

Abstract
The increasing potency of therapies that target the androgen receptor (AR) signalling axis has correlated with a rise in the proportion of patients with prostate cancer harbouring an adaptive phenotype, termed treatment-induced lineage crisis. This phenotype is characterized by features that include soft-tissue metastasis and/or resistance to standard anticancer therapies. Potent anticancer treatments might force cancer cells to evolve and develop alternative cell lineages that are resistant to primary therapies, a mechanism similar to the generation of multidrug-resistant microorganisms after continued antibiotic use. Herein, we assess the hypothesis that treatment-adapted phenotypes harbour reduced AR expression and/or activity, and acquire compensatory strategies for cell survival. We highlight the striking similarities between castration-resistant prostate cancer and triple-negative breast cancer, another poorly differentiated endocrine malignancy. Alternative treatment paradigms are needed to avoid therapy-induced resistance. Herein, we present a new clinical trial strategy designed to evaluate the potential of rapid drug cycling as an approach to delay the onset of resistance and treatment-induced lineage crisis in patients with metastatic castration-resistant prostate cancer.
Potent clinical suppression of androgen receptor (AR) signalling has been achieved with the pharmacological inhibitors abiraterone acetate1,2 and enzalutamide3,4 (drugs that were approved by the FDA in 2011 and 2014, respectively), resulting in significant survival benefits for men with metastatic castration-resistant prostate cancer (mCRPC). Emerging evidence suggests, however, that the prolonged therapeutic use of abiraterone and enzalutamide induces adaptive clinical phenotypes — including histological dedifferentiation and lineage alterations, such as treatment-induced neuroendocrine prostate cancer (t-NEPC)5 and treatment- induced epithelial-to-mesenchymal transition6,7 (t-EMT) (BOX 1). Such resistant phenotypes, in turn, might cause aggressive visceral metastases, a trend that has been reported with increasing prevalence in patients with prostate cancer who have received long-term androgen deprivation therapy (ADT)5,8–12. While the mechanisms by which treatment- adaptive pathologies arise are currently unclear, low levels of AR expression or activation (AR-lo) and low levels of prostate-specific antigen (PSA) secretion (PSA-lo) are two of the hallmarks of poorly differentiated and aggressive prostate cancer8,10,13–18. Herein, we propose the hypothesis that AR suppression by potent therapies facilitates a selective pressure on prostate cancer cells, whereby cells of a dedifferentiated and/or treatment- resistant lineage obtain a survival or proliferative advantage. The most well-known clinical and histopathological entity is probably neuroendocrine prostate cancer or prostate small-cell carcinoma. Similar to triple-negative breast cancer (TNBC), in which oestrogen receptor (ER) and progesterone receptor (PR) levels are low and HER2 is not amplified19, AR-lo-prostate tumours can acquire enhanced cellular plasticity (elevated stemness) that results in aggressive clinical features5,20. Such cancers exploit a variety of hyperactivated alternative oncogenic signalling mechanisms, thus warranting the consideration of novel treatment approaches for patients who have developed resistance. The ability of tumours to adapt to potent targeted therapies is analogous to the mechanism whereby infectious microorganisms become resistant to consecutive courses of antibiotic treatment, ultimately obtaining the status of a ‘superbug’ (REFS 21,22) — capable of growth in the presence of multiple types of antimicrobial agents. The precise mechanisms governing how targeted agents induce cellular and genetic plasticity are in the early stages of investigation, but the clinical observations resulting from the use of such targeted therapies suggest that lineage plasticity is a clinically relevant mechanism5,9.
Box 1. Neuroendocrine cells in prostate cancer.
Neuroendocrine tumours constitute a heterogeneous population for which classification is organ-dependent — even if they share common features, such as the expression or secretion of bioactive peptides. In the normal prostate, neuroendocrine cells are found at a low frequency and secrete neuropeptides and growth factors that support the structure and function of the neighbouring prostatic epithelium. By secreting such factors, either in an autocrine or paracrine manner, neuroendocrine cells can also enhance cancer growth arising from the prostatic epithelium to reduce sensitivity to androgen receptor (AR) targeted therapies.
Neuroendocrine cells can become cancerous, usually in the form of a de novo tumour occurring at very low frequencies (0.1% of diagnosed prostate cancers). Neuroendocrine prostate tumours are characterized as highly aggressive and metastatic, with low or negative AR signalling. Such tumours are typically lethal within 2 years of diagnosis.
In recent years, the number of patients presenting with visceral, bulky metastasis and having poor survival outcomes has substantially increased (about 25% of lethal prostate cancers). The prevalence of these cancers, and its correlation with the increased use of potent AR-targeted therapies5, have led to the designation of ‘treatment-related neuroendocrine prostate cancers’ (t-NEPCs)5. While not entirely understood, two general hypotheses address how t-NEPCs arise, which include the oncogenic transformation of normal neuroendocrine cells, and the transdifferentiation of adenocarcinomas to the neuroendocrine lineage owing to a series of genetic and epigenetic alterations. The sustained expression of markers known to be present in AR-positive adenocarcinoma (such as TMPRSS2-ERG) through the conversion to neuroendocrine phenotypes strongly supports an epithelial origin for most t-NEPCs.
Regardless of the mechanism of origin, patients with t-NEPCs are typically prescribed platinum-based chemotherapy as a consequence of the aggressive nature and poor response of these tumours to AR-targeted therapies. To reduce progression of treatment-induced neuroendocrine lineage crisis, cancer researchers should attempt to understand the genetic drivers of NEPC, including critical neuronal genes involved in lineage alterations, and those involved in the subsequent rapid, clonal expansion to a small cell-like cancer phenotype.
Increased research emphasis should be placed on the development of preclinical models, in which the effects of treatment-induced resistance can be tested directly, as well as on the development of alternative treatment strategies that could be used to prolong the survival of men with advanced-stage prostate cancer by delaying the onset of resistance and minimizing toxicities. We suggest a new treatment approach, the Prostate Cancer Intensive, Non-Cross Reactive Therapy protocol (PRINT), which is designed to explore the effectiveness of rapid cycling of drugs with different antitumour mechanisms in men with mCRPC, and has been applied to the design of a new clinical trial (NCT02903160)23. This ongoing trial could provide proof-of-principle for future treatment regimen approaches, such as collateral sensitivity, which are designed to delay the point at which prostate cancer cells become treatment-resistant. PRINT might also provide evidence that, with new treatment approaches, attention should be devoted not only to the use of novel drugs, but also to how and when such drugs are administered.
Treatment-induced resistance in mCRPC
The development of treatment resistance in cancer (for example, in mCRPC) can be paralleled with the development of antibiotic resistance in bacteria. Treatment-naive infections generally respond to a standard course of antibiotics, resulting in a reduced disease burden and recovery. Within the initial population, however, a small percentage of microorganisms have resistance mechanisms that are either acquired through genetic mutation or transferred from other bacteria24. Upon discontinued antibiotic use (or ‘drug holiday’), the infection can recur owing to expansion of the population of resistant clones. Subsequent treatment with a different antibiotic might then leads to the evolution of a strain with resistance to both antibiotics (FIG. 1a). Thus, while persistent antibiotic use often achieves initial clinical success, the generation of multidrug-resistant bacteria (also referred to as super-bugs) might be the ultimate result of persistent use of single agents21,22. Once drug exposure is discontinued, superbugs might then revert back to being drug-sensitive as a mechanism to avoid the fitness hurdles associated with resistance25 (FIG. 1a). Indeed, striking similarities exist between the ability of infectious organisms and cancer cells with dedifferentiated phenotypes to persist and adapt, despite the presence of potent therapeutic agents. Similarly to how superbugs are related to infectious disease, cancer cells are highly adaptable and can adapt phenotypically to survive under continuous drug exposures, ultimately leaving patients with few or no treatment options. While infectious diseases and most cancers operate on a different scale of progression and cannot be compared directly, the mechanisms of resistance rapidly outpace the development of effective drugs and treatment strategies in both situations.
Figure 1. Treatment-induced resistance and evolution to lineage crisis.
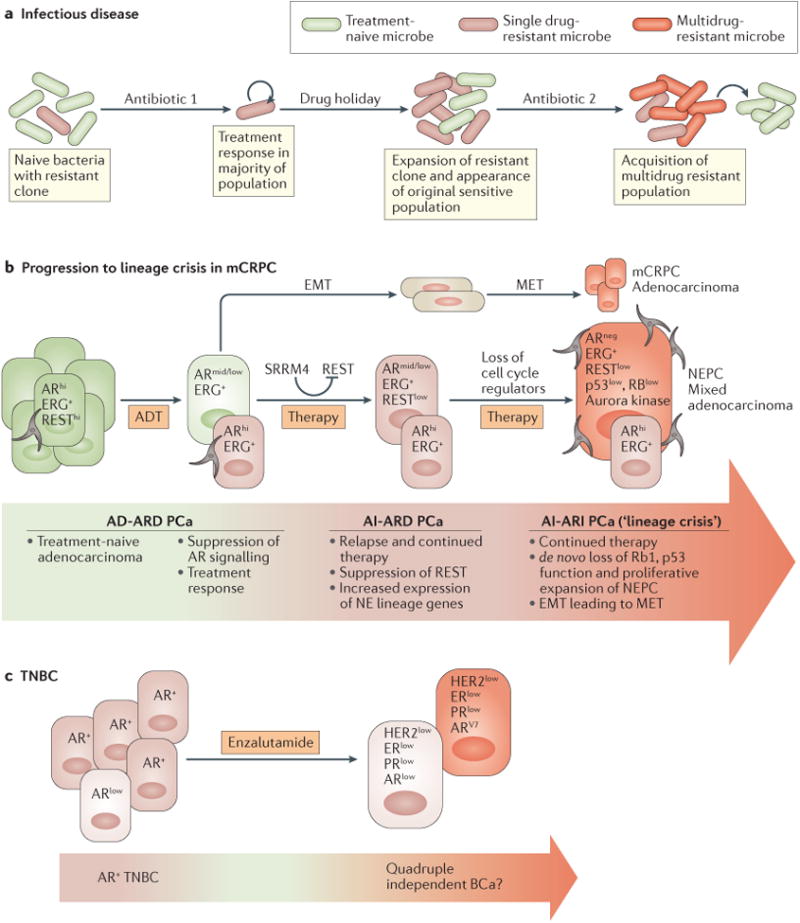
a | Treatment-naive bacterial populations are composed largely of antibiotic-sensitive subpopulations, but might contain small number of mutant bacteria harbouring genetically acquired mechanisms of resistance (left). Upon antibiotic treatment, widespread death typically occurs; however, upon drug removal or during a ‘drug holiday’, resistant bacterial clones expand, potentially taking over both sensitive and resistant populations. Additional antibiotic treatments can lead to further genetic alterations and to the selection of a population of multidrug resistant microorganisms, which would then have the option to increase fitness by becoming a drug-sensitive population. b | Treatment-naive prostate cancers are composed mostly of cells with high levels of androgen receptor (AR-hi) activity (left) for which radiation and/or androgen deprivation therapy (ADT) typically result in a pronounced treatment response that includes primary tumour regression and reduced prostate-specific antigen (PSA) secretion. Primary or metastatic foci can recur, often leading to expansion of the AR-hi tumour bulk and elevated serum PSA levels. Follow-up treatment can include androgen receptor (AR)-targeted therapies or chemotherapies that would drive cells to adapt by dedifferentiation, thus facilitating an increase in neuroendocrine (NE) gene expression. Continued ADT and taxane-based treatment ultimately selects populations that have undergone de novo loss-of-function mutations in key cell-cycle regulators. Ultimately, the accumulation of these genetic and epigenetic events leads to a clinically detectable lineage crisis. c | Triple-negative breast cancer (TNBC) is an undifferentiated disease characterized by low expression of oestrogen receptor (ER), progesterone receptor (PR), and an absence of HER2 amplification. BCa, breast cancer; EMT, epithelial-to-mesenchymal transition; mCRPC, metastatic castration-resistant prostate cancer; MET, mesenchymal-to-epithelial transition; NEPC, neuroendocrine prostate cancer; PCa, prostate cancer; REST, RE-1 silencing transcription factor; SRRM4, serine/arginine repetitive matrix protein 4.
