

Abstract
Background
Alcohol (ethanol) is an antinociceptive agent, working in part, by reducing sensitivity to painful stimuli. The transcription factor, Kruppel-like factor 11 (KLF11), a human diabetes-causing gene that also regulates the neurotransmitter-metabolic enzymes monoamine oxidase (MAOs), has recently been identified as an ethanol-inducible gene. However, its role in antinociception remains unknown. Consequently, we investigated the function of KLF11 in chronic ethanol-induced antinociception using a genetically engineered knockout mouse model.
Methods
Wild-type (Klf11+/+) and KLF11 knockout (Klf11−/−) mice were fed a liquid diet containing ethanol for 28 days with increasing amounts of ethanol from 0% up to a final concentration of 6.4%, representing a final diet containing 36% of calories primarily from ethanol. Control mice from both genotypes were fed liquid diet without ethanol for 28 days. The ethanol-induced antinociceptive effect was determined using the tail-flick test before and after ethanol exposure (on day 29). In addition, the enzyme activity and mRNA levels of MAO A and MAO B were measured by Real-time RT-PCR and enzyme assays, respectively.
Results
Ethanol produced an antinociceptive response to thermal pain in Klf11+/+ mice, as expected. In contrast, deletion of KLF11 in the Klf11−/− mice abolished the ethanol-induced antinociceptive effect. The mRNA and protein levels of KLF11were significantly increased in the brain prefrontal cortex of Klf11+/+ mice exposed to ethanol compared to control Klf11+/+ mice. Furthermore, MAO enzyme activities were affected differently in Klf11 wild-type versus Klf11 knockout mice exposed to chronic ethanol. Chronic ethanol intake significantly increased MAO-B activity in Klf1+/+ mice.
Conclusions
The data show KLF11 modulation of ethanol-induced antinociception. The KLF11-targeted MAO B enzyme, may contribute more significantly to ethanol-induced antinociception. Thus, this study revealed a new role for the KLF11 gene in the mechanisms underlying the antinociceptive effects of chronic ethanol exposure.
Keywords: Antinociceptive response, Kruppel-Like Factor 11 (Transforming Growth Factor-beta-Inducible Early Gene 2), Chronic Ethanol Intake, Mice, Monoamine Oxidase, Gene Knockout
INTRODUCTION
Ethanol has been used widely for relieving pain or inducing analgesia (reducing sensitivity to painful stimuli) (Campbell et al., 2006; He and Whistler, 2012; Sanchis-Segura et al., 2005); although the mechanism of its actions is not well understood. Ethanol-induced alterations in pain sensitivity may result in continuous and increased alcohol consumption to manage pain. This in turn, may contribute to alcohol abuse and addiction (Modesto-Lowe et al., 2007), as well as brain damage and other health problems (Brennan et al., 2005). In fact, disorders related to alcohol abuse are prevalent among patients with chronic pain. Therefore, the discovery of genes and pathways responsible for these effects are of significant clinical importance.
Ethanol affects many functions of the central nervous system and the peripheral nervous system, resulting in analgesia, sedation, hypnosis, memory disturbance, confusion, neurodegeneration, dependence and motor disturbance (Deitrich et al., 1989; Fadda and Rossetti, 1998; Lockridge et al., 2012). Recent studies have shown that the expression and/or activity of a variety of enzymes (Ou et al., 2010), neurotransmitter receptors (Krystal et al., 2003; Lockridge et al., 2012; Lovinger and White, 1991) and proteins (Lu et al., 2008), such as glyceraldehyde 3-phosphate dehydrogenase (Ou et al., 2010), monoamine oxidase B (Ou et al., 2011; Ou et al., 2010) and Kruppel-like factor 11 (Lu et al., 2008; Ou et al., 2011) are altered by ethanol. It is likely that the analgesic effects of ethanol are mediated through an interaction among these proteins.
Kruppel-like factor 11 (KLF11), a diabetes-causing gene in humans, is a transcription factor which belongs to the Sp/KLF-family (Bonnefond et al., 2011; Fernandez-Zapico et al., 2009; Garin et al., 2010). KLF11 has been shown to regulate genes involved in the metabolism of neurotransmitters, lipids, glucose and prostaglandins (Buttar et al., 2010; Cao et al., 2005; Ou et al., 2010). We have previously found that KLF11 mediates ethanol-induced brain cell death (Lu et al., 2008; Ou et al., 2011; Ou et al., 2009). In addition, KLF11 is a transcriptional activator for the metabolic enzyme monoamine oxidase (MAO) (Grunewald et al., 2012; Ou et al., 2004). MAO exists as two iso-enzymes, MAO A and MAO B. MAO A preferentially oxidizes catecholamines and serotonin, whereas MAO B preferentially oxidizes dopamine. Studies have documented an involvement of these neurotransmitter systems in the regulation of pain (Zubieta et al., 2003). For instance, increases in MAO A catecholamine metabolites have been proposed to be involved in pain perception and MAO A inhibitors have been shown to reduce pain perception (Dina et al., 2008).
Moreover, diabetes is associated with a significant damage in the central nervous system and peripheral nervous system, such as loss of feeling or feeling pain (Bierhaus et al., 2004). Therefore, these studies, combined with our own reports showing that KLF11 gene activation is also induced by ethanol (Lu et al., 2008; Ou et al., 2011) and stress (Grunewald et al., 2012), have led us to hypothesize that KLF11 functions as a genetic modifier in the pain response induced by chronic ethanol exposure. In the current study, we directly test this hypothesis using our previously described Klf11-knockout (Klf11−/−) mouse model.
MATERIALS AND METHODS
Animals
Adult male Klf11 homozygous knockout (Klf-−/−) mice were generated from the University of Washington, Seattle following standard homologous recombination techniques (Bonnefond et al., 2011; Song et al., 2002). The knockout mice were originally produced in a mixed C57BL/6 background and subsequently transferred to the Mayo Animal Facilities where they were crossed for more than 20 generations into a pure C57BL/6 background. In all of the experiments, male Klf11−/− animals were compared with age-matched male wild type Klf11+/+ littermates.
