi: 10.1038/ncomms1152. [DOI] [PubMed] [Google Scholar]
- 39.Toro M, Rump L, Cao G, Meng J, Brown E, Gonzalez-Escalona N. Simultaneous presence of insertion sequence excision enhancer and insertion sequence IS629 correlates with increased diversity and virulence in Shiga toxin-producing Escherichia coli. J Clin Microbiol. 2015;53(11):3466–3473. doi: 10.1128/JCM.01349-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dziva F, van Diemen PM, Stevens MP, Smith AJ, Wallis TS. Identification of Escherichia coli O157: H7 genes influencing colonization of the bovine gastrointestinal tract using signature-tagged mutagenesis. Microbiology. 2004;150(11):3631–3645. doi: 10.1099/mic.0.27448-0. [DOI] [PubMed] [Google Scholar]
- 41.Weiss A, Kortemeier D, Brockmeyer J. Biochemical characterization of the SPATE members EspPα and EspI. Toxins. 2014;6(9):2719–2731. doi: 10.3390/toxins6092719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tarr PI, Bilge SS, Vary JC, Jelacic S, Habeeb RL, Ward TR, Baylor MR, Besser TE. Iha: a novel Escherichia coli O157: H7 adherence-conferring molecule encoded on a recently acquired chromosomal island of conserved structure. Infect Immun. 2000;68(3):1400–1407. doi: 10.1128/IAI.68.3.1400-1407.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Benz I, Schmidt MA. Cloning and expression of an adhesin (AIDA-I) involved in diffuse adherence of enteropathogenic Escherichia coli. Infect Immun. 1989;57(5):1506–1511. doi: 10.1128/iai.57.5.1506-1511.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miethke M, Marahiel MA. Siderophore-based iron acquisition and pathogen control. Microbiol Mol Biol Rev. 2007;71(3):413–451. doi: 10.1128/MMBR.00012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stern A, Sorek R. The phage-host arms race: shaping the evolution of microbes. BioEssays. 2011;33(1):43–51. doi: 10.1002/bies.201000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dziva F, Mahajan A, Cameron P, Currie C, McKendrick IJ, Wallis TS, Smith DG, Stevens MP. EspP, a Type V-secreted serine protease of enterohaemorrhagic Escherichia coli O157: H7, influences intestinal colonization of calves and adherence to bovine primary intestinal epithelial cells. FEMS Microbiol Lett. 2007;271(2):258–264. doi: 10.1111/j.1574-6968.2007.00724.x. [DOI] [PubMed] [Google Scholar]
- 47.Tatsuno I, Horie M, Abe H, Miki T, Makino K, Shinagawa H, Taguchi H, Kamiya S, Hayashi T, Sasakawa C. toxB gene on pO157 of enterohemorrhagic Escherichia coli O157: H7 is required for full epithelial cell adherence phenotype. Infect Immun. 2001;69(11):6660–6669. doi: 10.1128/IAI.69.11.6660-6669.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoon JW, Lim JY, Park YH, Hovde CJ. Involvement of the Escherichia coli O157: H7 (pO157) ecf operon and lipid A myristoyl transferase activity in bacterial survival in the bovine gastrointestinal tract and bacterial persistence in farm water troughs. Infect Immun. 2005;73(4):2367–2378. doi: 10.1128/IAI.73.4.2367-2378.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grys TE, Siegel MB, Lathem WW, Welch RA. The StcE protease contributes to intimate adherence of enterohemorrhagic Escherichia coli O157:H7 to host cells. Infect Immun. 2005;73(3):1295–1303. doi: 10.1128/IAI.73.3.1295-1303.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Doughty S, Sloan J, Bennett-Wood V, Robertson M, Robins-Browne RM, Hartland EL. Identification of a novel fimbrial gene cluster related to long polar fimbriae in locus of enterocyte effacement-negative strains of enterohemorrhagic Escherichia coli. Infect Immun. 2002;70(12):6761–6769. doi: 10.1128/IAI.70.12.6761-6769.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Low AS, Holden N, Rosser T, Roe AJ, Constantinidou C, Hobman JL, Smith DG, Low JC, Gally DL. Analysis of fimbrial gene clusters and their expression in enterohaemorrhagic Escherichia coli O157: H7. Environ Microbiol. 2006;8(6):1033–1047. doi: 10.1111/j.1462-2920.2006.00995.x. [DOI] [PubMed] [Google Scholar]