Lineage crisis in mCRPC
Documenting the stage at which cancer treatments might induce adaptive phenotypes (or ‘lineage crisis’) is a clinical challenge, particularly when considering that such alterations might even occur in treatment-naive patients. In the clinical setting, the term lineage crisis can be applied to anaplastic or small-cell prostate carcinomas. This suggestion is supported by a report in which Aparicio et al.8 described that the presence of at least one of the following seven prospectively assessed criteria defines anaplastic prostate cancer: presence of a pure or mixed population of histologically defined small-cell prostate carcinoma cells; metastasis exclusively to visceral organs; a predominance of lytic-bone metastases (assessed radiographically); ‘bulky’ (≥5 cm) tumour masses; low serum PSA levels concurrent with a high number (>20) of bone metastases; the presence of neuroendocrine markers (histology assessment or in serum) plus elevated levels of serum lactate dehydro-genase (LDH) or carcinoembryonic antigen (CEA), or malignant hypercalcaemia; or a short time (≤6 months) to androgen-independent disease progression after starting ADT. These criteria were devised, in part, to guide the decision to perform additional biopsies when lineage crisis is suspected10,26. Beyond these criteria, the use of techniques such as genomic and/or transcriptional profiling of AR-related signatures and lineage markers directly from primary tumours or lesions, and urine sampling, have enabled the clinical documentation of lineage crisis27. Increasingly sensitive assays might also enable screening for the presence of neuroendocrine or EMT-like cells in blood samples from patients28.
For clinical oncologists to understand when and where lineage crisis is occurring, and how this progression can be halted, the mechanisms used by cancer cells to respond to the stress induced by the presence of persistent and potent therapies must be understood. We agree with the fundamental hypothesis that prostate cancer is a heterogeneous disease capable of drug-induced plasticity mediated by reduced AR expression or activity29–31. Most likely, this heterogeneity and adaptive lineage plasticity also accounts for disease states characterized by multidrug resistance, aggressive phenotypes and lethality (FIG. 1b). In the normal prostate, the AR has important roles in the initiation and maintenance of epithelial differentiation. Data from genetic studies in mice have shown that AR activation induces the secretion of critical paracrine signalling molecules (referred to as ‘andromedins’) involved in the maintenance of acinar differentiation and secretory function29–33. During cancer progression, however, the functional roles of the AR expand to control cellular differentiation, oncogenesis and tumour suppression34. Baseline and post-treatment changes in serum levels of PSA serve as an important biomarker of prognosis and tumour progression, but do not fully capture the varied survival outcomes of patients with mCRPC17,26,35,36. Indeed, NEPC is often characterized by the secretion of low levels of PSA and a poorly differentiated phenotype9. Most NEPCs typically have very low or no AR expression, and are thus poorly responsive to therapy and associated with unfavourable clinical outcomes37. The levels of AR expression and signalling also tend to be very low in visceral metastases compared with primary tumours17,38,39. In fact, the subset of patients with prostate cancer with the lowest levels of PSA (≤10 ng/ml reported)8 undergo the most- aggressive disease course, characterized by highly dedifferentiated tumour pathology14,16,40. We propose that dedifferentiated prostate tumours with low AR activity might represent a transition stage with a high susceptibility to lineage crisis similar to other undifferentiated malignancies, such as TNBC or undifferentiated acute myeloid leukaemia (AML). These clinical data8 suggest that, in certain contexts, differentiation markers (such as the AR) function as checkpoints in the progression to the most-aggressive forms of disease. Several compelling preclinical studies support this hypothesis. For example, upon reconstitution of AR expression, AR-negative, poorly differentiated human prostate cancer cells display more-differentiated phenotypes in culture41–44 and reduced migration45,46.
Treatment-induced prostate lineages
During hormone-dependent disease progression, the prostatic epithelium responds acutely to inhibition of AR signalling, resulting in tumour regression and lower PSA levels in serum. For example, RNA knockdown of AR expression in the human prostate cancer cell line LNCaP impedes ligand-independent AR activation and delays the progression of tumours subcutaneously induced in mice47. Conversely, transcriptomic profiling and tumour assessment in xenograft models of AR-lo LNCaP subpopulations isolated from the parental line revealed substantially increased elements of stemness, tumorigenesis and treatment resistance compared with either parental cells or subpopulations with high levels of AR expression (AR-hi)48. These data indicate that, although AR-targeted therapies can be effective anti-tumour strategies, they might ultimately lead not only to the expansion of adapted AR-hi cancer cells, but also to the expansion of AR-lo subpopulations with dedifferentiated phenotypes (FIG. 1b). Perhaps the strongest clinical evidence for a treatment-induced adaptive response is derived from measuring the presence of small-cell prostate carcinoma-related signatures before and after treatment9,18. Strikingly, some studies have reported a marked difference in the percentage of cells harbouring such signatures, ranging from 0.5–2% in patients with untreated prostate cancer to 25% in metastatic autopsy samples of lethal cancers of a prostatic origin49,50. Some analyses, however, have been restricted to a small number of matched samples from the same patient limiting the appreciation of intra-individual heterogeneity of disease50. In a study published in 2015, only 3% of patients with mCRPC (n = 150) had AR-negative tumours; however, 96% of the samples processed for this analysis were differentiated adenocarcinomas51. The results of analyses of the genomic landscapes of primary and metastatic prostate cancer samples published in the past 10 years show that, while a strong AR-expression signature is observed in prostate cancer samples from untreated patients, those from patients who received ADT have heterogeneous AR activity, particularly in metastatic lesions52–55. These clinical data might be in line with preclinical studies on AR-hi cells showing cross- resistance between AR-targeted therapies and docetaxel, through a mechanism involving nuclear AR transport56,57, even if clinical evidence on this mechanism remains limited58–60. Collectively, these analyses indicate that, during progression from hormone- naive to castration-resistant prostate cancer, overall AR activity often decreases52,54.
A challenge for the clinical detection of treatment-naive (occurring in de novo NEPC) and treatment-induced lineage alterations in prostate cancer comes from the marked overlap in results from the morphological and immunohistochemical profiling of many NEPCs. This difficulty is aggravated by the fact that visceral metastatic lesions of prostatic origin often have reduced or focal expression of markers of prostatic epithelial differentiation (such as PSA, glutamate car-boxipeptidase 2 (commonly referred to as PSMA) or solute carrier family 45 member 3 (commonly referred to as P501S))61,62. The use of high-resolution fluorescent in situ hybridization analysis has enabled the detection of persistent rearrangements of the gene encoding the transcriptional regulator ERG in visceral lesions of small-cell prostate cancers63, in treatment-resistant NEPCs64 or in concurrent adenocarcinomas and small-cell prostate cancers63,65–67. Thus, detection of ERG protein over-expression and/or TMPRSS2–ERG rearrangement in NEPCs provides strong evidence for lineage conversion from an initial AR-positive adenocarcinoma to NEPC. The detection of persistent ERG rearrangements and enrichment in ERG-related transcriptional signatures can also enable the evaluation of treatment-related lineage crisis subsequent to primary tumour resections in primary tumours, metastatic lesions, or even in urine samples in clinical settings27.
EMT is an additional form of lineage crisis. EMT phenotypes have been associated with reduced expression of AR in studies conducted both in mouse models68–70, and with human-derived samples6, results that further support the suggestion that AR-lo mCRPC-derived cells might represent transition states from which to acquire alternative therapy-induced lineages. Preclinical modelling has demonstrated that ADT induces both EMT and enhanced stemness in the mouse prostate and in xenograft tumours derived from the human cell line LuCaP35 (REF. 6). The results of animal studies, however, show that the combination of enzalutamide and cabazitaxel lead to the reversion of EMT-like phenotypes in EMT-driven prostate cancer models, to luminal and glandular cell-like differentiation7. Analysis of patient-derived tissue grafts isolated from those patients with high-risk primary prostate cancers indicate that ADT leads to increased expression of EMT markers in some, but not all patients71. In addition, a reciprocal negative feedback loop has been postulated to lead to reduced AR expression in cells that have undergone EMT6. In cultured cells, elevated stemness and reduced AR expression might be features of a transition state that is required while undergoing conversion to a neuroendocrine lineage (FIG. 1b). One of the challenges in determining the effects that primary and subsequent treatments have on the regulation of EMT-related gene signatures is to determine whether castration-resistant adenocarcinoma cells undergo trans-differentiation to an EMT lineage or whether pre-existing cells, such as an accumulation of t-NEPCs, are selected during treatment and subsequently acquire genetic and epigenetic modifications. Of note, the reverse process of mesenchymal-to-epithelial transition (MET) is poorly understood. Despite considerable evidence documenting the accumulation of cells in lineage crisis6,9,49,50,71 following first-line and subsequent treatments, the debate on the mechanisms responsible for such lineage crisis is still ongoing.
Mechanisms of lineage conversion
The accumulation of prostate cancer cells in NEPC lineage crisis is postulated to occur through three processes: first, lineage conversion (or transdifferentiation) of adenocarcinoma cells9; second, clonal selection72 and expansion of adenocarcinoma cells; and third, transformation of non-transformed or benign neuroendocrine cells20. Owing to the pronounced genetic similarities between adenocarcinoma and NEPC cells, the processes of transdifferentiation and/or clonal expansion are generally preferred, and have been addressed by several studies65–67,73,74. The ability of adenocarcinoma cells to originate as a neuroendocrine lineage implies that these cells have sufficient stem-like properties and an ability to proliferate in AR suppressive-conditions (FIG. 1b). Indeed, some investigators have suggested that the co-expression of CD44, a putative marker of stemness in the prostate75, with neuroendocrine lineage markers76 in clinical biopsy samples from patients with small-cell prostate carcinoma implies a stem-like cellular origin76. Preclinical studies by Owen Witte's group revealed that overexpression of N-Myc and Akt in benign, primary human basal cells led to NEPC and mixed adenocarcinoma formation upon transplantation in mice, compared with similarly transduced primary luminal cells77. These data, however, were not sufficient to determine whether luminal stem cells naturally lack the capacity to become NEPC cells or whether the conclusions of this study resulted from the technical limitations of the applied grafting assays78,79. Nevertheless, these data are consistent with the results of two consecutive analyses of samples from patients with prostate cancer, in which the genes encoding N-Myc and Aurora kinase A were found to be co-amplified in 40% of histologically defined NEPC and in ∼75% of patients with prostate cancer at different stages, who will develop clinically defined t-NEPCs, in contrast with only 5% co- amplification detected in patients with non-NEPC prostate cancer or in an unselected population64,80.
Almost all (>90%) NEPCs have high levels of genomic alterations, including loss of PTEN and mutations affecting RB1 and TP53 (REFS 74,81,82). These studies also indicate that small-cell prostate carcinomas harbour TP53 loss and CREBBP mutations concurrently with ERG rearrangements, indicating they are derived from a cell of prostatic origin expressing the AR74. NEPC cells are certainly present in untreated tumours and seldom undergo rapid proliferative expansion80,83; however, cancers from patients receiving extensive treatment are much more likely to undergo a stepwise process involving genetic and epigenetic modifications, ultimately becoming clonally derived t-NEPCs, than those from untreated patients5. The identification of de novo mutations in NEPCs74,82 (that is, those not detected in normal neuroendocrine cells) arising after treatment further supports the existence of the aforementioned stepwise process, which would have originated in either a clonal or polyclonal AR-hi and ERG-positive population of adenocarcinomas (FIG. 1b).
The process of transdifferentiation suggests the existence of certain cell populations that can convert or reprogram to a neuroendocrine lineage when suppression of AR signalling occurs, with the use of potent AR-targeting agents. Data are available indicating that androgen ablation or AR-targeted therapy can promote the rapid conversion of LNCaP cells to a neuroendocrine lineage in vitro84–86 and the rapid conversion of adenocarcinomas to NEPC in vivo87. Data from preclinical studies show that, in order to convert to a neuroendocrine lineage, epithelial cells likely require stemness determinants, such as CD44, and the presence of alterations in lineage- specific transcription factors. In non-neuronal cell lineages, the RE-1 silencing transcription factor (REST) normally binds to RE-1 sites in the promoters of neuronal genes to maintain suppression of these genes88. Conversely, in NEPC cells this repressive transcriptional function is lost, thereby allowing neuronal gene expression and subsequent epithelial-to-neuronal transdifferentiation88–91. Using whole transcriptome data obtained from prostate adenocarcinoma samples, t-NEPC samples and xenografts derived from treated patients, the loss of REST was shown to correlate with conversion to a NEPC-like signature90,91. Future in vivo and clinical studies will be required to determine whether the loss of REST function is sufficient for the onset of a NEPC transcriptional programme, and whether any genetic alteration conferring a proliferative advantage contributes to the maintenance of such transcriptional signatures.