Animal Care and Ethanol Feeding
All protocols for the animal experiments described in this study were carried out according to the Ethical Guidelines on Animal Experimentation and were approved by the Animal Usage Committee at the University of Mississippi Medical Center. Animals were housed in individual cages in a temperature- and humidity-controlled room with a 12:12-h light–dark cycle.
Forty male mice (20 wild type Klf11+/+ and 20 knockout Klf11−/− mice) were randomly assigned to ethanol or control groups (10 wild type and 10 knockout mice were fed ethanol-liquid diets; 10 wild type and 10 knockout mice were fed control-liquid diets without ethanol). The liquid ethanol diet (Dyets Inc., Bethlehem, PA) contained increasing amounts of ethanol as follows: no ethanol (0%) for days 1–3, followed by 1.0% for days 4–6, 2.0% for days 7–9, 3.0% for days 10–12, 4.0% for days 11–13, 5.0% for days 14–16, and finally, 6.4% for days 17–28. In the ethanol-fed groups, glucose was iso-calorically substituted with ethanol according to the Lieber–DeCarli diet formulae following the manufacturer’s instructions. The control groups were fed a glucose-liquid diet (without ethanol; #710027, Dyets) containing an equivalent amount of calories as in the ethanol diet (Ou et al., 2011).
The male mice were used in this study because the KLF11-targeted gene, MAO, has been shown more function in male mice than female mice (Brunner et al., 1993; Shih et al., 1999). The blood ethanol concentration of each of the mice was measured with an EnzyChrom Ethanol Assay Kit (Bioassay Systems, Hayward, CA) (Ou et al., 2010). The ethanol concentrations measured in this study (less than 80 mM) were similar to previously reported data (Jelic et al., 1998) and were within the physiological range observed in human alcoholics (Henriksen et al., 1997; Yao et al., 2001).
Tail-Flick Test
The analgesic effect of chronic ethanol exposure was determined using the tail-flick test. Mice were placed gently in a mouse restraint while the tail was placed directly under the radiant heat source of the apparatus (Columbus Instruments, Columbus, OH). The tail-flick response was elicited by applying a focused beam of radiant light to the dorsal surface of the tail, 1–1.5 cm from the tip. The latency of the tail-flick response was measured and defined as the time between the onset of the heat stimulus and voluntary tail withdrawal (Dai et al., 2008). The intensity of the heat stimulus was adjusted to yield a baseline response of 3–4 s. A cutoff time of 10 s was used to avoid tissue injury (Dai et al., 2008). The tests were conducted blinded in this study.
Sacrifice
All mice, in both the ethanol-diet and control-diet groups, were killed by decapitation the following morning after 28 days/4-weeks of exposure to the diet. The brain prefrontal cortex (PFC) was immediately dissected, removed and stored at −80°C until used. The PFC was chosen in our studies because the KLF11 (TIEG2)-mediated cell death pathway has been reported to be activated in the PFC of rats exposed to ethanol-liquid diets for 4 weeks (Ou et al., 2011). In addition, chronic alcohol use in humans has been associated with reductions in PFC volume (Paul et al., 2008).
Western Blot Analyses
The PFC from each mouse was homogenized on ice with a 0.5 ml solution containing 1 mM EDTA, 10 mM Tris–HCL (pH 7) and fresh protease inhibitors (1× Protease Inhibitor Cocktail, Sigma). The resulting homogenate was briefly stored on dry ice for 10 min, and subsequently thawed and centrifuged at 4°C (2800–3000 rpm) for 10 min in a microcentrifuge. Protein concentrations of the homogenized samples were determined using the BCA Protein Assay Kit (PIERCE, Rockford, IL). Thirty micrograms of total PFC protein for each rat were separated on 10.5% SDS–poly-acrylamide gels and transferred to PVDF membranes (Johnson et al., 2011; Ou et al., 2011). The mouse anti-TIEG2/KLF11 (1:500)) antibody (BD, Franklin Lakes, NJ) was used (Ou et al., 2011).
Quantitative Real-Time RT-PCR
Total RNA was isolated from the mouse PFC using TRIzol reagent (Invitrogen, Grand Island, NY; ~50 mg tissue/1 ml TRIzol) following the manufacturer’s instructions. The mRNA was then reverse-transcribed into cDNA using SuperScript® III Reverse Transcriptase (Invitrogen) (Ou et al., 2004; Ou et al., 2010). KLF11 mRNA levels for the ethanol-treated and untreated-control group of Klf11+/+ mice were analyzed using specific primers for the mouse KLF11 described: sense, 5′-ACAAGAAGTTCGCAAGGTCAGATG-3′ and antisense, 5′-CGAGCGTGCTTTGTCAGGTGGTCA-3′; for the mouse MAO A: sense, 5′-CCGCTCCTTTTCCATGCCTCTCAA-3 and antisense, 5′-CCCTTGTACCGCCCCTTGACTGAA-3′; for the mouse MAO B: sense, 5′-AATAAAGTTGAGCGGCTGATACAC-3′ and antisense, 5′-GGCCCATCTCATCCATTGTCC-3′.
Real-time PCR was performed with a SYBR supermix kit, and the 18S Ribosomal RNA primer was used as an internal control to avoid sample variations. The values were calculated as described previously (Ou et al., 2004).
MAO A and MAO B Enzyme Activity Assay
The brain frontal cortex was homogenized in assay buffer (50 mM sodium phosphate buffer). Approximately 100 μg of frontal cortex protein was incubated with 100 μM [14C]5-hydroxytryptamine (for MAO A) or 10 μM 14C-labeled phenylethylamine (for MAO B) (PerkinElmer Inc., Waltham, MA) in assay buffer at 37 °C for 20 min. The reaction was terminated by the addition of 100 μl of 6N HCl. The reaction product was then extracted with benzene/ethyl acetate (1:1) for MAO A or ethyl acetate/toluene (1:1) for MAO B. The organic phase containing the reaction product was extracted and mixed with liquid scintillation cocktail (Beckman Coulter, Inc., Danvers, MA). The radioactivity of the reaction product was quantified by liquid scintillation spectroscopy (Grunewald et al., 2012; Ou et al., 2010).