- 52.Stevens MP, Roe AJ, Vlisidou I, Van Diemen PM, La Ragione RM, Best A, Woodward MJ, Gally DL, Wallis TS. Mutation of toxB and a truncated version of the efa-1 gene in Escherichia coli O157: H7 influences the expression and secretion of locus of enterocyte effacement-encoded proteins but not intestinal colonization in calves or sheep. Infect Immun. 2004;72(9):5402–5411. doi: 10.1128/IAI.72.9.5402-5411.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wells TJ, Sherlock O, Rivas L, Mahajan A, Beatson SA, Torpdahl M, Webb RI, Allsopp LP, Gobius KS, Gally DL. EhaA is a novel autotransporter protein of enterohemorrhagic Escherichia coli O157: H7 that contributes to adhesion and biofilm formation. Environ Microbiol. 2008;10(3):589–604. doi: 10.1111/j.1462-2920.2007.01479.x. [DOI] [PubMed] [Google Scholar]
- 54.Laarmann S, Schmidt MA. The Escherichia coli AIDA autotransporter adhesin recognizes an integral membrane glycoprotein as receptor. Microbiology. 2003;149(7):1871–1882. doi: 10.1099/mic.0.26264-0. [DOI] [PubMed] [Google Scholar]
- 55.Barondess JJ, Beckwith J. bor gene of phage lambda, involved in serum resistance, encodes a widely conserved outer membrane lipoprotein. J Bacteriol. 1995;177(5):1247–1253. doi: 10.1128/jb.177.5.1247-1253.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De Groote MA, Ochsner UA, Shiloh MU, Nathan C, McCord JM, Dinauer MC, Libby SJ, Vazquez-Torres A, Xu Y, Fang FC. Periplasmic superoxide dismutase protects Salmonella from products of phagocyte NADPH-oxidase and nitric oxide synthase. Proc Natl Acad Sci. 1997;94(25):13997–14001. doi: 10.1073/pnas.94.25.13997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Castanie-Cornet M-P, Penfound TA, Smith D, Elliott JF, Foster JW. Control of acid resistance in Escherichia coli. J Bacteriol. 1999;181(11):3525–3535. doi: 10.1128/jb.181.11.3525-3535.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Uhlich GA. KatP contributes to OxyR-regulated hydrogen peroxide resistance in Escherichia coli serotype O157: H7. Microbiology. 2009;155(11):3589–3598. doi: 10.1099/mic.0.031435-0. [DOI] [PubMed] [Google Scholar]
- 59.Nishio M, Okada N, Miki T, Haneda T, Danbara H. Identification of the outer-membrane protein PagC required for the serum resistance phenotype in Salmonella enterica serovar Choleraesuis. Microbiology. 2005;151(3):863–873. doi: 10.1099/mic.0.27654-0. [DOI] [PubMed] [Google Scholar]
- 60.Kamada N, Chen GY, Inohara N, Núñez G. Control of pathogens and pathobionts by the gut microbiota. Nat Immunol. 2013;14(7):685–690. doi: 10.1038/ni.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rendón MA, Saldaña Z, Erdem AL, Monteiro-Neto V, Vázquez A, Kaper JB, Puente JL, Girón JA. Commensal and pathogenic Escherichia coli use a common pilus adherence factor for epithelial cell colonization. Proc Natl Acad Sci. 2007;104(25):10637–10642. doi: 10.1073/pnas.0704104104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.La Ragione RM, Best A, Woodward MJ, Wales AD. Escherichia coli O157: H7 colonization in small domestic ruminants. FEMS Microbiol Rev. 2009;33(2):394–410. doi: 10.1111/j.1574-6976.2008.00138.x. [DOI] [PubMed] [Google Scholar]
- 63.Naylor SW, Low JC, Besser TE, Mahajan A, Gunn GJ, Pearce MC, McKendrick IJ, Smith DG, Gally DL. Lymphoid follicle-dense mucosa at the terminal rectum is the principal site of colonization of enterohemorrhagic Escherichia coli O157: H7 in the bovine host. Infect Immun. 2003;71(3):1505–1512. doi: 10.1128/IAI.71.3.1505-1512.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fitzhenry R, Pickard D, Hartland E, Reece S, Dougan G, Phillips A, Frankel G. Intimin type influences the site of human intestinal mucosal colonisation by enterohaemorrhagic Escherichia coli O157: H7. Gut. 2002;50(2):180–185. doi: 10.1136/gut.50.2.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Girard F, Dziva F, van Diemen P, Phillips AD, Stevens MP, Frankel G. Adherence of enterohemorrhagic Escherichia coli O157, O26, and O111 strains to bovine intestinal explants ex vivo. Appl Environ Microbiol. 2007;73(9):3084–3090. doi: 10.1128/AEM.02893-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van Diemen PM, Dziva F, Stevens MP, Wallis TS. Identification of enterohemorrhagic Escherichia coli O26: H− genes required for intestinal colonization in calves. Infect Immun. 2005;73(3):1735–1743. doi: 10.1128/IAI.73.3.1735-1743.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kotewicz ML, Mammel MK, LeClerc JE, Cebula TA. Optical mapping and 454 sequencing of Escherichia coli O157: H7 isolates linked to the US 2006 spinach-associated outbreak. Microbiology. 2008;154(11):3518–3528. doi: 10.1099/mic.0.2008/019026-0. [DOI] [PubMed] [Google Scholar]