The alterations in AR gene signatures associated with NEPC, EMT and resistance to AR-targeted therapies strongly supports the idea that inhibition of AR signalling, and the subsequent selection for cellular populations with intermediate or low AR levels, is a requirement for treatment-induced lineage switch. Indeed, our hypothesis for the existence of a ‘checkpoint’ phenotype is in agreement with the results of a large-scale (n = 226) analysis of biopsy samples from patients with mCRPC obtained before and after treatment with enzalutamide or abiraterone. This clinical analysis not only showed that lethal treatment-induced NEPC is more common than previously appreciated, but also led to the identification of an intermediate histology phenotype (termed IAC phenotype) and distinct from small-cell carcinoma or adenocarcinoma18,92 (E. J. Small, personal communication). Consistent with these data, whole-exome sequencing of biopsy samples from patients with mCRPC revealed that a AR-lo status could be common to the clonal evolution of both prostate adenocarcinoma and NEPC, and that this evolutionary process might, in part, be epigenetically regulated15. Future preclinical studies should address whether the accumulation of a t-NEPC phenotype occurs through single or multiple processes, as well as the sequence of genetic and/or epigenetic changes that occur in the transition between AR-hi adenocarcinoma, IAC and AR-negative lineages, in order to further understand the aetiology of treatment-related lineage crisis.
Adaptive mechanisms of progression
The progressive loss of AR activity during progression of certain prostate cancers has prompted investigators to examine oncogenic signalling pathways providing compensatory prosurvival cues despite the pressure of potent antiproliferative agents or ADT. One such oncogene is the tyrosine kinase Src, which is a target of dasatinib93. The pathology assessment of prostates from Pten-null, Ar-null mice revealed enhanced cellular stemness, reduced differentiation and increased levels of activated Src compared with mice with Pten deletion only (D. J. Mulholland, unpublished work). Interestingly, the predictive signatures for the efficacy of dasatinib correlate well with CRPCs that are low or negative for AR activity52, an observation which parallels the increased Src expression and activity in breast cancers with low or negative ER expression94. Together, these observations suggest that dasatinib might be an effective treatment for patients with prostate tumours expressing low levels of AR signalling. The antitumour activity of dasatinib has been assessed in several phase II clinical trials of patients with mCRPC, showing promising results (in terms of objective response and disease control rates95,96), as well as in phase II97 and phase III98 randomized clinical trials. These controlled trials, however, did not meet their objectives of improved progression-free survival (PFS) and overall survival, potentially because patients were not stratified according to AR activity. Others have described an association between decreased AR activity and increased expression of Src kinase52,99,100 and sensitivity to dasatinib52 and thus, the evaluation of the levels of both proteins in patients before starting therapies with Src inhibitors is recommended. Preclinical studies have identified gene signatures that predict a response to dasatinib99, which can be evaluated using formalin-fixed paraffin-embedded tissue samples from patients74,101. Thus, given the enhancement of Src levels in AR-lo patients, clinical trials designed to stratify patients according to AR expression (AR-hi or AR-lo) might be an appropriate strategy to increase the predictive response to dasatinib and other similar tyro-sine kinase inhibitors. Such stratification processes would also enable patients with prostate cancer with low, or no AR expression to avoid receiving AR-targeted therapies, which have limited potential for success in these patients.
Oncogenic PI3K/Akt signalling can also provide compensatory proliferative cues in cells in the presence of AR inhibition. Results from our group102 and others103 demonstrated that AR signalling is not required for disease progression in prostate cancer cells of epithelial origin with no detectable PTEN expression or with activated PI3K/Akt signalling. Using genetic and pharmacological inhibition of either the AR or PI3K/Akt/mTOR signalling, reciprocal activation of these pathways was demonstrated102,103. Consistent with these results, subsequent studies have shown that treatment with enzalutamide can accelerate the progression of prostate cancer in PTEN-deficient mice104, and that the combined inhibition of both signalling axes had a synergistic antitumour effect in enzalutamide-resistant mouse models105. These results suggest that increased emphasis should be placed on understanding the oncogenic drivers of AR-lo versus AR-hi prostate cancer. Such studies should ideally be initiated in preclinical humanized animal models, designed to reproduce an AR-lo or AR-negative phenotype in an oncogenic signalling background, that are capable of growth in the absence of AR activation. These investigations would subsequently facilitate clinical stratification of patients to receive the most-appropriate targeted therapies.
Beyond receptor-tyrosine-kinase signalling, studies have begun to focus on the role of compensatory hormone nuclear receptor function, mostly in the context of enzalutamide resistance. The transcriptomic profile of the glucocorticoid receptor (GR) considerably overlaps with that of the AR, and has been implicated in mediating cell viability in mCRPC106. These observations suggest that a nuclear receptor ‘switch’ exists whereby drug-induced resistance promotes the transfer of proliferative signals to closely-related signalling pathways. Corticosteroids can provide palliative benefits, such as relief from pain and fatigue, as well as biochemical responses in patients with prostate cancer107, but data from a growing number of studies have revealed that the GR is overexpressed in PTEN-deficient and/or AR-deficient preclinical models106,108–110. The effect of corticosteroids on the outcomes of patients with mCRPC remain controversial111–113. Interestingly, data presented in 2015 also suggest that, in patients, GR overexpression could occur at the initial stages of cancer, thereby contributing to disease progression114. Despite this evidence, the efficacy of pharmacological GR inhibitors (such as mifepristone) in clinical settings remains unclear115, as their use might lead to potential complications related to low GR-binding specificity and even aberrant activation of AR signalling106,115,116. Such issues might be clarified when the results of an ongoing phase I/II clinical trial assessing the efficacy of enzalutamide–mifepristone combination treatments are reported (NCT02012296)117, as well as with the development of more-selective inhibitors of GR than mifepristone. Parallel increases in PR and ERα expression strongly support a role for enhanced epithelial ERα signalling in mCRPC progression118. ERβ is highly expressed in treatment-naive prostate cancers, including nodal and distant metastases, but is markedly reduced in about 40% of patients with mCRPC119. Similar to epithelial AR expression, a study published in 2015 suggests that epithelial ERβ can assume a tumour suppressive function in PTEN-deficient prostate cancer120. In the prostates of PTEN-deficient mice, epithelial deficiency of ERβ results in positive feedback signalling to promote disease progression and activation of BMI — a stemness factor implicated in prostate cancer initiation121. In clinical scoring, loss of ERβ expression during cancer progression is associated with increased polycomb complex protein BMI-1 expression122 and activation of EMT123. Interestingly, ERβ has also been reported to be a tumour suppressor in breast cancer124, whereas ERα seems to act differently125. Thus, while GR and ERα assume potentially oncogenic roles, ERβ and the AR function as gatekeepers to prevent the development of prostate cancer with a stem-like, dedifferentiated and aggressive phenotypes. These data suggest that the clinical targeting of the GR or ERα might be viable treatment options when stratified for selected patients who are no longer responsive to ADT.
The clinical progression of prostate cancer has been hypothesized to involve various stages of AR activity, from androgen-dependent–AR-dependent (AD–ARD) to androgen-independent–AR-dependent (AI–ARD), ultimately becoming androgen-independent– AR-independent (AI–ARI), a point at which a substantial portion of cancer cells might be undergoing a lineage crisis (FIG. 1b). If this hypothesis is confirmed, treatments for prostate cancer designed according to AR activity signature and disease stage would be more effective than the ‘one-drug-fits-all’ current approach. Basing decisions of treatment regimens on AR activity poses clinical challenges because prostate cancer is a heterogeneous disease demanding tailored treatment approaches52. Specifically, the assessment of AR activity in patients with metastatic disease might be the most straightforward option when the process is monoclonal in nature — that is, when one biopsy sample reflects the predominant clone, a hypothesis confirmed by the results of an analysis of patients who had received ADT and a taxane101. Intra-individual metastases, however, might undergo a monoclonal selection process attributable to aggressive treatments, which would account for the inconsistency between this and other reports documenting a multiclonal seeding process resulting in intra-individual molecular heterogeneity either in primary prostate tumours114 or metastases126. Despite these challenges, new developments in applied techniques (such as genomic and/or transcriptional profiling of AR signatures) and lineage markers might enable the advancement of clinical characterization of lineage crises because they can be used in clinical settings to evaluate treatment-induced lineage crises in formalin-fixed, paraffin-embedded primary or metastatic tumour samples74,127 and even in urine samples27. Increasingly sensitive assays can also be used to screen for the presence of neuroendocrine or EMT-like cells in patient blood samples28, thereby enabling more frequent sampling than many other sampling techniques. Collectively, these non-invasive procedures can satisfy the need for samples that are easier to obtain than tumour biopsy samples, while also being sufficiently sensitive to provide clinically useful information. In the future, we hope that appropriate treatment decisions will be made by determining AR activity in each individual patient with mCRPC and defining the pathways preferentially activated in AR-hi versus AR-lo cancers.
Lessons learned from other diseases
Triple-negative breast cancer
During tumour progression, endocrine tumours can dedifferentiate and converge both in phenotype and genetic signature even when from a different tissue type128,129. For example, TNBC is characterized by having low expression of ER and PR, and no HER2 amplification, and patients with this disease have the lowest 5-year survival rate of those with any invasive breast cancer, and few available treatment options19. Similar to AR-lo prostate tumours, TNBCs are poorly differentiated (with increased expression of stemness markers compared with other breast cancer subtypes)130,131. The existence of compensatory nuclear receptor activity facilitating disease progression in patients with certain breast cancers is another similarity with prostate cancer. For instance, substantial more AR expression can be observed in tumours from patients with ER-negative breast cancer (∼50%)132–134 or TNBC (one-third of patients)135–139, compared with ER-positive breast cancer. Accordingly, investigators have shown using enzalutamide or AR knockdown that non-luminal subtypes of TNBC have a strong dependency upon transcriptionally active AR140. AR targeting of cell lines of non-luminal subtypes using bicalutamide has also proven to be effective141. Curiously, some reports have correlated expression of AR in TNBC to a more favourable clinical prognosis138,142, possibly because AR-positive breast cancers are more differentiated than AR-negative cancers138. Another study showed an increase in levels of AR splice variants in MDA-MB-453, ER-negative breast cancer cells when treated with enzalutamide compared to untreated, or those treated with AR agonists further supporting a tumour promoting function of transcriptionally active AR143 (FIG. 1c). This maintained dependency upon a transcriptionally active AR in some TBNCs suggests that, similar to CRPC, nuclear hormone receptor switching confers a proliferation advantage to cancer cells and, therefore, could be exploited to treat patients with poorly differentiated disease. On the basis of such observations, a phase II trial demonstrated the benefits of targeting the AR by administering bicalutamide to 51 patients with AR-positive (>10% scored by immunohistochemistry), ER-negative or PR-negative metastatic breast cancer, with a 6-month clinical benefit rate of 19% (complete response, partial response or stable disease >6 months), and a PFS of 12 months134. These results provide a sound rationale for targeting the AR in patients with TNBC, and represent one of the most viable treatment strategies available for these patients. Similar to mCRPC, assaying AR activity levels in patients with TNBC might identify patients who should be treated with AR-targeted therapies. Nevertheless, one might speculate that patients with AR-positive TNBC could progress to a condition similar to castration resistance when subject to the continued therapeutic suppression of the AR and subsequent acquisition of AR-variant expression143. If this is the case, patients with TNBC receiving AR-targeted agents might conceivably progress to a state of complete dedifferentiation lacking ER, PR and AR activity and HER2 amplification (‘quadruple-negative breast cancer’)144. Currently, no functional evidence exists to support that TNBCs arise from hormone-receptor-positive breast cancers in a manner similar to AR-lo prostate cancers being derived from AR-positive prostate cancers, but the striking similarities between TNBCs and poorly differentiated prostate cancers suggest a convergence in terms of histopathological features and treatment management.
Infectious diseases
The development of drug resistance in patients with cancer is a challenging prospect, especially because the toxicity profile and quality of life associated with anti-cancer treatments have to be carefully balanced against the aggressiveness of such treatments when making clinical decisions. Given the success of the treatments for certain infectious diseases, such as multiresistant Clostridium difficile and the use of fidaxomicin21,145, we propose that such treatment strategies could be applied in antitumour therapies to alter the kinetics by which cancers progress to multidrug resistance. Resistance to infectious diseases is conventionally thought to arise after the continued use of a single treatment (FIG. 1). The results of a study using computational analysis of resistance in bacteria, however, indicate that approximately 70% of treatment sequences using two to four drugs result in resistance to the last antibiotic in the sequence146, thus underscoring the importance of how patients with cancer should be treated. Appropriate drug sequencing, however, might be achieved by understanding that drug resistance is regulated by Darwinian law — that is, therapeutic agents exert pressure on the microbial or cell populations most capable of survival. Drug resistance comes at a fitness cost and thus, treatment approaches that exploit this evolution might lead to more-beneficial outcomes147. Considerations should also be given to drug selection, dosage, treatment duration and combinations. The study of microorganisms offers remarkable advantages over preclinical mammalian cancer models in terms of understanding these parameters, particularly when considering the large number of treatment sequences and combinations that are feasible with even a minimal number of available drugs. The use of mathematical modelling and algorithms is perhaps the most efficient way to determine those drug sequences that constitute the ‘worst-drug-rule’ and ‘best-drug-rule’, which require information about drug targets, dosing, mechanism (cytotoxic versus cytostatic drug), toxicity, administration and exposure time148,149. Various elegant studies have resulted in the development of mathematical models that predict the optimal sequence of drugs with a maximum lethal effect in a scenario in which each individual drug would likely fail146. Mathematical algorithms that model microbial fitness have also been used to consider how antibiotic cycling can be used to avoid the continued selective pressure that would lead to the selection and expansion of a population with a resistant phenotype150. Such drug-cycling strategies have proven to be successful for the treatment of infections with bacteria and eukariotic parasites151–153.