Statistical Analyses
One-way ANOVA followed by Tukey’s post hoc test was used for analysis of the nociceptive responses among four groups (ethanol-fed and control Klf11+/+ mice; and ethanol-fed and control Klf1−/− mice). The data are reported as mean ± SEM, and a value of p<0.05 was considered statistically significant.
RESULTS
Genetic Inactivation of KLF11 Abolishes the Ethanol-Induced Antinociceptive Response
Ethanol-induced antinociception was determined using the tail-flick test by measuring the time between the onset of the heat stimulus and voluntary tail withdrawal (Dai et al., 2008). The response to a thermal pain stimulus was expressed as the percentage of the maximum possible effect (% MPE), which was calculated as [(T1 − T0)/(T2 − T0)] × 100. T0 and T1 were the tail-flick latencies before and after ethanol administration respectively, and T2 was the cutoff time (Dai et al., 2008). The test was conducted blinded and performed twice, before and after 4-weeks of ethanol exposure. Significant differences were found among the four test groups (F3, 36=12.97, p-value<0.0001). The percent of antinociception (%MPE) of wild type (Klf11+/+) mice fed with the control diet was 1.01%, indicating no significant change in their pain response. Conversely, the %MPE of wild type mice fed the ethanol diet was 16.12%, indicating their response to nociception was significantly reduced (by 16-fold) following ethanol exposure [Figure 1A, lanes 2 vs. 1; diff=−15.12, 95% CI (−22.29, −7.946); p<0.001]. The results demonstrate the well-known effect of ethanol in producing analgesia. However, the pain response latency, represented as %MPE, did not differ between the control and ethanol diet-fed Klf11−/− mice (Figure 1A, lanes 4 vs. 3), indicating a reduction in the antinociceptive effects of ethanol in the knockout mice. Moreover, the baseline nociceptive responses (before chronic ethanol feeding) were the same among each of the groups as shown in the Figure 1B. Thus, these results suggest that KLF11 acts as a genetic modifier of the antinociceptive effects of chronic ethanol exposure.
Figure 1.
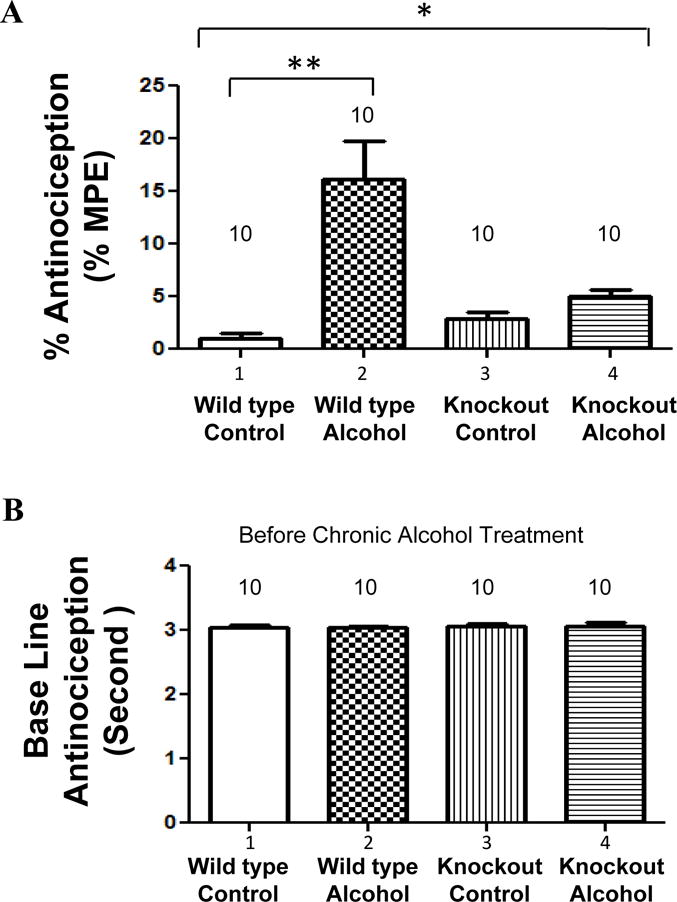
Effects of chronic ethanol intake on antinociceptive response (anti-pain effect) in the wild type (Klf11+/+) and KLF11 knockout (Klf11−/−) mice. Mice were fed an ethanol diet or control diet for 28 days. (A) Antinociceptive effects were determined in ethanol-fed and control-fed Klf11+/+ mice (lanes 2 vs. 1) as well as ethanol-fed and control-fed Klf1−/− mice (lanes 4 vs. 3) by the tail-flick test. The antinociceptive effects are expressed as a percentage of the maximum possible antinociceptive effect (% MPE). (B) The base line of the antinociceptive response was also determined in four groups of Klf11+/+ and Klf11−/− mice before chronic ethanol exposure. Data represent the mean ± SEM of 10 mice (n = 10) in each group. * p<0.0001 and **p<0.001
Chronic Ethanol Intake Significantly Increases Both mRNA and Protein Levels of KLF11 in Wild Type Mice
We have previously reported that the mRNA and protein levels of KLF11 (TIEG2) were significantly increased in the prefrontal cortex of rats that were fed a chronic ethanol diet for 4 weeks (Ou et al., 2011).Tthus, we determined whether the KLF11 levels were also elevated in the brain frontal cortex tissue from wild type Klf11+/+ mice.
KLF11 protein levels in wild type mice (Klf11+/+) were determined by Western blot analysis as shown in Figure 2. The results showed a significant increase in KLF11 in the prefrontal cortex of mice exposed to ethanol (Figure 2A). Statistical analysis of the western blotting using ANOVA revealed a significant difference among the four groups (F3, 36=77.53, p-value<0.0001). Ethanol increased the average protein level of KLF11 by ~1.7-fold compared to control mice (Figure 2A, lanes 2 vs. 1; p<0.01). β-actin was used as a loading control. As shown in Figure 2A, β-actin levels were not affected by ethanol.