- 68.Latreille P, Norton S, Goldman BS, Henkhaus J, Miller N, Barbazuk B, Bode HB, Darby C, Du Z, Forst S. Optical mapping as a routine tool for bacterial genome sequence finishing. BMC Genomics. 2007;8(1):321. doi: 10.1186/1471-2164-8-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Alikhan N-F, Petty NK, Zakour NLB, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12(1):1. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Toro M, Retamal P, Ayers S, Barreto M, Allard M, Brown EW, Gonzalez-Escalona N. Whole genome sequencing analysis of Salmonella Enteritidis isolated in Chile provides insights about possible transmission between gulls, poultry and humans. Appl Environ Microbiol. 2016:AEM. 01760–01716. [DOI] [PMC free article] [PubMed]
- 71.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4(4):406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 73.Rzhetsky A, Nei M. A simple method for estimating and testing minimum-evolution trees. Mol Biol Evol. 1992;9(5):945–967. [Google Scholar]
- 74.Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16(2):111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 75.Wirth T, Falush D, Lan R, Colles F, Mensa P, Wieler LH, Karch H, Reeves PR, Maiden MC, Ochman H. Sex and virulence in Escherichia coli: an evolutionary perspective. Mol Microbiol. 2006;60(5):1136–1151. doi: 10.1111/j.1365-2958.2006.05172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gonzalez-Escalona N, Toro M, Rump LV, Cao G, Nagaraja T, Meng J. Virulence gene profiles and clonal relationships of Escherichia coli O26: H11 isolates from feedlot cattle by whole genome sequencing. App Environ Microbiol. 2016:AEM. 00498–00416. [DOI] [PMC free article] [PubMed]
- 77.Joensen KG, Tetzschner AM, Iguchi A, Aarestrup FM, Scheutz F. Rapid and easy in silico serotyping of Escherichia coli using whole genome sequencing (WGS) data. J Clin Microbiol. 2015:JCM. 00008–00015. [DOI] [PMC free article] [PubMed]
- 78.Joensen KG, Scheutz F, Lund O, Hasman H, Kaas RS, Nielsen EM, Aarestrup FM. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J Clin Microbiol. 2014;52(5):1501–1510. doi: 10.1128/JCM.03617-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, Aarestrup FM, Larsen MV. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67(11):2640–2644. doi: 10.1093/jac/dks261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, Wishart DS. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016:gkw387. [DOI] [PMC free article] [PubMed]
- 81.Dhillon BK, Laird MR, Shay JA, Winsor GL, Lo R, Nizam F, Pereira SK, Waglechner N, McArthur AG, Langille MG. IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res. 2015:gkv401. [DOI] [PMC free article] [PubMed]
- 82.Siguier P, Pérochon J, Lestrade L, Mahillon J, Chandler M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006;34(suppl 1):D32–D36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Metadata and accession numbers of the 69 E. coli strains used for phylogenetic analysis. (XLSX 17 kb)
Dataset S1. SNP’s and SNP positions identified in the 1371 gene loci shared among the 69 E. coli genomes. Dataset S2. cgMLST for each of the 1371 identified gene loci shared among the 69 E. coli genomes. (XLSX 23678 kb)
Genomic dissimilarities between O145:H25 and other EHEC genomes. Table S2. Conservation of the EDL933-specific OIs in O145:H25 and the other EHEC strains. Table S3. Fimbrial gene cluster identified in the nine EHEC genomes, including nucleotide sequence identity. (XLSX 65 kb)
In silico MLST analysis of the nine EHEC strains. Table S2. Identified prophages and IEs in O145:H25. Table S3. IS elements present on EHEC chromosomes. Table S4. IS elements present on EHEC plasmids. Table S5. LEE- and non-LEE-encoded effectors present on EHEC chromosomes. (DOCX 59 kb)
Data Availability Statement
All data generated or analyzed during this study are included in this published article [and its supplementary information files].