For patients with advanced-stage prostate cancer, and with the exception of maintenance ADT, the classic treatment approach consists of administering a single therapeutic agent until resistance develops, followed by switching to a different agent (FIG. 2a). Through the use of potent new therapies, this approach might initially prove successful. Treatment resistance, however, will eventually occur and, for most patients, will lead to progression to more-aggressive disease. Thus, the application of drug cycling for the treatment of prostate cancer might be a well-suited alternative for patients with mCRPC — a heterogeneous and proliferative disease that is typically driven by multiple alterations in oncogenic signalling pathways. The application of rapid cycling of drugs with different mechanisms of action might reduce the expansion of lineages harbouring aggressive phenotypes. Rapid drug cycling (that is, several weeks per treatment) could also be an effective strategy to reduce the extent of cumulative toxicities thus, providing the option of performing repeated treatments (FIG. 2b). The duration of the exposure to any individual drug in a rapid cycling scheme is reduced and thus, determining whether exposure times are sufficient to achieve a clinical benefit is imperative.
Figure 2. Treatment cycling to prevent progression to treatment-induced mCRPC.
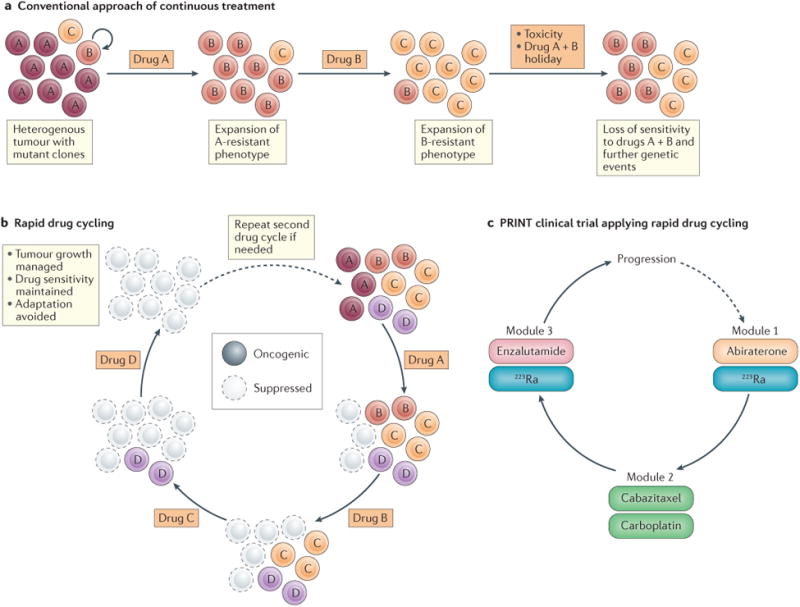
a | Prostate tumours are heterogeneous (left) but are often treated with drugs targeting a single oncogenic mechanism, until a clinical response is no longer obtainable. This approach can lead to toxicity, loss of drug sensitivity, and the accumulation of multidrug-resistant cancer cells. b | Rapid cycling of agents that target cancer cells might enable sustained drug sensitivity through different resistance mechanisms while, at the same time, controlling prostate cancer progression through the suppression of oncogenic signalling. Thus, the use of this approach can maintain drug sensitivity and continued growth inhibition. c | In the Prostate Cancer Intensive Non-crossreactive Therapy (PRINT) clinical trial23, men with metastatic castration-resistant prostate cancer (mCRPC) will receive different specific treatments in cyclical modules, each for 12 weeks. Module one is comprised of androgen receptor (AR)-targeted therapy (abiraterone) and α radiation (223Ra), module two consists of cytotoxic agents (cabazitaxel and/or carboplatin), and module three consists of androgen receptor (AR)-targeted therapy (enzalutamide) and α radiation (223Ra). Progression will be assessed after completion of modules one to three, with the option of performing additional cycles.
The PRINT protocol
The current therapeutic strategies for patients with mCRPC have one of several mechanisms of action: inhibition of AR signalling, cytotoxicity, bone-targeted toxicity and immunotherapy. Owing to the heterogeneity of metastatic prostate cancer154–156, single-agent treatment strategies are likely to potentiate the evolution of certain prostate cancer subclones into resistant lineages, which might no longer be responsive to the initial treatment. Thus, rapid switching of non-cross-reactive therapies might limit the selective pressure exerted by the maintained administration of a therapeutic agent157, enabling the risk of resistance to stabilize (or even decrease) in its absence. In addition to rapid cycling, combinations of agents from different therapeutic classes might yield superior long-term outcomes than those of monotherapies, because the probability of developing concurrent resistance towards multiple agents is expected to be lower than the probability of developing resistance to a single agent. Indeed, complex regimens that require alternating cycles of different drug combinations, such as hyper-CVAD (a dose-intensive and fractionated regimen using cyclophosphamide, vincristine, doxorubicin, and dexamethasone) for the treatment of acute lymphocytic leukaemia (ALL)158 and Burkett's lymphoma159, as well as the Stanford V regimen (combination of doxorubicin, vinblastine, mechlorethamine, vincristine, bleomycin, etoposide, and prednisone) for the treatment of advanced-stage bulky Hodgkin lymphoma160, have already been successfully implemented.
Owing to the intrinsic genetic instability of tumour cells, genetic mutation is one of the mechanisms proposed to account for the generation of treatment-resistant lineages. Of note, this hypothesis has been confirmed with the use of mathematical treatment models (such as the Goldie–Coldman hypothesis161). Thus, using different non-crossreactive agents in alternating cycles could improve the chances of treatment success. Multiple trials testing the Goldie–Coldman hypothesis have yielded mixed results. For example, small-cell lung cancer (SCLC) cells are sensitive to multiple chemotherapeutic agents, but patients with this disease quickly develop drug resistance and thus, the concurrent use of multiple drugs is limited because of toxicity. Some trials have showed limited or modest survival benefits for patients with SCLC using an alternating treatment regimen162–165, whereas others have failed to demonstrate any clinical benefits, possibly owing to the alternating regimen chosen and the size of the study population166. These early results, however, should not bias against designing and implementing new trials for different disease types taking advantage of more-potent and more-selective inhibitors than the agents used in these studies, such as the current FDA-approved drugs for treatment of patients with mCRPC.
The PRINT clinical trial (NCT02903160)23 was designed with the specific purpose of determining whether the rapid use of different agents specific for mCRPC (AR-targeting therapies, such as abiraterone acetate and enzalutamide, versus cytotoxic therapies versus radionuclides) can reduce treatment resistance and delay disease progression. The PRINT protocol is a phase II trial, in which investigators will evaluate the efficacy of rapidly cycling non-crossreactive therapies for patients who are newly diagnosed with mCRPC. This protocol aims to target different cancer-cell populations by switching between drug classes (five different agents), while maintaining the end goal of exposing the disease to multiple agents in a short period of time (three modules with 12 weeks per module). The individual toxicity profiles of most of the drugs used in this trial have been defined using large-scale phase III trials (755–1,200 patients per trial)1–4,167,168, but limited information is available about the toxicity profiles of these agents when combined in a rapid cycling regimen. This information will be obtained once the results of the PRINT clinical trial are compiled. Controlling treatment resistance not only depends on the duration of drug exposure but also on the selected drug sequencing. Thus, the PRINT trial contains three distinct treatment modules of two-drug combinations given in sequential order, that are rationally designed to optimize therapeutic dosing while reducing toxicity and minimizing therapeutic overlap (FIG. 2c). Modules one and three contain AR-targeted therapies operating through different mechanisms, such as abiraterone acetate, a CYP17 inhibitor1, and enzalutamide, which directly antagonizes AR expression and localization169. The toxicity of 223Ra, a radiopharmaceutical that targets bone metastases168, is not expected to substantially overlap with that of either abiraterone or enzalutamide and thus, 223Ra will be offered in combination with both agents to patients with osseous involvement. To minimize development of overlapping mechanisms of resistance between abiraterone and enzalutamide170–172, and to avoid other potential mechanisms of resistance (such as glucocorticoid receptor upregulation106) a combination of the cytotoxic agents cabazitaxel and carboplatin will be administered in module two — of note, this combination has demonstrated promising results in a phase II study and has a manageable toxicity pro-file173. The use of Sipuleucel-T174 before patient enrolment in the PRINT study will be permitted on the basis that immunotherapeutic agents require time to build an antitumour immune response, and sipuleucel-T is most effective in the patients with low-burden disease175. The primary end point of the PRINT trial is to evaluate the time to PSA progression, defined according to the Prostate Cancer Working Group criteria34, after the completion of all three treatment modules. Secondary end points include radiographic PFS, overall survival, PSA response and toxicity for each module. In parallel with the primary and secondary clinical objectives of this trial, an exploratory analysis will be conducted to evaluate the correlation of a peripheral whole-blood RNA-based signature with clinical outcome. Six genes define this signature176, which we found to be strongly associated with clinical prognosis. The expression of the AR–V7 splice variant, which increases in parallel with the development of resistance to AR-targeted agents in mCRPC177, will be assessed on circulating tumour cells before, during, and after the rapid cycling treatment regimen to better understand how different therapeutic agents affect AR-V7 expression and how those changes affect clinical outcomes.
The PRINT clinical trial (NCT02903160)23 should provide valuable data on the feasibility for cycling therapies to prevent resistance and prolong survival in patients with mCRPC. Evaluation of this trial will also inform on the value of future variations in this sequencing pattern, which could include incorporation of immunotherapy or new targeted therapies. The rationale used in the design of PRINT could also be applied to subsequent phase II randomized clinical trials. Whether rapid drug cycling will enable an enhancement of the cytotoxic effects of these drugs on tumours compared with current trial designs is unclear, but treatment cycling might have the desirable effect of controlling cellular proliferation and tumour growth. The efficacy of cytotoxic chemotherapeutic agents, such as cabazitaxel and/or carboplatin, mainly depends on the tumour's proliferative rate, potentially leaving dormant or non-proliferative cells intact. By design, PRINT also relies upon targeted agents such as abiraterone, enzalutamide and 223Ra, which should be effective in slowly proliferating and well-differentiated prostate cancer cells. Future approaches could also consider novel drug combinations, exploiting the potential to target multiple cellular lineages and mitotic rates. Drug cycling should continue to evolve and incorporate new drug combinations when possible, in order to combat treatment- induced cancer progression; although important consideration to the pairing and sequence of drugs used should be made in order to avoid the development of adverse events. One clinically viable strategy might be to combine cycling with rational combinations of drugs.
Collateral sensitivity
Our understanding of how cancers acquire resistance to standard-of-care therapies has increased considerably in the past decade, but very little, in comparison, is understood about how acquired resistance increases sensitivity to other drugs. This phenomenon, termed collateral sensitivity, can be considered a type of synthetic lethality and relies on the fact that drug resistance comes with a competitive fitness cost to cells178. The concepts of drug cycling and collateral sensitivity are different: while cycling relies on the suppression of oncogenic function through the rapid exchange of different drugs (FIG. 2), collateral sensitivity is designed to induce the maximum possible cytotoxic effects on cancer cells (FIG. 3). Treatment approaches based on the principle of collateral sensitivity are likely to be most effective when tumours contain both cell types that are maximally susceptible and resistant to the drug — that is, a heterogeneous cancer type such as mCRPC. Treatment-induced multidrug resistance often yields pleiotropic alterations and the mutation of a single gene can lead to multiple changes in phenotype. Indeed, up to 74% of treatment-resistant lines of bacteria were found to have enhanced sensitivities to one or more drugs179, indicating that treatment-resistant bacteria develop both resistance and sensitivity to other treatments. Moreover, while additive or synergistic antibiotic combinations might be beneficial from a treatment standpoint (for example, antibiogram and patient outcomes), such approaches cannot be used to predict the delayed onset of resistance. Conversely, collateral sensitivity can enable the prediction of decreased treatment resistance that occurs during concomitant drug exposure: for example, if drug A confers collateral sensitivity to drug B, then the combination of drugs A and B will decrease resistance compared to A or B alone, as shown on Escherichia coli180.