Figure 2.
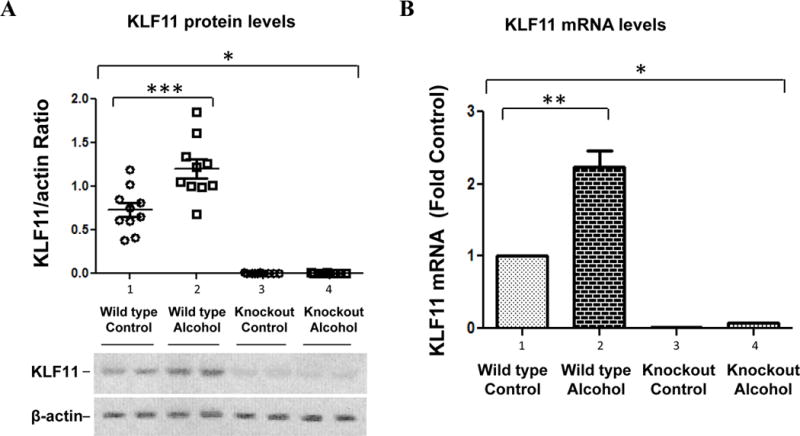
Effects of chronic ethanol intake on the expression of KLF11 in the brain prefrontal cortex of wild type (Klf11+/+) and knockout (Klf11−/−) mice. Mice were fed an ethanol diet or control diet for 28 days. (A) The protein levels of KLF11 in the prefrontal cortex were examined by Western blotting. The quantitative analysis of western blot result is shown on the top. Each KLF11 protein band was evaluated by the relative intensity (relative optical density × pixel area) of its autoradiographic band and normalized to the density of β-actin. The graph of the average optical density of KLF11/actin for the individual animals (open circles or open squares) and mean values (horizontal lines) are shown with 10 mice (n = 10) for each the control group (open circles) and the ethanol-fed group (open squares). The representative western blots (bottom) show the immunolabelling of KLF11 in the prefrontal cortex of 2 untreated Klf11+/+ controls, 2 ethanol-treated Klf11+/+, 2 untreated Klf1−/− controls and 2 ethanol-treated Klf11−/− mice. The anti-β-actin antibody was used as the loading controls. (B) The mRNA levels of KLF11 in the prefrontal cortex were determined by quantitative real-time RT-PCR. Data represent the mean ± SEM of 10 mice (n = 10) in each group. * p<0.0001, **p<0.001 and ***p<0.01
Similarly, KLF11 mRNA levels were determined by quantitative real-time RT-PCR (Figure 2B). Analysis of mRNA levels using ANOVA showed a significant difference among the four groups (F3, 36=92.64, p-value<0.0001). Our results demonstrated that the level of KLF11 mRNA was increased by more than 2-fold in the ethanol-treated group compared to the untreated control group of Klf11+/+ mice (Figure 2B, lanes 2 vs. 1; p<0.001).
KLF11 levels in the knockout mice (Klf11−/−) were also determined. As shown in Figure 2, both protein and mRNA levels of KLF11 in Klf11−/− mice could not be detected. (Figure 2, A and B, lanes 3 and 4 vs. lanes 1 and 2).
These results indicate that KLF11 is directly involved in modifying the pain response to ethanol. The role of KLF11 in ethanol-induce antinociception warrants further investigation, particularly for pain management of patients with a mutation in this transcription factor gene (Neve et al., 2005).
Chronic Ethanol Intake Alters Monoamine Oxidase Activity in the Prefrontal Cortex
KLF11 is a transcriptional activator for the neurotransmitter enzymes, MAO A (Grunewald et al., 2012) and MAO B (Ou et al., 2004). Although both KLF11 and MAO B levels have been found to be significantly increased in the brains of rats fed a chronic ethanol diet (Ou et al., 2011), the activity of these enzymes had not been tested in this mouse model. Therefore, we determined MAO enzymatic activity and mRNA levels in both wild type and knockout mice with or without chronic ethanol exposure.
The results showed that the mRNA levels of MAO A were significantly increased in mice exposed to chronic ethanol in both Klf11+/+ (Figure 3Aa, lanes 3 vs. 1; p<0.02) and Klf11−/− mice (Figure 3Aa, lanes 4 vs, 2; p<0.02). However, MAO A enzyme activity was not increased in Klf11+/+ mice exposed to ethanol (Figure 3Ab). Interestingly, MAO A activity was slightly increase in the ethanol-exposed Klf11−/− mice compared to control Klf11−/− mice (Figure 3Ab, lanes 4 vs. 2; p<0.07).
Figure 3.