Figure 3. Drug cycling with collateral sensitivity.
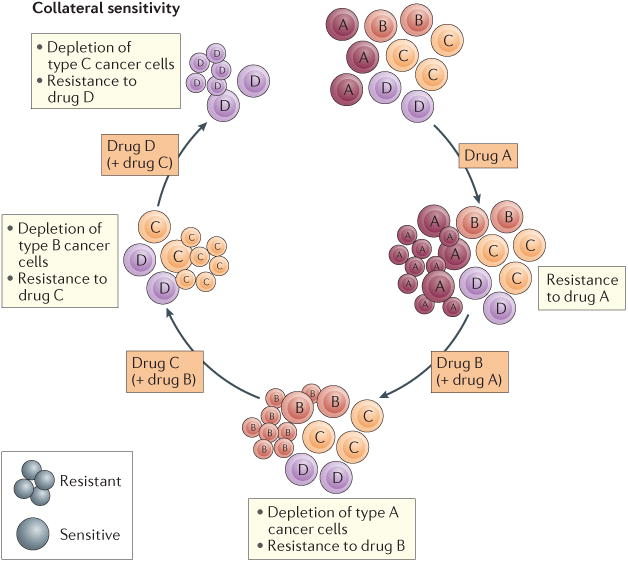
Drug cycling with collateral sensitivity uses rational drug-pairing to eradicate treatment-resistant cancer cells. Tumours that have become resistant to a drug should be rationally treated with a new combination of drugs. The application of consecutive cycles of complementary anticancer drugs could, in principle, be more effective than conventional single-agent or drug-pairing protocols that lack the benefits of collateral sensitivity. The cyclical application of drug partners working through collateral sensitivity could maximize cytotoxic effects while, at the same time, minimizing toxicities in patients.
We propose that, in order for clinical trials to have more-pronounced benefits in terms of patient survival, cooperative and rational drug design should be considered in the context of collateral sensitivity. For this drug phenomenon to be exploited, however, the partner drugs must have synergistic effects on cancer cells, and must have the ability to delay the onset of resistance181 (FIG. 3). One of the major challenges in adapting collateral sensitivity to the treatment of mCRPC would be the identification of compatible drug partners. In the treatment of infections, such screening studies can be readily tested179, but finding appropriate systems for the screening of candidate anticancer drug combinations is challenging. For example, in vitro studies in cancer cell lines have been repeatedly demonstrated to lack the capacity to predict the generation of multidrug resistance in clinical settings182. An overlap in drug response can exist between cell lines derived from different tissues182; therefore, other cell lines (such as breast or ovarian cancer-derived cell lines) might be used to screen for drug combinations with candidate collateral sensitivity in patients with mCRPC. ABC transporter B family member 2 (pgp-2), a drug efflux transporter that is expressed in most prostate cancer cell lines with increased expression in those that are taxane-resistant, is one of the primary targets for collateral- sensitivity-based treatment approaches183,184. Chemoresistance is a major treatment issue for patients with cancer, and this is aggravated by the observations that AR-targeting agents can promote cross-resistance to docetaxel56,185 — and, to a minor extent, to cabazitaxel185–188 — when initially selected on the basis of a low affinity to pgp-2 (REF. 189). Importantly, inhibition of ABC transporter B family member 1 (ABCB1) or pgp-2 by AR antagonists can resensitize in vitro and in vivo docetaxel-resistant prostate cancer cells190. These data indicate the possibility of treating advanced disease with potent pgp-2 inhibitors in combination with standard-of-care therapies to promote collateral sensitivity. Compensatory cell signalling often occurs upon therapeutic inhibition of targets in major prosurvival pathways. For example, inhibition of Akt in PTEN-deficient prostate cancer cells can lead to the activation of ERK–MAPK signalling through a feedback mechanism191,192. Thus, potential collateral sensitivity could be achieved through combined inhibition of PI3K and ERK — both of which are elevated in mCRPC68. Other potential combination approaches in prostate cancer include targeting both activating mutant PI3Kα and β isoforms, in conjunction with AR inhibition193.
Conclusions
Over the past ten years, we have witnessed a substantial increase in the number of anticancer drugs approved by the FDA. Such therapies increase the life expectancy of patients with mCRPC, but are not curative. The onset of resistance is one of the primary roadblocks to the extension of survival rates in most forms of cancer, and is likely related to the persistent use of a minimal number of highly potent drugs. The heterogeneity of mCRPC demands that treatments of this disease are also heterogeneous, potentially including modalities such as drug cycling and collateral sensitivity. Some key pathways associated with treatment-induced lineage crisis in mCRPC have been identified, but continued tumour growth is likely potentiated by cellular plasticity and compensatory oncogenic signalling, thus increasing the complexity of determining which therapeutic interventions should be applied to overcome the natural evolution to treatment-resistant phenotypes. Drug discovery is markedly expanding and should yield increased opportunities for rationale drug pairing in order to extend the survival of patients with mCRPC. Challenges also exist in identifying the appropriate preclinical model systems in which new anticancer drugs can be efficiently tested. Nevertheless, genomic sequencing and predictive algorithms might dramatically facilitate preclinical screening, thus fast-tracking the drugs that function best in a cooperative and minimally toxic manner in clinical settings.
Key points.
Potent androgen receptor (AR)-targeted therapies have increased survival rates for metastatic castration-resistant prostate cancer (mCRPC), but correlate with the emergence of ‘treatment-induced lineage crisis’ characterized by visceral and bulky metastases and low PSA secretion
In prostate cancer, lineage crisis can occur either in the form of treatment-induced neuroendocrine differentiation, which results in a neuroendocrine phenotype, or in the form of treatment-induced epithelial-to-mesenchymal transition
Regardless of the mechanism responsible for lineage crisis, a proposed common checkpoint that precedes such crisis is the loss of expression and/or activity of AR pathway (AR-lo prostate cancer)
Drug-cycling designs used to prevent multidrug resistance (or ‘superbugs’) in infectious diseases might delay treatment-induced lineage crisis in prostate cancer, owing to the partial similarities between both phenomena
The PRINT protocol is a phase II trial designed to alternate administration of FDA-approved drugs in rapid cycles of 3 months to prevent treatment-induced lineage crisis for mCRPC, which might provide a rationale for testing drug cycling in the setting of first-line treatment for mCRPC
Collateral sensitivity might result in increased cytotoxic effects compared with standard approaches for mCRPC (a heterogeneous disease); this treatment strategy uses synergistic drug pairs because drug resistance results in competitive fitness
Acknowledgments
The authors want to thank M. Galsky, J. Sfakianos, A. Wolfe and M. Ruscetti for constructive advice. This work was supported in part by a grant from the Fondation de France to G.R. and an NIH grant (R01CA197910) to D.J.M.
Footnotes
Author contributions: G.R. and D.J.M researched data and wrote the manuscript. All authors discussed the article's contents, revised and edited the manuscript before submission. B.C.L. and W.K.O. designed the PRINT clinical protocol.
Competing interests statement: The authors declare no competing interests.
References
- 1.de Bono JS, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ryan CJ, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138–148. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scher HI, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 4.Beer TM, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 2014;371:424–433. doi: 10.1056/NEJMoa1405095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beltran H, et al. Challenges in recognizing treatment-related neuroendocrine prostate cancer. J Clin Oncol. 2012;30:e386–e389. doi: 10.1200/JCO.2011.41.5166. [DOI] [PubMed] [Google Scholar]
- 6.Sun Y, et al. Androgen deprivation causes epithelial– mesenchymal transition in the prostate: implications for androgen-deprivation therapy. Cancer Res. 2012;72:527–536. doi: 10.1158/0008-5472.CAN-11-3004. [DOI] [PubMed] [Google Scholar]
- 7.Martin SK, et al. Multinucleation and mesenchymal-to-epithelial transition alleviate resistance to combined cabazitaxel and antiandrogen therapy in advanced prostate cancer. Cancer Res. 2016;76:912–926. doi: 10.1158/0008-5472.CAN-15-2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aparicio AM, et al. Platinum-based chemotherapy for variant castrate-resistant prostate cancer. Clin Cancer Res. 2013;19:3621–3630. doi: 10.1158/1078-0432.CCR-12-3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aparicio A, Tzelepi V. Neuroendocrine (small-cell) carcinomas: why they teach us essential lessons about prostate cancer. Oncology (Williston Park) 2014;28:831–838. [PubMed] [Google Scholar]
- 10.Beltran H, et al. Aggressive variants of castration- resistant prostate cancer. Clin Cancer Res. 2014;20:2846–2850. doi: 10.1158/1078-0432.CCR-13-3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang HT, et al. Neuroendocrine Prostate Cancer (NEPC) progressing from conventional prostatic adenocarcinoma: factors associated with time to development of NEPC and survival from NEPC diagnosis — a systematic review and pooled analysis. J Clin Oncol. 2014;32:3383–3390. doi: 10.1200/JCO.2013.54.3553. [DOI] [PubMed] [Google Scholar]
- 12.Tsao CK, Galsky MD, Oh WK. Is metastatic prostate cancer changing, and how will we know it? It's time for standard nomenclature for nonosseous metastases in clinical trials of patients with metastatic castration resistant prostate cancer. Eur Urol. 2014;66:184–185. doi: 10.1016/j.eururo.2013.12.061. [DOI] [PubMed] [Google Scholar]
- 13.Berglund RK, et al. Comparison of observed biochemical recurrence-free survival in patients with low PSA values undergoing radical prostatectomy and predictions of preoperative nomogram. Urology. 2009;73:1098–1103. doi: 10.1016/j.urology.2008.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McGuire BB, et al. Outcomes in patients with Gleason score 8–10 prostate cancer: relation to preoperative PSA level. BJU Int. 2012;109:1764–1769. doi: 10.1111/j.1464-410X.2011.10628.x. [DOI] [PubMed] [Google Scholar]
- 15.Beltran H, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22:298–305. doi: 10.1038/nm.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahal BA, Aizer AA, Efstathiou JA, Nguyen PL. Association of very low prostate-specific antigen levels with increased cancer-specific death in men with high-grade prostate cancer. Cancer. 2016;122:78–83. doi: 10.1002/cncr.29691. [DOI] [PubMed] [Google Scholar]
- 17.Pezaro CJ, et al. Visceral disease in castration-resistant prostate cancer. Eur Urol. 2014;65:270–273. doi: 10.1016/j.eururo.2013.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Small EJ, et al. Characterization of neuroendocrine prostate cancer (NEPC) in patients with metastatic castration resistant prostate cancer (mCRPC) resistant to abiraterone (Abi) or enzalutamide (Enz): Preliminary results from the SU2C/PCF/AACR West Coast Prostate Cancer Dream Team (WCDT) [abstract] J Clin Oncol. 2015;33(Suppl):5003. [Google Scholar]
- 19.Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13:674–690. doi: 10.1038/nrclinonc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Terry S, Beltran H. The many faces of neuroendocrine differentiation in prostate cancer progression. Front Oncol. 2014;4:60. doi: 10.3389/fonc.2014.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dolgin E. ‘Game changer’ antibiotic and others in works for superbug. Nat Med. 2011;17:10. doi: 10.1038/nm0111-10. [DOI] [PubMed] [Google Scholar]
- 22.Lowy FD. Secrets of a superbug. Nat Med. 2007;13:1418–1420. doi: 10.1038/nm1207-1418. [DOI] [PubMed] [Google Scholar]
- 23.US National Library of Medicine. 2016 doi: 10.1080/15360280801989377. ClinicalTrials.gov, https://www.clinicaltrials.gov/ct2/show/NCT02903160?term=NCT02903160&rank=1. [DOI] [PubMed]
- 24.Nikaido H. Multidrug resistance in bacteria. Annu Rev Biochem. 2009;78:119–146. doi: 10.1146/annurev.biochem.78.082907.145923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niederman MS. Appropriate use of antimicrobial agents: challenges and strategies for improvement. Crit Care Med. 2003;31:608–616. doi: 10.1097/01.CCM.0000050464.70382.D6. [DOI] [PubMed] [Google Scholar]
- 26.Gillessen S, et al. Management of patients with advanced prostate cancer: recommendations of the St Gallen Advanced Prostate Cancer Consensus Conference (APCCC) 2015. Ann Oncol. 2015;26:1589–1604. doi: 10.1093/annonc/mdv257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tomlins SA, et al. Urine TMPRSS2:ERG fusion transcript stratifies prostate cancer risk in men with elevated serum PSA. Sci Transl Med. 2011;3:94ra72. doi: 10.1126/scitranslmed.3001970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beltran H, et al. The initial detection and partial characterization of circulating tumor cells in neuroendocrine prostate cancer. Clin Cancer Res. 2016;22:1510–1519. doi: 10.1158/1078-0432.CCR-15-0137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simanainen U, et al. Disruption of prostate epithelial androgen receptor impedes prostate lobe-specific growth and function. Endocrinology. 2007;148:2264–2272. doi: 10.1210/en.2006-1223. [DOI] [PubMed] [Google Scholar]