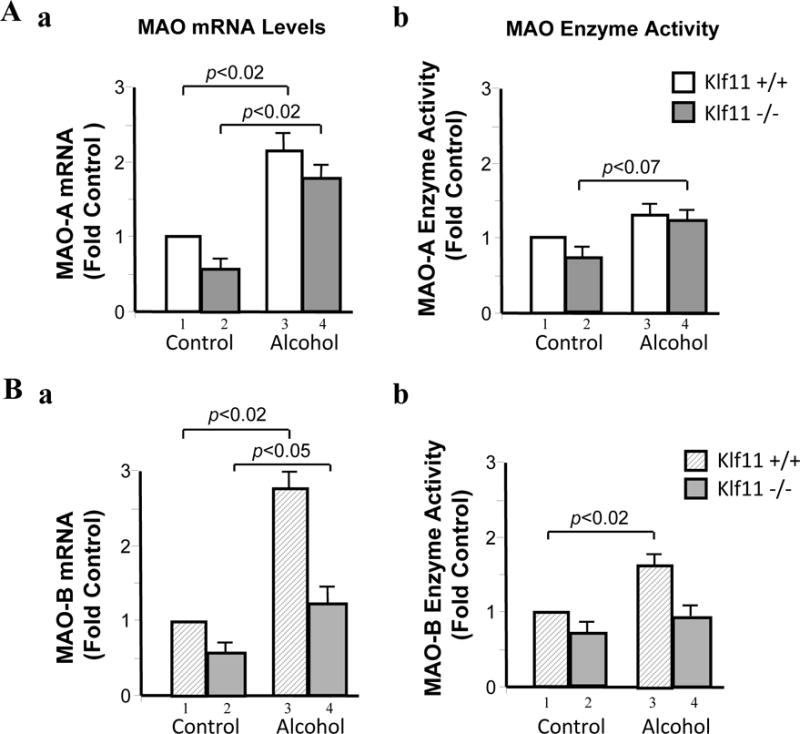
Effects of chronic ethanol intake on the expression of monoamine oxidase (MAO) in the brain prefrontal cortex of wild type (Klf11+/+) and knockout (Klf11−/−) mice. Mice were fed an ethanol diet or control diet for 28 days. (A) The mRNA (a) and enzyme activity (b) of MAO A and (B) the mRNA (a) and enzyme activity (b) of MAO B in the prefrontal cortex were examined by Real-time RT-PCR and catalytic activity assays, respectively. Data represent the mean ± SEM of 10 mice (n = 10) in each group. Aa, p<0.02 (lanes 3 vs. 1 and lanes 4 vs. 2); Ab, p<0.07 (lanes 4 vs. 2); Ba, p<0.02 (lanes 3 vs. 1) and p<0.05 (lanes 4 vs. 2); Bb, p<0.02 (lanes 3 vs. 1)
MAO B mRNA levels were also significantly increased in mice exposed to chronic ethanol in both Klf11+/+ (Figure 3Ba, lanes 3 vs, 1; p<0.02) and Klf11−/− mice (Figure 3Bb, lanes 4 vs, 2; p<0.05). Furthermore, MAO B enzyme activity was significantly increased in Klf11+/+ mice exposed to chronic ethanol (Figure 3Bb, lanes 3 vs, 1; p<0.02) as compared to control Klf11+/+ mice. However, unlike the effects of chronic ethanol on MAO A activity in Klf11−/− mice, MAO B activity in Klf11−/− mice fed a chronic ethanol diet did not differ from that of control Klf11−/− mice (Figure 3Bb, lanes 4 vs, 2). These results suggest that MAO B may play a role in ethanol-induced antinociception in the Klf11+/+ mice; whereas MAO A may contribute to the pain response in Klf11−/− mice chronically exposed to ethanol.
DISCUSSION
KLF11, a gene mutated in both juvenile and neonatal diabetes, has been reported to regulate genes with various functions, including the MAO genes responsible for the metabolism of neurotransmitters (Grunewald et al., 2012; Ou et al., 2004) and the dopamine D2 receptors gene (Seo et al., 2012). Moreover, KLF11 has also been found to mediate ethanol-induced neurotoxicity, leading to cell death in cell culture systems (Lu et al., 2008; Ou et al., 2011). However, the role of KLF11 in ethanol-induced antinociception remains unknown. Addressing this question is important, given that diabetes is associated with alterations in pain perception. Thus, chronic ethanol consumption could further compromise pain perception in patients who already suffer from impairments in this response due to diabetes. The current study sought to explore the modulatory effects of ethanol on the nociceptive response in mice with the KLF11 wild type or knockout gene. We first determined the antinociceptive effects of ethanol to thermal pain using the tail-flick test. Our data showed a significant difference in the wild type (Klf11+/+) mice fed the ethanol diets versus control diets. Chronic ethanol intake significantly increased the pain tolerance in wild type mice, consistent with many published observations (Campbell et al., 2007; Ikeda et al., 2002). Interestingly, chronic ethanol exposure did not abolish the pain response in Klf11-knockout (Klf11−/−) group when comparing to those fed a control diet. Moreover, this effect could not be explained by difference in body weight as a result of ethanol exposure because the Klf11−/− and Klf11+/+mice fed the chronic ethanol diet had similar reductions in body weight (data not shown). This is the first study to show that KLF11 may play an important role in ethanol-induced antinociception.
Subsequently, we investigated whether the KLF11-targeted MAO enzymes were altered given that monoamine neurotransmitter systems are involved in the regulation of nociceptive responses (Mathiesen et al., 2006; von Knorring et al., 1986; Zubieta et al., 2003). We found that chronic ethanol consumption slightly increased MAO A enzyme activity in Klf11−/− mice compared to control Klf11−/− mice.In contrast, MAO B enzyme activity in Klf1+/+ mice fed chronic ethanol was significantly increased compared to control Klf1+/+ mice. These data suggest that MAO B may play a role in ethanol-induced antinociception in wild type (Klf11+/+) mice and possibly contribute to ethanol abuse and addiction, whereas MAO A may be more involved in the pain response in knockout (Klf11−/−) mice exposed to chronic ethanol intake.
It is important to discuss potential molecular mechanisms underlying KLF11-mediated antinociception in our current mouse model. KLF11 is an activator for chronic ethanol-induced MAO B (Ou et al., 2011) and in our current study, ethanol also significantly increased MAO B activity in wild-type (Klf11+/+) mice, but not knockout mice (Klf11−/−). Therefore, the Klf11+/+ mice may have lower levels of dopamine due to the increase in MAO B activity which could potentially activate the opioid system as a compensatory response (George and Kertesz, 1985; George and Kertesz, 1987; Zubieta et al., 2003) and contribute to the antinociceptive effect of ethanol. On the other hand, the Klf11−/− mice may have higher levels of dopamine due to the decrease in MAO B activity, which could decrease the activity of opioid system and subsequently lead to a reduction in ethanol-induced antinociception (George and Kertesz, 1986; Zubieta et al., 2003).