- 30.Vander Griend DJ, Litvinov IV, Isaacs JT. Conversion of androgen receptor signaling from a growth suppressor in normal prostate epithelial cells to an oncogene in prostate cancer cells involves a gain of function in c-Myc regulation. Int J Biol Sci. 2014;10:627–642. doi: 10.7150/ijbs.8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu CT, et al. Increased prostate cell proliferation and loss of cell differentiation in mice lacking prostate epithelial androgen receptor. Proc Natl Acad Sci USA. 2007;104:12679–12684. doi: 10.1073/pnas.0704940104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xin L, et al. Progression of prostate cancer by synergy of AKT with genotropic and nongenotropic actions of the androgen receptor. Proc Natl Acad Sci USA. 2006;103:7789–7794. doi: 10.1073/pnas.0602567103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cunha GR, Chung LW, Shannon JM, Taguchi O, Fujii H. Hormone-induced morphogenesis and growth: role of mesenchymal– epithelial interactions. Recent Prog Horm Res. 1983;39:559–598. doi: 10.1016/b978-0-12-571139-5.50018-5. [DOI] [PubMed] [Google Scholar]
- 34.Niu Y, et al. Differential androgen receptor signals in different cells explain why androgen-deprivation therapy of prostate cancer fails. Oncogene. 2010;29:3593–3604. doi: 10.1038/onc.2010.121. [DOI] [PubMed] [Google Scholar]
- 35.Scher HI, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol. 2008;26:1148–1159. doi: 10.1200/JCO.2007.12.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scher HI, Morris MJ, Basch E, Heller G. End points and outcomes in castration-resistant prostate cancer: from clinical trials to clinical practice. J Clin Oncol. 2011;29:3695–3704. doi: 10.1200/JCO.2011.35.8648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang J, et al. Immunohistochemical characterization of neuroendocrine cells in prostate cancer. Prostate. 2006;66:1399–1406. doi: 10.1002/pros.20434. [DOI] [PubMed] [Google Scholar]
- 38.Fleischmann A, et al. Androgen receptors are differentially expressed in Gleason patterns of prostate cancer and down-regulated in matched lymph node metastases. Prostate. 2011;71:453–460. doi: 10.1002/pros.21259. [DOI] [PubMed] [Google Scholar]
- 39.Pouessel D, et al. Liver metastases in prostate carcinoma: clinical characteristics and outcome. BJU Int. 2007;99:807–811. doi: 10.1111/j.1464-410X.2006.06663.x. [DOI] [PubMed] [Google Scholar]
- 40.Falchook AD, et al. Stage at presentation and survival outcomes of patients with Gleason 8–10 prostate cancer and low prostate-specific antigen. Urol Oncol. 2016;34:119.e19–119.e26. doi: 10.1016/j.urolonc.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 41.Cinar B, et al. Androgen receptor mediates the reduced tumor growth, enhanced androgen responsiveness, and selected target gene transactivation in a human prostate cancer cell line. Cancer Res. 2001;61:7310–7317. [PubMed] [Google Scholar]
- 42.Bonaccorsi L, et al. Androgen receptor expression in prostate carcinoma cells suppresses α6β4 integrinmediated invasive phenotype. Endocrinology. 2000;141:3172–3182. doi: 10.1210/endo.141.9.7640. [DOI] [PubMed] [Google Scholar]
- 43.Chuu CP, Hiipakka RA, Fukuchi J, Kokontis JM, Liao S. Androgen causes growth suppression and reversion of androgen-independent prostate cancer xenografts to an androgen-stimulated phenotype in athymic mice. Cancer Res. 2005;65:2082–2084. doi: 10.1158/0008-5472.CAN-04-3992. [DOI] [PubMed] [Google Scholar]
- 44.Moehren U, et al. Wild-type but not mutant androgen receptor inhibits expression of the hTERT telomerase subunit: a novel role of AR mutation for prostate cancer development. FASEB J. 2008;22:1258–1267. doi: 10.1096/fj.07-9360com. [DOI] [PubMed] [Google Scholar]
- 45.Akashi T, Koizumi K, Nagakawa O, Fuse H, Saiki I. Androgen receptor negatively influences the expression of chemokine receptors (CXCR4, CCR1) and ligand-mediated migration in prostate cancer DU-145. Oncol Rep. 2006;16:831–836. [PubMed] [Google Scholar]
- 46.Joly-Pharaboz MO, et al. Inhibition of growth and induction of apoptosis by androgens of a variant of LNCaP cell line. J Steroid Biochem Mol Biol. 2000;73:237–249. doi: 10.1016/s0960-0760(00)00076-5. [DOI] [PubMed] [Google Scholar]
- 47.Cheng H, Snoek R, Ghaidi F, Cox ME, Rennie PS. Short hairpin RNA knockdown of the androgen receptor attenuates ligand-independent activation and delays tumor progression. Cancer Res. 2006;66:10613–10620. doi: 10.1158/0008-5472.CAN-06-0028. [DOI] [PubMed] [Google Scholar]
- 48.Qin J, et al. The PSA–/lo prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell Stem Cell. 2012;10:556–569. doi: 10.1016/j.stem.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mucci NR, Akdas G, Manely S, Rubin MA. Neuroendocrine expression in metastatic prostate cancer: evaluation of high throughput tissue microarrays to detect heterogeneous protein expression. Hum Pathol. 2000;31:406–414. doi: 10.1053/hp.2000.7295. [DOI] [PubMed] [Google Scholar]
- 50.Shah RB, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res. 2004;64:9209–9216. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 51.Robinson D, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mendiratta P, et al. Genomic strategy for targeting therapy in castration-resistant prostate cancer. J Clin Oncol. 2009;27:2022–2029. doi: 10.1200/JCO.2008.17.2882. [DOI] [PubMed] [Google Scholar]
- 53.Sharma NL, et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell. 2013;23:35–47. doi: 10.1016/j.ccr.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 54.Tomlins SA, et al. Integrative molecular concept modeling of prostate cancer progression. Nat Genet. 2007;39:41–51. doi: 10.1038/ng1935. [DOI] [PubMed] [Google Scholar]
- 55.Wang Q, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245–256. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Soest RJ, et al. Cross-resistance between taxanes and new hormonal agents abiraterone and enzalutamide may affect drug sequence choices in metastatic castration-resistant prostate cancer. Eur J Cancer. 2013;49:3821–3830. doi: 10.1016/j.ejca.2013.09.026. [DOI] [PubMed] [Google Scholar]
- 57.Zhu ML, et al. Tubulin-targeting chemotherapy impairs androgen receptor activity in prostate cancer. Cancer Res. 2010;70:7992–8002. doi: 10.1158/0008-5472.CAN-10-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mezynski J, et al. Antitumour activity of docetaxel following treatment with the CYP17A1 inhibitor abiraterone: clinical evidence for cross-resistance? Ann Oncol. 2012;23:2943–2947. doi: 10.1093/annonc/mds119. [DOI] [PubMed] [Google Scholar]
- 59.Schweizer MT, et al. The influence of prior abiraterone treatment on the clinical activity of docetaxel in men with metastatic castration-resistant prostate cancer. Eur Urol. 2014;66:646–652. doi: 10.1016/j.eururo.2014.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Bono JS, et al. Subsequent chemotherapy and treatment patterns after abiraterone acetate in patients with metastatic castration-resistant prostate cancer: post hoc analysis of COU-AA-302. Eur Urol. 2016 doi: 10.1016/j.eururo.2016.06.033. http://dx.doi.org/10.1016/j.eururo.2016.06.033. [DOI] [PMC free article] [PubMed]
- 61.Chakraborty PS, et al. Metastatic poorly differentiated prostatic carcinoma with neuroendocrine differentiation: negative on 68Ga-PSMA PET/CT. Clin Nucl Med. 2015;40:e163–e166. doi: 10.1097/RLU.0000000000000594. [DOI] [PubMed] [Google Scholar]
- 62.Sheridan T, Herawi M, Epstein JI, Illei PB. The role of P501S and PSA in the diagnosis of metastatic adenocarcinoma of the prostate. Am J Surg Pathol. 2007;31:1351–1355. doi: 10.1097/PAS.0b013e3180536678. [DOI] [PubMed] [Google Scholar]
- 63.Schelling LA, et al. Frequent TMPRSS2-ERG rearrangement in prostatic small cell carcinoma detected by fluorescence in situ hybridization: the superiority of fluorescence in situ hybridization over ERG immunohistochemistry. Hum Pathol. 2013;44:2227–2233. doi: 10.1016/j.humpath.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 64.Mosquera JM, et al. Concurrent AURKA and MYCN gene amplifications are harbingers of lethal treatment-related neuroendocrine prostate cancer. Neoplasia. 2013;15:1–10. doi: 10.1593/neo.121550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Williamson SR, et al. ERG-TMPRSS2 rearrangement is shared by concurrent prostatic adenocarcinoma and prostatic small cell carcinoma and absent in small cell carcinoma of the urinary bladder: evidence supporting monoclonal origin. Mod Pathol. 2011;24:1120–1127. doi: 10.1038/modpathol.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lotan TL, et al. ERG gene rearrangements are common in prostatic small cell carcinomas. Mod Pathol. 2011;24:820–828. doi: 10.1038/modpathol.2011.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guo CC, et al. TMPRSS2-ERG gene fusion in small cell carcinoma of the prostate. Hum Pathol. 2011;42:11–17. doi: 10.1016/j.humpath.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mulholland DJ, et al. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res. 2012;72:1878–1889. doi: 10.1158/0008-5472.CAN-11-3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ruscetti M, Quach B, Dadashian EL, Mulholland DJ, Wu H. Tracking and functional characterization of epithelial–mesenchymal transition and mesenchymal tumor cells during prostate cancer metastasis. Cancer Res. 2015;75:2749–2759. doi: 10.1158/0008-5472.CAN-14-3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ruscetti M, et al. HDAC inhibition impedes epithelial-mesenchymal plasticity and suppresses metastatic, castration-resistant prostate cancer. Oncogene. 2016;35:3781–3795. doi: 10.1038/onc.2015.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhao H, et al. Patient-derived tissue slice grafts accurately depict response of high-risk primary prostate cancer to androgen deprivation therapy. J Transl Med. 2013;11:199. doi: 10.1186/1479-5876-11-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weinstein MH, Partin AW, Veltri RW, Epstein JI. Neuroendocrine differentiation in prostate cancer: enhanced prediction of progression after radical prostatectomy. Hum Pathol. 1996;27:683–687. doi: 10.1016/s0046-8177(96)90398-6. [DOI] [PubMed] [Google Scholar]
- 73.Sauer CG, Roemer A, Grobholz R. Genetic analysis of neuroendocrine tumor cells in prostatic carcinoma. Prostate. 2006;66:227–234. doi: 10.1002/pros.20338. [DOI] [PubMed] [Google Scholar]
- 74.Grasso CS, et al. Integrative molecular profiling of routine clinical prostate cancer specimens. Ann Oncol. 2015;26:1110–1118. doi: 10.1093/annonc/mdv134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Leong KG, Wang BE, Johnson L, Gao WQ. Generation of a prostate from a single adult stem cell. Nature. 2008;456:804–808. doi: 10.1038/nature07427. [DOI] [PubMed] [Google Scholar]
- 76.Simon RA, et al. CD44 expression is a feature of prostatic small cell carcinoma and distinguishes it from its mimickers. Hum Pathol. 2009;40:252–258. doi: 10.1016/j.humpath.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 77.Lee JK, et al. N-Myc drives neuroendocrine prostate cancer initiated from human prostate epithelial cells. Cancer Cell. 2016;29:536–547. doi: 10.1016/j.ccell.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON. Identification of a cell of origin for human prostate cancer. Science. 2010;329:568–571. doi: 10.1126/science.1189992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stoyanova T, et al. Prostate cancer originating in basal cells progresses to adenocarcinoma propagated by luminal-like cells. Proc Natl Acad Sci USA. 2013;110:20111–20116. doi: 10.1073/pnas.1320565110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Beltran H, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011;1:487–495. doi: 10.1158/2159-8290.CD-11-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tan HL, et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin Cancer Res. 2014;20:890–903. doi: 10.1158/1078-0432.CCR-13-1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kadakia KC, et al. Comprehensive serial molecular profiling of an “N of 1” exceptional non-responder with metastatic prostate cancer progressing to small cell carcinoma on treatment. J Hematol Oncol. 2015;8:109. doi: 10.1186/s13045-015-0204-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Epstein JI, et al. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am J Surg Pathol. 2014;38:756–767. doi: 10.1097/PAS.0000000000000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cerasuolo M, et al. Neuroendocrine transdifferentiation in human prostate cancer cells: an integrated approach. Cancer Res. 2015;75:2975–2986. doi: 10.1158/0008-5472.CAN-14-3830. [DOI] [PubMed] [Google Scholar]