Chronic ethanol intake led to a slight increase in MAO A activity in Klf11−/− mice compared to Klf11+/+. Therefore, the increase in MAO A activity in Klf11−/− mice may play a role in the reduced antinociceptive effects of chronic ethanol exposure in Klf11−/− mice due to the increase in the production of the MAO A catecholamine metabolite 3,4-dihydroxyphenylglycolaldehyde (DOPEGAL) (Dina et al., 2008). DOPEGAL has been reported to produce hyperalgesia in a rat model given a short-term ethanol diet exposure and subsequent withdrawal (4-day fed/3-day off) (Dina et al., 2008). Since KLF11 has recently been reported to activate the expression of MAO A (Grunewald et al., 2012), the mechanism responsible for the slightly increase in MAO A activation in chronic ethanol-fed Klf11−/− mice in the current study needs to be further investigated. Nevertheless, other MAO A-transcriptional factors may also play a role in the chronic ethanol-induced increase in MAO A gene expression.
Another biochemical pathway that may also contribute to KLF11 modulation of the antinociceptive effects of ethanol is the prostaglandin E2 biosynthesis pathway. KLF11 has been reported to inhibit prostaglandin E2 synthesis (Buttar et al., 2010) and this pathway is involved in a broad range of physiological responses, including pain sensitivity. The Klf11−/− mice may have altered prostaglandin E2 biosynthetic activity, which may also contribute to the abolished antinociceptive effect in chronic ethanol-exposed Klf11−/− mice.
Additionally, it is possible that KLF11 assists the development of diabetes-associated damage to the peripheral system given that KLF11 is a transcriptional activator for MAO A and MAO B which are also expressed in the peripheral nervous system. Consequently, the diabetic risk gene, KLF11, may also be involved in diabetes associated peripheral neuropathy through the increased production of neurotransmitter metabolites of MAO, such as DOPEGAL. For example, alterations in KLF11 associated with diabetes may play a role in the damage to the peripheral nervous system by MAO leading to the sensation of numbness (loss of pain perception) or increased pain found in distal extremities. Based on our current results, it appears that MAO B may be more involved in the antinociceptive effects of ethanol whereas MAO A may be more involved in pain perception. However, this speculation requires further investigation.
In summary, we investigated the role of KLF11 in ethanol-induced antinociception using a KLF11-knockout (Klf11−/−) mouse model in which the mice were given a liquid diet with or without ethanol for 28 days. The antinociceptive effect of ethanol was significantly increased in wild type (Klf11+/+) mice. In contrast, chronic ethanol consumption did not produce the expected antinociceptive effect in the KLF11-knockout (Klf11−/−) mice. Furthermore, chronic ethanol exposure led to differential effects in KLF11-targeted MAO enzyme activities in Klf11 wild-type versus Klf11 knockout mice. Chronic ethanol intake slightly increased MAO A enzyme activity in Klf11−/− mice, but significantly increased MAO B enzyme activity in Klf1+/+ mice. This suggests that MAO A contributes more to the nociceptive response in Klf11−/− mice, but MAO B contributes more to alcohol-induced antinociception in Klf11+/+ mice. Therefore, KLF11 may play a key role in regulating ethanol-induced antinociceptive reponse and may serve as a novel target in further understanding the pain control system in alcohol abuse and addiction.
Acknowledgments
This research was supported by National Institutes of Health Grant (NIH/NIAAA) R01AA020103, Public Health Service Grants P20 RR 017701 and an Intramural Research Support grant from the University of Mississippi Medical Center.
References
- Bonnefond A, Lomberk G, Buttar N, Busiah K, Vaillant E, Lobbens S, Yengo L, Dechaume A, Mignot B, Simon A, Scharfmann R, Neve B, Tanyolac S, Hodoglugil U, Pattou F, Cave H, Iovanna J, Stein R, Polak M, Vaxillaire M, Froguel P, Urrutia R. Disruption of a novel Kruppel-like transcription factor p300-regulated pathway for insulin biosynthesis revealed by studies of the c.-331 INS mutation found in neonatal diabetes mellitus. J Biol Chem. 2011;286:28414–24. doi: 10.1074/jbc.M110.215822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan PL, Schutte KK, Moos RH. Pain and use of alcohol to manage pain: prevalence and 3-year outcomes among older problem and non-problem drinkers. Addiction. 2005;100:777–86. doi: 10.1111/j.1360-0443.2005.01074.x. [DOI] [PubMed] [Google Scholar]
- Brunner HG, Nelen M, Breakefield XO, Ropers HH, van Oost BA. Abnormal behavior associated with a point mutation in the structural gene for monoamine oxidase A. Science. 1993;262:578–80. doi: 10.1126/science.8211186. [DOI] [PubMed] [Google Scholar]
- Buttar NS, DeMars CJ, Lomberk G, Rizvi S, Bonilla-Velez J, Achra S, Rashtak S, Wang KK, Fernandez-Zapico ME, Urrutia R. Distinct role of Kruppel-like factor 11 in the regulation of prostaglandin E2 biosynthesis. J Biol Chem. 2010;285:11433–44. doi: 10.1074/jbc.M109.077065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell VC, Taylor RE, Tizabi Y. Antinociceptive effects of alcohol and nicotine: involvement of the opioid system. Brain Res. 2006;1097:71–7. doi: 10.1016/j.brainres.2006.04.054. [DOI] [PubMed] [Google Scholar]