- 85.Burchardt T, et al. Transdifferentiation of prostate cancer cells to a neuroendocrine cell phenotype in vitro and in vivo. J Urol. 1999;162:1800–1805. [PubMed] [Google Scholar]
- 86.Zhang XQ, et al. Receptor protein tyrosine phosphatase alpha signaling is involved in androgen depletion-induced neuroendocrine differentiation of androgen-sensitive LNCaP human prostate cancer cells. Oncogene. 2003;22:6704–6716. doi: 10.1038/sj.onc.1206764. [DOI] [PubMed] [Google Scholar]
- 87.Lin D, et al. High fidelity patient-derived xenografts for accelerating prostate cancer discovery and drug development. Cancer Res. 2014;74:1272–1283. doi: 10.1158/0008-5472.CAN-13-2921-T. [DOI] [PubMed] [Google Scholar]
- 88.Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell. 2005;121:645–657. doi: 10.1016/j.cell.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 89.Zhang X, et al. SRRM4 expression and the loss of REST activity may promote the emergence of the neuroendocrine phenotype in castration-resistant prostate cancer. Clin Cancer Res. 2015;21:4698–4708. doi: 10.1158/1078-0432.CCR-15-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lapuk AV, et al. From sequence to molecular pathology, and a mechanism driving the neuroendocrine phenotype in prostate cancer. J Pathol. 2012;227:286–297. doi: 10.1002/path.4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li Y, et al. SRRM4 drives neuroendocrine transdifferentiation of prostate adenocarcinoma under androgen receptor pathway inhibition. Eur Urol. 2016 doi: 10.1016/j.eururo.2016.04.028. http://dx.doi.org/10.1016/j.eururo.2016.04.028. [DOI] [PubMed]
- 92.Aggarwal RR, et al. Persistence of androgen receptor (AR) expression in patients (pts) with small cell prostate cancer (SCPC): Preliminary results from the SU2C/PCF/AACR West Coast Prostate Cancer Dream Team (WCDT) J Clin Oncol. 2016;34(Suppl. 2S):288. [Google Scholar]
- 93.Puls LN, Eadens M, Messersmith W. Current status of SRC inhibitors in solid tumor malignancies. Oncologist. 2011;16:566–578. doi: 10.1634/theoncologist.2010-0408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chu I, et al. Src promotes estrogen-dependent estrogen receptor α proteolysis in human breast cancer. J Clin Invest. 2007;117:2205–2215. doi: 10.1172/JCI21739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yu EY, et al. Once-daily dasatinib: expansion of phase II study evaluating safety and efficacy of dasatinib in patients with metastatic castration-resistant prostate cancer. Urology. 2011;77:1166–1171. doi: 10.1016/j.urology.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Araujo JC, et al. Dasatinib combined with docetaxel for castration-resistant prostate cancer: results from a phase 1–2 study. Cancer. 2012;118:63–71. doi: 10.1002/cncr.26204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Spreafico A, et al. A randomized phase II study of cediranib alone versus cediranib in combination with dasatinib in docetaxel resistant, castration resistant prostate cancer patients. Invest New Drugs. 2014;32:1005–1016. doi: 10.1007/s10637-014-0106-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Araujo JC, et al. Docetaxel and dasatinib or placebo in men with metastatic castration-resistant prostate cancer (READY): a randomised, double-blind phase 3 trial. Lancet Oncol. 2013;14:1307–1316. doi: 10.1016/S1470-2045(13)70479-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang XD, et al. Identification of candidate predictive and surrogate molecular markers for dasatinib in prostate cancer: rationale for patient selection and efficacy monitoring. Genome Biol. 2007;8:R255. doi: 10.1186/gb-2007-8-11-r255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Siu MK, et al. Androgen receptor regulates SRC expression through microRNA-203. Oncotarget. 2016;7:25726–25741. doi: 10.18632/oncotarget.8366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kumar A, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med. 2016;22:369–378. doi: 10.1038/nm.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mulholland DJ, et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011;19:792–804. doi: 10.1016/j.ccr.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Carver BS, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19:575–586. doi: 10.1016/j.ccr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jia S, et al. Opposing effects of androgen deprivation and targeted therapy on prostate cancer prevention. Cancer Discov. 2013;3:44–51. doi: 10.1158/2159-8290.CD-12-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Toren P, et al. Combination AZD5363 with enzalutamide significantly delays enzalutamide-resistant prostate cancer in preclinical models. Eur Urol. 2015;67:986–990. doi: 10.1016/j.eururo.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 106.Arora VK, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155:1309–1322. doi: 10.1016/j.cell.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ndibe C, Wang CG, Sonpavde G. Corticosteroids in the management of prostate cancer: a critical review. Curr Treat Options Oncol. 2015;16:6. doi: 10.1007/s11864-014-0320-6. [DOI] [PubMed] [Google Scholar]
- 108.Isikbay M, et al. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm Cancer. 2014;5:72–89. doi: 10.1007/s12672-014-0173-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yemelyanov A, et al. Differential targeting of androgen and glucocorticoid receptors induces ER stress and apoptosis in prostate cancer cells: a novel therapeutic modality. Cell Cycle. 2012;11:395–406. doi: 10.4161/cc.11.2.18945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pham LK, et al. Contextual effect of repression of bone morphogenetic protein activity in prostate cancer. Endocr Relat Cancer. 2013;20:861–874. doi: 10.1530/ERC-13-0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Scher HI, et al. Impact of on-study corticosteroid use on efficacy and safety in the phase III AFFIRM study of enzalutamide (ENZA), an androgen receptor inhibitor [abstract] J Clin Oncol. 2013;31(Suppl. 6):6. [Google Scholar]
- 112.Morgan CJ, Oh WK, Naik G, Galsky MD, Sonpavde G. Impact of prednisone on toxicities and survival in metastatic castration-resistant prostate cancer: a systematic review and meta-analysis of randomized clinical trials. Crit Rev Oncol Hematol. 2014;90:253–261. doi: 10.1016/j.critrevonc.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 113.Montgomery B, et al. Impact of baseline corticosteroids on survival and steroid androgens in metastatic castration-resistant prostate cancer: exploratory analysis from COU-AA-301. Eur Urol. 2015;67:866–873. doi: 10.1016/j.eururo.2014.06.042. [DOI] [PubMed] [Google Scholar]
- 114.Efstathiou E, et al. Biological heterogeneity in localized high-risk prostate cancer (LHRPC) from a study of neoadjuvant abiraterone acetate plus leuprolide acetate (LHRHa) versus LHRHa [abstract] J Clin Oncol. 2015;33(Suppl):5005. [Google Scholar]
- 115.Sartor O, Parker CC, de Bono J. Reappraisal of glucocorticoids in castrate-resistant prostate cancer. Asian J Androl. 2014;16:666. doi: 10.4103/1008-682X.133314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Klokk TI, et al. Ligand-specific dynamics of the androgen receptor at its response element in living cells. Mol Cell Biol. 2007;27:1823–1843. doi: 10.1128/MCB.01297-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.US National Library of Medicine. 2016 doi: 10.1080/15360280801989377. ClinicalTrials.gov, https://www.clinicaltrials.gov/ct2/show/NCT02012296?term=NCT02012296&rank=1. [DOI] [PubMed]
- 118.Omoto Y, Iwase H. Clinical significance of estrogen receptor β in breast and prostate cancer from biological aspects. Cancer Sci. 2015;106:337–343. doi: 10.1111/cas.12613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fixemer T, Remberger K, Bonkhoff H. Differential expression of the estrogen receptor beta (ERβ) in human prostate tissue, premalignant changes, and in primary, metastatic, and recurrent prostatic adenocarcinoma. Prostate. 2003;54:79–87. doi: 10.1002/pros.10171. [DOI] [PubMed] [Google Scholar]
- 120.Mak P, et al. Prostate tumorigenesis induced by PTEN deletion involves estrogen receptor beta repression. Cell Rep. 2015;10:1982–1991. doi: 10.1016/j.celrep.2015.02.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lukacs RU, Memarzadeh S, Wu H, Witte ON. Bmi-1 is a crucial regulator of prostate stem cell self-renewal and malignant transformation. Cell Stem Cell. 2010;7:682–693. doi: 10.1016/j.stem.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Goel HL, et al. VEGF/neuropilin-2 regulation of Bmi-1 and consequent repression of IGF-IR define a novel mechanism of aggressive prostate cancer. Cancer Discov. 2012;2:906–921. doi: 10.1158/2159-8290.CD-12-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mak P, et al. ERβ impedes prostate cancer EMT by destabilizing HIF-1α and inhibiting VEGF-mediated snail nuclear localization: implications for Gleason grading. Cancer Cell. 2010;17:319–332. doi: 10.1016/j.ccr.2010.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yuan B, et al. A phosphotyrosine switch determines the antitumor activity of ERβ. J Clin Invest. 2014;124:3378–3390. doi: 10.1172/JCI74085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jia M, Dahlman-Wright K, Gustafsson JA. Estrogen receptor alpha and beta in health and disease. Best Pract Res Clin Endocrinol Metab. 2015;29:557–568. doi: 10.1016/j.beem.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 126.Gundem G, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520:353–357. doi: 10.1038/nature14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Manson-Bahr D, et al. Mutation detection in formalin-fixed prostate cancer biopsies taken at the time of diagnosis using next-generation DNA sequencing. J Clin Pathol. 2015;68:212–217. doi: 10.1136/jclinpath-2014-202754. [DOI] [PubMed] [Google Scholar]
- 128.Sole X, et al. Biological convergence of cancer signatures. PLoS ONE. 2009;4:e4544. doi: 10.1371/journal.pone.0004544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hoadley KA, et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;158:929–944. doi: 10.1016/j.cell.2014.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Honeth G, et al. The CD44+/CD24– phenotype is enriched in basal-like breast tumors. Breast Cancer Res. 2008;10:R53. doi: 10.1186/bcr2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363:1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 132.Garay JP, Park BH. Androgen receptor as a targeted therapy for breast cancer. Am J Cancer Res. 2012;2:434–445. [PMC free article] [PubMed] [Google Scholar]
- 133.Niemeier LA, Dabbs DJ, Beriwal S, Striebel JM, Bhargava R. Androgen receptor in breast cancer: expression in estrogen receptor-positive tumors and in estrogen receptor-negative tumors with apocrine differentiation. Mod Pathol. 2010;23:205–212. doi: 10.1038/modpathol.2009.159. [DOI] [PubMed] [Google Scholar]
- 134.Gucalp A, et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic breast cancer. Clin Cancer Res. 2013;19:5505–5512. doi: 10.1158/1078-0432.CCR-12-3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Proverbs-Singh T, Feldman JL, Morris MJ, Autio KA, Traina TA. Targeting the androgen receptor in prostate and breast cancer: several new agents in development. Endocr Relat Cancer. 2015;22:R87–R106. doi: 10.1530/ERC-14-0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Collins LC, et al. Androgen receptor expression in breast cancer in relation to molecular phenotype: results from the Nurses' Health Study. Mod Pathol. 2011;24:924–931. doi: 10.1038/modpathol.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mrklic I, Pogorelic Z, Capkun V, Tomic S. Expression of androgen receptors in triple negative breast carcinomas. Acta Histochem. 2013;115:344–348. doi: 10.1016/j.acthis.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 138.Thike AA, et al. Loss of androgen receptor expression predicts early recurrence in triple-negative and basal-like breast cancer. Mod Pathol. 2014;27:352–360. doi: 10.1038/modpathol.2013.145. [DOI] [PubMed] [Google Scholar]