- Campbell VC, Taylor RE, Tizabi Y. Effects of selective opioid receptor antagonists on alcohol-induced and nicotine-induced antinociception. Alcohol Clin Exp Res. 2007;31:1435–40. doi: 10.1111/j.1530-0277.2007.00432.x. [DOI] [PubMed] [Google Scholar]
- Cao S, Fernandez-Zapico ME, Jin D, Puri V, Cook TA, Lerman LO, Zhu XY, Urrutia R, Shah V. KLF11-mediated repression antagonizes Sp1/sterol-responsive element-binding protein-induced transcriptional activation of caveolin-1 in response to cholesterol signaling. J Biol Chem. 2005;280:1901–10. doi: 10.1074/jbc.M407941200. [DOI] [PubMed] [Google Scholar]
- Dai X, Brunson CD, Rockhold RW, Loh HH, Ho IK, Ma T. Gender differences in the antinociceptive effect of tramadol, alone or in combination with gabapentin, in mice. J Biomed Sci. 2008;15:645–51. doi: 10.1007/s11373-008-9252-0. [DOI] [PubMed] [Google Scholar]
- Deitrich RA, Dunwiddie TV, Harris RA, Erwin VG. Mechanism of action of ethanol: initial central nervous system actions. Pharmacol Rev. 1989;41:489–537. [PubMed] [Google Scholar]
- Dina OA, Khasar SG, Alessandri-Haber N, Bogen O, Chen X, Green PG, Reichling DB, Messing RO, Levine JD. Neurotoxic catecholamine metabolite in nociceptors contributes to painful peripheral neuropathy. Eur J Neurosci. 2008;28:1180–90. doi: 10.1111/j.1460-9568.2008.06425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadda F, Rossetti ZL. Chronic ethanol consumption: from neuroadaptation to neurodegeneration. Prog Neurobiol. 1998;56:385–431. doi: 10.1016/s0301-0082(98)00032-x. [DOI] [PubMed] [Google Scholar]
- Fernandez-Zapico ME, van Velkinburgh JC, Gutierrez-Aguilar R, Neve B, Froguel P, Urrutia R, Stein R. MODY7 gene, KLF11, is a novel p300-dependent regulator of Pdx-1 (MODY4) transcription in pancreatic islet beta cells. J Biol Chem. 2009;284:36482–90. doi: 10.1074/jbc.M109.028852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garin I, Edghill EL, Akerman I, Rubio-Cabezas O, Rica I, Locke JM, Maestro MA, Alshaikh A, Bundak R, del Castillo G, Deeb A, Deiss D, Fernandez JM, Godbole K, Hussain K, O’Connell M, Klupa T, Kolouskova S, Mohsin F, Perlman K, Sumnik Z, Rial JM, Ugarte E, Vasanthi T, Johnstone K, Flanagan SE, Martinez R, Castano C, Patch AM, Fernandez-Rebollo E, Raile K, Morgan N, Harries LW, Castano L, Ellard S, Ferrer J, Perez de Nanclares G, Hattersley AT. Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. Proc Natl Acad Sci U S A. 2010;107:3105–10. doi: 10.1073/pnas.0910533107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George SR, Kertesz M. Dopamine receptors regulate Met-enkephalin content in pituitary. Brain Res. 1985;334:187–9. doi: 10.1016/0006-8993(85)90586-4. [DOI] [PubMed] [Google Scholar]
- George SR, Kertesz M. Met-enkephalin-like immunoreactivity in neurointermediate pituitary is decreased by DA receptor stimulation. Peptides. 1986;7:277–81. doi: 10.1016/0196-9781(86)90225-1. [DOI] [PubMed] [Google Scholar]
- George SR, Kertesz M. Met-enkephalin concentrations in striatum respond reciprocally to alterations in dopamine neurotransmission. Peptides. 1987;8:487–92. doi: 10.1016/0196-9781(87)90014-3. [DOI] [PubMed] [Google Scholar]
- Grunewald M, Johnson S, Lu D, Wang Z, Lomberk G, Albert PR, Stockmeier CA, Meyer JH, Urrutia R, Miczek KA, Austin MC, Wang J, Paul IA, Woolverton WL, Seo S, Sittman DB, Ou XM. Mechanistic role for a novel glucocorticoid-KLF11 (TIEG2) pathway in stress-induced monoamine oxidase A expression. J Biol Chem. 2012 doi: 10.1074/jbc.M112.373936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Whistler JL. Chronic ethanol consumption in rats produces opioid antinociceptive tolerance through inhibition of mu opioid receptor endocytosis. PLoS One. 2012;6:e19372. doi: 10.1371/journal.pone.0019372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen JH, Gronbaek M, Moller S, Bendtsen F, Becker U. Carbohydrate deficient transferrin (CDT) in alcoholic cirrhosis: a kinetic study. J Hepatol. 1997;26:287–92. doi: 10.1016/s0168-8278(97)80043-8. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Kobayashi T, Kumanishi T, Yano R, Sora I, Niki H. Molecular mechanisms of analgesia induced by opioids and ethanol: is the GIRK channel one of the keys? Neurosci Res. 2002;44:121–131. doi: 10.1016/s0168-0102(02)00094-9. [DOI] [PubMed] [Google Scholar]
- Jelic P, Shih MF, Taberner PV. Diurnal variation in plasma ethanol levels of TO and CBA mice on chronic ethanol drinking or ethanol liquid diet schedules. Psychopharmacology (Berl) 1998;138:143–50. doi: 10.1007/s002130050656. [DOI] [PubMed] [Google Scholar]
- Johnson S, Stockmeier CA, Meyer JH, Austin MC, Albert PR, Wang J, May WL, Rajkowska G, Overholser JC, Jurjus G, Dieter L, Johnson C, Sittman DB, Ou XM. The reduction of R1, a novel repressor protein for monoamine oxidase a, in major depressive disorder. Neuropsychopharmacology. 2011;36:2139–48. doi: 10.1038/npp.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, Petrakis IL, Mason G, Trevisan L, D’Souza DC. N-methyl-D-aspartate glutamate receptors and alcoholism: reward, dependence, treatment, and vulnerability. Pharmacol Ther. 2003;99:79–94. doi: 10.1016/s0163-7258(03)00054-8. [DOI] [PubMed] [Google Scholar]