- 139.Safarpour D, Pakneshan S, Tavassoli FA. Androgen receptor (AR) expression in 400 breast carcinomas: is routine AR assessment justified? Am J Cancer Res. 2014;4:353–368. [PMC free article] [PubMed] [Google Scholar]
- 140.Barton VN, et al. Multiple molecular subtypes of triple-negative breast cancer critically rely on androgen receptor and respond to enzalutamide in vivo. Mol Cancer Ther. 2015;14:769–778. doi: 10.1158/1535-7163.MCT-14-0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lehmann BD, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Peters KM, et al. Androgen receptor expression predicts breast cancer survival: the role of genetic and epigenetic events. BMC Cancer. 2012;12:132. doi: 10.1186/1471-2407-12-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hickey TE, et al. Expression of androgen receptor splice variants in clinical breast cancers. Oncotarget. 2015;6:44728–44744. doi: 10.18632/oncotarget.6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Barton VN, et al. Androgen receptor biology in triple negative breast cancer: a case for classification as AR+ or quadruple negative disease. Horm Cancer. 2015;6:206–213. doi: 10.1007/s12672-015-0232-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Louie TJ, et al. Fidaxomicin versus vancomycin for Clostridium difficile infection. N Engl J Med. 2011;364:422–431. doi: 10.1056/NEJMoa0910812. [DOI] [PubMed] [Google Scholar]
- 146.Nichol D, et al. Steering evolution with sequential therapy to prevent the emergence of bacterial antibiotic resistance. PLoS Comput Biol. 2015;11:e1004493. doi: 10.1371/journal.pcbi.1004493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gatenby RA, Silva AS, Gillies RJ, Frieden BR. Adaptive therapy. Cancer Res. 2009;69:4894–4903. doi: 10.1158/0008-5472.CAN-08-3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Goldie JH, Coldman AJ. Quantitative model for multiple levels of drug resistance in clinical tumors. Cancer Treat Rep. 1983;67:923–931. [PubMed] [Google Scholar]
- 149.Day RS. Treatment sequencing, asymmetry, and uncertainty: protocol strategies for combination chemotherapy. Cancer Res. 1986;46:3876–3885. [PubMed] [Google Scholar]
- 150.Goulart CP, et al. Designing antibiotic cycling strategies by determining and understanding local adaptive landscapes. PLoS ONE. 2013;8:e56040. doi: 10.1371/journal.pone.0056040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kublin JG, et al. Reemergence of chloroquine-sensitive Plasmodium falciparum malaria after cessation of chloroquine use in Malawi. J Infect Dis. 2003;187:1870–1875. doi: 10.1086/375419. [DOI] [PubMed] [Google Scholar]
- 152.Martinez JA, et al. Comparison of antimicrobial cycling and mixing strategies in two medical intensive care units. Crit Care Med. 2006;34:329–336. doi: 10.1097/01.ccm.0000195010.63855.45. [DOI] [PubMed] [Google Scholar]
- 153.Bennett KM, et al. Implementation of antibiotic rotation protocol improves antibiotic susceptibility profile in a surgical intensive care unit. J Trauma. 2007;63:307–311. doi: 10.1097/TA.0b013e318120595e. [DOI] [PubMed] [Google Scholar]
- 154.Cheng L, et al. Evidence of independent origin of multiple tumors from patients with prostate cancer. J Natl Cancer Inst. 1998;90:233–237. doi: 10.1093/jnci/90.3.233. [DOI] [PubMed] [Google Scholar]
- 155.Macintosh CA, Stower M, Reid N, Maitland NJ. Precise microdissection of human prostate cancers reveals genotypic heterogeneity. Cancer Res. 1998;58:23–28. [PubMed] [Google Scholar]
- 156.Liu W, et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat Med. 2009;15:559–565. doi: 10.1038/nm.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Madan RA, Pal SK, Sartor O, Dahut WL. Overcoming chemotherapy resistance in prostate cancer. Clin Cancer Res. 2011;17:3892–3902. doi: 10.1158/1078-0432.CCR-10-2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kantarjian HM, et al. Results of treatment with hyper-CVAD, a dose-intensive regimen, in adult acute lymphocytic leukemia. J Clin Oncol. 2000;18:547–561. doi: 10.1200/JCO.2000.18.3.547. [DOI] [PubMed] [Google Scholar]
- 159.Thomas DA, et al. Outcome with the hyper-CVAD regimens in lymphoblastic lymphoma. Blood. 2004;104:1624–1630. doi: 10.1182/blood-2003-12-4428. [DOI] [PubMed] [Google Scholar]
- 160.Horning SJ, et al. Assessment of the Stanford V regimen and consolidative radiotherapy for bulky and advanced Hodgkin's disease: Eastern Cooperative Oncology Group pilot study E1492. J Clin Oncol. 2000;18:972–980. doi: 10.1200/JCO.2000.18.5.972. [DOI] [PubMed] [Google Scholar]
- 161.Goldie JH, Coldman AJ. A mathematic model for relating the drug sensitivity of tumors to their spontaneous mutation rate. Cancer Treat Rep. 1979;63:1727–1733. [PubMed] [Google Scholar]
- 162.Havemann K, et al. Alternating versus sequential chemotherapy in small cell lung cancer. A randomized German multicenter trial. Cancer. 1987;59:1072–1082. doi: 10.1002/1097-0142(19870315)59:6<1072::aid-cncr2820590605>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 163.Ettinger DS, et al. A randomized comparison of standard chemotherapy versus alternating chemotherapy and maintenance versus no maintenance therapy for extensive-stage small-cell lung cancer: a phase III study of the Eastern Cooperative Oncology Group. J Clin Oncol. 1990;8:230–240. doi: 10.1200/JCO.1990.8.2.230. [DOI] [PubMed] [Google Scholar]
- 164.Evans WK, et al. Superiority of alternating non-cross-resistant chemotherapy in extensive small cell lung cancer. A multicenter, randomized clinical trial by the National Cancer Institute of Canada. Ann Intern Med. 1987;107:451–458. doi: 10.7326/0003-4819-107-4-451. [DOI] [PubMed] [Google Scholar]
- 165.Wampler GL, Heim WJ, Ellison NM, Ahlgren JD, Fryer JG. Comparison of cyclophosphamide, doxorubicin, and vincristine with an alternating regimen of methotrexate, etoposide, and cisplatin/cyclophosphamide, doxorubicin, and vincristine in the treatment of extensive-disease small-cell lung carcinoma: a Mid-Atlantic Oncology Program study. J Clin Oncol. 1991;9:1438–1445. doi: 10.1200/JCO.1991.9.8.1438. [DOI] [PubMed] [Google Scholar]
- 166.Feld R, et al. Canadian multicenter randomized trial comparing sequential and alternating administration of two non-cross-resistant chemotherapy combinations in patients with limited small-cell carcinoma of the lung. J Clin Oncol. 1987;5:1401–1409. doi: 10.1200/JCO.1987.5.9.1401. [DOI] [PubMed] [Google Scholar]
- 167.de Bono JS, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376:1147–1154. doi: 10.1016/S0140-6736(10)61389-X. [DOI] [PubMed] [Google Scholar]
- 168.Parker C, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013;369:213–223. doi: 10.1056/NEJMoa1213755. [DOI] [PubMed] [Google Scholar]
- 169.Tran C, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Loriot Y, et al. Antitumour activity of abiraterone acetate against metastatic castration-resistant prostate cancer progressing after docetaxel and enzalutamide (MDV3100) Ann Oncol. 2013;24:1807–1812. doi: 10.1093/annonc/mdt136. [DOI] [PubMed] [Google Scholar]
- 171.Noonan KL, et al. Clinical activity of abiraterone acetate in patients with metastatic castration-resistant prostate cancer progressing after enzalutamide. Ann Oncol. 2013;24:1802–1807. doi: 10.1093/annonc/mdt138. [DOI] [PubMed] [Google Scholar]
- 172.Schrader AJ, et al. Enzalutamide in castration-resistant prostate cancer patients progressing after docetaxel and abiraterone. Eur Urol. 2014;65:30–36. doi: 10.1016/j.eururo.2013.06.042. [DOI] [PubMed] [Google Scholar]
- 173.Corn PG, et al. A multi-institutional randomized phase II study ( NCT01505868) of cabazitaxel (CAB) plus or minus carboplatin (CARB) in men with metastatic castration-resistant prostate cancer (mCRPC) [abstract] J Clin Oncol. 2015;33(Suppl):5010. [Google Scholar]
- 174.Kantoff PW, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 175.Schellhammer PF, et al. Lower baseline prostate-specific antigen is associated with a greater overall survival benefit from sipuleucel-T in the Immunotherapy for Prostate Adenocarcinoma Treatment (IMPACT) trial. Urology. 2013;81:1297–1302. doi: 10.1016/j.urology.2013.01.061. [DOI] [PubMed] [Google Scholar]
- 176.Ross RW, et al. A whole-blood RNA transcript-based prognostic model in men with castration-resistant prostate cancer: a prospective study. Lancet Oncol. 2012;13:1105–1113. doi: 10.1016/S1470-2045(12)70263-2. [DOI] [PubMed] [Google Scholar]
- 177.Antonarakis ES, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371:1028–1038. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Gagneux S, et al. The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science. 2006;312:1944–1946. doi: 10.1126/science.1124410. [DOI] [PubMed] [Google Scholar]
- 179.Imamovic L, Sommer MO. Use of collateral sensitivity networks to design drug cycling protocols that avoid resistance development. Sci Transl Med. 2013;5:204ra132. doi: 10.1126/scitranslmed.3006609. [DOI] [PubMed] [Google Scholar]
- 180.Munck C, Gumpert HK, Wallin AI, Wang HH, Sommer MO. Prediction of resistance development against drug combinations by collateral responses to component drugs. Sci Transl Med. 2014;6:262ra156. doi: 10.1126/scitranslmed.3009940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lehar J, et al. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat Biotechnol. 2009;27:659–666. doi: 10.1038/nbt.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Gillet JP, Varma S, Gottesman MM. The clinical relevance of cancer cell lines. J Natl Cancer Inst. 2013;105:452–458. doi: 10.1093/jnci/djt007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.O'Neill AJ, et al. Characterisation and manipulation of docetaxel resistant prostate cancer cell lines. Mol Cancer. 2011;10:126. doi: 10.1186/1476-4598-10-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zhang B, et al. Telomere and microtubule targeting in treatment-sensitive and treatment-resistant human prostate cancer cells. Mol Pharmacol. 2012;82:310–321. doi: 10.1124/mol.111.076752. [DOI] [PubMed] [Google Scholar]
- 185.van Soest RJ, et al. Targeting the androgen receptor confers in vivo cross-resistance between enzalutamide and docetaxel, but not cabazitaxel, in castration-resistant prostate cancer. Eur Urol. 2015;67:981–985. doi: 10.1016/j.eururo.2014.11.033. [DOI] [PubMed] [Google Scholar]
- 186.Al Nakouzi N, et al. Cabazitaxel remains active in patients progressing after docetaxel followed by novel androgen receptor pathway targeted therapies. Eur Urol. 2015;68:228–235. doi: 10.1016/j.eururo.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 187.van Soest RJ, et al. The influence of prior novel androgen receptor targeted therapy on the efficacy of cabazitaxel in men with metastatic castration-resistant prostate cancer. Eur J Cancer. 2015;51:2562–2569. doi: 10.1016/j.ejca.2015.07.037. [DOI] [PubMed] [Google Scholar]
- 188.Pezaro CJ, et al. Activity of cabazitaxel in castration-resistant prostate cancer progressing after docetaxel and next-generation endocrine agents. Eur Urol. 2014;66:459–465. doi: 10.1016/j.eururo.2013.11.044. [DOI] [PubMed] [Google Scholar]
- 189.Mita AC, et al. Phase I and pharmacokinetic study of XRP6258 (RPR 116258A), a novel taxane, administered as a 1-hour infusion every 3 weeks in patients with advanced solid tumors. Clin Cancer Res. 2009;15:723–730. doi: 10.1158/1078-0432.CCR-08-0596. [DOI] [PubMed] [Google Scholar]
- 190.Zhu Y, et al. Antiandrogens inhibit ABCB1 efflux and ATPase activity and reverse docetaxel resistance in advanced prostate cancer. Clin Cancer Res. 2015;21:4133–4142. doi: 10.1158/1078-0432.CCR-15-0269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Carracedo A, Baselga J, Pandolfi PP. Deconstructing feedback-signaling networks to improve anticancer therapy with mTORC1 inhibitors. Cell Cycle. 2008;7:3805–3809. doi: 10.4161/cc.7.24.7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Carracedo A, Pandolfi PP. The PTEN–PI3K pathway: of feedbacks and cross-talks. Oncogene. 2008;27:5527–5541. doi: 10.1038/onc.2008.247. [DOI] [PubMed] [Google Scholar]
- 193.Schwartz S, et al. Feedback suppression of PI3Kα signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kβ. Cancer Cell. 2015;27:109–122. doi: 10.1016/j.ccell.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]