- Lockridge A, Romero G, Harrington J, Newland B, Gong Z, Cameron A, Yuan LL. Timing-dependent reduction in ethanol sedation and drinking preference by NMDA receptor co-agonist d-serine. Alcohol. 2012;46:389–400. doi: 10.1016/j.alcohol.2011.11.004. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, White G. Ethanol potentiation of 5-hydroxytryptamine3 receptor-mediated ion current in neuroblastoma cells and isolated adult mammalian neurons. Mol Pharmacol. 1991;40:263–70. [PubMed] [Google Scholar]
- Lu D, Johnson C, Johnson S, Tazik S, Ou XM. The Neuroprotective Effect of Antidepressant Drug via Inhibition of TIEG2-MAO B Mediated Cell Death. Drug Discov Ther. 2008;2:289–295. [PMC free article] [PubMed] [Google Scholar]
- Mathiesen O, Imbimbo BP, Hilsted KL, Fabbri L, Dahl JB. CHF3381, a N-methyl-D-aspartate receptor antagonist and monoamine oxidase-A inhibitor, attenuates secondary hyperalgesia in a human pain model. J Pain. 2006;7:565–74. doi: 10.1016/j.jpain.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Modesto-Lowe V, Brooks D, Freedman K, Hargus E. Addiction and chronic pain: diagnostic and treatment dilemmas. Conn Med. 2007;71:139–44. [PubMed] [Google Scholar]
- Neve B, Fernandez-Zapico ME, Ashkenazi-Katalan V, Dina C, Hamid YH, Joly E, Vaillant E, Benmezroua Y, Durand E, Bakaher N, Delannoy V, Vaxillaire M, Cook T, Dallinga-Thie GM, Jansen H, Charles MA, Clement K, Galan P, Hercberg S, Helbecque N, Charpentier G, Prentki M, Hansen T, Pedersen O, Urrutia R, Melloul D, Froguel P. Role of transcription factor KLF11 and its diabetes-associated gene variants in pancreatic beta cell function. Proc Natl Acad Sci U S A. 2005;102:4807–12. doi: 10.1073/pnas.0409177102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou XM, Chen K, Shih JC. Dual functions of transcription factors, transforming growth factor-beta-inducible early gene (TIEG)2 and Sp3, are mediated by CACCC element and Sp1 sites of human monoamine oxidase (MAO) B gene. J Biol Chem. 2004;279:21021–8. doi: 10.1074/jbc.M312638200. [DOI] [PubMed] [Google Scholar]
- Ou XM, Johnson C, Lu D, Johnson S, Paul IA, Austin MC, Iyo AH, Miguel-Hidalgo JJ, Luo J, Bell RL, Grunewald M, Wang J, Sittman DB. Ethanol Increases TIEG2-MAO B Cell Death Cascade in the Prefrontal Cortex of Ethanol-Preferring Rats. Neurotox Res. 2011 doi: 10.1007/s12640-010-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou XM, Lu D, Johnson C, Chen K, Youdim MB, Rajkowska G, Shih JC. Glyceraldehyde-3-Phosphate Dehydrogenase-Monoamine Oxidase B-Mediated Cell Death-Induced by Ethanol is Prevented by Rasagiline and 1-R-Aminoindan. Neurotox Res. 2009;16:148–59. doi: 10.1007/s12640-009-9064-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou XM, Stockmeier CA, Meltzer HY, Overholser JC, Jurjus GJ, Dieter L, Chen K, Lu D, Johnson C, Youdim MB, Austin MC, Luo J, Sawa A, May W, Shih JC. A novel role for glyceraldehyde-3-phosphate dehydrogenase and monoamine oxidase B cascade in ethanol-induced cellular damage. Biol Psychiatry. 2010;67:855–63. doi: 10.1016/j.biopsych.2009.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul CA, Au R, Fredman L, Massaro JM, Seshadri S, Decarli C, Wolf PA. Association of alcohol consumption with brain volume in the Framingham study. Arch Neurol. 2008;65:1363–7. doi: 10.1001/archneur.65.10.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchis-Segura C, Grisel JE, Olive MF, Ghozland S, Koob GF, Roberts AJ, Cowen MS. Role of the endogenous opioid system on the neuropsychopharmacological effects of ethanol: new insights about an old question. Alcohol Clin Exp Res. 2005;29:1522–7. doi: 10.1097/01.alc.0000174913.60384.e8. [DOI] [PubMed] [Google Scholar]
- Seo S, Lomberk G, Mathison A, Buttar N, Podratz J, Calvo E, Iovanna J, Brimijoin S, Windebank A, Urrutia R. Kruppel-like factor 11 differentially couples to histone acetyltransferase and histone methyltransferase chromatin remodeling pathways to transcriptionally regulate dopamine D2 receptor in neuronal cells. J Biol Chem. 2012;287:12723–35. doi: 10.1074/jbc.M112.351395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih JC, Ridd MJ, Chen K, Meehan WP, Kung MP, Seif I, De Maeyer E. Ketanserin and tetrabenazine abolish aggression in mice lacking monoamine oxidase A. Brain Res. 1999;835:104–12. doi: 10.1016/s0006-8993(99)01478-x. [DOI] [PubMed] [Google Scholar]
- Song CZ, Keller K, Murata K, Asano H, Stamatoyannopoulos G. Functional interaction between coactivators CBP/p300, PCAF, and transcription factor FKLF2. J Biol Chem. 2002;277:7029–36. doi: 10.1074/jbc.M108826200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Knorring L, Almay BG, Haggendal J, Johansson F, Oreland L, Wetterberg L. Discrimination of idiopathic pain syndromes from neurogenic pain syndromes and healthy volunteers by means of clinical rating, personality traits, monoamine metabolites in CSF, serum cortisol, platelet MAO and urinary melatonin. Eur Arch Psychiatry Neurol Sci. 1986;236:131–8. doi: 10.1007/BF00380940. [DOI] [PubMed] [Google Scholar]
- Yao Z, Zhang J, Dai J, Keller ET. Ethanol activates NFkappaB DNA binding and p56lck protein tyrosine kinase in human osteoblast-like cells. Bone. 2001;28:167–73. doi: 10.1016/s8756-3282(00)00425-7. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Heitzeg MM, Smith YR, Bueller JA, Xu K, Xu Y, Koeppe RA, Stohler CS, Goldman D. COMT val158met genotype affects mu-opioid neurotransmitter responses to a pain stressor. Science. 2003;299:1240–3. doi: 10.1126/science.1078546. [DOI] [PubMed] [Google Scholar]